

Abstract
The spliceosome is a dynamic ribonucleoprotein (RNP) machine that catalyzes the removal of introns in the two transesterification steps of eukaryotic pre-mRNA splicing. Here we used single molecule fluorescence resonance energy transfer to monitor the distance of the 5′ splice site (5′SS) and branchpoint (BP) of pre-mRNA in affinity-purified spliceosomes stalled by a mutation in the DExD/H-box helicase Prp2 immediately prior to the first splicing step. Addition of recombinant Prp2 together with NTP and protein cofactor Spp2 rearranges the spliceosome-substrate complex to reversibly explore conformations with proximal 5′SS and BP that accommodate chemistry. Addition of Cwc25 then strongly biases this equilibrium towards the proximal conformation, promoting efficient first-step splicing. The spliceosome thus functions as a biased Brownian ratchet machine where a helicase unlocks thermal fluctuations subsequently rectified by a cofactor “pawl”, a principle possibly widespread among the many helicase-driven RNPs.
INTRODUCTION
Introns are removed by the spliceosome, a large ribonucleoprotein complex, in a two-step transesterification process. In the first step, the 2′OH of the branchpoint adenosine (BP) attacks the phosphodiester bond at the 5′ splice site (5′SS), releasing the 5′ exon and creating the branched, lariat structure; in the second step, the 3′ hydroxyl of this exon attacks the phosphodiester bond at the 3′ splice site (3′SS), releasing the lariat intron and creating the spliced mRNA product1. The most conspicuous feature of this enzyme is that it lacks a preformed catalytic core, which is created in a stepwise fashion, beginning with the assembly of the U1 and U2 snRNPs at the 5′SS and BP, respectively, to form the pre-spliceosome (A complex)2. The U4–U6.U5 tri-snRNP then binds to create the mature spliceosome (B complex)2. Notably, however, U1 and U4 snRNPs must be removed before catalysis, creating first the activated Bact complex, and after additional rearrangements, the catalytically active B* complex. The resulting post-first-step C complex then undergoes further remodeling required for the second step of splicing and the formation of mature mRNA2.
The highly dynamic process of spliceosome assembly and catalysis is guided by a set of RNA-dependent ATPases of the DExD/H-box helicase family, which collectively function to insure the fidelity of splicing3. A major experimental challenge has been to understand the precise conformational rearrangements of RNA and protein that accompany each ATP-dependent step. DExD/H-box helicase Prp2 is required for the first chemical step of splicing and recent proteomic analyses of the Bact, B* and C complexes revealed that its action results in the destabilization of the U2 snRNP-associated proteins SF3a and SF3b4–7. An attractive hypothesis is that SF3b sequesters the BP adenosine8 to prevent a premature attack on the 5′SS, and thus the ATP-dependent action of Prp2, together with its cofactor Spp2, would be required to initiate catalysis. In a biochemical tour-de-force, successful reconstitution of both steps of splicing with the addition of recombinantly expressed proteins to immunopurified splicing complexes has been demonstrated6,9,10. In particular, first-step chemistry could be achieved with the addition of ATP, Prp2, Spp2 and Cwc25, a small heat-stable factor splicing factor6.
We set out to investigate the roles of Prp2, Spp2, and Cwc25 in activating the spliceosome for the first step of splicing, developing an approach that couples the purification of specific splicing complexes with single molecule fluorescence resonance energy transfer (FRET). We used the resulting single molecule pull-down FRET (SiMPull11-FRET) technique to analyze a functional Bact complex assembled on a pre-mRNA with fluorophores near the scissile bonds. By assembling this complex in an extract with a temperature-sensitive allele of Prp2 (prp2-1)12, the Bact spliceosome is stalled and can catalyze the first step of splicing only via formation of first the B* complex upon addition of ATP, Prp2 and Spp2 and, second, the C complex upon further addition of Cwc25. Using SiMPull-FRET, we show that ATP-dependent action of Prp2 along with its cofactor Spp2 unlocks reversible switching of the intron between splicing-active and –inactive conformations. Cwc25 then rectifies this thermal Brownian ratcheting by stabilizing the conformation in which the 5′SS and BP are in close proximity, driving the equilibrium toward catalysis.
RESULTS
Purification of Bact complex with FRET labeled Ubc4 pre-mRNA
It has been known for almost 25 years that a prp2-1 yeast splicing extract can be heat-inactivated12. In this extract, the spliceosome is fully assembled but cannot carry out the first step of splicing. The immature prp2-1 spliceosome purified by gradient centrifugation (Bact) was shown to proceed through the first step of splicing only upon the addition of Prp2p and a heat stable factor(s)9. More recently, Lührmann and colleagues have repeated this experiment with purified Prp2, Spp2 and the (now identified) heat stable factor Cwc256. Such a purified system is ideal for exploring substrate dynamics using single molecule FRET. To this end, we constructed a yeast strain containing the prp2-1 mutation and a Tandem Affinity Purification (TAP) tag derivative of one of the NTC components, Cef1, known to be present in the spliceosome at this stage5,6. This approach allowed us to purify the stalled Bact complex via the TAP tag using biotin-IgG bound to streptavidin.
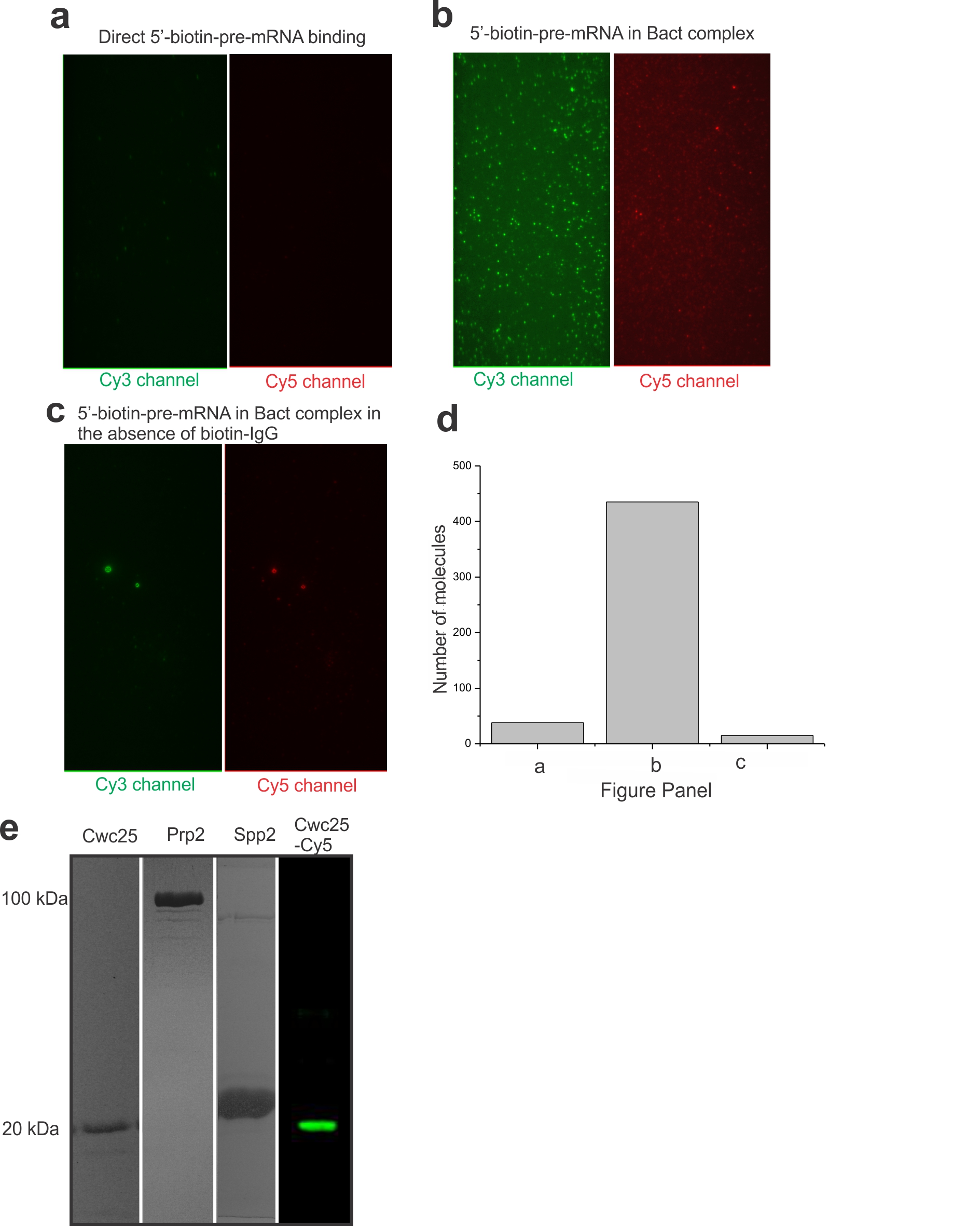
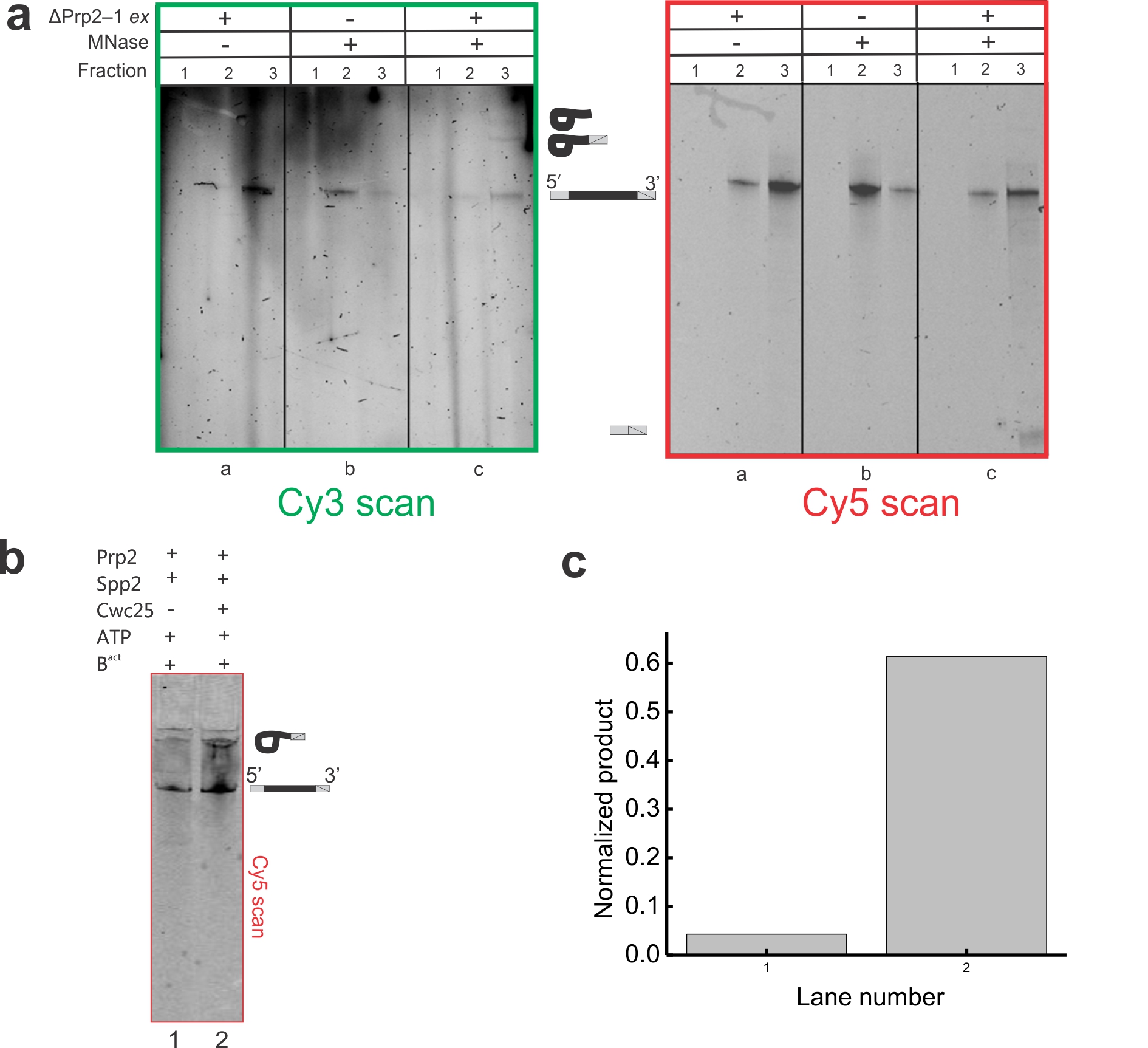
The Ubc4 pre-mRNA used in this study was synthesized chemically and labeled with the FRET donor Cy3 +6 nucleotides downstream from the BP adenosine and with the FRET acceptor Cy5 −7 nucleotides upstream from the 5′SS (Supplementary Table 1). Splicing reactions were assembled for 30 min using heat-inactivated Prp2-1 extract containing the fluorophore labeled pre-mRNA in the presence of 2 mM ATP. We bound the pre-catalytic Bact spliceosome either to streptavidin coated magnetic beads for biochemical analysis or to a PEG-passivated slide coated with streptavidin, biotin-IgG, and excess free biotin for single molecule analysis (Fig. 1a). Free biotin was added to block any biotin binding sites not associated with biotin-IgG and prevent direct binding of the 5′ biotinylated pre-mRNA to the slide. We observed at least 11-fold reduced surface binding of the pre-mRNA by itself compared to the Cef1-TAP-tagged Bact complex containing the pre-mRNA (Supplementary Figs. 1a, 1b and 1d). We detected similarly minimal non-specific binding of the tagged complex upon omission of biotin-IgG (Supplementary Fig. 1c) or when instead using a TAP-tag on protein Prp4, recently shown to be absent from the Bact complex (data not shown)5. To verify that our purification yielded functional Bact pre-spliceosome, we added micrococcal nuclease (MNase) treated (and thus RNA-free) whole cell extract to the stalled bead-purified Bact complex and incubated for 40 min at 23 °C under splicing conditions. We found that both steps of splicing were reconstituted (Supplementary Fig. 2), demonstrating that only proteins are needed to chase Bact into splicing the substrate. In addition, we bead-purified the Bact complex and found that the recombinant proteins Prp2 and Spp2 (Supplementary Fig. 1) are sufficient to yield a low level of first-step splicing products (Fig. 1b), slightly above that observed for the previously characterized actin pre-mRNA6 (Supplementary Fig. 2). Additionally, when Cwc25 was added, a 2- to 10-fold enhancement of first-step splicing was observed (Fig. 1b). Our fluorophore labeled Ubc4 construct showed a first-step splicing efficiency ranging from 12–40% when incubated at 23 °C for 30–40 min, within two-fold of that of other labeled and unlabeled Ubc4 constructs13. Taken together, these experiments establish that FRET labeling the pre-mRNA substrate is compatible with the expected assembly and splicing activity of the immunopurified Bact complex, paving the way for SiMPull-FRET interrogation.
Figure 1.

The SiMPull-FRET approach used here to interrogate active splicing complexes. (a) Schematic showing the affinity purified Bact complex immobilized to a streptavidin coated quartz slide via biotinylated-IgG. The green and red circles on the pre-mRNA represent Cy3 and Cy5 fluorophores, respectively. (b) A 15% urea-polyacrylamide gel scanned using a variable mode Typhoon imager shows pre-mRNA and first-step products (the top panel shows intron-lariat, the mid panel shows pre-mRNA, and the bottom panel shows the 5′-exon, in each case rendered using an overlay of the Cy3 and Cy5 scans). Error bars indicate the standard deviation obtained from triplicate experimental sets.
Bact complex bound pre-mRNA exhibits a static low-FRET state
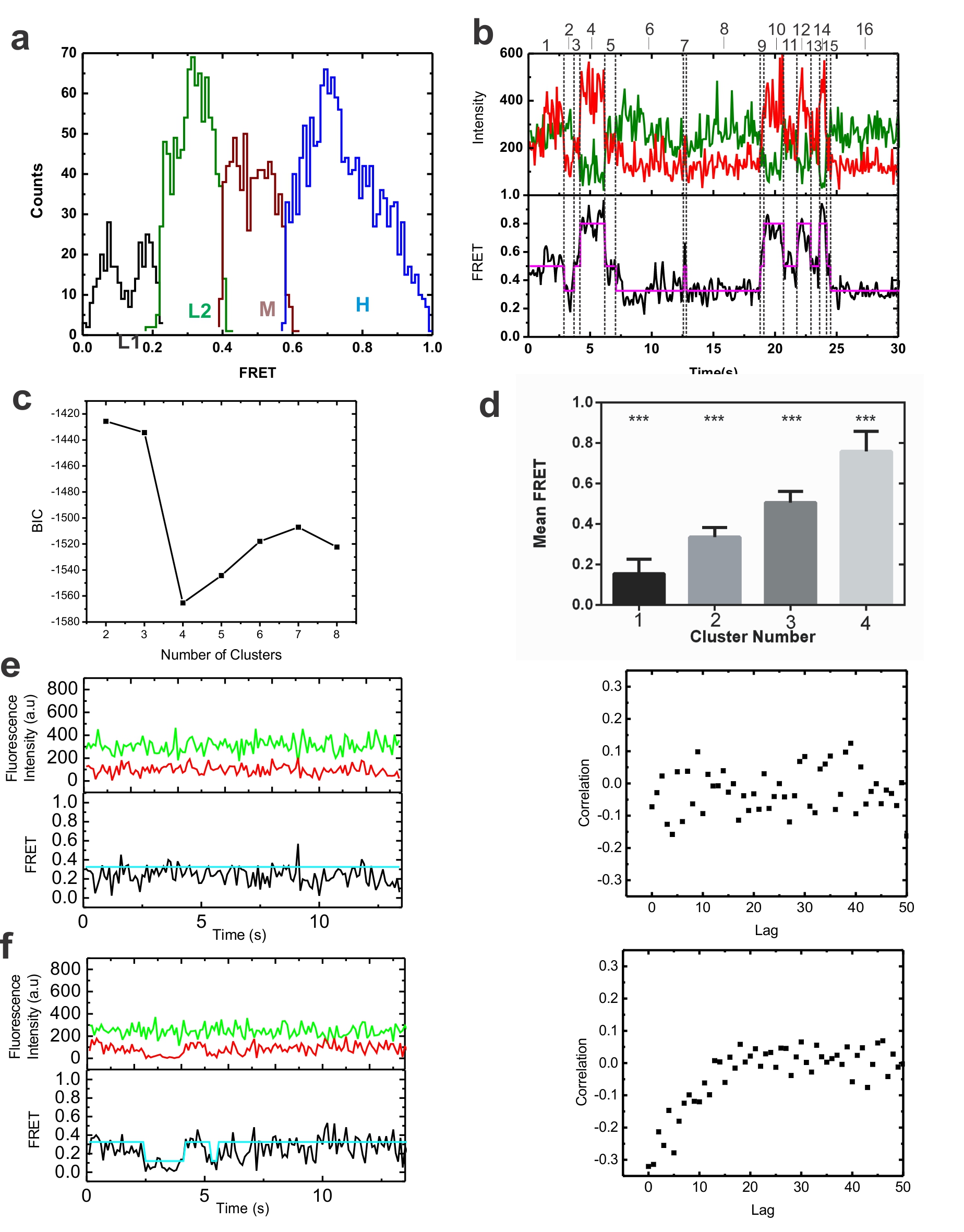
To obtain mechanistic insight into pre-mRNA splice site juxtaposition during the Prp2-driven restructuring of the Bact complex into the catalytically activate B* complex, we carried out SiMPull-FRET on the slide-bound Bact complex (Fig. 1a) in standard splicing buffer. After verifying that each selected pre-mRNA molecule contained a single Cy3 and Cy5 fluorophore, FRET histograms of the FRET values collected from 297 molecules over the first 100 video frames (at 100 ms time resolution) indicated a single Gaussian distribution with an average FRET value of 0.3±0.15 (Fig 2a). To objectively find the underlying FRET states given the background noise inherent to any single molecule experiment, we employed Hidden Markov Modeling (HMM) as described14. Using a K-means approach, we then clustered the HMM assigned states using all the experimental conditions used in this study into four macro states with FRET values of 0.0–0.23 (state L1), 0.23–0.42 (L2), 0.42–0.60 (M), and 0.6–1.0 (H) (Supplementary Fig. 3 and Supplementary Table 2). The dominant behavior in the Bact complex is a static low-FRET state L2 (Fig. 2b). Transition Occupancy Density Plots (TODPs), which are scaled to emphasize the transitions found to be most common among a molecule population14, indicate that the static L2 state represents the only behavior in ~52% of all Bact molecules (Fig. 2c and Supplementary Table 3). In addition, molecules in this state have few transitions (Fig. 2b, HMM fit, cyan line). To test for dynamics too fast for detection by HMM, we performed cross-correlation analysis between the donor and acceptor trajectories of each molecule and in the resulting scatter around zero found no evidence for rapid transitions (Fig. 2b and Supplementary Fig. 3). Although splice site recognition begins in the splicing cycle as early as the commitment complex15 our results suggest that the 5′SS and BP in the Bact complex are kept stably apart, likely not yet close enough for splicing chemistry to occur.
Figure 2.

In the Prp2-stalled Bact complex, the pre-mRNA is predominantly restricted to a static low FRET state. (a) FRET probability distribution of the raw single molecule FRET trajectories from the purified Bact complex. (b) Representative time traces of the Bact complex with raw donor (Cy3, green), acceptor (Cy5, red), and FRET (black) trajectories and their idealized HMM (cyan). The right panel of each molecule is the corresponding cross-correlation of donor and acceptor intensities (c) Transition Occupancy Density Plot (TODP) showing the fraction of Bact complex molecules that either lack a transition and thus lie on the diagonal (dotted white line) or transition from one indicated FRET state to another. L1, L2, M, and H refer to the four states resulting from clustering analysis.
Prp2 mediates NTP-dependent remodeling of the pre-mRNA
The ATPase action of Prp2 has been shown to catalyze a large conformational change that activates the spliceosome for the first step of splicing5,6,9. Spliceosomal binding of Prp2 is dependent on its interaction with the G patch domain of its cofactor protein Spp216,17. The addition of Prp2, Spp2 and ATP transforms the pre-catalytic Bact complex into the catalytically active, distinctly sedimenting B* complex and results in low levels of first-step splicing6. To investigate the role of Prp2 in pre-mRNA remodeling during this step, we incubated the Bact complex assembled on the slide surface with Prp2, Spp2 and 2 mM ATP (henceforth referred to as B* complex condition). B* complex conditions resulted in a substantial shift in the FRET histogram toward a new ~45% population with a mean FRET value of 0.71±0.01, diminishing the lone 0.33±0.01 FRET distribution observed for the Bact complex (Fig. 3a). In contrast to the predominantly static L2 state of the Bact complex, molecules under B* conditions show dynamic (reversible) excursions to high-FRET states, indicating that the 5′SS and BP can now reach the close proximity required for first-step chemistry. More specifically, the B* condition comprises the L2, M and H FRET states, where the H state is accessed from either the L2 or M states (Fig. 3b). TODP plots show that only ~11% of molecules retain the static L2 state characteristic of the Bact complex, while ~39% of molecules exhibit at least one L2→H transition (Fig. 3c and Supplementary Table 3). Notably, transitions into the H state are short-lived in a majority of molecules (Fig. 3b). However, 12% of molecules show a static high-FRET state (Fig. 3c), as expected if they transitioned through the low levels of first-step splicing observed under B* conditions, upon which the labeled 5′SS and BP become covalently linked (Fig. 1b). To verify that this static high-FRET state corresponds to the pre-mRNA substrate configuration after the first step of splicing, we trapped this configuration using a dominant negative Prp16 mutant (Prp16DN, K379A)5,18,19 added to the (non-heat inactivated) Prp2-1, Cef1TAP yeast extract in the presence of 2 mM ATP. This protocol is expected to enrich for the post-first-step C complex, which was then immobilized on the slide surface, washed and imaged. The resulting histogram showed a dramatic enrichment to ~76% of a high-FRET population with a mean FRET value of 0.7±0.01 (Supplementary Fig. 4 and Supplementary Table 3). TODP analysis revealed that ~70% of all molecules adopt the same static high-FRET state first observed under B* conditions, strongly supporting the notion that these molecules indeed have undergone the first chemical step of splicing.
Figure 3.

Upon the addition of ATP, Prp2 and Spp2, the pre-mRNA is able to explore splice site proximity. (a) FRET probability distribution of the raw single molecule FRET trajectories upon the addition of Prp2, Spp2, and ATP to the Bact complex, leading to formation of the B* complex. (b) Representative single molecule FRET time trajectories from the B* condition with the raw donor (Cy3, green), acceptor (Cy5, red), FRET (black) trajectories, and idealized HMM models (cyan). (c) TODP generated from the idealized HMM for molecules in the B* condition. L1, L2, M, or H refers to the four states resulting from clustering analysis. (d) FRET probability densities generated from molecules in Bact incubated with various combinations of components required for formation of B* (Prp2, Spp2, and ATP). In addition, Bact was incubated with Prp2, Spp2, and one of two NTP analogs, non-hydrolysable AMP-PNP or UTP.
Prp2 can directly bind a region in the pre-mRNA downstream of the BP adenosine, even in the absence of ATP20,21. To ask whether Prp2 alone can induce the observed pre-mRNA remodeling, we omitted either Prp2 or Spp2 from our B* conditions and found the resulting FRET histograms to be indistinguishable from those of the starting Bact complex (Fig. 3d, average FRET value of 0.31±0.16 and 0.33±0.13, respectively). Next, we studied the role of ATP in the remodeling of the Bact complex. For most spliceosomal DExD/H-box helicases, both ATP dependent and independent roles have been proposed22–26. Prp2 in particular has been shown to cause extensive conformational remodeling of the spliceosome in the absence of ATP, whereas the displacement of SF3b is ATP dependent5. To ask whether the pre-mRNA remodeling observed here requires ATP, the Bact complex was incubated with Prp2 and Spp2 in the absence of ATP, leading again to no appreciable change in the FRET histogram (Fig. 3d, average FRET value of 0.33±0.12). When the non-hydrolysable ATP analog AMPPNP was used instead of ATP, the FRET histogram was again similar (average FRET value of 0.37±0.11) to that of the Bact complex, with no notable excursion to higher FRET states, showing that ATP hydrolysis is required for these excursions to occur (Fig. 3d). (We note that the minor upwards shift observed in the histogram may be due to binding of AMPPNP to Prp2, resulting in a slight conformational change.) Finally, the DExD/H box helicases involved in spliceosomal reorganization are either integral components of snRNPs or extrinsic components like Prp2. Mass spectrometry studies have shown that the stalled Bact complex contains stoichiometric amounts of the DExD/H-box helicase Brr2, the integral component of the U5 snRNP responsible for U4:U6 unwinding27. Direct interactions between Prp2 and the C-terminus of Brr2 have recently been discovered21, suggesting a possible role for Brr2 in first-step catalytic activation. To test this possibility, we exploited the fact that Brr2 is a strict ATPase28 whereas Prp2 is a broad NTPase, and supplemented the Bact complex under B* conditions with UTP instead of ATP. The resulting FRET efficiency histogram is clearly distinct from that of Bact and overlays well with that of the ATP-mediated B* condition with just a slightly less efficient shift towards the higher FRET population (Fig. 3d). This lower efficiency is consistent with the slightly (by ~2-fold) reduced activity of Prp2 in the presence of NTPs other than ATP29. Collectively, these results indicate that the NTP-driven helicase activity of Prp2 in complex with its activator Spp2 causes a large structural reorganization of the pre-mRNA that allows the distal 5′SS and BP of the Bact complex to reversibly access proximal conformations, which in turn enable first-step splicing.
Cwc25 enhances first-step splicing by H state stabilization
Although the pre-mRNA is remodeled by the NTPase action of Prp2–Spp2, it does not undergo efficient first-step catalysis. To further enhance first-step splicing efficiency, addition of Cwc25 to the Bact complex incubated with Prp2, Spp2 and ATP is necessary (Fig. 1b). Cwc25 was identified as one of a group of proteins complexed with Cef1–Ntc85 of the NTC complex30. To determine its role in remodeling of the pre-mRNA, we performed SiMPull-FRET on the purified Bact complex supplemented with Prp2, Spp2, ATP and Cwc25 (henceforth referred to as C complex conditions). This resulted in a FRET histogram with an enhanced ~73% population with a mean FRET value of 0.75±0.01 (Fig. 4a). We found the FRET states under C complex conditions to be the same as those under B* conditions, with the L2→H and M→H transitions prevalent; however, the occupancy in the H state is considerably enhanced under C conditions (Fig. 4b). TODP analysis revealed the fraction of molecules displaying at least one L2→H and M→H transition to be similar under B* and C conditions, whereas the static H state occupancy (35%) is ~3-fold increased (Fig. 4c and Supplementary Table 3). This shift is similar in magnitude to the enhancement in first-step splicing that Cwc25 induces (Fig. 1b), consistent with the static H state representing the pre-mRNA in the C complex after the first chemical step of splicing. Post-synchronized histograms (PSHs) created by aligning the HMM-fitted traces to start at the M state show that the molecules under C conditions both transition more frequently to the H state and exhibit a higher residence time once in the H state (Fig. 5a). A similar comparison of transitions starting at the H state further emphasizes the stabilization of this state by Cwc25 under C conditions (Fig. 5a). To rule out that a change in photostabilty of molecules in the C complex affects the relative prevalence of the H state, we analyzed the average photobleaching time under Bact, B* and C conditions and found them to be comparable (Supplementary Table 4). To quantitatively characterize the effects of Cwc25 on the conversion of the B* to the C complex, we plotted the cumulative dwell times for the forward and backward L2→H and M→H transitions under both conditions and fit them with double-exponential functions (Supplementary Fig. 4). A comparison of the weighted average rate constant for the L2→H transition showed similar forward and backward rate constants under both conditions, yielding equivalent equilibrium constants Keq = kforward/kbackward of ~0.80 (Supplementary Fig. 4e). In contrast, the presence of Cwc25 accelerates the forward and reduces the backward rate constant of the observed M→H transition, leading to a Keq that is ~3-fold more favorable for the dynamic H state under C compared to B* conditions (Fig. 5b). Notably, the molecules in the static H state, which is a result of the chemical bond formed after first-step catalysis, do not contribute to this kinetic effect. State dwell times cannot be calculated for such molecules, which are only in one state of poorly defined duration during the entirety of our observation window. The effect of static H molecules is therefore more appropriately represented by the increase in molecules of high FRET on the TODP diagonal (compare Figs. 3c and 4c). We also note that both dynamic and static H state molecules, however, do contribute to the enhanced high-FRET peak of the histogram in Fig. 4a.
Figure 4.

Under C complex conditions, the pre-mRNA accesses dynamic and stabilized high-FRET states. (a) FRET probability distribution of the raw single molecule FRET trajectories upon the addition of Prp2, Spp2, Cwc25, and ATP to the Bact complex. (b) Representative single molecule FRET trajectories of molecules from the C complex condition with the raw donor (Cy3, green), acceptor (Cy5, red), FRET (black) trajectories, and idealized HMM models (cyan). (c) TODP generated from the idealized HMM for molecules in the C complex condition. L1, L2, M, or H refers to the four states resulting from clustering analysis.
Figure 5.

Cwc25 enhances the first step of splicing by stabilizing the H state. (a) A comparison of the aggregate molecular behavior before (B* condition) and after (C condition) Cwc25 addition through post-synchronized histograms (PSH) with all trajectories synchronized to start from either the M (top) or H state (bottom). (b) A comparison of the rate constants of the observable transitions under B* and C conditions. The thickness of arrows corresponds to the relative rate constants. (c) Representative single molecule FRET trajectories showing transition dynamics from the same molecules imaged before (B* condition) and after (C condition) Cwc25 addition with the raw donor (Cy3, green), acceptor (Cy5, red), and FRET (black) trajectories. The axis breaks represent 10 min of incubation after Cwc25 addition.
To show that the same pre-mRNA molecule can be converted from B* to C complex, we observed the same field of view before and after shifting from B* conditions (excluding Cwc25) to C conditions (including Cwc25) and incubating for 10 min in the dark. Before the dark period, molecules were dynamically shuttling between the L2 and H states. A subset of molecules were observable after the dark period and of those ~50% shifted to the stabilized H state (Fig. 5c and Supplementary Table 5). Taken together, our results suggest that Cwc25 acts kinetically to stabilize the catalytically favorable conformation, thereby effecting an enhancement of the first chemical step of splicing.
Cwc25 dynamically interacts near the BP upon B* formation
Previous studies have shown that Cwc25 binds stably to the spliceosome following Prp2 mediated SF3a–b destabilization7. It seems likely that Cwc25 enhances first-step chemistry by binding to the pre-mRNA since mutation at the BP abolishes this interaction31. Additionally, Cwc25 has been recently shown to crosslink near the BP of the pre-mRNA32. To directly observe the binding of Cwc25 to the pre-mRNA we labeled the protein’s C-terminus with Cy5 fluorophore. The Cy5 near the 5′SS of the pre-mRNA was pre-bleached so that the pre-mRNA had a single fluorescent Cy3 label near the BP. We tested the activity of the Cy5-tagged Cwc25 using our bead pull-down assay and found it to be fully functional. SiMPull-FRET experiments were then carried out with Cwc25-Cy5 added to the Bact complex with and without the addition of Prp2, Spp2 and ATP (Fig. 6a). We observed repeated binding and dissociation of Cwc25-Cy5 and resulting FRET with the BP under B* condition (Fig. 6b and 6d, B* condition). From the FRET distribution of these binding events, centered around 0.37±0.03, we estimate that Cwc25 binds within FRET distance of the Cy3–Cy5 pair (<100 Å), roughly ~52 Å from the BP Adenosine (Fig. 6b). The lower FRET peak centered around 0.15 represents background signal. By contrast, there was little observable FRET between Cwc25 and the pre-mRNA BP in the absence of Prp2–Spp2 and ATP (Fig. 6c and 6e, Bact condition). We conclude that Cwc25 activates the spliceosome for the first step by dynamically binding to the pre-mRNA near the branch point.
Figure 6.

Prp2 mediated spliceosome remodeling creates a binding site for Cwc25 near the BP. (a) Schematic of our SiMPull experiment to test for binding of Cy5-tagged Cwc25 near the BP of spliceosome-associated pre-mRNA containing a single active Cy3 fluorophore. (b) FRET probability distribution of the raw single molecule FRET trajectories under B* conditions (in the presence of Prp2-mediated remodeling). (c) FRET probability distribution of the raw single molecule FRET trajectories under Bact conditions (in the absence of Prp2-mediated remodeling). (d) Representative single molecule FRET trajectories showing the binding and associated FRET between the Cy5 on Cwc25 and the Cy3 near the pre-mRNA BP under B* conditions with the raw donor (Cy3, green), acceptor (Cy5, red), and FRET (black) trajectories. Arrows indicate binding events with close proximity to the BP. (e) Representative single molecule FRET trajectory showing the absence of FRET between the Cy5 on Cwc25 and the Cy3 near the pre-mRNA BP under Bact conditions with the raw donor (Cy3, green), acceptor (Cy5, red), and FRET (black) trajectories.
DISCUSSION
Here we have combined single molecule FRET between fluorophores attached near the 5′SS and BP of the pre-mRNA substrate with affinity purification into a technique we term SiMPull-FRET to study the spliceosomal Bact complex stalled by heat inactivation of Prp2. Stepwise addition of ATP and the recombinant proteins Prp2, Spp2 and Cwc25 revealed the role of each factor in pre-mRNA remodeling as summarized in Fig. 7. We find that the pre-mRNA remains in the static low-FRET L2 state of the Bact complex (which for clarity we term L2act) with distal 5′SS and BP until the activation by Prp2–Spp2 in the presence of ATP produces the B* complex. Prp2-mediated hydrolysis of ATP (or UTP) in this step weakens the binding of some seven proteins, including SF3a and SF3b4–7 that bind the pre-mRNA upstream and downstream of the BP adenosine, presumably preventing its premature nucleophilic attack on the 5′SS. Accordingly, the B*-associated low-FRET state (L2*) allows the pre-mRNA to transiently and reversibly visit two new states of either mid- (M*) or high-FRET (H*) and more proximal 5′SS and BP. First-step splicing now proceeds with low efficiency, leading to post-catalytic C complex formation signified by a static high-FRET state (HC). This finding indicates that the increased proximity of the reactive sites is sufficient for catalysis. However, reaction chemistry is greatly enhanced by the addition of Cwc25, which binds the pre-mRNA substrate near the BP and slows particularly the rate constant of the high- to mid-FRET transition, leading to a longer dwell time in the pre-catalytic, stabilized FRET state HC-pre. In turn, this event leads to enhanced progression to the static high-FRET state associated with the post-catalytic C complex (HC) (Fig. 7).
Figure 7.

Model for the conformational mechanism of first-step splicing. The 5′SS and BP of the pre-mRNA in the Bact complex reside predominantly in the static distal L2act conformation with low FRET (i.e., high Cy3 and low Cy5 fluorescence, indicated by the green and red circles, respectively). Upon ATP hydrolysis and conversion into the B* complex, Prp2 along with its cofactor Spp2 unlocks the B*-associated low-FRET state L2* to reversibly sample the mid- and high-FRET (spatially proximal) conformations M* and H* (and HC-pre under C complex conditions). Cwc25 binds near the BP of the pre-mRNA, thus reducing the rate constant of the high- to mid-FRET transition and enhancing first-step chemistry, upon which the pre-mRNA adopts the static high-FRET state HC.
Our data show that before the action of Prp2, Spp2, and ATP, the spliceosome keeps the reactive sites of the pre-mRNA strictly apart. This observation is consistent with and refines a recent report that suggested that stable splice site juxtaposition occurs some time after the NTC assembles on the pre-mRNA33. Furthermore, it has previously been speculated that the catalytically activated B* complex, may shift back and forth between inactive and active conformations6. We have presented direct evidence for this hypothesis by showing that only in the B* state dynamic excursions between low- and high-FRET states become observable, and only the high-FRET state places the reactive 5′SS and BP in close enough proximity for subsequent catalysis, correlated with the appearance of the static high-FRET state HC. The same authors also proposed that Cwc25 binding may shift the equilibrium between inactive and active conformations towards the latter, which we directly observe and assign to a marked increase of the dwell time in the active conformation HC-pre with proximal 5′SS and BP.
The behavior of the spliceosome resembles that of a classical biased Brownian ratchet machine that draws path directionality from the random thermal fluctuations, which it constantly experiences, through a form of directional “rectification” or “biasing”34,35. In fact, the ribosome has been described as a biased Brownian ratchet machine36–38 and our previous single molecule FRET probing of pre-mRNA dynamics in whole yeast cell extract suggested that the spliceosome, like the ribosome, works close to thermal equilibrium13. We therefore propose that the ATP-driven helicase activity of Prp2–Spp2 acts to remove SF3a–SF3b as an impediment to the intrinsic thermal fluctuations of the spliceosome-substrate complex, while Cwc25 provides directionality to the reaction pathway by then acting as a “pawl” to stabilize the catalytically competent conformation. Perhaps the closest known analogy is found in translocation of the ribosome, where the random conformational ratcheting between the two ribosomal subunits at thermal equilibrium is rectified by GTP-bound EF-G in conjunction with the intercalation of a conserved two-nucleotide 16S ribosomal RNA “pawl” into the mRNA that appears to prevent it from ratcheting back39. We note that alternation of reversible thermal motion and irreversible NTP hydrolysis steps is thought to form the basis for repeated proofreading by the ribosome40, and may do so for the spliceosome. Future studies will likely further illuminate the molecular mechanisms of these events.
Finally, DExD/H-box helicases such as Prp2 are widespread enzymes that participate in many aspects of RNA processing41,42. In general, they are thought to use ATP hydrolysis to remodel RNA and RNP complexes by binding, unwinding and releasing the RNA. Unlike previous single molecule approaches13,33,43–45, our SiMPull-FRET approach has not only allowed us to unveil the dependence of pre-mRNA ratcheting on the NTPase activity of Prp2, which is then kinetically biased by Cwc25 binding, but to do so in a well-controlled purified system. Biased Brownian ratcheting may be widespread among helicase-driven RNPs, and SiMPull-FRET will allow us to test this hypothesis further.
ONLINE METHODS
Affinity purification of the Bact complex
Extracts were prepared from a prp2-1 cef1-TAP yeast strain (ATCC 201388: MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ046), heated at 37 °C for 40 min to inactivate Prp2 and stall the spliceosome at the Bact complex. In a final volume of 135 μl, 40% (v/v) of this heat treated extract was incubated with ~50 pmoles FRET labeled Ubc4 pre-mRNA13 in the presence of 2 mM ATP in splicing buffer (8 mM HEPES-KOH, pH 7.0, 2 mM MgCl2, 0.08 mM EDTA, 60 mM Ki(PO4), 20 mM KCl, 8% (v/v) glycerol, 3% (w/v) PEG, 0.5 mM DTT) and incubated at 23 °C for 35 min. For biochemical experiments, streptavidin-coated magnetic beads (Dynabeads® MyOne™ Streptavidin C1, Invitrogen) were handled as per the manufacturer’s recommendation. For each splicing reaction, 200 μl of the suspended beads were equilibrated in 200 μl of T50 buffer (50 mM Tris-HCl, pH 7.5, 50 mM NaCl). An equal volume of 0.5 mg/ml biotin-IgG (ZyMAX™ Rabbit Anti-Mouse IgG (H+L) - BT (ZyMAX™ Grade)) in T50 was added and incubated in a tube rotator at 23°C for 30 min. The beads were then pulled down using a magnet and the supernatant was discarded. To block any streptavidin not bound by biotin-IgG, the beads were incubated with excess free biotin at 1.5 mg/ml in T50 buffer in a tube rotator at 23°C for 20 min. After equilibration in splicing buffer, the independently assembled splicing reaction were added and incubated in a tube rotator for 30 min at 23°C to allow the protein A of the Cef1-TAP tag in the spliceosome complex to bind the biotin-IgG. Upon removal of the supernatant, the beads were further washed three times with buffer A (20 mM HEPES-KOH, pH 7.9, 120 mM KCl, 0.01% NP40, 1.5 mM MgCl2, 5% (v/v) glycerol) and once with splicing buffer to further purify the Bact complex. The reactions are scaled up for reconstitution reactions pursued in parallel and split at this step. Prp2, Spp2 and Cwc25 were added at 90–120 nM final concentration in splicing buffer in the presence or absence of 2 mM ATP or AMPPNP or UTP and incubated in the tube rotator for 30–40 min for various levels of reconstitution. RNA was isolated and products of splicing were analyzed on a denaturing, 7 M urea, 15% polyacrylamide gel and scanned on a Typhoon variable mode imager (GE Healthcare). Normalized product for Ubc4 was calculated by taking the amount of free 5′ exon and dividing that by the total amount of pre-mRNA and free 5′ exon. Normalized product for Actin was calculated by taking the amount of lariat intermediate and dividing that by the total amount of pre-mRNA and lariat intermediate.
Cloning, expression and purification of splicing factor proteins
The full-length PRP2 gene was PCR-amplified and ligated into plasmid pRSETA (Invitrogen) with a C-terminal hexahistidine tag. The N-terminally truncated form of SPP2 (coding for amino acids 37-185) containing a C-terminal hexahistidine tag, and the full-length Cwc25 gene were obtained from Reinhard Lührmann (Max Planck Institute for Biophysical Chemistry, Germany). Cwc25 was subcloned into a pRSETa plasmid containing a single cysteine residue and hexahistidine tag at the C-terminus. The constructs were then transformed into Escherichia coli strain Rossetta II (Novagen). Cultures were grown in 2–4 l of TB medium and induced with 125 μM IPTG. Cultures were then incubated at 20 °C for 18 h. Cells were harvested, washed, and the pellets stored at −80 °C.
Purification of Cwc25-His and Spp2-His was performed as described6. Protein purity was confirmed by 16% SDS-PAGE (Supplementary Fig. 1) and proteins were either first fluorescently labeled or directly aliquoted, flash frozen in liquid nitrogen, and stored at −80 °C. Protein concentrations were determined by Bradford assay and measurement at A280. His-tagged Prp2 obtained from E. coli cell lysate was purified as described47 Protein purity was confirmed by 10% SDS-PAGE (Supplementary Fig. 1) and the final product aliquoted, flash frozen in liquid nitrogen, and stored at −80 °C. Protein concentrations were determined by Bradford assay and measurement at A280.
Single molecule FRET of purified spliceosomal complexes
For the single molecule FRET experiments on affinity-purified complexes, we prepared slides using previously published procedures14. In short, the surface of a quartz slide was amino functionalized, PEGylated and reacted with 0.2 mg/ml streptavidin in T50 buffer for 15 min at 23°C. 100 μl of 0.5 mg/ml biotin-IgG in T50 was flowed onto the slide and incubated for 20 min, followed by free biotin at 1.5 mg/ml in T50 buffer for 15 min. Bact spliceosomal complexes were assembled and stalled as described above by incubation of FRET labeled Ubc4 pre-mRNA with heat treated prp2-1 cef1-TAP yeast splicing extract in splicing buffer supplemented with 2 mM ATP and an oxygen scavenger system (OSS) composed of protocatechuate dioxygenase, protocatechuate and Trolox. These complexes were then flowed onto the slide surface and incubated for 15–20 min to allow the Cef-1-TAP on the spliceosome to bind biotin-IgG. The slide surface was washed rigorously and reconstituted (in the presence of OSS) as described for the biochemical purification and incubated for 10–40 min before acquiring data. A home-built prism-based TIRF microscope was used to collect data as described13,48,49. To obtain FRET data, we directly excited the Cy3 donor near the BP adenosine with a 532-nm laser, and we recorded emission by Cy3 and Cy5 fluorophores at 100-ms time resolution using an intensified CCD camera (I-Pentamax, Princeton Instruments).
Fluorescent labeling of Cwc25 and distance estimation from FRET
The single-cysteine mutant of Cwc25 was labeled with Cy5-maleimide (GE Healthcare). Labeling was performed using 0.150 μmol of Cwc25 in storage buffer and 0.5 mg of dye containing 10 μM reducing agent Tris(2-carboxyethyl)phosphine (TCEP) (Sigma). Reactions were incubated at 23°C for 1 h followed by overnight at 4 °C. Free dye was removed by re-purification of protein on a Ni2+ column and dialysis back into storage buffer. The degree of labeling was determined using GE Healthcare’s protocol and was found to be 70%. Protein functionality was confirmed using an ensemble pull down assay as described above. The fluorophore distance, R, and the apparent FRET efficiency, Eapp, were calculated as described48,50,51 from the equations Eapp = c[1 + (R/R0)6]−1, where c = 0.69, R0 = 54 Å, and . ϕ and η signify the fluorophores quantum yields and detector channel efficiencies, respectively. The donor and acceptor intensities ICy3 and ICy5, respectively, were corrected for leakage of donor photons into the acceptor channel.
Single Molecule Data Analysis
Cross-correlation analysis was carried out utilizing customized MATLAB scripts with built-in xcorr function. Time lags for the cross-correlation ranged from 0–5 s (0–50 frames of each 0.1 s integration time). Quality control for the raw smFRET trajectories obtained from the experimental conditions were performed as described14. Histograms for data sets measuring pre-mRNA dynamics were constructed by sampling 100 frames of data from each molecule. Histograms for Cwc25-Cy5 and BP-Cy3 FRET experiments were constructed by sampling the entire length of data. Hidden Markov Modeling (HMM) was performed on trajectories utilizing the vbFRET software suite14. Each trajectory was individually fit with models ranging from 1–5 states with the optimal number of states determined by the vbFRET algorithm. The inherent experimental variations of the FRET signal between single molecules leads to a slightly different state assignment for similar states across different molecules. A K-means clustering approach was therefore performed in MATLAB to group similar states into larger macro states (L1, L2, M and H). A matrix cataloging the HMM assigned FRET state, raw FRET level, and difference in donor and acceptor intensities for each HMM derived event was utilized as input for the K-means algorithm (Supplementary Figure 3 and Supplementary Table 2). Four macro states were identified whose boundaries were used to re-assign the original HMM idealized FRET states. The number of macro states was determined using the Bayesian Information Criterion (BIC) as a model selection tool. K-means and BIC have been used previously to group and accurately determine the number of clusters in single molecule data52–54. The BIC score was calculated in MATLAB as follows:
where LLF is the log-likelihood function, NumParams the number of parameters, and NumObs the number of observations. The number of clusters (K) was varied from 2–8. The lowest BIC score was achieved for k = 4 (Supplementary Figure 3c). Once identified, the clusters were tested for similarity using the multi-comparison Tukey test in the software package PRISM-GraphPad. The results of this analysis indicate that there is a extremely significant difference (p<0.001) for the pairwise comparison of all four clusters (Supplementary Figure 3d).
Transition Occupancy Density Plots (TODPs) were used to plot the fraction of molecules that contain any given HMM transition at least once14. Molecules that did not exhibit any transitions were plotted along the diagonal at their respective positions. For kinetic rate calculations, Transition Density Plots (TDPs) that are scaled by the number of times a transition occurs irrespective of how many molecules exhibit that transition were used as described14. A cumulative histogram scatter plot was then fit with a double-exponential association equation in MicroCal Origin (Supplementary Figure 4). A weighted average (kw,observed) of the two rate constants from the double-exponential fits was calculated based on the amplitude values of the exponential equation. To correct for bias introduced by the limited observation window used to measure dwell times, the measured kw,observed values were corrected by subtracting the photobleaching rate constant and the reciprocal of the observation window to yield kw,actual as described55. Equilibrium constants (Keq) were calculated by taking a ratio of the forward and backward rate constants for a set of state-to-state transitions. Post-synchronized histograms (PSHs) were constructed by synchronizing individual FRET events to the time where one of the macro-states (M, H) was achieved. The scale bar represents the fraction of FRET events which exhibit a certain FRET state at a given time.
METHODS
Methods and any associated references are available in the online version of the paper at http://www.nature.com/nsmb/.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
The authors wish to thank R. Lührmann (Max Planck Institute for Biophysical Chemistry, Göttingen, Germany) and R. J. Lin (Fujian Medical University, Fuzhou, China) for generously supplying expression plasmids for Spp2, Cwc25 and Prp2; H. Hadjivassiliou and A. Price (University of California, San Francisco) for providing Cy5-body labeled actin pre-mRNA substrate; D. Semlow and J. P. Staley (University of Chicago, Illinois) for providing the dominant negative Prp16 protein. The authors acknowledge funding through NIH grant R01 GM098023 to N.G.W. and J.A. and NIH grant R01 GM021119 to C.G.
Footnotes
AUTHOR CONTRIBUTIONS
R.K., M.R.B. and M.L.K. performed the biochemical and single molecule experiments and performed data analysis. M.L.K. cloned, expressed and labeled the proteins. R.K., M.R.B., M.L.K., J.A., C.G. and N.G.W. wrote the manuscript.
References
- 1.Brody E, Abelson J. The “spliceosome”: yeast pre-messenger RNA associates with a 40S complex in a splicing-dependent reaction. Science. 1985;228:963–7. doi: 10.1126/science.3890181. [DOI] [PubMed] [Google Scholar]
- 2.Wahl MC, Will CL, Luhrmann R. The Spliceosome: Design Principles of a Dynamic RNP Machine. Cell. 2009;136:701–718. doi: 10.1016/j.cell.2009.02.009. [DOI] [PubMed] [Google Scholar]
- 3.Staley JP, Guthrie C. Mechanical devices of the spliceosome: motors, clocks, springs, and things. Cell. 1998;92:315–26. doi: 10.1016/s0092-8674(00)80925-3. [DOI] [PubMed] [Google Scholar]
- 4.Chen JH, Lin RJ. The yeast PRP2 protein, a putative RNA-dependent ATPase, shares extensive sequence homology with two other pre-mRNA splicing factors. Nucleic Acids Res. 1990;18:6447. doi: 10.1093/nar/18.21.6447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lardelli RM, Thompson JX, Yates JR, 3rd, Stevens SW. Release of SF3 from the intron branchpoint activates the first step of pre-mRNA splicing. RNA. 2010;16:516–28. doi: 10.1261/rna.2030510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Warkocki Z, et al. Reconstitution of both steps of Saccharomyces cerevisiae splicing with purified spliceosomal components. Nat Struct Mol Biol. 2009;16:1237–U50. doi: 10.1038/nsmb.1729. [DOI] [PubMed] [Google Scholar]
- 7.Ohrt T, et al. Prp2-mediated protein rearrangements at the catalytic core of the spliceosome as revealed by dcFCCS. RNA. 2012;18:1244–1256. doi: 10.1261/rna.033316.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Berglund JA, Rosbash M, Schultz SC. Crystal structure of a model branchpoint-U2 snRNA duplex containing bulged adenosines. RNA. 2001;7:682–91. doi: 10.1017/s1355838201002187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim SH, Lin RJ. Spliceosome activation by PRP2 ATPase prior to the first transesterification reaction of pre-mRNA splicing. Mol Cell Biol. 1996;16:6810–9. doi: 10.1128/mcb.16.12.6810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chiu YF, et al. Cwc25 Is a Novel Splicing Factor Required after Prp2 and Yju2 To Facilitate the First Catalytic Reaction. Mol Cell Biol. 2009;29:5671–5678. doi: 10.1128/MCB.00773-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jain A, et al. Probing cellular protein complexes using single-molecule pull-down. Nature. 2011;473:484–U322. doi: 10.1038/nature10016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vijayraghavan U, Company M, Abelson J. Isolation and characterization of pre-mRNA splicing mutants of Saccharomyces cerevisiae. Genes Dev. 1989;3:1206–16. doi: 10.1101/gad.3.8.1206. [DOI] [PubMed] [Google Scholar]
- 13.Abelson J, et al. Conformational dynamics of single pre-mRNA molecules during in vitro splicing. Nat Struct Mol Biol. 2010;17:504–12. doi: 10.1038/nsmb.1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Blanco M, Walter NG. Analysis of complex single-molecule FRET time trajectories. Methods Enzymol. 2010;472:153–78. doi: 10.1016/S0076-6879(10)72011-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Black DL. Finding splice sites within a wilderness of RNA. RNA. 1995;1:763–71. [PMC free article] [PubMed] [Google Scholar]
- 16.Silverman EJ, et al. Interaction between a G-patch protein and a spliceosomal DEXD/H-box ATPase that is critical for splicing. Mol Cell Biol. 2004;24:10101–10110. doi: 10.1128/MCB.24.23.10101-10110.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roy J, Kim K, Maddock JR, Anthony JG, Woolford JL. The Final Stages of Spliceosome Maturation Require Spp2p That Can Interact with the Deah Box Protein Prp2p and Promote Step-1 of Splicing. RNA. 1995;1:375–390. [PMC free article] [PubMed] [Google Scholar]
- 18.Schwer B, Guthrie C. A Conformational Rearrangement in the Spliceosome Is Dependent on Prp16 and Atp Hydrolysis. Embo Journal. 1992;11:5033–5039. doi: 10.1002/j.1460-2075.1992.tb05610.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hotz HR, Schwer B. Mutational analysis of the yeast DEAH-box splicing factor Prp16. Genetics. 1998;149:807–15. doi: 10.1093/genetics/149.2.807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Teigelkamp S, McGarvey M, Plumpton M, Beggs JD. The splicing factor PRP2, a putative RNA helicase, interacts directly with pre-mRNA. EMBO J. 1994;13:888–97. doi: 10.1002/j.1460-2075.1994.tb06332.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu HL, Cheng SC. The Interaction of Prp2 with a Defined Region of the Intron Is Required for the First Splicing Reaction. Mol Cell Biol. 2012;32:5056–5066. doi: 10.1128/MCB.01109-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Newnham CM, Query CC. The ATP requirement for U2 snRNP addition is linked to the pre-mRNA region 5′ to the branch site. RNA. 2001;7:1298–1309. doi: 10.1017/s1355838201010561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Perriman R, Barta I, Voeltz GK, Abelson J, Ares M. ATP requirement for Prp5p function is determined by Cus2p and the structure of U2 small nuclear RNA. Proc Natl Acad Sci USA. 2003;100:13857–13862. doi: 10.1073/pnas.2036312100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schwer B, Gross CH. Prp22, a DExH-box RNA helicase, plays two distinct roles in yeast pre-mRNA splicing. EMBO J. 1998;17:2086–2094. doi: 10.1093/emboj/17.7.2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Company M, Arenas J, Abelson J. Requirement of the RNA helicase-like protein PRP22 for release of messenger RNA from spliceosomes. Nature. 1991;349:487–93. doi: 10.1038/349487a0. [DOI] [PubMed] [Google Scholar]
- 26.Schwer B. A conformational rearrangement in the spliceosome sets the stage for prp22-dependent mRNA release. Mol Cell. 2008;30:743–754. doi: 10.1016/j.molcel.2008.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bartels C, Klatt C, Luhrmann R, Fabrizio P. The ribosomal translocase homologue Snu114p is involved in unwinding U4/U6 RNA during activation of the spliceosome. EMBO rep. 2002;3:875–880. doi: 10.1093/embo-reports/kvf172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Raghunathan PL, Guthrie C. RNA unwinding in U4/U6 snRNPs requires ATP hydrolysis and the DEIH-box splicing factor Brr2. Curr Biol. 1998;8:847–55. doi: 10.1016/s0960-9822(07)00345-4. [DOI] [PubMed] [Google Scholar]
- 29.Kim SH. The purified yeast pre-mRNA splicing factor PRP2 is an RNA-dependent NTPase. EMBO J. 1992;11:2319–2326. doi: 10.1002/j.1460-2075.1992.tb05291.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ohi MD, et al. Proteomics analysis reveals stable multiprotein complexes in both fission and budding yeasts containing Myb-related Cdc5p/Cef1p, novel pre-mRNA splicing factors, and snRNAs. Mol Cell Biol. 2002;22:2011–2024. doi: 10.1128/MCB.22.7.2011-2024.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tseng CK, Liu HL, Cheng SC. DEAH-box ATPase Prp16 has dual roles in remodeling of the spliceosome in catalytic steps. RNA. 2011;17:145–154. doi: 10.1261/rna.2459611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen HC, Tseng CK, Tsai RT, Chung CS, Cheng SC. Link of NTR-mediated spliceosome disassembly with DEAH-box ATPases Prp2, Prp16, and Prp22. Mol Cell Biol. 2013;33:514–25. doi: 10.1128/MCB.01093-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Crawford DJ, Hoskins AA, Friedman LJ, Gelles J, Moore MJ. Single-molecule colocalization FRET evidence that spliceosome activation precedes stable approach of 5′ splice site and branch site. Proc Natl Acad Sci USA. 2013;110:6783–8. doi: 10.1073/pnas.1219305110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cordova NJ, Ermentrout B, Oster GF. Dynamics of single-motor molecules: the thermal ratchet model. Proc Natl Acad Sci USA. 1992;89:339–43. doi: 10.1073/pnas.89.1.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Astumian RD. Thermodynamics and kinetics of a Brownian motor. Science. 1997;276:917–22. doi: 10.1126/science.276.5314.917. [DOI] [PubMed] [Google Scholar]
- 36.Spirin AS. How Does a Scanning Ribosomal Particle Move along the 5′-Untranslated Region of Eukaryotic mRNA? Brownian Ratchet Model. Biochemistry. 2009;48:10688–10692. [Google Scholar]
- 37.Frank J, Gonzalez RL., Jr Structure and dynamics of a processive Brownian motor: the translating ribosome. Annu Rev Biochem. 2010;79:381–412. doi: 10.1146/annurev-biochem-060408-173330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rodnina MV, Wintermeyer W. The ribosome as a molecular machine: the mechanism of tRNA-mRNA movement in translocation. Biochem Soc Trans. 2011;39:658–62. doi: 10.1042/BST0390658. [DOI] [PubMed] [Google Scholar]
- 39.Zhou J, Lancaster L, Donohue JP, Noller HF. Crystal structures of EF-G-ribosome complexes trapped in intermediate states of translocation. Science. 2013;340:1236086. doi: 10.1126/science.1236086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Blanchard SC, Gonzalez RL, Kim HD, Chu S, Puglisi JD. tRNA selection and kinetic proofreading in translation. Nat Struct Mol Biol. 2004;11:1008–14. doi: 10.1038/nsmb831. [DOI] [PubMed] [Google Scholar]
- 41.Tanner NK, Linder P. DExD/H box RNA helicases: from generic motors to specific dissociation functions. Mol Cell. 2001;8:251–62. doi: 10.1016/s1097-2765(01)00329-x. [DOI] [PubMed] [Google Scholar]
- 42.Jankowsky E. RNA helicases at work: binding and rearranging. Trends Biochem Sci. 2011;36:19–29. doi: 10.1016/j.tibs.2010.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Robertson KL, Yu L, Armitage BA, Lopez AJ, Peteanu LA. Fluorescent PNA probes as hybridization labels for biological RNA. Biochemistry. 2006;45:6066–74. doi: 10.1021/bi052050s. [DOI] [PubMed] [Google Scholar]
- 44.Crawford DJ, Hoskins AA, Friedman LJ, Gelles J, Moore MJ. Visualizing the splicing of single pre-mRNA molecules in whole cell extract. RNA. 2008;14:170–9. doi: 10.1261/rna.794808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hoskins AA, et al. Ordered and dynamic assembly of single spliceosomes. Science. 2011;331:1289–95. doi: 10.1126/science.1198830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ghaemmaghami S, et al. Global analysis of protein expression in yeast. Nature. 2003;425:737–41. doi: 10.1038/nature02046. [DOI] [PubMed] [Google Scholar]
- 47.Edwalds-Gilbert G, et al. Dominant negative mutants of the yeast splicing factor Prp2 map to a putative cleft region in the helicase domain of DExD/H-box proteins. RNA. 2000;6:1106–19. doi: 10.1017/s1355838200992483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pereira MJB, et al. Single VS ribozyme molecules reveal dynamic and hierarchical folding toward catalysis. J Mol Biol. 2008;382:496–509. doi: 10.1016/j.jmb.2008.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ditzler MA, Rueda D, Mo JJ, Hakansson K, Walter NG. A rugged free energy landscape separates multiple functional RNA folds throughout denaturation. Nucleic Acids Res. 2008;36:7088–7099. doi: 10.1093/nar/gkn871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sabanayagam CR, Eid JS, Meller A. Using fluorescence resonance energy transfer to measure distances along individual DNA molecules: Corrections due to nonideal transfer. J Chem Phys. 2005;122:061103. doi: 10.1063/1.1854120. [DOI] [PubMed] [Google Scholar]
- 51.Cosa G, et al. Secondary structure and secondary structure dynamics of DNA hairpins complexed with HIV-1 NC protein. Biophys J. 2004;87:2759–67. doi: 10.1529/biophysj.104.043083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hwang H, Kim H, Myong S. Protein induced fluorescence enhancement as a single molecule assay with short distance sensitivity. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:7414–7418. doi: 10.1073/pnas.1017672108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Uphoff S, et al. Monitoring multiple distances within a single molecule using switchable FRET. Nature Methods. 2010;7:831–U90. doi: 10.1038/nmeth.1502. [DOI] [PubMed] [Google Scholar]
- 54.Ishioka T. Extended K-means with an Efficient Estimation of the Number of Clusters. Lecture Notes in Computer Science. 2000;1983:17–22. [Google Scholar]
- 55.Rueda D, et al. Single-molecule enzymology of RNA: essential functional groups impact catalysis from a distance. Proc Natl Acad Sci USA. 2004;101:10066–71. doi: 10.1073/pnas.0403575101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.