

Abstract
The Insulin-like Growth Factor Receptor (IGF-IR) has been implicated in a number of human tumors, including breast cancer. Data from human breast tumors has demonstrated that IGF-IR is over-expressed and hyper-phosphorylated. Additionally, microarray analysis has shown that IGF-I treatment of MCF7 cells leads to a gene signature comprised of induced and repressed genes, which correlated with luminal B tumors. FOXA1, a forkhead family transcription factor, has been shown to be crucial for mammary ductal morphogenesis, similar to IGF-IR, and expressed at high levels in luminal subtype B breast tumors. Here, we investigated the relationship between FOXA1 and IGF-I action in breast cancer cells. We show that genes regulated by IGF-I are enriched for FOXA1 binding sites, and knock down of FOXA1 blocked the ability of IGF-I to regulate gene expression. IGF-I treatment of MCF7 cells increased the half-life of FOXA1 protein and this increase in half-life appeared to be dependent on canonical IGF-I signal transduction through both MAPK and AKT pathways. Finally, knock down of FOXA1 led to a decreased ability of IGF-I to induce proliferation and protect against apoptosis. Together, these results demonstrate that IGF-I can increase the stability of FOXA1 protein expression and place it as a critical mediator of IGF-I regulation of gene expression and IGF-I-mediated biological responses.
Keywords: IGF-I, FOXA1, Breast Cancer, Gene Regulation
INTRODUCTION
The Insulin-like Growth Factor Receptor (IGF-IR) is a key target in the treatment of breast cancer. The importance of IGF-IR in cancer is indicated by the numerous inhibitors of IGF-IR that have been developed and are currently under clinical investigation. These various inhibitors include monoclonal antibodies as well as tyrosine kinase inhibitors that target the IGF-IR in various forms of cancer, including but not limited to breast cancer [Weroha and Haluska, 2008]. Preclinical data has shown that IGF-IR is necessary for transformation of embryonic mouse fibroblasts and that IGF-IR is hyperactive and over-expressed in many breast cancers [Law et al., 2008; Sell et al., 1993; Surmacz, 2000; Werner and Bruchim, 2009]. Furthermore, development of two different mouse models, one over-expressing a constitutively active IGF-IR and the other expressing a doxycycline-inducible IGF-IR model, showed that over-expression and hyper-activation of the IGF-IR leads to a decrease in the time to tumor formation as well as an increase in the frequency of tumor development [Carboni et al., 2005; Jones et al., 2007].
The IGF-IR is a receptor tyrosine kinase, which is activated upon the binding of either IGF-I or IGF-II. This leads to auto-phosphorylation of the intracellular kinase domains and subsequent downstream signal transduction. The activation of the downstream signaling cascade leads primarily to the activation of the canonical signaling cascades involving phosphatidylinositol 3-kinase (PI3K)/AKT and the mitogen-activated protein kinase (MAPK) pathways [Dearth et al., 2007]. Activation of these canonical IGF-I signal transduction pathways results in the two predominant biological functions of IGF-IR activity, cellular proliferation and inhibition of apoptosis [LeRoith and Roberts, 2003; Surmacz, 2000]. We have previously shown that activation of the IGF-IR signal transduction pathway, following treatment with IGF-I, leads to the regulation of a particular gene signature, consisting of induced and repressed genes, in MCF7 breast cancer cells [Creighton et al., 2008]. This IGF-I gene signature was shown to correlate with poor patient prognosis. While this data revealed a potentially relevant and important role for this IGF-I gene signature, it is currently unclear how IGF-IR signal transduction regulates this subset of genes.
Forkhead box A1 (FOXA1) is a member of the FOXA subfamily of winged-helix/forkhead box transcription factors. FOXA1 has been shown to function as a ‘pioneer factor’, binding to promoters and enhancers enabling chromatin access for other transcription factors [Carroll et al., 2005; Cirillo et al., 2002]. Conversely, FOXA1 has also been shown to function as a repressor of transcription by out-competing FOXA2 for forkhead binding sites [Duncan et al., 1998]. The competition between FOXA1 and FOXA2 to bind to forkhead binding sites is mediated by the ratio of the two transcription factors relative to each other, and this ratio is moderated by insulin signaling [Duncan et al., 1998]. Additionally, the c. elegan homologue of FOXA1, DAF-16, has previously been shown to be downstream of and regulated by the insulin receptor homologue, DAF-2 [Ogg et al., 1997]. These data imply that FOXA1 can be regulated by insulin signaling and that FOXA1 can serve as an activator and a repressor of transcription. Although current literature only reveals a role for insulin-mediated signal transduction in the regulation of FOXA1, it is important to note that insulin and IGF-I share a high degree of sequence homology as well as the ability to regulate the same downstream signaling cascades, in particular activation of PI3K and MAPK pathways [Pollak, 2008; Zapf et al., 1984]. Additionally, it has recently been shown that IGF-I and insulin, through activation of the AKT signaling cascade, can both mediate the nuclear localization and transcriptional activity of FOXO3a, another member of the forkhead box family [Jag et al., 2009; Wilk et al., 2011; Zheng et al., 2000].
Previous studies have shown high expression of FOXA1 in certain subtypes of breast cancer. Immunohistochemical staining revealed that high FOXA1 expression correlated with luminal subtype A and B tumors [Badve et al., 2007; Sircoulomb et al., 2010]. Similarly, the IGF-I gene signature, discovered following IGF-I treatment of MCF7 cells, also correlated with subtype luminal B tumors, suggesting a correlation between expression of FOXA1 and IGF-IR activation in particular tumor subtypes [Creighton et al., 2008]. Furthermore, FOXA1 null mammary glands revealed a defect in the ability for the ductal tree to invade the fat pad as well as an inability to form terminal end buds during mammary gland development [Bernardo et al., 2010]. IGF-IR null mammary glands had a similar phenotype, in which terminal end bud formation was severely defective. Cell proliferation was significantly decreased as was the ability of the ductal outgrowths to invade the fat pad [Bonnette and Hadsell, 2001].
Although evidence suggests that there may be a relationship between FOXA1 and canonical IGF-I-mediated signal transduction, currently there is no data to support this hypothesis. Herein, we report that FOXA1 is required for IGF-I-mediated regulation of target genes, as well as IGF-I-induced cell proliferation and cell survival in MCF-7 breast cancer cells. In two other breast cancer cell line models, FOXA1 was partially or not required for IGF action. Additionally, we show that IGF-I is able to increase the stability of FOXA1 at the protein level by activation of IGF-I-mediated canonical signal transduction through MAPK and AKT. These data provide the first evidence that FOXA1 is necessary for IGF-I activity in a subset of breast cancer cells.
MATERIALS AND METHODS
Cell Culture
MCF-7 human breast cancer cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM; Cellgro) supplemented with 5% characterized fetal bovine serum (HyClone), 100 IU/ml penicillin, 100 µg/ml streptomycin, and 0.25 µg/ml amphotericin B (antibiotics and antimycotic from Cellgro). BT20 and MDA-MB-134 human breast cancer cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM; Cellgro) supplemented with 10% characterized fetal bovine serum (HyClone), 100 IU/ml penicillin, 100 µg/ml amphotericin B (antibiotics and antimycotic from Cellgro). Prior to ligand treatment for all experiments, cells were starved under serum-free conditions for at least one day. Serum-free medium (SFM) consists of phenol red-free improved minimal essential medium (IMEM; Invitrogen), 100 IU/ml penicillin, 100 µg/ml streptomycin, 0.25 µg/ml amphotericin B, 10mM HEPES (Invitrogen), 1µg/mL transferrin (Sigma), and 1µg/mL fibronectin (BD Biosciences). For ligand stimulation, cells were treated with 100ng/mL insulin-like growth factor I (IGF-I; Novozymes). For untreated control samples, cells were simply maintained in SFM. In some experiments, cells were pre-treated for 30min with a chemical inhibitor prior to ligand stimulation. Inhibitors used include the PI3K inhibitor LY-294002 (20µM; Calbiochem), the MEK inhibitor U0126 (10µM; Calbiochem), the PKC inhibitor Gö6983 (0.25µM; Calbiochem), the AKT inhibitor 124005 (5µM; Calbiochem) and inhibitor of protein synthesis Cycloheximide (20ng/mL;Sigma).
RNA Extraction and qRT-PCR
Total RNA was isolated using the RNeasy RNA isolation kit (Qiagen) as recommended by the supplier. Triplicate RNA samples were prepared for each treatment group. RNA was reverse transcribed using SuperScript II reverse transcriptase (Invitrogen) in accordance with manufacturer’s instructions. The PCR reaction was then carried out on an ABI 7500 fast real-time thermocycler (Applied Biosystems) using the SYBR green master mix (Applied Biosystems) and 150nM each of both the forward and reverse primers. The cycling conditions were 50°C for 2min, 95°C for 10min, followed by 40 cycles at 95°C for 15s and 60°C for 30s. Primer Express 3.0 software (Applied Biosystems) was used to design all of the primers. The sequences of the primers used are listed in the supplementary material Table S1. The fold change for each gene was calculated using the cycle threshold (ΔΔCT) method as previously described [Livak and Schmittgen, 2001], and data are represented as ligand-mediated fold change over SFM, unless otherwise stated. For each sample, real-time quantitative reverse transcription-PCRs (qRT-PCRs) were done in triplicate for both the genes of interest and the reference gene (β-actin) to normalize for input cDNA.
Immunoblotting
Proteins were resolved on 8% SDS-PAGE and transferred to nitrocellulose. The membrane was blocked in 5% milk dissolved in phosphate-buffered saline + 0.1% Tween 20 (PBST) for 1hr at room temperature. Primary antibodies used include anti-FOXA1 (AbCam ; ab55178), anti-MAP Kinase 1/2 (Millipore; 06-182), anti-phospho-p44/42 MAPK (Thr202/Tyr204) (Cell Signaling Technology; 9101), anti-Akt (Cell Signaling Technology; 9272), anti-phospho-Akt (Ser473) (Cell Signaling Technology; 9271), anti-IGF-I Receptor β (Cell Signaling Technology; 3027) and anti-β-actin (Sigma; A5441). All total antibodies were diluted 1:500 in PBST + 5% milk and all phospho antibodies were diluted 1:500 in PBST and incubated overnight at 4°C. The β-actin antibody was diluted 1:5000 in PBST + 5% milk and incubated overnight at 4°C. After washing three times for 5min with PBST, the membranes were incubated with anti-mouse or anti-rabbit secondary antibodies conjugated to either IRDye700 or IRDye800 (Rockland) for 1hr at room temperature. Secondary antibodies were diluted 1:5000 in PBST + 5% milk. After incubation, membranes were washed three times for 5min with PBST, and the signal was visualized using the Odyssey imaging system (Li-Cor Biosciences).
siRNA
MCF-7 cells were transfected with 50nM small interfering RNA (siRNA) using DharmaFECT1 (Thermo Fisher Scientific). The siRNAs used were as follows: ON-TARGETplus SMARTpool non-specific siRNA (Dharmacon), ON-TARGETplus SMARTpool FOXA1 siRNA (Dharmacon) and ON-TARGETplus SMARTpool IGF-IR siRNA (Dharmacon).
MTS Assay
MCF-7 cells were plated in 60mm dishes. Approximately 48hr after, cells were transfected with either FOXA1 siRNA or non-specific control siRNA. The next day, cells were split and re-plated in a 96-well plate. Cells were then incubated in serum free medium for 24 hours. Cells were then either maintained in SFM or treated with IGF-I (100ng/mL). IGF-I was added every 3 days to the medium. Cell growth was assessed by MTS assay (Promega) on days 0, 2, 5 and 7. The experiment was performed with biological septuplicates.
Annexin V Apoptosis Assay
This experiment was carried out by using the Vybrant Apoptosis Assay kit #6 (Invitrogen) according to the manufacturer’s specifications. MCF7 cells were either maintained in serum-free medium or in the presence of IGF-I (100ng/mL) for 4 days. Cells were then harvested and re-suspended in 100µl 1× annexin binding buffer. 5µl of Biotin-X-annexin V (20µg/mL) and 1µl Alexa Fluor350 streptavidin (1mg/mL) solution were added to the cell suspension and incubated on ice for 30min. After the incubation period the cells were suspended in 1x binding buffer to which 1µl of propidium iodide (1mg/mL) was added. Samples were analyzed by flow cytometry.
Stable Cell Lines
MCF7 cells were transduced with lentiviral particles (produced by the CBASS core of Baylor College of Medicine) containing pGIPZ shRNAmir-GFP to FOXA1 (Open Biosystems - V2LHS 16780 and V2LHS 16813). Non silencing shRNAmir-GFP was transduced as a control. Stably integrated cells were selected by adding 5µg/mL puromycin (Invitrogen) to the culture medium for 5–6 days. These pools of colonies were selected and screened for FOXA1 expression by q-RT-PCR and Western blot analysis.
Immunofluorescence and Confocal Microscopy
Cells were grown on glass coverslips in 6-well plates and fixed in 4% paraformaldehyde. Following permeabilization with Triton X-100, cells were blocked with 5% horse serum. Primary antibodies were diluted in 1% horse serum solution and cells were incubated with antibody overnight at 4°C. Primary antibodies used include FOXA1 (Abcam; ab55178). Secondary antibodies were conjugated to AlexaFluor 488 (Invitrogen). Nuclear counterstain was performed using DAPI (Vector Laboratories). Proteins were visualized by confocal microscopy (Leica Microsystems; CTR 6500).
RESULTS
IGF-I regulated genes in MCF7 cells are enriched for FOXA1 binding sites
IGF-IR and FOXA1 have both been shown to be highly expressed in luminal B breast cancer, and necessary for mammary gland development. Additionally, IGF-I signal transduction has been shown to regulate activation and localization of other forkhead box transcription factors, such as FOXO3a. However, a role for FOXA1 in IGF-I signal transduction and IGF-I biology remains to be elucidated. To begin to establish a role for FOXA1 in the IGF-I-mediated regulation of target genes, we performed in silico analysis of target genes that were either strongly induced or repressed following IGF-I (100ng/mL) treatment for 3hr of MCF7 cells, and publicly available FOXA1 ChIP-on-chip data [Carroll et al., 2006]. The top 50 IGF-I-induced (>2-fold, p-value<0.05) and the top 50 IGF-I-repressed (<0.5-fold, p-value<0.05) genes from the microarray previously reported from our lab [Creighton et al., 2008], were analyzed for FOXA1 binding in the sequence ranging from −50kB to the transcriptional start site, and within the intronic sequences of the gene. This analysis revealed that 38% (95% CI 28.5% to 48.3%) of the IGF-I-regulated genes had FOXA1 binding sites, compared to 16% (95% CI 9.4% to 24.7%) of a set of 100 random non-IGF-I-regulated genes, and this difference was significant (p=0.007) (Fig. 1A).
Figure 1. FOXA1 is necessary for IGF-I-mediated regulation of target gene expression.
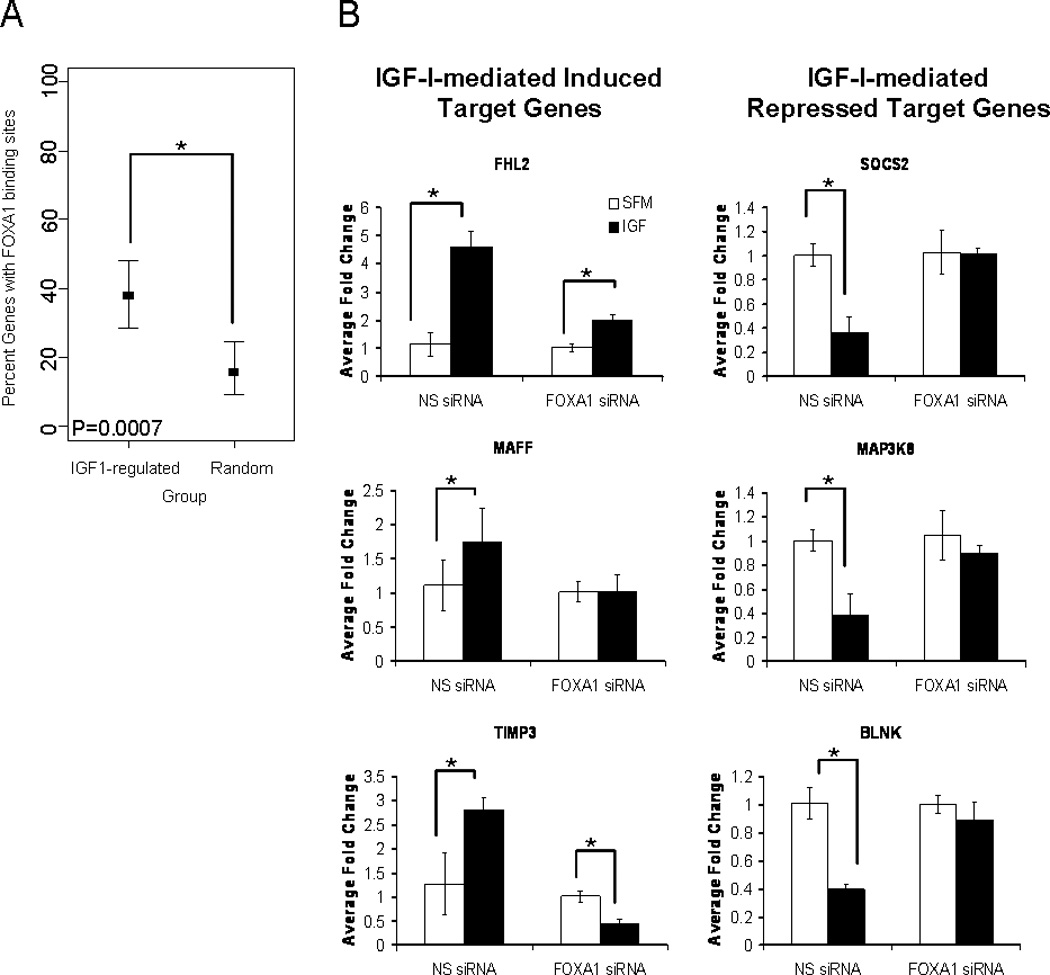
A) In silico analysis was performed comparing the top 50 IGF-I induced and top 50 IGF-I-repressed genes from a microarray previously reported from our lab with publically available FOXA1 ChIP-on-chip data. FOXA1 binding was assessed in the regions ranging from the transcriptional start site to −50kB and intronic sequences of each gene. A Fisher’s Exact Test was used to determine the significance and the confidence interval. B) MCF7 cells were transfected with non-specific siRNA (NS siRNA) or siRNA against FOXA1 (FOXA1 siRNA) followed by either maintenance in serum-free medium or stimulation with IGF-I (100ng/mL). Q-RT-PCR was used to calculate relative mRNA expression as fold change compared to serum-free conditions. The data are an average of three biological replicates ± SEM. A t-test analysis was performed in which the IGF-I treated group was compared to the serum-free group (* p<0.05).
While IGF-I regulated genes were enriched for FOXA1 binding sites, we also found that FOXA1 was necessary for IGF-I-mediated induction and repression of target genes. MCF7 cells were transiently transfected with either non-specific pooled siRNA or FOXA1 pooled siRNA and subsequently treated with IGF-I (100ng/mL; ~12.5nM) for 3hr. Quantitative real-time RT-PCR was then performed on representative target genes to assess the effect of knock down of FOXA1 on the IGF-I-mediated regulation of these genes (Fig. 1B). IGF-I treatment of MCF7 cells, in the presence of non-specific siRNA, confirmed induction or repression of target genes which had been shown to be induced or repressed by the microarray. Knockdown of FOXA1 led to a significant decrease in the ability of IGF-I to induce expression of these target genes. Similarly, this transient knockdown of FOXA1 also significantly decreased the ability of IGF-I to repress the expression of target genes. Interestingly, knockdown of FOXA1 in the estrogen receptor negative cell line, BT20, led to a relief of repression of IGF-I target genes, but had no effect on IGF-I-induced target genes (Fig. S1). Furthermore, knockdown of FOXA1 in MDA-134 breast cancer cells had no effect on the ability for IGF-I to regulate target gene expression. Clearly, these experiments do not test the relevance of FOXA1 in the regulation of the genes that were moderately (<2-fold induced or > 0.5-fold repressed) regulated by IGF-I, but it does suggest that genes that are strongly regulated by IGF-I are enriched for FOXA1 binding and that there is a necessary role for FOXA1 in the IGF-I-regulation of these target genes. Furthermore, the data from BT20 and MDA-134 cell lines suggests that the role of FOXA1 in IGF-I-mediated regulation may be cell line specific and most likely requires additional transcriptional machinery for induction and repression of IGF-I target genes.
Expression of IGF-IR is regulated by FOXA1
IGF-IR has previously been shown to be an estrogen receptor (ER)-regulated gene in MCF7 cells [Maor et al., 2006]. Additionally, FOXA1 is known to be a ‘pioneer factor’ that can open regions of chromatin and expose ER binding sites, allowing for the recruitment of ER to these binding sites [Cirillo et al., 2002]. We next addressed whether the inability of IGF-I to alter gene expression following knock down of FOXA1 was simply due to a decrease in IGF-IR expression, and subsequent decrease in IGF-I signal transduction. Transient knock down of FOXA1 in MCF7 cells led to a decrease in expression of both IGF-IR protein and mRNA (Fig. 2A, 2B). However, we found that following knockdown of FOXA1, ER protein expression was not altered. We tested if FOXA1 regulation of IGF-IR required ER by knocking down FOXA1 in C4–12 cells, an ER-negative MCF7 cell line. Following transient knockdown of FOXA1, we observed a decrease in IGF-IR protein expression in the absence of ER expression, suggesting that FOXA1 is able to regulate IGF-IR expression independently of ER (Fig. 2C). It is worth noting that there was a greater decrease in FOXA1 expression following transient transfection of C4–12 cells than MCF7 cells. This is most likely due to the fact that there is a lower endogenous level of FOXA1 expression in C4–12 cells, therefore less siRNA is required to silence expression of FOXA1 than in MCF7 cells. Furthermore, this discrepancy maybe due to the variability in transfection efficiency between two different cell lines. We next knocked down FOXA1 in MCF7 cells and treated with IGF-I (100ng/mL) for 30, 60 and 180 minutes to assess if canonical IGF-I downstream signaling was affected by the decrease in FOXA1 expression. Again, knockdown of FOXA1 led to a decrease in IGF-IR expression, but interestingly, there was no attenuation of the canonical downstream signaling cascades (Fig. 2D). As predicted, IGF-I treatment of MCF7 cells led to increased phosphorylation of AKT and ERK1/2. Following knockdown of FOXA1, IGF-I-induced activation of AKT and MAPK was intact and just as robust as in the control IGF-I treated cells (Fig. 2D). To examine this further, we directly reduced IGF-IR levels by transiently knocking down IGF-IR expression with siRNA in MCF7 cells, followed by treatment with IGF-I (100ng/mL) (Fig. 2E). The transient knockdown of IGF-IR decreased the expression of IGF-IR to a similar level as was observed after transient knock down of FOXA1, however, when the cells transiently transfected with IGF-IR siRNA were treated with IGF-I, the expression of target genes was unaffected. This suggests that the decreased level of IGF-IR that we have observed following knockdown of FOXA1, has not reached a threshold critical for observing a loss of IGF-IR signaling. These data reveal a novel observation of the ability of FOXA1 to regulate IGF-IR expression, but interestingly this down-regulation of IGF-IR does not appear to affect the ability of IGF-I to activate downstream signal transduction, namely AKT and MAPK, or regulate target genes. This suggests that there is indeed a role for FOXA1 in the IGF-I-mediated regulation of target genes and that the results shown in figure 1B are not a result of a FOXA1-mediated down-regulation of IGF-I signal transduction.
Figure 2. Knockdown of FOXA1 leads to a decrease in IGF-IR expression, independently of estrogen receptor, but does not inhibit IGF-IR signal transduction.
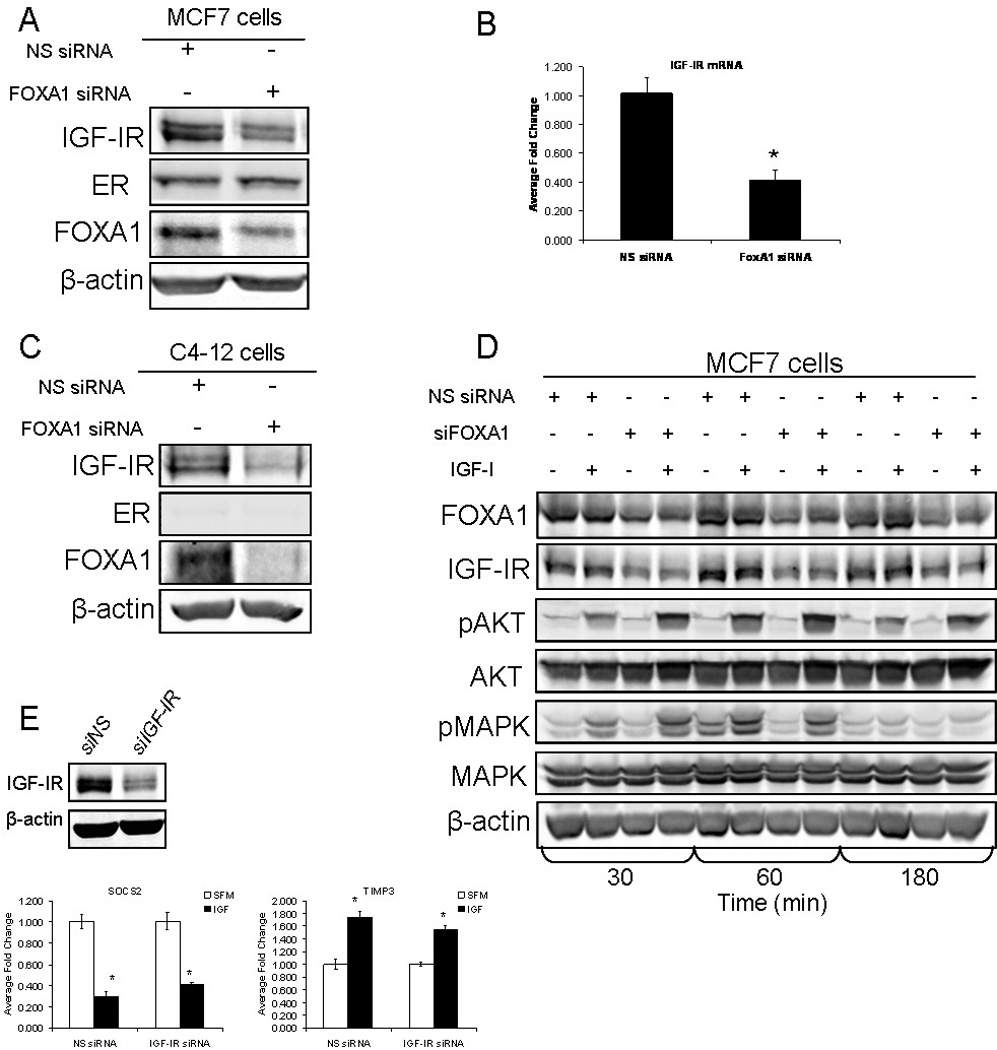
A) MCF7 cells were either transfected with non-specific siRNA (NS siRNA) or siRNA targeted to FOXA1 (FOXA1 siRNA). Cells were then lysed and protein was harvested, followed by western blot analysis as indicated. B) MCF7 cells were either transfected with NS siRNA, FOXA1 siRNA or IGF-IR siRNA. Cells were then lysed and RNA was isolated. Q-RT-PCR analysis was used to calculate relative mRNA expression as fold change compared to NS siRNA. The data are an average of three biological replicates ± SEM. A t-test analysis was performed comparing the NS siRNA group to the FOXA1 siRNA group (* p<0.05). C) C4–12 cells were transfected with either NS siRNA or FOXA1 siRNA. Cells were then lysed and protein was harvested, followed by western blot analysis as indicated. D) MCF7 cells were either transfected with NS siRNA or FOXA1 siRNA and either maintained in serum-free medium or treated with IGF-I (100ng/mL) for 30, 60 and 180 minutes. Cells were then lysed and protein was harvested, followed by western blot analysis as indicated. E) MCF7 cells were either transfected with NS siRNA or IGF-IR siRNA (siIGF-IR) followed by treatment with IGF-I (100ng/mL). Cells were then lysed and RNA was isolated. Q-RT-PCR analysis was used to calculate relative mRNA expression as fold change compared to NS siRNA. The data are an average of three biological replicates ± SEM. A t-test analysis was performed comparing the IGF-I treated group with the serum-free group (* p<0.05).
IGF-I stabilizes the expression level of FOXA1 protein
IGF-I has previously been shown to have an effect on the activation and localization of FOXO3a, but no data exists on regulation of FOXA1. We thus examined if IGF-I had an effect on the expression or localization of FOXA1. MCF7 cells were maintained in serum-free medium overnight and then treated with IGF-I (100ng/mL) for time course of 30, 60 and 240 minutes. Immunofluorescent staining for FOXA1 revealed that IGF-I treatment didn’t alter FOXA1 localization. FOXA1 is predominately a nuclear protein, and following IGF-I treatment at all time points, FOXA1 remained located in the nucleus (Fig. S2). Furthermore, IGF-I treatment had no inductive effect on FOXA1 mRNA levels, but interestingly, IGF-I treatment led to a stabilization of FOXA1 protein levels (Fig. 3A, 3B). Cells without IGF-I treatment had a decreased level of FOXA1 expression by 12 hours, and maintained this low level of basal expression throughout the time course. Conversely, cells treated with IGF-I maintained similar or slightly elevated FOXA1 expression compared to the control sample, over the time course ranging from 12 to 48 hours. Furthermore, to test whether the stabilization of FOXA1 is an IGF-I-specific event and not due to activation of the related insulin receptor, we maintained MCF7 cells in either serum-free medium or treated them with IGF-I at a concentration of either 10, 50 or 100ng/mL (Fig. 3C). The data from this experiment revealed that the IGF-I was able to stabilize FOXA1 even when used at 50ng/ml, however no response was seen at 10ng/ml which is presumably below a threshold needed for response. These data suggested that IGF-I stabilized the expression of FOXA1 protein. To test this directly, MCF7 cells were maintained in serum-free medium overnight and then pre-treated with cycloheximide (CHX) to inhibit new protein translation. Following 30 minutes of pre-treatment, cells were either maintained in CHX alone or stimulated with IGF-I (100ng/mL) in the continued presence of CHX, over a time course spanning from 4 to 24 hours. As predicted, CHX treatment led to a steady decline in the expression of FOXA1 protein and revealed a half-life of approximately 4–6 hours. Interestingly treatment with CHX and IGF-I stabilized FOXA1 protein expression and the half-life was increased to approximately 8–12 hours (Fig. 3D). Moreover, these results were confirmed in BT20 cells in which we also observed an IGF-I-induced stabilization of FOXA1 protein expression (Fig. 3E). These experiments reveal that IGF-I does not regulate FOXA1 localization or cause an induction of FOXA1 expression, but it does stabilize FOXA1 protein by increasing its half-life.
Figure 3. IGF-I increases the half-life of FOXA1 protein in MCF7 Cells.
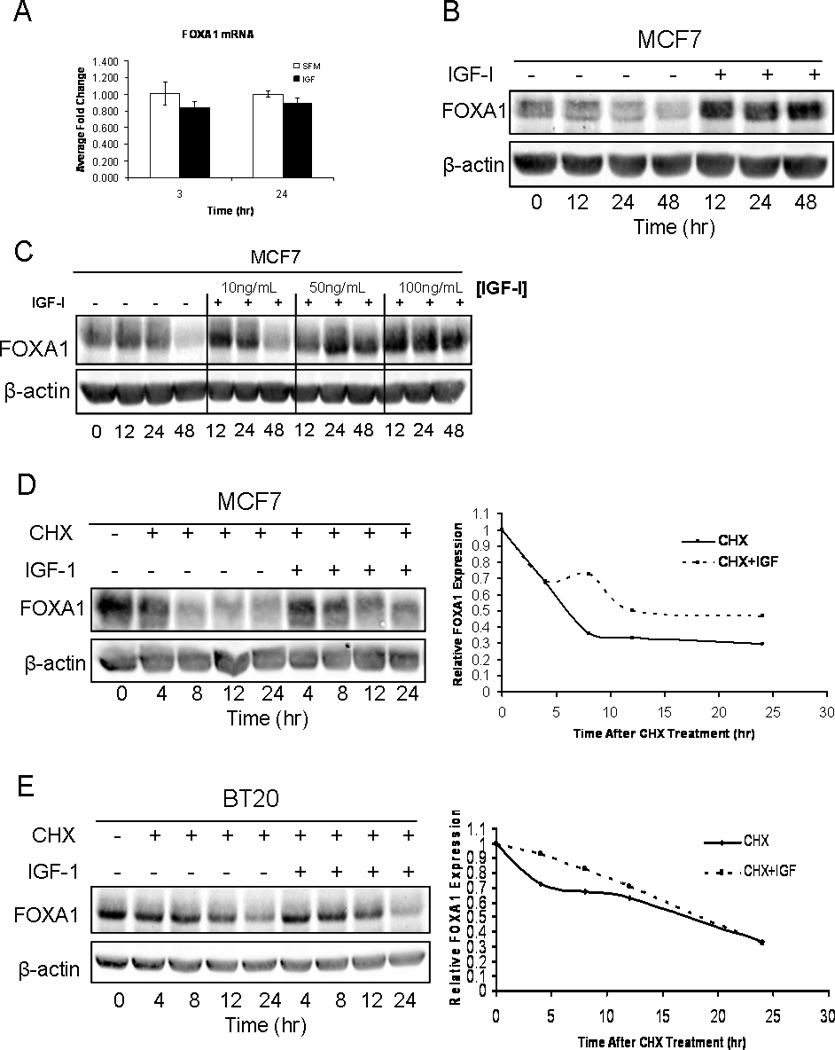
A) MCF7 cells were either maintained in serum-free medium or treated with IGF-I (100ng/mL) for 3 or 24 hours. Cells were then lysed and RNA was harvested. Q-RT-PCR analysis was used to calculate relative mRNA expression as fold change compared to serum-free conditions. The data are an average of three biological replicates ± SEM. A t-test analysis was performed in which the IGF-I treated group was compared to the serum-free group (* p<0.05). B) MCF7 cells were either maintained in serum-free medium or treated with IGF-I (100ng/mL) for 0, 12, 24 or 48 hours. Cells were then lysed, protein was harvested, followed by western blot analysis as indicated. C) MCF7 cells were either maintained in serum-free medium or treated with 10ng/mL, 50ng/mL or 100ng/mL of IGF-I for 0, 12, 24 or 48 hours. Cells were then lysed, protein was harvested, followed by western blot analysis as indicated. D) MCF7 cells were either maintained in the presence of cycloheximide (CHX) alone, or CHX in combination with IGF-I (100ng/mL) for 0, 12, 24 or 48hours. Cells were lysed and protein was harvested followed by western blot analysis. Blots represent prototypical examples of experiments replicated at least three times. Quantitative data (OD, optical density) for D is shown in the right panel. E) BT20 cells were either maintained in the presence of cycloheximide (CHX) alone, or CHX in combination with IGF-I (100ng/mL) for 0, 12, 24 or 48hours. Cells were lysed and protein was harvested followed by western blot analysis. Quantitative data (OD, optical density) for E is shown in the right panel.
IGF-I-mediated stabilization of FOXA1 is dependent on activation of IGF-I canonical signal transduction through both AKT and MAPK
The two canonical downstream signaling cascades involved in IGF-I signal transduction result in activation of both MAPK and AKT. Therefore, we assessed the role of these signaling pathways in the IGF-I-mediated stabilization of FOXA1. To accomplish this MCF7 cells were maintained in serum-free medium overnight and then pre-treated with CHX or inhibitor plus CHX for 30 minutes. Inhibitors used consisted of the MAPK inhibitor U0126 (10µM), the PI3K inhibitor LY294002 (20µM) and the PKC inhibitor Gö6983 (.025µM). Following the 30 minute pre-treatment, cells were either maintained in CHX alone or administered IGF-I (100ng/mL), in the presence of CHX or CHX plus the inhibitor. In the absence of any inhibitor, IGF-I stabilized the expression of FOXA1 protein. However, inhibition of MAPK prevented IGF-I treatment from stabilizing the expression of FOXA1 protein (Fig. 4A). Furthermore, blockade of PI3K activation, with LY294002, also prevented the ability of IGF-I to stabilize the expression level of FOXA1 protein (Fig. 4B). Moreover, an AKT-specific inhibitor, 124005, also attenuated IGF-I-mediated stabilization of FOXA1 protein expression, further confirming a role for AKT in the IGF-regulated stabilization of FOXA1 (Fig. S3). Conversely, attenuation of PKC activation had no effect on the ability of IGF-I to stabilize the expression of FOXA1 (Fig. 4C). Cells treated with PKC inhibitor revealed a level of FOXA1 expression that overlapped with the expression of FOXA1 in cells that had been treated with IGF-I and CHX in the absence of the inhibitor. These experiments do not rule out all possible signaling cascades involved in IGF-I signal transduction, but it does address the major downstream pathways involved. The data from these experiments suggest that the IGF-I-induced stabilization of FOXA1 is dependent on IGF-I canonical activation of MAPK and AKT, but does not directly involve activation of PKC.
Figure 4. IGF-I increases the half-life of FOXA1 protein by signaling through AKT and ERK 1/2.
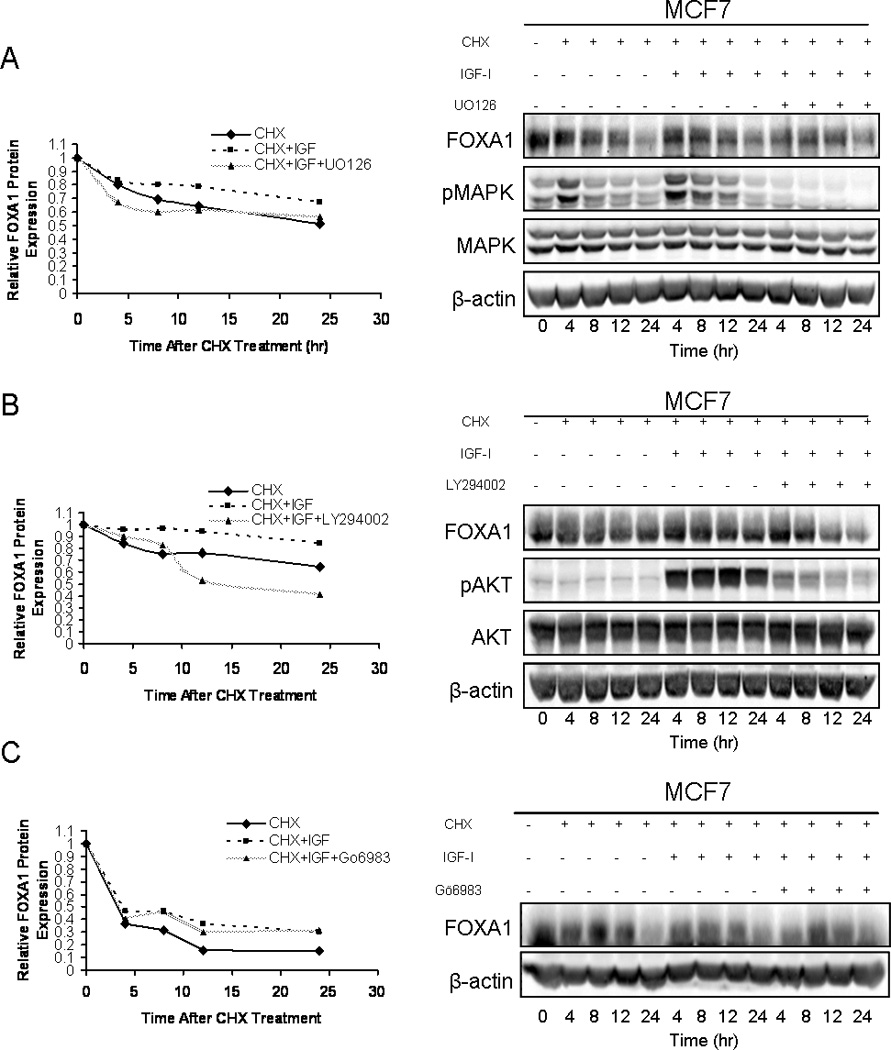
MCF7 cells were either treated with cycloheximide (CHX)(20ng/mL), CHX in combination with IGF-I (100ng/mL), or CHX, IGF-I and the MAPK inhibitor U0126 (10µM) (A), PI3K inhibitor LY294002 (20µM) (B), or the PKC inhibitor Gö6983 (0.25µM) (C). Cells were then lysed, protein was harvested and western blot analysis was performed as indicated. Blots represent prototypical examples of experiments replicated at least three times. Quantitative data (OD, optical density) for A, B and C is shown in the left panels.
FOXA1 is necessary for IGF-I-induced cell proliferation and cell survival in MCF7 cells
In light of the importance of FOXA1 in the ability of IGF-I to regulate gene expression, and the ability of IGF-I to regulate the stability of FOXA1, we sought to determine the importance of FOXA1 in IGF-I-mediated biological functions. We transiently transfected MCF7 cells with either non-specific pooled siRNA or pooled siRNA targeted to FOXA1. Following knock down of FOXA1 expression, cells were maintained in serum-free medium overnight and subsequently treated with IGF-I (100ng/mL) the following day. The cells were maintained in either serum-free medium or in the presence of IGF-I for seven days, with fresh IGF-I being added every 3 days, and the rate of proliferation was measured via an MTS assay, with readings being taken at 0, 2, 5 and 7 days. As expected, cells transiently transfected with non-specific siRNA and subsequently treated with IGF-I showed an increase in proliferation versus cells transiently transfected with non-specific siRNA and maintained in serum-free medium. Conversely, cells transiently transfected with siRNA targeting FOXA1 and treated with IGF-I showed a significantly decreased induction of proliferation when compared with cells transiently transfected with siRNA targeted to FOXA1 and maintained in serum-free medium (Fig. 5A). This data suggests that there is a necessary role for FOXA1 in the ability of IGF-I to induce cell proliferation. At day 7 the rate of proliferation induced by IGF-I in the cells in which FOXA1 had been transiently knocked down was increasing at an accelerated rate, but this is most likely a consequence of the transient nature of the FOXA1 knockdown and that the efficacy of the siRNA may be diminishing over time.
Figure 5. FOXA1 plays a crucial role in IGF-I functional biology.
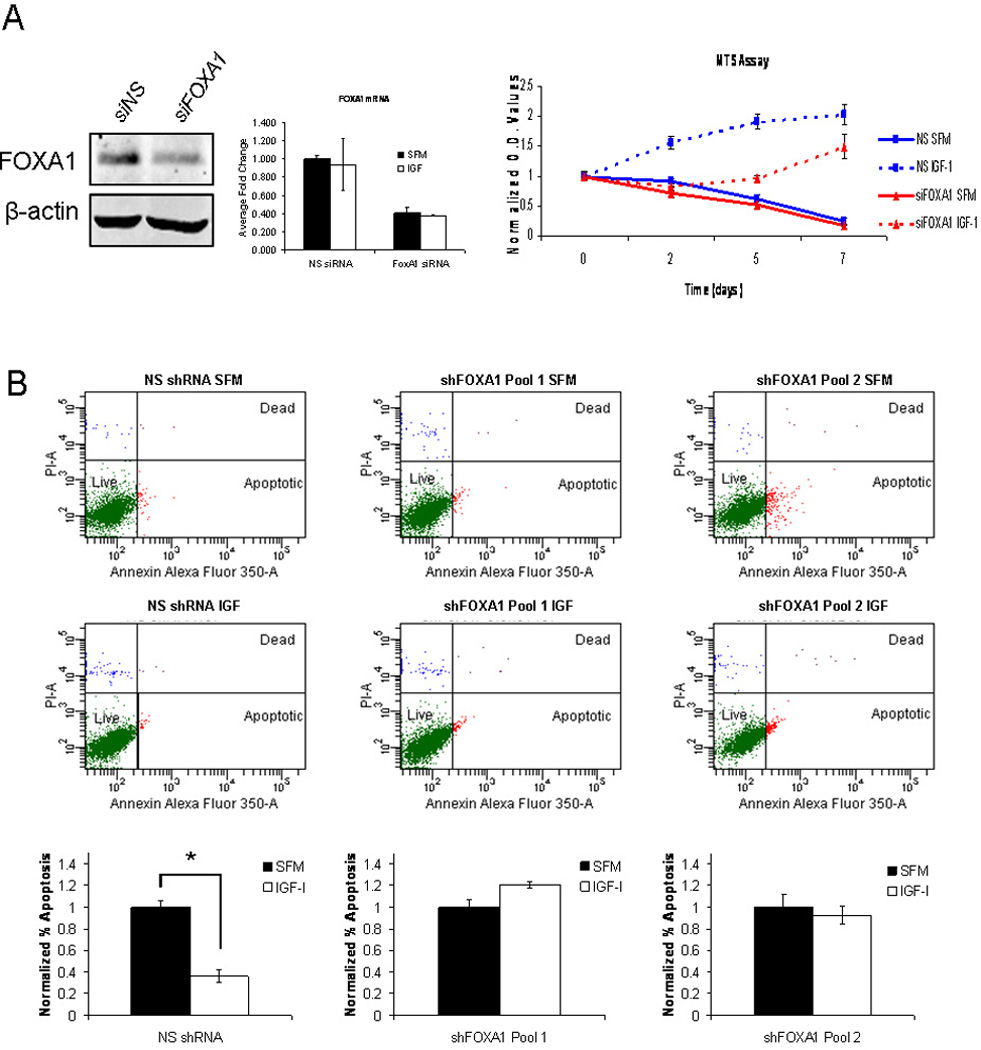
A) MCF7 cells were transiently transfected with FOXA1 siRNA (red lines) or non-specific siRNA (blue lines) and incubated in either serum-free medium (solid lines) or medium containing IGF-I (100ng/mL) (dashed lines). Cell growth was assessed by MTS assay on days 0, 2, 5 and 7. The assay was performed with biological septuplicates and each point represents the average value ± SEM. B) Cells stably expressing either non-specific shRNA or one of two shRNAs targeted to FOXA1 were cultured in serum-free medium and then either maintained in serum-free medium or treated with IGF-I (100ng/mL) for 4 days. Apoptosis was determined using an Annexin V apoptosis assay. The data are an average of three biological replicates ± SEM. A t-test analysis was performed in which normalized percentage of apoptosis in serum-free conditions was compared to IGF-I treated conditions for non-specific shRNA, shFOXA1 clone1 and shFOXA1 clone2 (* p<0.05).
We also wanted to assess the role of FOXA1 in the ability of IGF-I to protect cells against the induction of apoptosis. To accomplish this we created three different cell lines, one stably expressing a non-targeting shRNA and two that stably expressed two different shRNAs targeted to FOXA1 (shFOXA1 clone 1 and shFOXA1 clone 2), which resulted in a stable knockdown of FOXA1 expression (Fig. S2). These cells were either maintained in serum-free medium or treated with IGF-I (100ng/mL) for 4 days and then stained with Annexin V Alexa Fluor 350 and propidium iodide (PI), cell sorted and assessed for percentage of apoptotic cells. In cells stably expressing non-targeting shRNA, IGF-I stimulation led to a significant decrease in the percentage of apoptotic cells when compared to cells maintained in serum-free medium. Interestingly, in the two cell lines that stably expressed shRNA targeted to FOXA1, IGF-I stimulation was unable to decrease the percentage of apoptotic cells compared to cells maintained in serum-free medium (Fig. 5B). The reduced percentage of apoptosis following stable knockdown of FOXA1 was observed for both stable cell lines, which express two distinct shRNAs. Together, this data suggests that FOXA1 is a necessary component in the ability of IGF-I to induce a cell survival mechanism in vitro.
DISCUSSION
Currently, there have been no reports of a regulatory relationship between IGF-I and FOXA1, but previous studies have shown that insulin signaling can regulate members of the forkhead box family including FOXA1, FOXA2, FOXO1 and FOXO3a [Duncan et al., 1998; Howell and Stoffel, 2009; Matsuzaki et al., 2003; Ogg et al., 1997; Wolfrum et al., 2003]. Additionally, IGF-I has been shown to regulate forkhead member FOXO3a [Wilk et al., 2011]. Therefore, we predicted that FOXA1 may be important in the regulation of IGF-I target genes and that IGF-I may be able to regulate FOXA1, due to the redundancy in insulin and IGF-I action and signal transduction pathways [Pollak, 2008; Zapf et al., 1984].
Previous studies have shown that insulin and IGF-I have the ability to regulate the transcriptional activity of forkhead family members, resulting in either activation or repression of forkhead target genes [Duncan et al., 1998; Jag et al., 2009; Wilk et al., 2011; Wolfrum et al., 2003]. In this study, we have reported a novel role for FOXA1 as a mediator of IGF-I-regulated target gene expression (Fig. 1). Through in silico analysis, we showed that 38% of IGF-I-regulated genes (strongly induced or repressed) were enriched for FOXA1 binding sites. Moreover, transient knock down of FOXA1 in MCF7 cells revealed that both the IGF-I-mediated repression, as well as the IGF-I-mediated induction of target genes was inhibited. Furthermore, two of the IGF-I-repressed genes that were shown to require FOXA1, SOCS2 and BLNK, have been shown to be tumor suppressor genes and two of the IGF-induced genes that were shown to require FOXA1, FHL2 and MAFF. are involved in cell division and gene transcription, respectively [Blank, 2008; Flemming et al., 2003; Haffner et al., 2007; Martin et al., 2007]. These results confirmed a necessary role for FOXA1 in the IGF-I-mediated regulation of target genes that are involved in proliferation and survival. Interestingly, in BT20 cells, knockdown of FOXA1 only relieved IGF-I-mediated repression of target genes, but had no effect on IGF-I-induced target genes. Conversely, in MDA-MB-134 cells, knockdown of FOXA1 had no consistent effect on either IGF-I-induced or repressed target genes (Fig. S1). Taken together, the data from these cell lines suggests that the need for FOXA1 in IGF-I-mediated regulation of target genes may be cell line dependent. This may further be due to the availability of additional transcriptional co-regulators that work in conjunction with FOXA1 which may not be expressed at sufficient levels, or at all, in specific cell lines. While the evidence from our studies supports a role for FOXA1, it does not rule out the involvement of co-regulators or other transcription factors, such as ERα, which have previously been shown to be regulated by IGF-I [Becker et al., 2011]. Although many transcription factors act as either an activator or repressor of transcription, FOXA1 has been shown to act as both an activator and a repressor of transcription. The ability of FOXA1 to act as a repressor has been shown to be mediated by insulin, which shares a great deal of overlap with IGF-IR signal transduction, suggesting that IGF-I-signal transduction may be able to result in FOXA1 functioning as a transcriptional repressor [Carroll et al., 2005; Cirillo et al., 2002; Duncan et al., 1998; Pollak, 2008; Zapf et al., 1984]. Our data suggest that FOXA1 can indeed serve as a repressor as well as an activator of gene transcription in the presence of IGF-I. This dual functionality is most likely regulated by varying recruitment of either transcriptional co-activators or co-repressors, although establishing which co-regulators are involved in this process remains to be elucidated.
Members of the forkhead family have been shown in previous studies, to undergo various post-translational modifications resulting in diverse functions. A previous study showed that FOXA1 can undergo acetylation which attenuates the binding of FOXA1 to its regulatory elements [Kohler and Cirillo, 2010]. Additionally, IGF-I has been shown to phosphorylate FOXO3a, through activation of AKT, resulting in the inhibition of its transcriptional activity and insulin has been shown to induce phosphorylation of FOXO1 resulting in proteasomal degradation [Matsuzaki et al., 2003; Zheng et al., 2000]. Therefore, we sought to determine whether IGF-I had the ability to regulate FOXA1. We found that IGF-I had no inductive or repressive effect on FOXA1 mRNA expression, but rather stabilized FOXA1 protein expression. This was true in both MCF7, and to a lesser extent, BT20 breast cancer cell lines. Furthermore, in MCF7 cells, the stabilization of FOXA1 was shown to be IGF-I-specific. This was evident by the ability of IGF-I to stabilize FOXA1 at a physiological concentration of 100ng/mL (~12.5nM), to a lesser degree at 50 ng/mL (~6.3nM) and IGF-I was unable to stabilize FOXA1 at 10ng/mL (Fig. 3). Together, these data suggest that IGF-I is capable of post-transcriptionally, and more than likely, post-translationally regulating FOXA1 protein expression. The IGF-I-mediated stabilization of FOXA1 was accomplished through canonical IGF-I-induced activation of MAPK and AKT (Fig. 4 & S3). This IGF-I activation of MAPK and AKT may lead to phosphorylation of FOXA1, which can then serve as a signal for other molecules to post-translationally modify FOXA1, resulting in its increased half-life. Insulin signaling has been shown to lead to phosphorylation of FOXO1, through PI3K activation, which leads to ubiquitination and subsequent proteasomal degradation [Matsuzaki et al., 2003]. While our proposed mechanism (Fig. 6) results in a prolonged half-life and not a more rapid degradation, the principle behind the mechanism may be the same, but is yet to be established.
Figure 6. A mechanism detailing the role of FOXA1 in IGF-I Activity.
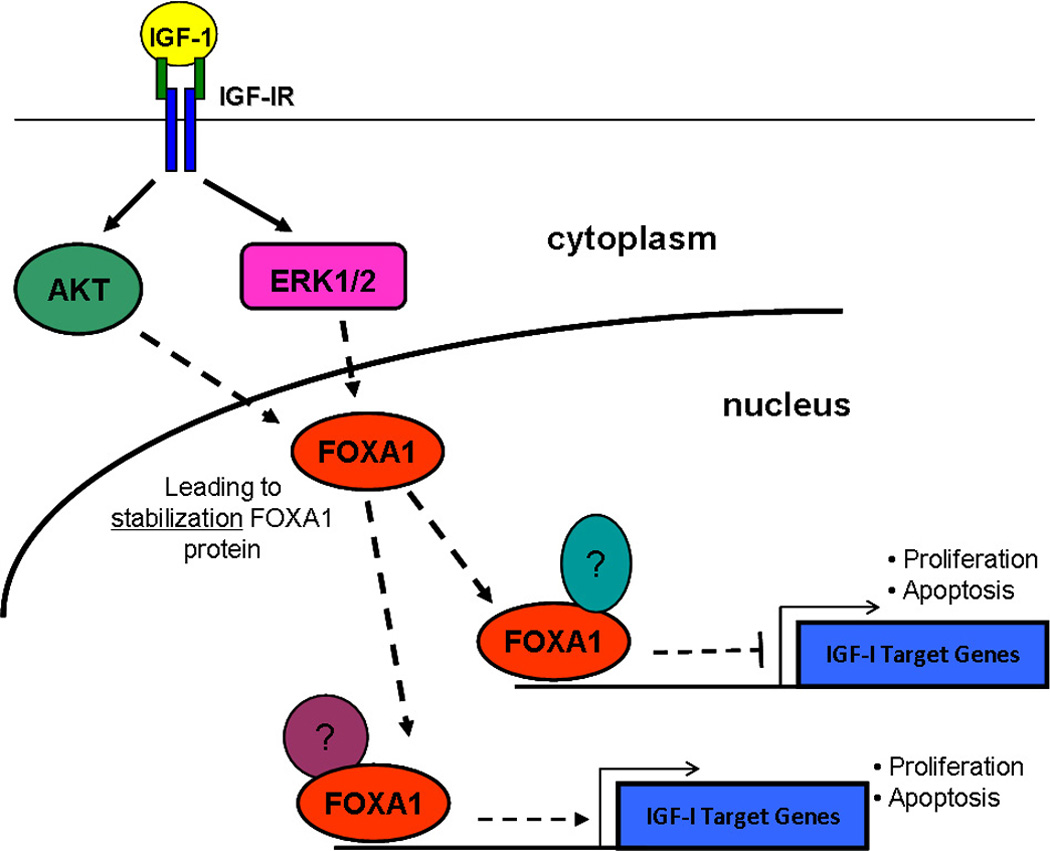
IGF-I treatment of MCF7 cells leads to an increase in the half-life of FOXA1 protein, via activation of MAPK and AKT. FOXA1 is also a crucial component of the IGF-I-mediated regulation of target genes as well as IGF-I-induced cellular proliferation and cell survival.
Cellular proliferation and protection against cell death are the two predominant biological consequences of IGF-I signal transduction [Chitnis et al., 2008]. IGF-I target genes have been shown to be involved in promoting the cell cycle and cell division [Creighton et al., 2008]. Additionally, FOXA1 has been shown to be crucial for estrogen-mediated proliferation in MCF7 cells [Carroll et al., 2005; Laganiere et al., 2005]. Therefore we assessed the importance of FOXA1 in IGF-I-mediated cellular proliferation. We observed a significant decrease in the ability of IGF-I to induce cellular proliferation in MCF7 cells following transient knockdown of FOXA1 (Fig 5A). This suggests that not only is FOXA1 a crucial component in the ability of IGF-I to regulate target genes, but that there are far reaching biological consequences of this role for FOXA1 in IGF-I signal transduction. It has also been previously reported that in vivo loss of FOXA1 in mammary epithelial cells leads to an increase in apoptosis during mammary gland development [Bernardo et al.]. It is reasonable to hypothesize that FOXA1 may also be important in regulating apoptosis in transformed mammary epithelial cells; therefore, we sought to determine if there was a role for FOXA1 in the cell survival mechanism induced by IGF-I in MCF7 cells (Fig. 5B). We were able to confirm that IGF-I was able to protect against induction of apoptosis, but following stable knockdown of FOXA1, this IGF-I-induced protective effect was lost. These data suggest a crucial and necessary role for FOXA1 in IGF-I functional biology.
FOXA1 has been shown to be important in breast cancer as well as mammary gland development, two areas where IGF-IR has also been shown to be highly relevant [Badve et al., 2007; Bernardo et al., 2010; Bonnette and Hadsell, 2001; Law et al., 2008; Pollak, 2008; Stratikopoulos et al., 2008]. A regulatory relationship between the two has never been established, but we now describe a necessary and crucial role for FOXA1 in IGF-I-regulation of target genes, as well as IGF-I functional biology. Furthermore, we also report that IGF-I is capable of stabilizing the expression of FOXA1 by activation of canonical IGF-I signal transduction cascades. This stabilization may lead to an increased ability for FOXA1 to act as a transcriptional regulator of IGF-I target genes, ultimately affecting the biological output of IGF-I signal transduction. Moreover, previous data has shown a ligand-dependent interaction between FOXA1 and ERα, showing that FOXA1 is capable of interacting with other transcription factors to regulate gene expression [Schuur et al., 2001]. Therefore, it led us to propose that there may be particular co-regulators that FOXA1 works in conjunction with to either induce gene expression or repress gene expression, even though we currently have no data to support this hypothesis (Fig. 6). With the emerging importance of IGF-IR as a target for cancer therapy, it becomes ever more important to better understand the nuances of IGF-I signal transduction and the mechanisms involved in IGF-I-mediated regulation of target genes. Establishing key mediators of this pathway could lead to development of other targeted therapies that may be able to be used in conjunction with current IGF-IR targeted therapies as a treatment modality in various forms of human cancer.
Supplementary Material
BT20 and MDA134 cells were transfected with non-specific siRNA (NS siRNA) or siRNA against FOXA1 (FOXA1 siRNA) followed by either maintenance in serum-free medium or stimulation with IGF-I (100ng/mL). Q-RT-PCR was used to calculate relative mRNA expression as fold change compared to serum-free conditions. The data are an average of three biological replicates ± SEM.
MCF7 cells were either maintained in serum-free medium or treated with IGF-I (100ng/mL) for 30, 60 or 240 minutes. Cells were then fixed, permeabilized and stained with an antibody directed against FOXA1. Following fluorescently-conjugated secondary antibody staining, confocal microscopy was performed.
MCF7 cells were either treated with cycloheximide (CHX)(20ng/mL), CHX in combination with IGF-I (100ng/mL), or CHX, IGF-I and the AKT specific inhibitor 124005 (5µM). Cells were then lysed, protein was harvested and western blot analysis was performed as indicated. Quantitative data (OD, optical density) is shown in the left panel.
RNA and protein was harvested from stable cell lines and subjected to q-RT-PCR and western blot analysis as indicated.
All sequences are listed in the 5’ – 3’ direction.
ACKNOWLEDGMENTS
This work was supported in part by the Training Program in Molecular Endocrinology (NIH DK07696)(AP), and grants from the National Institutes of Health R01CA94118 (AVL) and P30CA58183 (AVL). We would like to thank Dr. Michael T. Lewis and Dr. Larry Donehower for their help in reviewing this manuscript and Dr. Sue Hilsenbeck for her help with biostatistical analysis.
Footnotes
All work was performed at Baylor College of Medicine, Houston, Texas 77030
Conflict of Interest Notice
A.S.P, A.J.C and A.V.L. have no conflicts of interest.
REFERENCES
- Badve S, Turbin D, Thorat MA, Morimiya A, Nielsen TO, Perou CM, Dunn S, Huntsman DG, Nakshatri H. FOXA1 expression in breast cancer--correlation with luminal subtype A and survival. Clin Cancer Res. 2007;13:4415–4421. doi: 10.1158/1078-0432.CCR-07-0122. [DOI] [PubMed] [Google Scholar]
- Becker MA, Ibrahim YH, Cui X, Lee AV, Yee D. The IGF pathway regulates ERalpha through a S6K1-dependent mechanism in breast cancer cells. Mol Endocrinol. 2011;25:516–528. doi: 10.1210/me.2010-0373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardo GM, Lozada KL, Miedler JD, Harburg G, Hewitt SC, Mosley JD, Godwin AK, Korach KS, Visvader JE, Kaestner KH, Abdul-Karim FW, Montano MM, Keri RA. FOXA1 is an essential determinant of ERalpha expression and mammary ductal morphogenesis. Development. 2010;137:2045–2054. doi: 10.1242/dev.043299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blank V. Small Maf proteins in mammalian gene control: mere dimerization partners or dynamic transcriptional regulators? J Mol Biol. 2008;376:913–925. doi: 10.1016/j.jmb.2007.11.074. [DOI] [PubMed] [Google Scholar]
- Bonnette SG, Hadsell DL. Targeted disruption of the IGF-I receptor gene decreases cellular proliferation in mammary terminal end buds. Endocrinology. 2001;142:4937–4945. doi: 10.1210/endo.142.11.8500. [DOI] [PubMed] [Google Scholar]
- Carboni JM, Lee AV, Hadsell DL, Rowley BR, Lee FY, Bol DK, Camuso AE, Gottardis M, Greer AF, Ho CP, Hurlburt W, Li A, Saulnier M, Velaparthi U, Wang C, Wen ML, Westhouse RA, Wittman M, Zimmermann K, Rupnow BA, Wong TW. Tumor development by transgenic expression of a constitutively active insulin-like growth factor I receptor. Cancer Res. 2005;65:3781–3787. doi: 10.1158/0008-5472.CAN-04-4602. [DOI] [PubMed] [Google Scholar]
- Carroll JS, Liu XS, Brodsky AS, Li W, Meyer CA, Szary AJ, Eeckhoute J, Shao W, Hestermann EV, Geistlinger TR, Fox EA, Silver PA, Brown M. Chromosome-wide mapping of estrogen receptor binding reveals long-range regulation requiring the forkhead protein FoxA1. Cell. 2005;122:33–43. doi: 10.1016/j.cell.2005.05.008. [DOI] [PubMed] [Google Scholar]
- Carroll JS, Meyer CA, Song J, Li W, Geistlinger TR, Eeckhoute J, Brodsky AS, Keeton EK, Fertuck KC, Hall GF, Wang Q, Bekiranov S, Sementchenko V, Fox EA, Silver PA, Gingeras TR, Liu XS, Brown M. Genome-wide analysis of estrogen receptor binding sites. Nat Genet. 2006;38:1289–1297. doi: 10.1038/ng1901. [DOI] [PubMed] [Google Scholar]
- Chitnis MM, Yuen JS, Protheroe AS, Pollak M, Macaulay VM. The type 1 insulin-like growth factor receptor pathway. Clin Cancer Res. 2008;14:6364–6370. doi: 10.1158/1078-0432.CCR-07-4879. [DOI] [PubMed] [Google Scholar]
- Cirillo LA, Lin FR, Cuesta I, Friedman D, Jarnik M, Zaret KS. Opening of compacted chromatin by early developmental transcription factors HNF3 (FoxA) and GATA-4. Mol Cell. 2002;9:279–289. doi: 10.1016/s1097-2765(02)00459-8. [DOI] [PubMed] [Google Scholar]
- Creighton CJ, Casa A, Lazard Z, Huang S, Tsimelzon A, Hilsenbeck SG, Osborne CK, Lee AV. Insulin-like growth factor-I activates gene transcription programs strongly associated with poor breast cancer prognosis. J Clin Oncol. 2008;26:4078–4085. doi: 10.1200/JCO.2007.13.4429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dearth RK, Cui X, Kim HJ, Hadsell DL, Lee AV. Oncogenic transformation by the signaling adaptor proteins insulin receptor substrate (IRS)-1 and IRS-2. Cell Cycle. 2007;6:705–713. doi: 10.4161/cc.6.6.4035. [DOI] [PubMed] [Google Scholar]
- Duncan SA, Navas MA, Dufort D, Rossant J, Stoffel M. Regulation of a transcription factor network required for differentiation and metabolism. Science. 1998;281:692–695. doi: 10.1126/science.281.5377.692. [DOI] [PubMed] [Google Scholar]
- Flemming A, Brummer T, Reth M, Jumaa H. The adaptor protein SLP-65 acts as a tumor suppressor that limits pre-B cell expansion. Nat Immunol. 2003;4:38–43. doi: 10.1038/ni862. [DOI] [PubMed] [Google Scholar]
- Haffner MC, Petridou B, Peyrat JP, Revillion F, Muller-Holzner E, Daxenbichler G, Marth C, Doppler W. Favorable prognostic value of SOCS2 and IGF-I in breast cancer. BMC Cancer. 2007;7:136. doi: 10.1186/1471-2407-7-136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell JJ, Stoffel M. Nuclear export-independent inhibition of Foxa2 by insulin. J Biol Chem. 2009;284:24816–24824. doi: 10.1074/jbc.M109.042135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jag UR, Zavadil J, Stanley FM. Insulin acts through FOXO3a to activate transcription of plasminogen activator inhibitor type 1. Mol Endocrinol. 2009;23:1587–1602. doi: 10.1210/me.2008-0421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones RA, Campbell CI, Gunther EJ, Chodosh LA, Petrik JJ, Khokha R, Moorehead RA. Transgenic overexpression of IGF-IR disrupts mammary ductal morphogenesis and induces tumor formation. Oncogene. 2007;26:1636–1644. doi: 10.1038/sj.onc.1209955. [DOI] [PubMed] [Google Scholar]
- Kohler S, Cirillo LA. Stable chromatin binding prevents FoxA acetylation, preserving FoxA chromatin remodeling. J Biol Chem. 2010;285:464–472. doi: 10.1074/jbc.M109.063149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laganiere J, Deblois G, Lefebvre C, Bataille AR, Robert F, Giguere V. From the Cover: Location analysis of estrogen receptor alpha target promoters reveals that FOXA1 defines a domain of the estrogen response. Proc Natl Acad Sci U S A. 2005;102:11651–11656. doi: 10.1073/pnas.0505575102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law JH, Habibi G, Hu K, Masoudi H, Wang MY, Stratford AL, Park E, Gee JM, Finlay P, Jones HE, Nicholson RI, Carboni J, Gottardis M, Pollak M, Dunn SE. Phosphorylated insulin-like growth factor-i/insulin receptor is present in all breast cancer subtypes and is related to poor survival. Cancer Res. 2008;68:10238–10246. doi: 10.1158/0008-5472.CAN-08-2755. [DOI] [PubMed] [Google Scholar]
- LeRoith D, Roberts CT., Jr The insulin-like growth factor system and cancer. Cancer Lett. 2003;195:127–137. doi: 10.1016/s0304-3835(03)00159-9. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Maor S, Mayer D, Yarden RI, Lee AV, Sarfstein R, Werner H, Papa MZ. Estrogen receptor regulates insulin-like growth factor-I receptor gene expression in breast tumor cells: involvement of transcription factor Sp1. J Endocrinol. 2006;191:605–612. doi: 10.1677/joe.1.07016. [DOI] [PubMed] [Google Scholar]
- Martin BT, Kleiber K, Wixler V, Raab M, Zimmer B, Kaufmann M, Strebhardt K. FHL2 regulates cell cycle-dependent and doxorubicin-induced p21Cip1/Waf1 expression in breast cancer cells. Cell Cycle. 2007;6:1779–1788. doi: 10.4161/cc.6.14.4448. [DOI] [PubMed] [Google Scholar]
- Matsuzaki H, Daitoku H, Hatta M, Tanaka K, Fukamizu A. Insulin-induced phosphorylation of FKHR (Foxo1) targets to proteasomal degradation. Proc Natl Acad Sci U S A. 2003;100:11285–11290. doi: 10.1073/pnas.1934283100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogg S, Paradis S, Gottlieb S, Patterson GI, Lee L, Tissenbaum HA, Ruvkun G. The Fork head transcription factor DAF-16 transduces insulin-like metabolic and longevity signals in C. elegans. Nature. 1997;389:994–999. doi: 10.1038/40194. [DOI] [PubMed] [Google Scholar]
- Pollak M. Insulin and insulin-like growth factor signalling in neoplasia. Nat Rev Cancer. 2008;8:915–928. doi: 10.1038/nrc2536. [DOI] [PubMed] [Google Scholar]
- Schuur ER, Loktev AV, Sharma M, Sun Z, Roth RA, Weigel RJ. Ligand-dependent interaction of estrogen receptor-alpha with members of the forkhead transcription factor family. J Biol Chem. 2001;276:33554–33560. doi: 10.1074/jbc.M105555200. [DOI] [PubMed] [Google Scholar]
- Sell C, Rubini M, Rubin R, Liu JP, Efstratiadis A, Baserga R. Simian virus 40 large tumor antigen is unable to transform mouse embryonic fibroblasts lacking type 1 insulin-like growth factor receptor. Proc Natl Acad Sci U S A. 1993;90:11217–11221. doi: 10.1073/pnas.90.23.11217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sircoulomb F, Bekhouche I, Finetti P, Adelaide J, Ben Hamida A, Bonansea J, Raynaud S, Innocenti C, Charafe-Jauffret E, Tarpin C, Ben Ayed F, Viens P, Jacquemier J, Bertucci F, Birnbaum D, Chaffanet M. Genome profiling of ERBB2-amplified breast cancers. BMC Cancer. 2010;10:539. doi: 10.1186/1471-2407-10-539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stratikopoulos E, Szabolcs M, Dragatsis I, Klinakis A, Efstratiadis A. The hormonal action of IGF1 in postnatal mouse growth. Proc Natl Acad Sci U S A. 2008;105:19378–19383. doi: 10.1073/pnas.0809223105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surmacz E. Function of the IGF-I receptor in breast cancer. J Mammary Gland Biol Neoplasia. 2000;5:95–105. doi: 10.1023/a:1009523501499. [DOI] [PubMed] [Google Scholar]
- Werner H, Bruchim I. The insulin-like growth factor-I receptor as an oncogene. Arch Physiol Biochem. 2009;115:58–71. doi: 10.1080/13813450902783106. [DOI] [PubMed] [Google Scholar]
- Weroha SJ, Haluska P. IGF-1 receptor inhibitors in clinical trials--early lessons. J Mammary Gland Biol Neoplasia. 2008;13:471–483. doi: 10.1007/s10911-008-9104-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilk A, Urbanska K, Yang S, Wang JY, Amini S, Del Valle L, Peruzzi F, Meggs L, Reiss K. Insulin-like growth factor-I-forkhead box O transcription factor 3a counteracts high glucose/tumor necrosis factor-alpha-mediated neuronal damage: implications for human immunodeficiency virus encephalitis. J Neurosci Res. 2011;89:183–198. doi: 10.1002/jnr.22542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfrum C, Besser D, Luca E, Stoffel M. Insulin regulates the activity of forkhead transcription factor Hnf-3beta/Foxa-2 by Akt-mediated phosphorylation and nuclear/cytosolic localization. Proc Natl Acad Sci U S A. 2003;100:11624–11629. doi: 10.1073/pnas.1931483100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zapf J, Schmid C, Froesch ER. Biological and immunological properties of insulin-like growth factors (IGF) I and II. Clin Endocrinol Metab. 1984;13:3–30. doi: 10.1016/s0300-595x(84)80006-7. [DOI] [PubMed] [Google Scholar]
- Zheng WH, Kar S, Quirion R. Insulin-like growth factor-1-induced phosphorylation of the forkhead family transcription factor FKHRL1 is mediated by Akt kinase in PC12 cells. J Biol Chem. 2000;275:39152–39158. doi: 10.1074/jbc.M002417200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
BT20 and MDA134 cells were transfected with non-specific siRNA (NS siRNA) or siRNA against FOXA1 (FOXA1 siRNA) followed by either maintenance in serum-free medium or stimulation with IGF-I (100ng/mL). Q-RT-PCR was used to calculate relative mRNA expression as fold change compared to serum-free conditions. The data are an average of three biological replicates ± SEM.
MCF7 cells were either maintained in serum-free medium or treated with IGF-I (100ng/mL) for 30, 60 or 240 minutes. Cells were then fixed, permeabilized and stained with an antibody directed against FOXA1. Following fluorescently-conjugated secondary antibody staining, confocal microscopy was performed.
MCF7 cells were either treated with cycloheximide (CHX)(20ng/mL), CHX in combination with IGF-I (100ng/mL), or CHX, IGF-I and the AKT specific inhibitor 124005 (5µM). Cells were then lysed, protein was harvested and western blot analysis was performed as indicated. Quantitative data (OD, optical density) is shown in the left panel.
RNA and protein was harvested from stable cell lines and subjected to q-RT-PCR and western blot analysis as indicated.
All sequences are listed in the 5’ – 3’ direction.