

Abstract
The ability to respond to various intracellular and/or extracellular stresses allows the organism to adapt to changing environmental conditions and drives evolution. It is now well accepted that a progressive decline of the efficiency of stress response pathways occurs with aging. In this context, a correct proteostasis is essential for the functionality of the cell, and its dysfunction has been associated with protein aggregation and age-related degenerative diseases. Complex response mechanisms have evolved to deal with unfolded protein stress in different subcellular compartments and their moderate activation translates into positive effects on health. In this review, we focus on the mitochondrial unfolded protein response (UPRmt), a response to proteotoxic stress specifically in mitochondria, an organelle with a wide array of fundamental functions, most notably the harvesting of energy from food and the control of cell death. We compare UPRmt with the extensively characterized cytosolic heat shock response (HSR) and the unfolded protein response in endoplasmic reticulum (UPRER), and discuss the current knowledge about UPRmt signaling pathways as well as their potential involvement in physiology.
KEY WORDS: Aging, Proteostasis, Mitochondria, Chaperones, Signaling
Introduction – reaction to stress and adaptive response
One of the keystones driving evolution is the capacity to detect, and respond and adapt to various stressors in the environment through the development of defensive mechanisms at a cellular level that protect the entire organism and maintain its capacity to grow and reproduce (Badyaev, 2005). A large body of evidence has established that stress adaptation and aging are intimately linked (Kourtis and Tavernarakis, 2011). The concept that stress responses and the aging process share common mechanisms arose initially from studies in model organisms, where the identification of molecular pathways that regulate aging – insulin/insulin-like growth factor (IGF), sirtuins, target of rapamycin (TOR), AMP-activated kinase (AMPK) – highlighted that intrinsic induction of stress defense programs and the resulting adaptation can increase life expectancy (Haigis and Yankner, 2010). As a case in point, downregulation of insulin/IGF-1 signaling promotes stress defense and increases lifespan from worms to mice (Holzenberger et al., 2003; Kenyon et al., 1993; Kimura et al., 1997). Over time, it became clear that applying moderate levels of stress could trigger beneficial and adaptive stress defense pathways, allowing longer life (Gems and Partridge, 2008); this is now known as a ‘hormetic stress response’ and drew the interest of the scientific community for its potential application in the pharmacological management of age-related diseases. Illustrative of such a hormetic stress response is the reaction to the low dose energy stress induced by caloric restriction, which prolongs lifespan in a wide array of species from yeast to primates (Mair and Dillin, 2008).
Maintenance of protein homeostasis, also known as ‘proteostasis’, which refers to all the cellular pathways that govern the production, folding and degradation of proteins, is essential for cellular and organismal functionality and survival. Complex pathways to ensure proteostasis in different subcellular compartments, defined as unfolded protein responses (UPRs), have evolved in the cytosol, endoplasmic reticulum and mitochondria, which are finely coordinated and require a close communication with the nucleus. Dysfunction of proteostasis is associated with protein aggregation and is a feature of several age-related diseases (Kikis et al., 2010). While the unfolded protein response in the endoplasmic reticulum (UPRER) and the cytosolic heat shock response (HSR) have been extensively studied (Vabulas et al., 2010; Walter and Ron, 2011), the mitochondrial unfolded protein response (UPRmt) was more recently discovered and sheds light on the complex relationships between this organelle and the nucleus (Haynes and Ron, 2010). In this review we discuss the complex molecular mechanisms of proteotoxic stress responses in different cellular compartments with a focus on the UPRmt and its role in health and life-span regulation.
UPRs
Proteostasis in the cytosol and the endoplasmic reticulum
Most cellular proteins are folded directly after translation in the cytosol with the assistance of chaperones, whereas membrane and secreted proteins fold and mature in the endoplasmic reticulum. Specific response mechanisms help the cell to cope with the accumulation of misfolded proteins in these cellular compartments and include the irreducible steps of detection of cellular stress, transduction of the stress signal to the nucleus, triggering of the defense programs and restoration of cellular homeostasis. Upon sustained stress, when response mechanisms fail to restore homeostasis, apoptotic pathways may be activated as a last line of defense to ensure survival of the multicellular organism.
The HSR manages denatured proteins in the cytosol (Morimoto, 2011; Vabulas et al., 2010). The major player is the transcription factor heat shock factor 1 (HSF1), which is activated during cellular stress and induces the transcription of chaperones and other protective genes. Under normal conditions HSF1 is bound to the cytosolic chaperones heat shock protein (HSP)90 and HSP70 in a monomeric inactive form. When the load of unfolded proteins increases, these chaperones will dissociate from HSF1 and be recruited to unfolded proteins. HSF1 then trimerizes and translocates to the nucleus, where it is post-translationally modified and activates the transcription of HSPs.
UPRER is mediated by three transmembrane sensors: inositol requiring element 1 (IRE1), PKR-like endoplasmic reticulum (ER) kinase (PERK) and activating transcription factor 6 (ATF6), which are retained in an inactive state by the chaperone BiP/GRP78 (Walter and Ron, 2011). During ER stress, released and activated sensors elicit the following main responses to unfolded protein stress. PERK phosphorylates and thereby inhibits the eukaryotic translation initiation factor 2α (eIF2α) to prevent further protein synthesis, ATF6 is proteolytically cleaved and translocates to the nucleus to induce the transcription of ER chaperones, while IRE1 splices the mRNA of XBP1, leading to a functional transcription factor which consequently activates genes involved in misfolded protein export and degradation. Prolonged UPRER induces apoptosis via several pathways downstream of PERK, ATF6 and IRE1, including c-Jun N-terminal kinase (JNK), transcription factor C/EBP homologous protein (CHOP) and B-cell lymphoma 2 (BCL2) family proteins, which activate proapoptotic factors, leading to the final execution of cell death (Szegezdi et al., 2006).
Proteostasis in mitochondria, the UPRmt
Mitochondria originated from endosymbiosis of eukaryotic progenitor cells with proteobacteria, which provided their host with the advantage of efficient ATP production via oxidative phosphorylation (OXPHOS) (Gray et al., 2001). Through evolution, mitochondria became well-integrated organelles interconnected with other cell compartments and able to quickly adapt to changing conditions. Mitochondria perform a number of crucial functions in the cell, which include energy harvesting, programmed cell death, regulation of Ca2+ levels and biosynthesis or metabolism of lipids, amino acids and iron sulfur (Fe-S) clusters (Nunnari and Suomalainen, 2012).
The mitochondrial proteome is composed of ~1500 proteins, encoded by both mitochondrial and nuclear genomes (Pagliarini et al., 2008). Whereas 13 essential proteins of the electron transport chain (ETC) together with part of the required machinery for their synthesis are encoded by the mitochondrial DNA (Anderson et al., 1981), the vast majority of mitochondrial proteins are encoded in the nucleus and must be imported from the cytoplasm. As mitochondria contain separate matrix and intermembrane space (IMS) compartments, enclosed by inner and outer membranes, which complicates protein import, elaborate targeting and sorting machineries exist to deliver the proteins to their correct mitochondrial destinations (Neupert and Herrmann, 2007). Most mitochondrial matrix proteins such as metabolic enzymes and intermembrane (IM)-tethered matrix-facing ETC proteins are imported as precursors with an N-terminal matrix-targeting sequence (MTS), which is recognized by the mitochondrial translocation machinery (Neupert and Herrmann, 2007). Precursor proteins are transported across mitochondrial membranes by translocase of outer membrane (TOM) and translocase of inner membrane (TIM) complexes in a largely unfolded state. Upon translocation into the mitochondria, the MTS is cleaved and proteins undergo chaperone-assisted folding, followed, if needed, by assembly into multiprotein complexes (Neupert and Herrmann, 2007).
There are two major mitochondrial chaperone systems that facilitate protein folding in the matrix: mtHSP70, part of presequence translocase-associated import-motor (PAM) complex, which directly folds the incoming proteins, and multimeric HSP60–HSP10 machinery in the matrix (Chacinska et al., 2009). Complementary to the action of chaperones to maintain proteostasis, protein quality control (PQC) proteases, specific for each mitochondrial compartment, recognize and degrade the proteins that fail to fold or assemble properly (Tatsuta and Langer, 2008). In this context, the UPRmt senses the perturbations that overload its quality control network capacities, and activates the transcription of nuclear-encoded protective genes in order to re-establish mitochondrial homeostasis (Haynes and Ron, 2010).
UPRmt signaling
As the mitochondrion is an organelle enclosed by a double membrane, the signal transmission from the unfolded mitochondrial proteins to the nucleus cannot be achieved using a system similar to HSR and UPRER, which is regulated via interactions between signaling proteins and chaperones. The best-described mitochondria-to-nucleus signal transduction pathway is the retrograde response (RTG), which is used for metabolic adaptations in response to decreased mitochondrial activity in yeast (Liu and Butow, 2006). However, the RTG does not modulate the expression of mitochondrial chaperones, suggesting that UPRmt has a distinct regulatory mechanism (Haynes and Ron, 2010).
While UPRmt was first discovered in mammals (Martinus et al., 1996), the molecular mechanism involved has been more extensively studied in the model organism Caenorhabditis elegans (Pellegrino et al., 2013). Worm strains, expressing a GFP reporter fused to promoters of the mitochondrial chaperones HSP-6 and HSP-60 (homologs of mammalian mtHSP70 and HSP60, respectively), were used to evaluate the activation of the UPRmt pathway. The temperature-sensitive zc32 C. elegans strain, which conditionally activates UPRmt, has been isolated by ethyl methanesulfonate (EMS) mutagenesis, but its molecular target remains obscure (Benedetti et al., 2006). However, in most studies, specific manipulations that all trigger proteotoxic stress in the mitochondria have been used to induce UPRmt experimentally. Paraquat treatment leading to excessive production of reactive oxygen species (ROS) activates UPRmt (Runkel et al., 2013). Additionally, UPRmt can be induced by inactivation of multiple genes implicated in the mitochondrial protein-handling machinery, such as by RNAi of PQC protease paraplegin/spg-7, as well as other proteins involved in importing, sorting and processing of the mitochondrial proteome (Yoneda et al., 2004). Importantly, disruption of stoichiometric balance between components of OXPHOS complexes I, III, IV and V, which are encoded by both mitochondrial and nuclear genomes, activates UPRmt. First, interference with the expression of mitochondrial genome-encoded ETC components, either by depletion of mtDNA using ethidium bromide (Yoneda et al., 2004) or by inhibition of mtDNA translation by doxycycline treatment and RNAi of mitochondrial ribosomal proteins (Houtkooper et al., 2013), all trigger the response. Likewise, downregulation of nuclear-encoded cco-1 (complex IV), isp-1 (complex III) and clk-1 (ubiquinone synthesis) also induces UPRmt (Baker et al., 2012; Durieux et al., 2011). These manipulations deplete single components of particular OXPHOS complexes and presumably overload the mitochondrial chaperones with their respective partner proteins, which cannot be assembled into multiprotein complexes.
UPRmt signaling in C. elegans
UPRmt signaling originates in the matrix of the mitochondria, when the misfolded or unassembled proteins accumulate and overload the capacity of the chaperones. Consequently, the excessive proteins are digested into short peptides of 6–30 amino acids in length by the CLPP-1 proteolytic complex (Fig. 1) (Haynes et al., 2007). These peptides are transported by an inner membrane-spanning ATP-binding cassette (ABC) transporter HAF-1 from the matrix into the IMS and then presumably diffuse into the cytoplasm (Haynes et al., 2010). It has recently been suggested that the released peptides weaken mitochondrial import. Consequently, the main UPRmt regulator, the leucine zipper transcription factor ATFS-1 is able to translocate to the nucleus and activate the response genes (Nargund et al., 2012). ATFS-1, which contains both nuclear localization/export (NLS) and mitochondrial targeting sequences (MTS) acts as a link between the mitochondria and nucleus in UPRmt signaling (Fig.1). Under basal conditions, ATFS-1 is imported into the mitochondrial matrix and degraded by the LON protease (Nargund et al., 2012). Because of impaired protein import into the mitochondria during proteotoxic stress, ATFS-1 cannot be targeted to the mitochondria for degradation. As a result, ATFS-1 translocates to the nucleus, where it activates a broad protective transcriptional program.
Fig. 1.

Overview of the mitochondrial unfolded protein response (UPRmt) signaling in Caenorhabditis elegans. Chaperones heat shock protein (HSP)-6, HSP-60 and the protease CLPP-1 perform protein quality control in the mitochondria. When unfolded, misfolded or unassembled proteins accumulate in the matrix, CLPP-1 cleaves the proteins into short peptides, which are exported by the transporter HAF-1 into the cytoplasm and thereby inhibit mitochondrial protein import. As a result, the transcription factor ATFS-1 cannot be imported into mitochondria for degradation, and therefore translocates to the nucleus, where, presumably in a complex with small ubiquitin-like protein UBL-5 and homeodomain-containing transcription factor DVE-1, it activates the transcription of stress response genes, which reconstitute mitochondrial homeostasis. In a complementary pathway, reactive oxygen species (ROS), generated by oxidative phosphorylation (OXPHOS) complexes (I–V) under stress, activate GCN-2 kinase, which inhibits cytosolic protein translation by phosphorylation of eukaryotic translation initiation factor 2α (eIF2α), thus reducing the protein-folding load in the mitochondria. TIM, translocase of inner membrane complex; TOM, translocase of outer membrane complex; NLS, nuclear localization/export sequence; MTS, mitochondrial targeting sequence; mt, mitochondria.
In the nucleus, ATFS-1 induces mitochondrial protein quality control by transcriptional activation of chaperone and protease genes (Nargund et al., 2012). In addition, expression of proteins involved in mitochondrial import, ROS detoxification and protection against mitochondrial dysfunction are all induced by ATFS-1, which ultimately reconstitutes the mitochondrial homeostasis (Nargund et al., 2012). Moreover, a few glycolysis genes are upregulated by ATFS-1, which indicates that UPRmt is able to shift the global cell metabolism from respiration to glycolysis for ATP production when mitochondria are stressed (Nargund et al., 2012). Two additional proteins are considered to play a role in UPRmt transcriptional regulation: small ubiquitin-like protein UBL-5 and homeodomain-containing transcription factor DVE-1 (Benedetti et al., 2006; Haynes et al., 2007). Upon mitochondrial stress the expression of UBL-5 is induced and both UBL-5 and DVE-1 translocate into the nucleus (Fig.1). The two proteins form a complex, which associates with the promoter of the mitochondrial chaperone hsp-60 gene and activates its transcription. Presumably the UBL-5 and DVE-1 complex cooperates with ATFS-1 to activate chaperone expression, but the mechanism of this interaction has not been investigated. As the closest DVE-1 homologs in mammals are the global chromatin organizers SATB1 and SATB2 (Dobreva et al., 2003; Yasui et al., 2002) the DVE-1/UBL-5 complex in C. elegans might mediate chromatin remodeling in order to facilitate ATFS-1 access to chaperone promoters.
The crosstalk between UPRmt and UPRER in C. elegans has recently been identified (Baker et al., 2012). Inhibition of cytosolic translation by phosphorylation of eIF2α is one of the main responses during ER stress mediated by PERK (Harding et al., 1999). Downregulation of protein translation in the cytosol was also detected during mitochondrial proteotoxic stress (Baker et al., 2012). Within the context of UPRmt, general control non-repressed 2 (GCN-2) kinase and the GLC7-like phosphatase (GSP-1) alter the phosphorylation status of eIF2α (Fig. 1). Under mitochondrial unfolded protein stress, GCN-2 phosphorylates eIF2α and thus inhibits protein translation in the cytoplasm, which sequentially reduces the folding load on mitochondrial chaperones (Baker et al., 2012). The GCN-2-mediated phosphorylation was shown to be dependent on ROS generated in dysfunctional mitochondria. As ATFS-1 and HAF-1 activities are not required for eIF2α phosphorylation, GCN-2 is defined as a player of a parallel complementary protective pathway in UPRmt regulation. Additionally, phosphoinositide 4-kinase (PIFK-1) has been suggested to act in the signaling of both UPRmt and UPRER activated by ROS inducer paraquat; however, its mechanism of action in these pathways has not yet been investigated (Runkel et al., 2013).
UPRmt signaling in mammals
In mammals, the UPRmt signaling mechanism has been investigated in cell culture models by ethidium bromide treatment (Martinus et al., 1996) and overexpression of aggregation-prone mutant protein ornithine transcarbamylase (OTC) targeted to the mitochondrial matrix (Zhao et al., 2002). Several components of the pathway, such as the mitochondrial chaperones and the quality control protease ClpP were shown to be conserved from C. elegans. However, downstream signaling steps and transcriptional regulation of UPRmt remain not well understood.
In response to unfolded protein stress in mammalian cells, the expression of the nuclear encoded mitochondrial chaperones HSP60, HSP10 and mtDnaJ, and the protease ClpP is induced in a transient manner, the extent of which correlates with the level of unfolded proteins in mitochondria (Zhao et al., 2002). In addition, mitochondrial proteases YME1L1 and PMPCB, the import component TIMM17A and the enzymes NDUFB2, endonuclease G and thioredoxin 2 are all upregulated during UPRmt (Aldridge et al., 2007). Bioinformatics analysis identified conserved regulatory elements in promoters of these UPRmt-responsive genes (Fig. 2). One of these elements was shown to be bound by a heterodimer of leucine zipper transcription factors CHOP and C/EBPβ, leading to UPRmt-responsive gene induction (Zhao et al., 2002). CHOP is induced during mitochondrial proteotoxic stress; however, it is also activated during UPRER and is well known for its role in mediating apoptotic cell death under conditions of extreme ER stress (McCullough et al., 2001), questioning its specificity in UPRmt regulation. Interestingly, both CHOP and C/EBPβ contain an AP-1 binding site in their promoter, which is required for UPRmt but not for induction of genes in UPRER (Horibe and Hoogenraad, 2007). The transcription factor c-Jun binds to this AP-1 site (Weiss et al., 2003), suggesting regulation of UPRmt by the JNK pathway (Fig. 2) (Jaeschke et al., 2006). In the proximity of the CHOP binding site, two mitochondrial unfolded protein response elements (MURE1 and MURE2) (Aldridge et al., 2007; Zhao et al., 2002) are present in promoters of UPRmt-responsive genes (Fig. 2). Possibly, as yet unidentified transcription factors or regulators binding to MUREs could in part mediate the UPRmt specificity. Similar to the role of GCN-2 in C. elegans (Baker et al., 2012), dsRNA-activated protein kinase (PKR), also known as eukaryotic translation initiation factor 2-α kinase 2 (EIF2AK2), mediates phosphorylation of eIF2α, thus attenuating protein translation in the cytosol during UPRmt in mammalian systems (Fig. 2) (Rath et al., 2012). Interestingly, PKR together with ClpP is also required for the activation of c-Jun, the transcriptional regulator of CHOP (Rath et al., 2012).
Fig. 2.
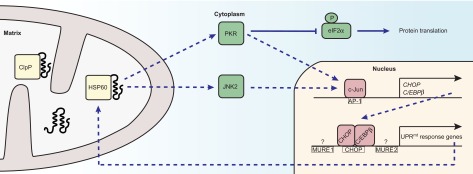
Overview of the mitochondrial matrix UPRmt in mammals. Upon mitochondrial stress, c-Jun N-terminal kinase (JNK)2 and dsRNA-activated protein kinase (PKR) contribute to the activation of transcription factor c-Jun, which binds to AP-1 elements and activates transcription of the transcription factors CHOP and C/EBPβ. Dimers of CHOP and C/EBPβ then bind to the CHOP element on the promoter of UPRmt genes that encode mitochondrial protein quality control proteins, such as HSP60 and ClpP, and mitochondrial import components. Two other conserved elements in promoters of UPRmt response genes, MURE1 and MURE2, are presumably bound by factors that have not yet been identified. PKR inhibits cytosolic protein translation by mediating phosphorylation of eIF2α.
Recent findings show the presence of a separate UPR from the ‘classical’ matrix UPRmt, which senses unfolded proteins specifically in the mitochondrial IMS (Fig. 3). This type of UPRmt induces a protective program, which is not dependent on CHOP (Papa and Germain, 2011). IMS protein quality control is different from that of matrix proteins and is performed in two steps. First, misfolded proteins, targeted to the IMS, are ubiquitinated and degraded by the 26S proteasome in the cytoplasm. Second, if any unfolded and excessive proteins still enter the IMS, they are eliminated by the protease HTRA2 (also known as Omi) (Radke et al., 2008). Overexpression of aggregate-forming IMS proteins results in ROS overproduction and protein kinase B (AKT) phosphorylation, which induces the activation of estrogen receptor α (ERα) (Fig. 3) (Papa and Germain, 2011). ERα then boosts the protein quality control in IMS by increasing the levels of HTRA2 and the activity of the proteasome (Fig. 3). In addition, ERα activation enhances the transcription of the nuclear respiratory factor 1 (NRF1), which further protects mitochondria and prevents loss of mitochondrial potential.
Fig. 3.

Overview of the response to unfolded proteins in the intermembrane space (IMS) in mammals. ROS, produced by stressed mitochondria, activate protein kinase B (AKT), which consequently phosphorylates estrogen receptor α (ERα). Activated ERα then induces IMS protein quality control by increasing the activity of proteasome 26S and inducing the transcription of IMS protease HTRA2 and mitochondrial regulator NRF1.
Mitochondrial unfolded protein response in aging
A gradual decrease of mitochondrial function occurs during aging, as demonstrated by numerous studies using simple organisms as well as mammalian models (Pulliam et al., 2012). Indeed, a global RNAi screen in C. elegans identified that genes important for mitochondrial function stand out as the most important in affecting the duration of lifespan (Lee et al., 2003). Furthermore, mutations of mitochondrial ETC components in various organisms, such as yeast, C. elegans, Drosophila melanogaster and mouse were shown to increase their lifespan (Copeland et al., 2009; Dell'Agnello et al., 2007; Dillin et al., 2002; Feng et al., 2001; Lapointe et al., 2009; Liu et al., 2005). Interestingly, the lifespan of C. elegans can only be improved if ETC inhibition occurs no later than during the L3/L4 larval stage of development (Dillin et al., 2002), when massive mitochondrial proliferation takes place (Tsang and Lemire, 2002). This suggested existence of a regulatory system, which inspects mitochondrial activity early in life and through an adaptive response, establishes the rate of aging, which persists during the whole life, presumably through epigenetic changes (Dillin et al., 2002). It has recently been shown that knockdown of complex IV component cco-1 only during larval development triggers UPRmt, which is maintained into adulthood and is associated with longevity (Durieux et al., 2011). This effect is tissue specific, based on findings that inhibition of ETC specifically in intestine or neurons, but not in muscle, is sufficient to increase longevity in a UPRmt-dependent fashion, as evidenced by the requirement for the UPRmt component UBL-5. Interestingly, ETC impairment in neurons can activate UPRmt in the worm intestine, which suggests the existence of a released extracellular ‘mitokine’, which can migrate from the producing cell type and activate UPRmt in distant receiving tissues (Durieux et al., 2011). The neuronally produced mitokine can spread the adaptive stress response to other tissues, preparing the entire worm against the harmful insult.
We recently showed that activation of UPRmt correlates with longevity both in C. elegans and in mice (Houtkooper et al., 2013). Using the BXD isogenic genetic reference population of mice with a high level of genetic variability in their genome (Peirce et al., 2004), we mapped a quantitative trait locus for mouse lifespan on chromosome 2. The expression of mitochondrial ribosomal protein S5 (Mrps5), encoded in this region, strongly correlated with longevity, with a 50% reduction of Mrps5 expression in the BXD strains causing a lifespan extension of ~250 days. In C. elegans, mrps-5 RNAi increased lifespan by more than 50% and maintained health, revealing a strong causality between this gene and aging regulation (Houtkooper et al., 2013).
Knockdown of mrps-5 expression reduces the translation of mtDNA-encoded ETC components, which results in a mito-nuclear protein imbalance between mtDNA and nuclear DNA-encoded OXPHOS complex components. This imbalance is perceived as mitochondrial proteotoxic stress, which subsequently activates UPRmt and prolongs lifespan. This hypothesis is consistent with previous findings that only RNAi against OXPHOS complexes I, III, IV and V, which are encoded by both genomes, can extend lifespan (Dillin et al., 2002), while mutations in complex II proteins, encoded by only nuclear DNA, result in a shortened rather than extended lifespan (Kuang and Ebert, 2012). In accordance to the results using C. elegans, Mrps5 as well as genes involved in OXPHOS associate with Hsp60 in a close expression correlation network in the mouse BXD lines, suggesting that mito-nuclear imbalance leading to UPRmt and potentially beneficial effects on lifespan may be conserved across species. Correspondingly, RNAi of most of the specific UPRmt components in C. elegans leads to deficient mitochondria and premature aging phenotypes (Table 1).
Table 1.
Effects of downregulation of UPRmt signaling components on mitochondrial morphology, health and longevity in Caenorhabditis elegans

We subsequently used antibiotics – doxycycline and chloramphenicol – that specifically target bacterial translation and thus also inhibit mitochondrial translation, to pharmacologically mimic the reduction of mrps-5. Treatment of both C. elegans and mouse hepatocyte cell cultures with these antibiotics triggered mito-nuclear imbalance, leading to a robust UPRmt and an increased lifespan in C. elegans (Houtkooper et al., 2013). Interestingly, exposing worms and cells to the well-known longevity compounds rapamycin and resveratrol, which are mitochondrial biogenesis inducers and, differently from mrps-5 and ETC downregulation, increase mitochondrial respiration, also induced UPRmt (Houtkooper et al., 2013). Moreover, treatment with NAD+ boosters, such as nicotinamide riboside (NR) and poly(ADP-ribose) polymerase inhibitors (PARPi), which stimulate mitochondrial biogenesis via the sirtuin 1 (SIRT1) pathway, also induced a mito-nuclear protein imbalance resulting in UPRmt in mammalian cells and in C. elegans (Mouchiroud et al., 2013). In line with the beneficial and protective effects of UPRmt, the stimulation of the SIRT1 pathway also increased worm lifespan (Mouchiroud et al., 2013). This suggests that UPRmt, triggered by different mitochondrial stresses, i.e. by increasing protein-folding workload during biogenesis or by mito-nuclear proteostatic imbalance, could be the underlying mechanism leading to increased longevity.
These results highlight the importance of UPRmt in the control of mitochondrial function across multiple species. Furthermore, if the C. elegans data can be extended to mammals, one can speculate that health and lifespan may be improved by using pharmacological agents to activate UPRmt.
Conclusions
Our understanding of UPRmt has so far already provided useful information about the communication between mitochondria and nucleus, and has contributed to deciphering the role of mitochondria in lifespan regulation from simple model organisms to mammals. However, further clarification of the full complexity of the UPRmt pathway and its role in physiology and aging will pose a scientific challenge for years to come.
First, the identification of possible functional mammalian homologs of the key elements that constitute the UPRmt in the worm (e.g. the nature of the mitochondrial peptides, ATFS-1 and HAF-1) would allow a better description and mechanistic understanding of the UPRmt pathway in mammals. It will also be important to identify how UPRmt is specified and cross-talks with similar defense mechanisms against proteotoxic stress in other cellular compartments, such as the cytoplasmic HSR or UPRER; as discussed above, certain signaling nodes are common between all these pathways. The physiological role of UPRmt in development, health, disease and aging should likewise be determined. We need to evaluate whether, similar to the worm, UPRmt is a developmentally confined pathway in mammals. Mapping the precise periods in mammalian development where this response may be essential is therefore of utmost importance. Furthermore, the physiological conditions in which this pathway is solicited, e.g. specific diet, exercise, stress and their possible contribution to improving health, should be determined. Also of note is that UPRmt can be activated at a distance. This involves secreting cells (e.g. neurons) that produce an endocrine signal, which activates their cognate receptor in the receiving cells, so that they also can mount an adaptive response. Identifying the mitokine signal–receptor tandem in the worm model and ultimately in mammals is hence expected to be a hot area of future research. Finally, defining the mechanisms that help the cell to memorize and maintain the beneficial adaptive response through epigenetic or mitogenetic modifications and imprinting will be essential.
While these outstanding issues are being addressed, we should not forget that this pathway could provide therapeutic opportunities that may help us to manage some of the most pervasive diseases that are associated with aging, such as metabolic diseases, cancer and neurodegenerative diseases. Drawing parallels with the beneficial impact of UPRmt in C. elegans, targeting this pathway via nutritional and/or pharmaceutical approaches could hence also improve health and lifespan in higher organisms and eventually help to manage some of the common age-related diseases.
Acknowledgements
We thank the members of the Auwerx lab and Andy Dillin (UC Berkeley) for discussions.
FOOTNOTES
Competing interests
J.A. is a scientific founder and SAB member of Mitokyne, a company that develops treatments for mitochondrial disease.
Funding
The work in the laboratory of the authors is supported by grants of the Ecole Polytechnique Fédérale de Lausanne, the EU Ideas program (Sirtuins; ERC-2008-AdG-231138), The Velux Stiftung, National Institutes of Health (NIH) [1R01HL106511 and R01AG043930], and the Swiss National Science Foundation [SNF 31003A-140780 and CRSII3-136201]. J.A. is the Nestlé Chair in Energy Metabolism. Deposited in PMC for release after 12 months.
References
- Aldridge J. E., Horibe T., Hoogenraad N. J. (2007). Discovery of genes activated by the mitochondrial unfolded protein response (mtUPR) and cognate promoter elements. PLoS ONE 2, e874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson S., Bankier A. T., Barrell B. G., de Bruijn M. H., Coulson A. R., Drouin J., Eperon I. C., Nierlich D. P., Roe B. A., Sanger F., et al. (1981). Sequence and organization of the human mitochondrial genome. Nature 290, 457-465 [DOI] [PubMed] [Google Scholar]
- Badyaev A. V. (2005). Stress-induced variation in evolution: from behavioural plasticity to genetic assimilation. Proc. Biol. Sci. 272, 877-886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker B. M., Nargund A. M., Sun T., Haynes C. M. (2012). Protective coupling of mitochondrial function and protein synthesis via the eIF2α kinase GCN-2. PLoS Genet. 8, e1002760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedetti C., Haynes C. M., Yang Y., Harding H. P., Ron D. (2006). Ubiquitin-like protein 5 positively regulates chaperone gene expression in the mitochondrial unfolded protein response. Genetics 174, 229-239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chacinska A., Koehler C. M., Milenkovic D., Lithgow T., Pfanner N. (2009). Importing mitochondrial proteins: machineries and mechanisms. Cell 138, 628-644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland J. M., Cho J., Lo T., Jr, Hur J. H., Bahadorani S., Arabyan T., Rabie J., Soh J., Walker D. W. (2009). Extension of Drosophila life span by RNAi of the mitochondrial respiratory chain. Curr. Biol. 19, 1591-1598 [DOI] [PubMed] [Google Scholar]
- Dell'Agnello C., Leo S., Agostino A., Szabadkai G., Tiveron C., Zulian A., Prelle A., Roubertoux P., Rizzuto R., Zeviani M. (2007). Increased longevity and refractoriness to Ca2+-dependent neurodegeneration in Surf1 knockout mice. Hum. Mol. Genet. 16, 431-444 [DOI] [PubMed] [Google Scholar]
- Dillin A., Hsu A. L., Arantes-Oliveira N., Lehrer-Graiwer J., Hsin H., Fraser A. G., Kamath R. S., Ahringer J., Kenyon C. (2002). Rates of behavior and aging specified by mitochondrial function during development. Science 298, 2398-2401 [DOI] [PubMed] [Google Scholar]
- Dobreva G., Dambacher J., Grosschedl R. (2003). SUMO modification of a novel MAR-binding protein, SATB2, modulates immunoglobulin mu gene expression. Genes Dev. 17, 3048-3061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durieux J., Wolff S., Dillin A. (2011). The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell 144, 79-91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J., Bussière F., Hekimi S. (2001). Mitochondrial electron transport is a key determinant of life span in Caenorhabditis elegans. Dev. Cell 1, 633-644 [DOI] [PubMed] [Google Scholar]
- Gems D., Partridge L. (2008). Stress-response hormesis and aging: ‘that which does not kill us makes us stronger’. Cell Metab. 7, 200-203 [DOI] [PubMed] [Google Scholar]
- Gray M. W., Burger G., Lang B. F. (2001). The origin and early evolution of mitochondria. Genome Biol. 2, 1018.1-1018.5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haigis M. C., Yankner B. A. (2010). The aging stress response. Mol. Cell 40, 333-344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding H. P., Zhang Y., Ron D. (1999). Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 397, 271-274 [DOI] [PubMed] [Google Scholar]
- Haynes C. M., Ron D. (2010). The mitochondrial UPR – protecting organelle protein homeostasis. J. Cell Sci. 123, 3849-3855 [DOI] [PubMed] [Google Scholar]
- Haynes C. M., Petrova K., Benedetti C., Yang Y., Ron D. (2007). ClpP mediates activation of a mitochondrial unfolded protein response in C. elegans. Dev. Cell 13, 467-480 [DOI] [PubMed] [Google Scholar]
- Haynes C. M., Yang Y., Blais S. P., Neubert T. A., Ron D. (2010). The matrix peptide exporter HAF-1 signals a mitochondrial UPR by activating the transcription factor ZC376.7 in C. elegans. Mol. Cell 37, 529-540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzenberger M., Dupont J., Ducos B., Leneuve P., Géloën A., Even P. C., Cervera P., Le Bouc Y. (2003). IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature 421, 182-187 [DOI] [PubMed] [Google Scholar]
- Horibe T., Hoogenraad N. J. (2007). The chop gene contains an element for the positive regulation of the mitochondrial unfolded protein response. PLoS ONE 2, e835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houtkooper R. H., Mouchiroud L., Ryu D., Moullan N., Katsyuba E., Knott G., Williams R. W., Auwerx J. (2013). Mitonuclear protein imbalance as a conserved longevity mechanism. Nature 497, 451-457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaeschke A., Karasarides M., Ventura J. J., Ehrhardt A., Zhang C., Flavell R. A., Shokat K. M., Davis R. J. (2006). JNK2 is a positive regulator of the cJun transcription factor. Mol. Cell 23, 899-911 [DOI] [PubMed] [Google Scholar]
- Kenyon C., Chang J., Gensch E., Rudner A., Tabtiang R. (1993). A C. elegans mutant that lives twice as long as wild type. Nature 366, 461-464 [DOI] [PubMed] [Google Scholar]
- Kikis E. A., Gidalevitz T., Morimoto R. I. (2010). Protein homeostasis in models of aging and age-related conformational disease. Adv. Exp. Med. Biol. 694, 138-159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura K. D., Tissenbaum H. A., Liu Y., Ruvkun G. (1997). daf-2, an insulin receptor-like gene that regulates longevity and diapause in Caenorhabditis elegans. Science 277, 942-946 [DOI] [PubMed] [Google Scholar]
- Kimura K., Tanaka N., Nakamura N., Takano S., Ohkuma S. (2007). Knockdown of mitochondrial heat shock protein 70 promotes progeria-like phenotypes in Caenorhabditis elegans. J. Biol. Chem. 282, 5910-5918 [DOI] [PubMed] [Google Scholar]
- Kourtis N., Tavernarakis N. (2011). Cellular stress response pathways and ageing: intricate molecular relationships. EMBO J. 30, 2520-2531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuang J., Ebert P. R. (2012). The failure to extend lifespan via disruption of complex II is linked to preservation of dynamic control of energy metabolism. Mitochondrion 12, 280-287 [DOI] [PubMed] [Google Scholar]
- Lapointe J., Stepanyan Z., Bigras E., Hekimi S. (2009). Reversal of the mitochondrial phenotype and slow development of oxidative biomarkers of aging in long-lived Mclk1+/− mice. J. Biol. Chem. 284, 20364-20374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S. S., Lee R. Y., Fraser A. G., Kamath R. S., Ahringer J., Ruvkun G. (2003). A systematic RNAi screen identifies a critical role for mitochondria in C. elegans longevity. Nat. Genet. 33, 40-48 [DOI] [PubMed] [Google Scholar]
- Liu Z., Butow R. A. (2006). Mitochondrial retrograde signaling. Annu. Rev. Genet. 40, 159-185 [DOI] [PubMed] [Google Scholar]
- Liu X., Jiang N., Hughes B., Bigras E., Shoubridge E., Hekimi S. (2005). Evolutionary conservation of the clk-1-dependent mechanism of longevity: loss of mclk1 increases cellular fitness and lifespan in mice. Genes Dev. 19, 2424-2434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mair W., Dillin A. (2008). Aging and survival: the genetics of life span extension by dietary restriction. Annu. Rev. Biochem. 77, 727-754 [DOI] [PubMed] [Google Scholar]
- Martinus R. D., Garth G. P., Webster T. L., Cartwright P., Naylor D. J., Høj P. B., Hoogenraad N. J. (1996). Selective induction of mitochondrial chaperones in response to loss of the mitochondrial genome. Eur. J. Biochem. 240, 98-103 [DOI] [PubMed] [Google Scholar]
- McCullough K. D., Martindale J. L., Klotz L. O., Aw T. Y., Holbrook N. J. (2001). Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol. Cell. Biol. 21, 1249-1259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimoto R. I. (2011). The heat shock response: systems biology of proteotoxic stress in aging and disease. Cold Spring Harb. Symp. Quant. Biol. 76, 91-99 [DOI] [PubMed] [Google Scholar]
- Mouchiroud L., Houtkooper R. H., Moullan N., Katsyuba E., Ryu D., Cantó C., Mottis A., Jo Y.-S., Viswanathan M., Schoonjans K., et al. (2013). The NAD+/sirtuin pathway modulates longevity through activation of mitochondrial UPR and FOXO signaling. Cell 154, 430-441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nargund A. M., Pellegrino M. W., Fiorese C. J., Baker B. M., Haynes C. M. (2012). Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science 337, 587-590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neupert W., Herrmann J. M. (2007). Translocation of proteins into mitochondria. Annu. Rev. Biochem. 76, 723-749 [DOI] [PubMed] [Google Scholar]
- Nunnari J., Suomalainen A. (2012). Mitochondria: in sickness and in health. Cell 148, 1145-1159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagliarini D. J., Calvo S. E., Chang B., Sheth S. A., Vafai S. B., Ong S. E., Walford G. A., Sugiana C., Boneh A., Chen W. K., et al. (2008). A mitochondrial protein compendium elucidates complex I disease biology. Cell 134, 112-123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papa L., Germain D. (2011). Estrogen receptor mediates a distinct mitochondrial unfolded protein response. J. Cell Sci. 124, 1396-1402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peirce J. L., Lu L., Gu J., Silver L. M., Williams R. W. (2004). A new set of BXD recombinant inbred lines from advanced intercross populations in mice. BMC Genet. 5, 7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrino M. W., Nargund A. M., Haynes C. M. (2013). Signaling the mitochondrial unfolded protein response. Biochim. Biophys. Acta 1833, 410-416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulliam D.A., Bhattacharya A., Van Remmen H. (2012) Mitochondrial dysfunction in aging and longevity: a causal or protective role? Antioxid. Redox. Signal. 19, 1373-1387 [DOI] [PubMed] [Google Scholar]
- Radke S., Chander H., Schäfer P., Meiss G., Krüger R., Schulz J. B., Germain D. (2008). Mitochondrial protein quality control by the proteasome involves ubiquitination and the protease Omi. J. Biol. Chem. 283, 12681-12685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rath E., Berger E., Messlik A., Nunes T., Liu B., Kim S. C., Hoogenraad N., Sans M., Sartor R. B., Haller D. (2012). Induction of dsRNA-activated protein kinase links mitochondrial unfolded protein response to the pathogenesis of intestinal inflammation. Gut 61, 1269-1278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Runkel E. D., Liu S., Baumeister R., Schulze E. (2013). Surveillance-activated defenses block the ROS-induced mitochondrial unfolded protein response. PLoS Genet. 9, e1003346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szegezdi E., Logue S. E., Gorman A. M., Samali A. (2006). Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 7, 880-885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatsuta T., Langer T. (2008). Quality control of mitochondria: protection against neurodegeneration and ageing. EMBO J. 27, 306-314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang W. Y., Lemire B. D. (2002). Mitochondrial genome content is regulated during nematode development. Biochem. Biophys. Res. Commun. 291, 8-16 [DOI] [PubMed] [Google Scholar]
- Vabulas R. M., Raychaudhuri S., Hayer-Hartl M., Hartl F. U. (2010). Protein folding in the cytoplasm and the heat shock response. Cold Spring Harb. Perspect. Biol. 2, a004390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter P., Ron D. (2011). The unfolded protein response: from stress pathway to homeostatic regulation. Science 334, 1081-1086 [DOI] [PubMed] [Google Scholar]
- Weiss C., Schneider S., Wagner E. F., Zhang X., Seto E., Bohmann D. (2003). JNK phosphorylation relieves HDAC3-dependent suppression of the transcriptional activity of c-Jun. EMBO J. 22, 3686-3695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasui D., Miyano M., Cai S., Varga-Weisz P., Kohwi-Shigematsu T. (2002). SATB1 targets chromatin remodelling to regulate genes over long distances. Nature 419, 641-645 [DOI] [PubMed] [Google Scholar]
- Yoneda T., Benedetti C., Urano F., Clark S. G., Harding H. P., Ron D. (2004). Compartment-specific perturbation of protein handling activates genes encoding mitochondrial chaperones. J. Cell Sci. 117, 4055-4066 [DOI] [PubMed] [Google Scholar]
- Zhao Q., Wang J., Levichkin I. V., Stasinopoulos S., Ryan M. T., Hoogenraad N. J. (2002). A mitochondrial specific stress response in mammalian cells. EMBO J. 21, 4411-4419 [DOI] [PMC free article] [PubMed] [Google Scholar]