

Summary
Aims
Stroke causes both brain inflammation and immunodepression. Mild‐to‐moderate hypothermia is known to attenuate brain inflammation, but its role in stroke‐induced immunodepression (SIID) of the peripheral immune system remains unknown. This study investigated the effects in rats of moderate intra‐ischemic hypothermia on SIID and brain inflammation.
Methods
Stroke was induced in rats by permanent distal middle cerebral artery occlusion combined with transient bilateral common carotid artery occlusion, while body temperature was reduced to 30°C. Real‐time PCR, flow cytometry, in vitro T‐cell proliferation assays, in vivo delayed‐type hypersensitivity (DTH) reaction and confocal microscopy were used to study SIID and brain inflammation.
Results
Brief intra‐ischemic hypothermia helped maintain certain leukocytes in the peripheral blood and spleen and enhanced T‐cell proliferation in vitro and delayed‐type hypersensitivity in vivo, suggesting that hypothermia reduces SIID. In contrast, in the brain, brief intra‐Ischemic hypothermia inhibited mRNA expression of anti‐inflammatory cytokine IL‐10 and proinflammatory mediators INF‐γ, TNF‐α, IL‐2, IL‐1β and MIP‐2. Brief intra‐Ischemic hypothermia also attenuated the infiltration of lymphocytes, neutrophils (MPO + cells) and macrophages (CD68+ cells) into the ischemic brain, suggesting that hypothermia inhibited brain inflammation.
Conclusions
Brief intra‐ischemic hypothermia attenuated SIID and protected against acute brain inflammation.
Keywords: Focal cerebral ischemia, Hypothermia, Immunodepression, Inflammation, Leukocytes
Introduction
Mild (32–33°C) and moderate (28–30°C) hypothermia have been shown to improve neurologic function in patients suffering cardiac arrest from ventricular fibrillation 1, 2 and to reduce mortality and disability in neonate hypoxic–ischemic encephalopathy 3, 4. Although hypothermia's effectiveness for focal stroke treatment remains unproven, several clinical trials have shown the feasibility of applying hypothermia to ischemic stroke 5, 6. A critical barrier to clinical translation, however, is the potential exacerbation of stroke‐induced immunodepression (SIID).
Stroke‐induced immunodepression is a major cause of death in stroke patients. It is characterized by T‐cell depletion in peripheral blood, an inhibited T‐cell response to antigen stimulation, and a suppressed response to the delayed‐type hypersensitivity (DTH) skin test 7, 8, 9, 10, 11, 12. SIID research dates back 40 years 13 when it was found that death after stroke was frequently associated with non‐neurologic diseases such as pneumonia, pulmonary embolism, and urinary tract infections. Only recently has stroke treatment research shifted focus from brain injury to SIID and its underlying mechanisms in the peripheral immune organs 8, 9, 10, 12, 14.
Induced mild‐to‐moderate hypothermia in the ischemic brain inhibits local inflammation 15, 16, 17 and is believed to contribute to neuroprotection 18. Hypothermia by itself is known to cause immunosuppression 19, 20, but its effect on SIID deserves further study. In fact, SIID in animals coexists with local brain inflammation, as brain inflammation occurs immediately after stroke while SIID follows a few hours later and peaks at 2–3 days 9. Moreover, the degree of SIID correlates with the severity of brain inflammation and infarct size 21. The cause and effect relationship between brain inflammation and SIID is not clearly understood, but it is likely that SIID is a compensatory response of the immune system to reduce brain inflammation and thus neuronal injury. We investigated the effects of brief intra‐ischemic hypothermia on SIID and brain inflammation and found that it is safe, ameliorates brain inflammation and attenuates SIID.
Materials and methods
Focal Ischemia and Induced Hypothermia
All protocols were approved by the Stanford Institutional Animal Care and Use Committee and conducted according to the NIH Guide for Care and Use of Laboratory Animals. Experiments were designed and conducted based on STAIR protocols criteria or suggestions when applicable 22. Focal cerebral ischemia in male Sprague Dawley rats (280–320 g) was generated as described previously 23, 24. Animals were randomly assigned to different groups. Anesthesia was induced by 5% isoflurane and maintained with 2–3% isoflurane during surgery. Core body temperatures were monitored with a rectal probe and were maintained at either 37 or 30°C throughout the experiment. The distal middle cerebral artery (MCA) was exposed and cauterized permanently above the rhinal fissure. The bilateral common carotid arteries (CCA) were occluded for 1 h. Moderate hypothermia (30°C) was induced by spraying 100% alcohol on the rat body; temperature was controlled by a heating pad underneath the rat combined with an overhead light above the rat, as described previously 23. Body temperature was adjusted to 30 ± 0.5°C (at 10 min before ischemia onset) and maintained for 1 h during CCA occlusion. After CCA release, the wound was sutured and the body temperature was increased to 37 ± 0.5°C 23.
Infarct Volume Assessment
Infarct volume was evaluated 72 h post‐stroke as described previously 25, 26. Brains were sectioned coronally into five slices, stained with 2% 2,3,5‐triphenyletrazolium chloride (TTC) for 30 min and fixed in 4% paraformaldehyde overnight. The infarct area in each section was measured using the National Institutes of Health Image program (Image J 1.37v, National Institutes of Health, Bethesda, MD, USA) by a blinded observer. Infarction volume is the sum of infarction areas multiplied by slice thickness. To correct for edema, infarct volume was normalized and expressed as a percentage of the contralateral hemisphere according to the formula: {area of contralateral cortex − (area of contralateral cortex − [area of ipsilateral cortex − area of infarct cortex])/area of contralateral cortex × 100%}, as described in our previous study 23.
Quantitative RT‐PCR
Real‐time reverse transcription PCR (QRT‐PCR) was used to determine mRNA expression levels of cytokines, chemokines, and other inflammatory mediators in the ischemic brain. Rats were perfused transcardially with 200‐mL cold phosphate‐buffered saline (PBS, pH = 7.2). Ischemic brains were collected 5, 24, and 72 h after stroke onset. Total mRNA was extracted from the tissues using the TRIzol reagent (Invitrogen, Carlsbad, CA, USA), and 2 μg of total mRNA was reverse‐transcribed using a RT‐PCR kit (SuperScript™ III First‐Strand Synthesis System for RT‐PCR) with Oligo‐dT primer according to the manufacturer's instructions (Invitrogen). Quantitative real‐time PCR was performed using SYBR‐GreenER™ qPCR SuperMix for the iCycler (Invitrogen). Reactions were performed in triplicate in a spectrofluorometric thermal cycler (iCycler, iQ5 Multicolor Real‐time PCR Detection; Bio‐Rad Laboratories, Hercules, CA, USA). Gene‐specific primers were the following: IL‐1β, forward: 5′‐CACCTCTCAAGCAGAGCACAG‐3′ and reverse: GGGTTCCATGGTGAAGTCAAC; TNF‐α, forward: 5′‐AAATGGGCTCCCTCTCATCATCAGTTC‐3 and reverse: TCTGCTTGGTGGTTTGCTACGAC; IL‐4, forward: 5′‐GCAACAAGGAACACCACGG‐3 and reverse: AAGCACGGAGGTACATCACGT; IL‐10, forward: 5′‐CAGTCAGCCAGACCCACAT‐3 and reverse: GCTCCACTGCCTTGCTTT; IFN‐γ, forward: 5′‐CACGCCGCGTCTTGGT‐3 and reverse: TCTAGGCTTTCAATGAGTGTGCC; MIP‐2, forward: 5′‐TGCCTGACGACCCTACCAA‐3 and reverse: TCACCGTCAAGCTCTGGATGT; COX‐2, forward: 5′‐AAGGGAGTCTGGAACATTGTGAAC‐3 and reverse: CAAATGTGATCTGGACGTCAACA; iNOS, forward: 5′‐GCATCCCAAGTACGAGTGGT‐3 and reverse: GAAGTCTCGGACTCCAATCTC; GAPDH, forward: 5′‐GTATTGGGCGCCTGGTCACC‐3 and reverse: CGCTCCTGGAAGATGGTGATGG. GAPDH was amplified in parallel as the internal control. Each reaction volume was 25 μL. PCRs were performed at 50°C for 2 min, 95°C for 8 min, followed by 40 cycles of 95°C for 15 seconds, and 60°C for 1 min. Data were collected and analyzed with the provided application software (Bio‐Rad iQ5; Bio‐Rad Laboratories). mRNA expression levels are expressed as folds compared with the mRNA levels in naive animals.
Isolation of Mononuclear Leukocytes from the Brain, Blood, and Spleen
To determine how hypothermia affects the population of mononuclear leukocytes in the brain and peripheral organs, rats were deeply anesthetized, and heparinized peripheral blood was collected 72 h after stroke onset. The rats were then perfused transcardially with 200‐mL cold PBS, and the brain and spleen were harvested. Mononuclear leukocytes were isolated from these organs, which are briefly described later. (1) Peripheral blood mononuclear cells (PBMCs) were isolated using the Ficoll‐Paque PLUS kit (GE Healthcare, Pittsburgh, PA, USA) according to the manufacturer's instructions. In brief, 5 mL of whole blood cell was diluted with RPMI 1640 and layered onto the Ficoll‐Paque PLUS solution. Cells were centrifuged at 500 g for 30 min at RT with the brake off, then aspirated from the interphase, transferred to a new tube, and washed with RPMI 1640. Cells were resuspended in RPMI 1640 and counted cells using the method of trypan blue exclusion. (2) Brain mononuclear leukocytes were collected as described by Lim et al. 27. Both ipsilateral and contralateral cortices were dissected and homogenized with the FACS buffer (F‐PBS, PBS with 1% FBS, and 0.1% sodium azide). After centrifugation, the cells were resuspended in 7 mL FACS buffer, completely mixed with 3 mL 90% Percoll, and 1 mL of 70% Percoll (in PBS) was loaded under the cell suspension. The cell suspension was then centrifuged at 600 g for 30 min at 4°C, and leukocytes at the interphase were collected for FACS analysis. (3) To isolate splenic mononuclear leukocytes, spleens were chopped into small pieces, homogenized, filtered through a 70‐μm strainer, and centrifuged at 150 g for 5 min. After red blood cells were lysed using ACK Lysing buffer (GIBCO, Invitrogen, Carlsbad, CA, USA), the cells were resuspended in RPMI1640 medium for FACS analyses.
FACS Analysis of Leukocyte Subsets
We used three‐color (FITC, PE, PerCP) fluorescence flow cytometry analyses to phenotype leukocytes as described previously 28. In brief, cell suspensions were placed into a FACS tube and washed with F‐PBS (PBS with 1% FCS and 0.1% NaN3). All labeling procedures were carried out on ice. Conjugated antibodies TCR‐FITC, NKRP‐RPE, SIRP‐Biotin, CD8a‐PE, CD4‐Biotin, and CD45RA were added to each tube and incubated on ice for 20 min. Cells were washed with F‐PBS and centrifuged (200 g) for 5 min. After discarding the supernatant, cells were incubated with a secondary antibody (e.g., 1 μL streptavidin [SA]‐Cy5PE) on ice for 20 min. After a final wash, the cells were ready for flow cytometry analysis. For intracellular Foxp3 staining, cells were resuspended in 1 mL Fix/Perm working solution and incubated at RT for 30 min in the dark. After two washes with permeabilization buffer, Foxp3‐FITC antibodies were added, and cells were incubated at RT for 30 min in the dark. After a final wash with permeabilization buffer, cells were resuspended in F‐PBS for flow cytometry analysis. All primary antibodies were IgG proteins purchased from AbD Serotec (Kidlington, Oxford, UK), except CD8a‐PE (BD Pharmingen, San Diego, CA, USA) and Foxp3‐FITC (eBioscience, San Diego, CA, USA). Stained cells were suspended in 400 μL of F‐PBS, and data were acquired on a BD FACScan flow cytometer using CellQuest software (Becton Dickinson, San Jose, CA, USA) and analyzed using FlowJo software. Leukocyte subsets were identified as B cells as CD45R+TCR−, T cells as TCR+NKRP1−, CD4 T cells as CD4+TCR+, CD8 T cells as CD8+TCR+, NKT cells as NKRP1+TCR+, NK cells as NKRP1+TCR−, monocytes as SIRP+TCR−, and Treg cells as CD4+CD25+FoxP3+. All experiments were replicated at least three times on different days with gated populations based on the mAbs and isotype‐matched negative controls.
In Vitro T‐Cell Proliferation Assay
Splenocytes were prepared 72 h after stroke onset. Mononuclear leukocytes at 2 × 105 cells/well in 96‐well flat‐bottom tissue culture plates were incubated at 37°C and 5% CO2 for 66 h in the presence of 1, 2.5 or 5 μg/mL T‐cell mitogen concanavalin A (ConA; Sigma‐Aldrich, Santa Clara, CA, USA). [3H]‐thymidine (PerkinElmer, Waltham, MA, USA) was then added to the wells and incubated for 18 h. Cells were harvested onto a glass fiber filter (PerkinElmer) and the amount of radioactivity was counted in a 1250 beta‐plate scintillation reader (PerkinElmer). Results are expressed as counts per minute (cpm). Each sample was in triplicate in each plate and repeated at least three times at different days.
Tissue Confocal Immunofluorescent Microscopy Study
Rats were euthanized 72 h after stroke onset with an overdose of isoflurane and perfused with cold PBS, followed by 4% paraformaldehyde in PBS. The brains were removed and post‐fixed for 48 h in 4% paraformaldehyde in PBS and cut into 50 μm sections. Immunofluorescent staining was carried out on free‐floating sections under moderate shaking. All washes and incubations were carried out in 0.1 M PBS (pH 7.4) containing 0.3% triton X‐100. Sections were incubated for 1 h with a blocking solution of 0.1 M PBS containing 0.3% Triton X‐100 and 5% equine serum. After the washes were completed, sections were incubated with either mouse anti‐rat CD68 mAb (a marker for reactive macrophages/microglia, 1:200, MCA341GA; AbD Serotec) or a rabbit anti‐human myeloperoxidase (MPO) antibody (which cross‐reacts with rats, 1:50, #A0398; Dako North America, Inc, Carpinteria, CA, USA) at 4°C overnight. Sections were washed and incubated with Alexa 488‐conjugated goat anti‐mouse secondary antibody to detect CD68 or an Alexa 488‐conjugated goat anti‐rabbit secondary antibody to detect MPO for 2 h at RT (1:200; Invitrogen). Sections were then washed and mounted on glass slides using VECTASHIELD® mounting medium with 4′,6‐diamidino‐2‐phenylindole (DAPI; Vector Laboratories, Burlingame, CA, USA). A negative control without primary antibodies was performed in parallel.
Morphometric Analysis
The expression of CD68 or MPO was investigated using the optical fractionator method on epifluorescent photomicrographs (Zeiss Axiovert inverted scanning microscope; Carl Zeiss Microscopy, Jena, Germany) covering a total of 0.16 mm2 (400 μm × 400 μm). For each animal, three sections (0.20–0.70 mm relative to bregma) were chosen, from which the number of immunoreactive cells for both CD68 and MPO in the ischemic region was counted using Image J software (NIH), and an average number of each animal was calculated. All counts were performed blind to the investigator on coded sections.
DTH Reaction
A 2 mg/mL solution of ovalbumin (OVA; Thermo Fisher Scientific, Waltham, MA, USA) in saline was mixed with complete Freund's adjuvant H37Ra (CFA; Pierce, Rockford, IL, USA) to form an emulsion. The mixture (200 μL) was injected subcutaneously at the base of the tail to stimulate an immune response in the rats. At 14 days, the rats were subjected to stroke. At 1 day post‐stroke, an OVA solution (20 μL of 1 mg/mL) was injected intradermally into the pinna of the ears. Inflammation was indicated by redness and swelling at the injection site. At 48–72 h after the second OVA injection, the inflamed area was measured by a caliper three times to determine the mean size in a blinded pattern.
Statistical Analysis
Data are presented as mean ± SEM. Differences were considered significant at P values <0.05. The t‐test was used to compare the means of two groups. One‐way analysis of variance (ANOVA) was used to compare mean differences in multiple groups followed by the Bonferroni post hoc tests using Prism 5 (GraphPAD Software for Science, San Diego, CA, USA).
Results
We first confirmed that intra‐ischemic hypothermia significantly reduced infarct size (Figure 1), which is consistent with our previous study 23.
Figure 1.
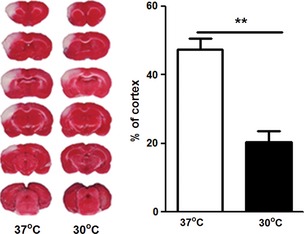
Hypothermia reduced infarct size. (Left) Representative images for infarction stained by 2,3,5‐triphenyletrazolium chloride from ischemic brains at 37 and 30°C. (Right) Bar graph showing average infarct size. Rats were subjected to 1 h of bilateral common carotid artery occlusion combined with permanent distal middle cerebral artery occlusion. Moderate hypothermia (30°C) was induced before stroke onset and maintained for 1 h. Animals were euthanized 3 days after stroke for infarction measurement. ** between the two indicated groups, P < 0.01, n = 7/group.
The Effects of Intra‐Ischemic Hypothermia on Gene Expression of Inflammatory Cytokines in the Brain
Using qRT‐PCR, we first examined the effects of hypothermia on gene expression in inflammatory cytokines and other mediators in the ischemic brain at 5, 24, and 72 h after stroke (Figure 2). Sham surgery did not significantly alter gene expression while normothermic stroke did increase Th1‐type cytokine gene expression, including INF‐γ, TNF‐α, and IL‐2, and the Th2‐type cytokine of IL‐10, as well as macrophage inflammatory factors IL‐1β, MIP‐2, and iNOS. Hypothermia, however, inhibited all of the above at one or more time points. Although the Th2 anti‐inflammtory cytokine IL‐4 and the inflammatory factor COX‐2 increased after stroke, their rates were neither significant nor affected by hypothermia (Figure 2).
Figure 2.
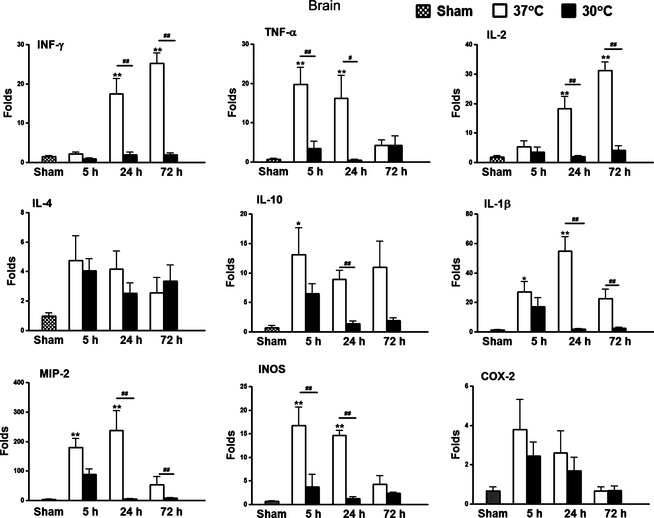
Effects of hypothermia on gene expression in the brain after stroke. Tissues were collected from animals with sham surgery but without stroke and 5, 24, and 72 h after stroke with or without hypothermia. QRT‐PCR was performed to detect gene expression of INF‐γ, TNF‐α, IL‐2, IL‐4, IL‐10, IL‐1β, MIP‐2, iNOS, and COX‐2. Folds of gene expression were normalized to the levels in naive animals. *,** versus sham, P < 0.05, 0.01, respectively, #,##, P < 0.05, 0.01, respectively, between the two indicated groups, n = 5–7/group.
Hypothermia Inhibited Leukocyte Infiltration and Inflammation into the Ischemic Brain after Stroke
Without hypothermia, FACS analyses showed a significant increase in mononuclear leukocyte infiltration into the ischemic brain 3 days after stroke. Gating was used to analyze lymphocyte subsets as described previously 28, observing increases in all major mononuclear leukocyte subsets but significant changes only in total T cells, CD4 T cells, and monocytes/macrophages.
Hypothermia reversed this trend in all subsets of mononuclear leukocytes, with the most significant reductions in T cells, CD4 T cells, B cells, and monocytes/macrophages (Figure 3). Consistent with this, hypothermia also suppressed the expression of MPO and CD68, reflecting the activity of neutrophils and macrophages, respectively (Figure 4).
Figure 3.
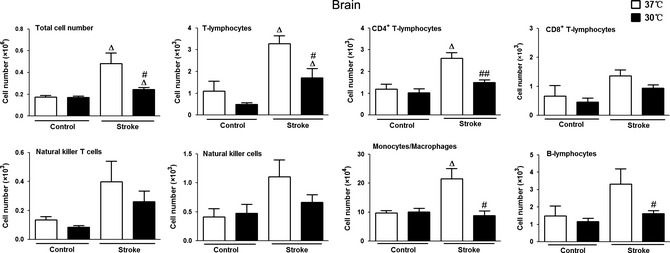
Effects of hypothermia on mononuclear leukocyte distribution in the ischemic brain. Ischemic brain hemispheres were collected 3 days after stroke and total mononuclear leukocytes were extracted for FACS analyses. Data from the nonischemic hemispheres were used as controls. Δ versus corresponding controls (the contralateral, nonischemic hemisphere) at 37 or 30°C, P < 0.05; #,## 30°C versus 37°C, between two stroke groups (ischemic hemispheres), P < 0.05, 0.01, respectively, n = 5/group.
Figure 4.
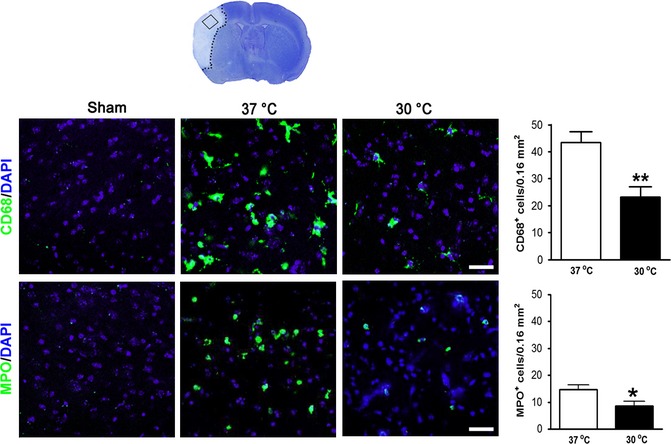
Hypothermia blocks inflammation in the brain. Brain tissues were harvested 3 days after stroke. Positive cells were counted from three coronal brain sections, and a region of interest was identified on each section. Immunoreactive cells are shown by merged images of 4′,6‐diamidino‐2‐phenylindole staining (blue) and immunofluorescence staining using a 40× objective lens by laser scanning confocal microscope. Top panel: representative immunostaining of CD68 and bar graph showing statistical results. Bottom panel: representative immunostaining of myeloperoxidase and bar graph showing statistical results. *,** versus 37°C, P < 0.05, 0.01, respectively, Scale bar: 20 μm, n = 5/group.
Intra‐Ischemic Hypothermia Inhibits SIID
Next, we studied whether stroke results in immunodepression and how hypothermia affects it. SIID is manifested by reduced numbers of total mononuclear leukocytes and their subsets. At 72 h post‐stroke, we counted all mononuclear leukocytes as well as individual subsets in naive, sham surgery and stroke rats, with and without hypothermia.
Sham surgery, with or without hypothermia, did not significantly change total numbers of mononuclear leukocytes or individual subsets in the peripheral blood. Normothermic stroke, however, resulted in significant reductions of total mononuclear leukocytes, T cells, CD4, CD8, and B cells compared to naive rats or rats with sham surgery. These reductions were inhibited by hypothermia (Figure 5A). In the spleen, normothermic stroke resulted in significant reductions in total mononuclear leukocytes and individual subsets. Hypothermia significantly attenuated these reductions in total mononuclear leukocytes, T cells, CD4, CD8, and NKT cells (Figure 5B).
Figure 5.
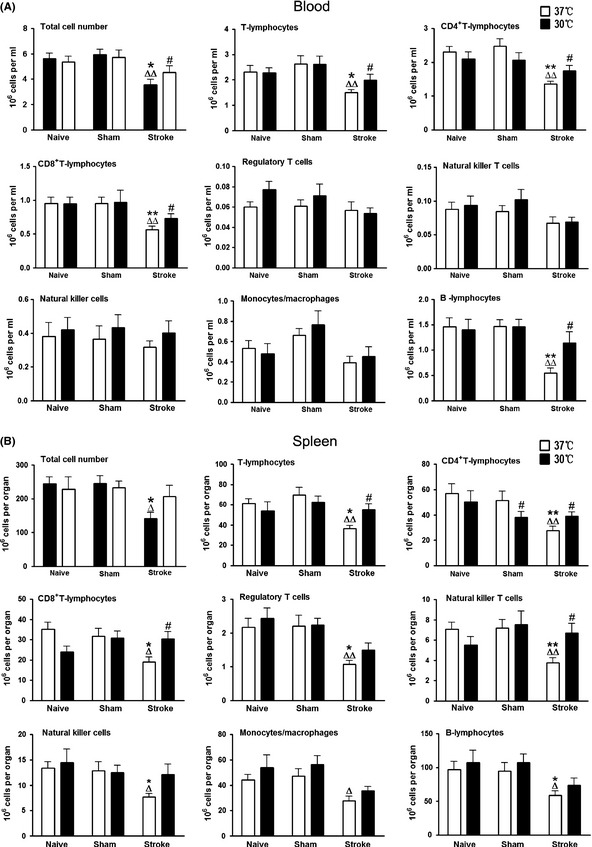
Effects of hypothermia on mononuclear leukocyte populations in the peripheral organs after stroke. Absolute numbers of total mononuclear leukocytes, T cells, CD4 and CD8 T cells, Treg cells, NK cell, NKT cells, monocytes/macrophages and B cells, were analyzed in different peripheral organs 3 days post‐stroke. A total of six groups were studied. For naive animals, sham surgery and stroke were not performed, but animals received anesthesia and body temperature were maintained at 37 or 30°C for the same duration as stroke rats. For sham surgery, rats received sham surgery without stroke and body temperature were maintained at 37 or 30°C for the same duration as other groups receiving stroke. For stroke groups, animals were subjected to focal ischemia and allowed to survive for 72 h. Absolute cell numbers were calculated by multiplying the percentage of each cell type with total cell numbers in an analyzed sample, and adjusted to a number in 1 mL of blood, and a number in each organ. (A) Cell numbers in the peripheral blood. Total numbers of peripheral blood mononuclear cells and each subset of lymphocytes and monocytes/macrophages are shown, n = 9–12/group. (B) Cell numbers in the spleen. Total numbers of splenocytes in each spleen and each subset of mononuclear leukocytes are shown, n = 9–13/group. *, ** versus naive, Δ, ΔΔ versus sham, #, ## 30°C versus 37°C, P < 0.05, 0.01, respectively.
The in vitro assay for ConA‐stimulated T‐cell proliferation is commonly used to evaluate T‐cell immunoreactivity. Results using ConA stimulation showed that sham surgery at 37 or 30°C yielded a slight yet insignificant reduction in splenic T‐cell proliferation compared to naive rats (Figure 6A). No difference was observed between sham surgery at 37 or 30°C, suggesting that hypothermia alone did not affect T‐cell function in vitro. Normothermic stroke significantly inhibited splenic T‐cell proliferation and this inhibition was attenuated by hypothermia (Figure 6A). This result is supported by the in vivo DTH assay, which is commonly used to evaluate T‐cell‐mediated immunity in vivo. OVA challenges resulted in ear redness and increased ear thickness in naive and sham surgery rats. Ear thickness was reduced in rats receiving normothermic stroke, however, and this effect was significantly attenuated by hypothermic treatment (Figure 6B).
Figure 6.
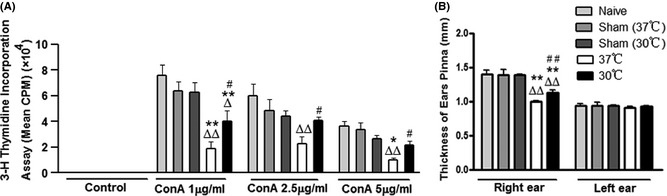
Effects of hypothermia on immunoreaction after stroke. (A) In vitro assay for T‐cell proliferation after stroke. Splenocytes were prepared 72 h after stroke from four groups of animals, including naive, sham surgery, normothermic, and hypothermic animals. The cultured splenocytes were stimulated with 0, 1, 2.5, and 5 μg/mL concanavalin A. The cells were incubated for 66 h at 37°C and 5% CO 2. [3H]‐Thymidine was added to the culture and incubated for 18 h. Cells were harvested onto a glass fiber filter. The amount of radioactivity that integrated into divided lymphocytes was measured by a beta‐plate scintillation reader. Results are expressed as counts per minute (cpm). *,** versus naive, Δ , ΔΔ versus sham, # , ## 30°C versus 37°C, P < 0.05, 0.01, respectively, n = 5 wells/experiment, repeat five times. (B) In vivo delayed‐type hypersensitivity test to measure immune response in naive rats, rats receiving sham surgery, or stroke with or without hypothermia. The antigen, ovalbumin (OVA), was injected subcutaneously at the base of the tail. Stroke was induced 14 days after immunization, and 1 day after stroke, rats were challenged intradermally with an OVA solution in the pinna of the ears. The inflamed area was measured three times to get the mean value at 48–72 h after the challenge. *,** versus naive, Δ , ΔΔ, versus sham, # , ##, 30°C versus 37°C, P < 0.05, 0.01, respectively, n = 5/group.
Discussion
We report the first evidence that brief intra‐ischemic moderate hypothermia robustly attenuated SIID and demonstrate that hypothermia improved T‐cell function, attenuated spleen atrophy, and helped maintain total numbers of mononuclear leukocytes and individual subsets in the peripheral blood and spleen. Hypothermia's inhibitory effect on SIID also correlated with its protective effects on infarct size, indicated by reductions in proinflammatory cytokine expression, macrophage activity, and infiltration of neutrophils and lymphocytes into the brain.
We found that normothermic stroke caused a strong proinflammatory response in the brain, and this response correlated with robust SIID. This is consistent with previous studies that show bidirectional interactions between the injured brain and the peripheral immune system after stroke. On one hand, the peripheral immune system can increase local brain inflammation via recruitment and infiltration of circulating neutrophils 29, 30, macrophages 31 and T cells 32, thus exacerbating ischemic injury. On the other hand, stroke can cause peripheral immune suppression 8, 9, which may result in post‐stroke infection and mortality 18.
Stroke‐induced immunodepression is manifested by a reduction in lymphocyte activation and spleen atrophy 8, 9, which is supported by our current study. The underlying mechanisms of SIID, however, are not completely understood. All peripheral lymphoid organs are innervated by sympathetic nerves, and lymphocytes express adrenergic receptors 33. Previous studies have shown that brain injury induced by stroke activates the sympathetic nerve pathway through the secretion of catecholamines, which inhibits lymphocyte function and causes immunosuppression 34. Stroke may also affect the hypothalamus–pituitary–adrenal axis, leading to glucocorticoid secretion and immunosuppression 18. Most recently, invariant natural killer T cells (iNKT) in the liver were found to play a crucial role in stroke‐induced immunosuppression 35. Stroke results in adrenergic nerve activity in the liver, which alters the behavior of iNKT cells. iNKT cells normally promote immunity, but iNKT cells after stroke produce more antiinflammatory IL‐10, which leads to immunosuppression 35.
The primary goal of this study was to investigate whether stroke and hypothermia act synergistically to worsen SIID. Stroke increases infection rates in patients and hypothermia by itself causes immunosuppression 19, 20. Thus, it is possible that stroke and hypothermia together could exacerbate immunosuppression. We show that hypothermia unexpectedly and robustly reduced peripheral immunosuppression induced by stroke. However, this is not surprising for two possible reasons. First, in our study, intra‐ischemic hypothermia robustly reduced infarct size, which could result in less immunosuppression. As suggested by previous studies, the degree of SIID may be determined by infarct size. The larger the infarct size, the more severe the immunosuppression and, conversely, the smaller infarct size, the less severe the immunosuppression 21. Second, in our study hypothermia lasted only 60 min. Hypothermia without stroke in the control group did not significantly change mononuclear leukocyte populations. In most studies showing the immunosuppressive properties of hypothermia, however, hypothermia was administered from 10 h to a few days 19, 20. Thus, long‐term hypothermia, but not short‐term hypothermia, induced immunosuppression. Our results suggest that brief intra‐ischemic hypothermia did not cause immunosuppression. Rather, it reduced immunosuppression when it attenuated brain injury. Future studies should address if long‐term hypothermia and stroke act synergistically on immunosuppression.
One could speculate that less SIID as seen with hypothermia would result in a stronger immune response, thus promote inflammation and lead to a worsened infarction. However, in our study, suppression of SIID did not peak until 3 days after stroke, which is when the infarction had matured. Moreover, hypothermia did not activate the immune system but recovered it to normal baseline levels. Therefore, it is unlikely that the reduced inhibition of SIID observed with hypothermia would result in a worsened infarction.
Our methods face two clinical limitations. First, our use of moderate hypothermia poses a higher risk of side effects than does mild hypothermia, whose clinical relevance is already known. Second, our use of intra‐ischemic hypothermia rather than post‐ischemic hypothermia is applicable only to those patients who receive surgical intervention in which a stroke is destined to occur. Future studies of mild and post‐ischemic hypothermia are therefore warranted.
In conclusion, our study provides strong evidence to support the translation of intra‐ischemic hypothermia to the clinic for patients in whom stroke is predictable. One major concern of hypothermia is its side effect of inhibiting immune function, which can lead to infection and increased mortality. Our study suggests that brief intra‐ischemic hypothermia can robustly attenuate brain infarction without worsening stroke‐induced immune suppression. Moreover, it may improve stroke‐induced immune dysfunction.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgments
The authors thank Ms. Cindy H. Samos and Felicia Beppu for manuscript assistance. This study was supported by NIH R01NS27292‐03 (GKS/HZ), AHA grant in aid and 1R01NS064136‐01 (HZ).
The first two authors equally contributed to this article.
References
- 1. Hypothermia‐after‐Cardiac‐Arrest‐Study‐Group . Mild therapeutic hypothermia to improve the neurologic outcome after cardiac arrest. N Engl J Med 2002;346:549–556. [DOI] [PubMed] [Google Scholar]
- 2. Bernard SA, Gray TW, Buist MD, Jones BM, Silvester W, Gutteridge G, et al. Treatment of comatose survivors of out‐of‐hospital cardiac arrest with induced hypothermia. N Engl J Med 2002;346:557–563. [DOI] [PubMed] [Google Scholar]
- 3. Gluckman PD, Gunn AJ, Wyatt JS. Hypothermia for neonates with hypoxic‐ischemic encephalopathy. N Engl J Med 2006;354:1643–1645; author reply ‐5. [PubMed] [Google Scholar]
- 4. Shankaran S, Laptook AR, Ehrenkranz RA, Tyson JE, McDonald SA, Donovan EF, et al. Whole‐body hypothermia for neonates with hypoxic‐ischemic encephalopathy. N Engl J Med 2005;353:1574–1584. [DOI] [PubMed] [Google Scholar]
- 5. den Hertog H, van der Worp B, van Gemert M, Dippel D. Therapeutic hypothermia in acute ischemic stroke. Expert Rev Neurother 2007;7:155–164. [DOI] [PubMed] [Google Scholar]
- 6. Krieger DW, Yenari MA. Therapeutic hypothermia for acute ischemic stroke: What do laboratory studies teach us? Stroke 2004;35:1482–1489. [DOI] [PubMed] [Google Scholar]
- 7. Czlonkowska A, Cyrta B, Korlak J. Immunological observations on patients with acute cerebral vascular disease. J Neurol Sci 1979;43:455–464. [DOI] [PubMed] [Google Scholar]
- 8. Offner H, Subramanian S, Parker SM, Wang C, Afentoulis ME, Lewis A, et al. Splenic atrophy in experimental stroke is accompanied by increased regulatory T cells and circulating macrophages. J Immunol 2006;176:6523–6531. [DOI] [PubMed] [Google Scholar]
- 9. Offner H, Vandenbark AA, Hurn PD. Effect of experimental stroke on peripheral immunity: CNS ischemia induces profound immunosuppression. Neuroscience 2009;158:1098–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Haeusler KG, Schmidt WU, Fohring F, Meisel C, Helms T, Jungehulsing GJ, et al. Cellular immunodepression preceding infectious complications after acute ischemic stroke in humans. Cerebrovasc Dis 2008;25:50–58. [DOI] [PubMed] [Google Scholar]
- 11. Meisel C, Prass K, Braun J, Victorov I, Wolf T, Megow D, et al. Preventive antibacterial treatment improves the general medical and neurological outcome in a mouse model of stroke. Stroke 2004;35:2–6. [DOI] [PubMed] [Google Scholar]
- 12. Sarrafzadeh A, Schlenk F, Meisel A, Dreier J, Vajkoczy P, Meisel C. Immunodepression after aneurysmal subarachnoid hemorrhage. Stroke 2011;42:53–58. [DOI] [PubMed] [Google Scholar]
- 13. Brown M, Glassenberg M. Mortality factors in patients with acute stroke. JAMA 1973;224:1493–1495. [PubMed] [Google Scholar]
- 14. Mergenthaler P, Dirnagl U, Meisel A. Pathophysiology of stroke: Lessons from animal models. Metab Brain Dis 2004;19:151–167. [DOI] [PubMed] [Google Scholar]
- 15. Yenari MA, Han HS. Influence of hypothermia on post‐ischemic inflammation: Role of nuclear factor kappa B (NFkappaB). Neurochem Int 2006;49:164–169. [DOI] [PubMed] [Google Scholar]
- 16. Zhao H, Steinberg GK, Sapolsky RM. General versus specific actions of mild‐moderate hypothermia in attenuating cerebral ischemic damage. J Cereb Blood Flow Metab 2007;27:1879–1894. [DOI] [PubMed] [Google Scholar]
- 17. Maier CM, Ahern K, Cheng ML, Lee JE, Yenari MA, Steinberg GK. Optimal depth and duration of mild hypothermia in a focal model of transient cerebral ischemia: Effects on neurologic outcome, infarct size, apoptosis, and inflammation. Stroke 1998;29:2171–2180. [DOI] [PubMed] [Google Scholar]
- 18. Meisel C, Schwab JM, Prass K, Meisel A, Dirnagl U. Central nervous system injury‐induced immune deficiency syndrome. Nat Rev Neurosci 2005;6:775–786. [DOI] [PubMed] [Google Scholar]
- 19. Qadan M, Gardner SA, Vitale DS, Lominadze D, Joshua IG, Polk HC Jr. Hypothermia and surgery: Immunologic mechanisms for current practice. Ann Surg 2009;250:134–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lee SL, Battistella FD, Go K. Hypothermia induces T‐cell production of immunosuppressive cytokines. J Surg Res 2001;100:150–153. [DOI] [PubMed] [Google Scholar]
- 21. Hug A, Dalpke A, Wieczorek N, Giese T, Lorenz A, Auffarth G, et al. Infarct volume is a major determiner of post‐stroke immune cell function and susceptibility to infection. Stroke 2009;40:3226–3232. [DOI] [PubMed] [Google Scholar]
- 22. Albers GW, Goldstein LB, Hess DC, Wechsler LR, Furie KL, Gorelick PB, et al. Stroke Treatment Academic Industry Roundtable (STAIR) recommendations for maximizing the use of intravenous thrombolytics and expanding treatment options with intra‐arterial and neuroprotective therapies. Stroke 2011;42:2645–2650. [DOI] [PubMed] [Google Scholar]
- 23. Zhao H, Shimohata T, Wang JQ, Sun G, Schaal DW, Sapolsky RM, et al. Akt contributes to neuroprotection by hypothermia against cerebral ischemia in rats. J Neurosci 2005;25:9794–9806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xiong X, Gu L, Zhang H, Xu B, Zhu S, Zhao H. The protective effects of T cell deficiency against brain injury are ischemic model‐dependent in rats. Neurochem Int 2013;62:265–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Han RQ, Ouyang YB, Xu L, Agrawal R, Patterson AJ, Giffard RG. Postischemic brain injury is attenuated in mice lacking the beta2‐adrenergic receptor. Anesth Analg 2009;108:280–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Xiong X, Barreto GE, Xu L, Ouyang YB, Xie X, Giffard RG. Increased brain injury and worsened neurological outcome in interleukin‐4 knockout mice after transient focal cerebral ischemia. Stroke 2011;42:2026–2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lim JK, Obara CJ, Rivollier A, Pletnev AG, Kelsall BL, Murphy PM. Chemokine receptor Ccr2 is critical for monocyte accumulation and survival in West Nile virus encephalitis. J Immunol 2011;186:471–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gu L, Xiong X, Wei D, Gao X, Krams S, Zhao H. T cells contribute to stroke‐induced lymphopenia in rats. PLoS One 2013;8:e59602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Weston RM, Jones NM, Jarrott B, Callaway JK. Inflammatory cell infiltration after endothelin‐1‐induced cerebral ischemia: Histochemical and myeloperoxidase correlation with temporal changes in brain injury. J Cereb Blood Flow Metab 2007;27:100–114. [DOI] [PubMed] [Google Scholar]
- 30. Hiraga N, Adachi N, Liu K, Nagaro T, Arai T. Suppression of inflammatory cell recruitment by histamine receptor stimulation in ischemic rat brains. Eur J Pharmacol 2007;557:236–244. [DOI] [PubMed] [Google Scholar]
- 31. Gelderblom M, Leypoldt F, Steinbach K, Behrens D, Choe CU, Siler DA, et al. Temporal and spatial dynamics of cerebral immune cell accumulation in stroke. Stroke 2009;40:1849–1857. [DOI] [PubMed] [Google Scholar]
- 32. Schroeter M, Jander S, Witte OW, Stoll G. Local immune responses in the rat cerebral cortex after middle cerebral artery occlusion. J Neuroimmunol 1994;55:195–203. [DOI] [PubMed] [Google Scholar]
- 33. Elenkov IJ, Wilder RL, Chrousos GP, Vizi ES. The sympathetic nerve–an integrative interface between two supersystems: The brain and the immune system. Pharmacol Rev 2000;52:595–638. [PubMed] [Google Scholar]
- 34. Prass K, Meisel C, Hoflich C, Braun J, Halle E, Wolf T, et al. Stroke‐induced immunodeficiency promotes spontaneous bacterial infections and is mediated by sympathetic activation reversal by poststroke T helper cell type 1‐like immunostimulation. J Exp Med 2003;198:725–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wong CH, Jenne CN, Lee WY, Leger C, Kubes P. Functional innervation of hepatic iNKT cells is immunosuppressive following stroke. Science 2011;334:101–105. [DOI] [PubMed] [Google Scholar]