

Abstract
Thyroid carcinoma is the most common endocrine malignancy, and although the disease generally has an excellent prognosis, therapeutic options are limited for patients not cured by surgery and radioiodine. Thyroid carcinomas commonly contain one of a small number of recurrent genetic mutations. The identification and study of these mutations has led to a deeper understanding of the pathophysiology of this disease and is providing new approaches to diagnosis and therapy. Papillary thyroid carcinomas usually contain an activating mutation in the RAS cascade, most commonly in BRAF and less commonly in RAS itself or through gene fusions that activate RET. A chromosomal translocation that results in production of a PAX8-PPARG fusion protein is found in follicular carcinomas. Anaplastic carcinomas may contain some of the above changes as well as additional mutations. Therapies that are targeted to these mutations are being used in patient care and clinical trials.
Keywords: AKT, anaplastic, BRAF, follicular, NTRK1, papillary, PAX8-PPARG, PTEN, RAS, RET, thyroid carcinoma
1. Introduction
Cancer is a consequence of accumulated genetic alterations; understanding how these changes effect the disease state offers the hope of developing new and better treatments, and may provide improved prognostic information for the individual patient. Thyroid cancer is the most common endocrine malignancy and more than 1800 US deaths are estimated to result from thyroid carcinomas annually (Cancer of the Thyroid - SEER Stat Fact Sheets; Tuttle et al., 2010). We offer below a review of genetic abnormalities commonly encountered in sporadic thyroid carcinomas. This review will be focused on carcinomas of the major thyroid cell type, the thyroid hormone-producing follicular cell. It will not discuss medullary thyroid carcinomas, which are tumors of the calcitonin-producing parafollicular C cells.
Thyroid carcinomas are classified by their histological appearance in a schema largely affirmed by more recent molecular data. Differentiated thyroid carcinomas (DTC) are the most common and contain cells which retain many features of normal thyrocytes. DTCs are further divided into papillary and follicular thyroid carcinomas (PTC and FTC, respectively). Anaplastic thyroid carcinomas (ATC) are completely undifferentiated, aggressive, therapy resistant, and usually fatal cancers (Cooper et al., 2009). Recurrent mutations in thyroid carcinomas are predominantly associated with specific tumor histologies and are implicated in disease etiology (Fig. 1 and Table 1).
Fig. 1.
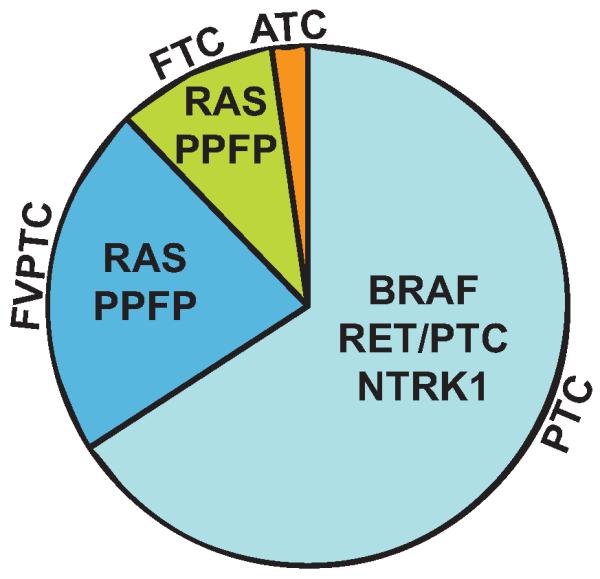
Pie chart showing the most common histological types of thyroid carcinoma and the most common mutations and gene fusions within each type. ATC, anaplastic thyroid carcinoma; FTC, follicular thyroid carcinoma; FVPTC, follicular variant papillary thyroid carcinoma; PPFP, PAX8-PPARG fusion protein; PTC, papillary thyroid carcinoma. Recurring mutations in ATC are not shown.
Table 1.
Detailed listing of gene mutations associated with sporadic thyroid carcinomas
| Gene | Mutations | Activating or Inactivating | Common associations |
|---|---|---|---|
| AKT1 | G49A | Activating | Poorly differentiated carcinomas. |
| ALK | Exon 23 | Activating | Poorly differentiated and anaplastic carcinomas. |
| BRAF | V600E, K601E, AKAP9 gene fusion | Activating | Papillary and anaplastic carcinomas. V600E is by far most common and is over-represented in tall cell variant, uncommon in follicular variant. AKAP9-BRAF is over-represented in radiation induced papillary carcinomas. |
| CTNNB1 | Exon 3 | Activating | Poorly differentiated and anaplastic carcinomas. |
| IDH1 | Exon 4 | Abnormal activity | Identified in a minor fraction of most types of thyroid carcinoma. |
| NDUFA13 (GRIM19) | various | Inactivating | Hurthle cell carcinoma. |
| NTRK1 | Gene fusions with TPM3, TPR, and TFG | Activating | Infrequently found in papillary carcinoma. |
| PIK3CA | Exons 9, 20 | Activating | Follicular, poorly differentiated, and anaplastic carcinomas. |
| PPARG | Gene fusion with PAX8, rarely with CREB3L2 | Context dependent | Follicular carcinomas and follicular variant of papillary carcinomas. |
| PTEN | Deletion or inactivating mutation | Inactivating | Follicular, poorly differrentiated and anaplastic carcinomas. |
| RAS | NRAS Q61, HRAS Q61, KRAS G12, G13 | Activating | Follicular, follicular variant of papillary, poorly differentiated and anaplastic carcinomas. NRAS mutations are most common, KRAS are least common. |
| RET | Gene fusions with CCDC6 (RET/PTC1), NCOA4 (RET/PTC3), and others | Activating | Papillary carcinomas. Gene fusions result in constitutively active RET kinase domain. RET/PTC1 and RET/PTC3 are over-represented in radiation induced thyroid cancer. |
| TP53 | Exons 5–8 | Inactivating | Poorly differentiated and anaplastic carcinomas. |
2. Papillary thyroid carcinoma
Papillary thyroid carcinomas account for more than 85% of DTCs. PTCs are etymologically derived from the replacement of normal thyroid follicular structures by papillae - several layers of neoplastic thyroid epithelial cells lining a fibrous core. Presently PTCs are identified by certain characteristic nuclear features such as larger, clearer nuclei with inclusion bodies of cytoplasm and nuclear grooves (Baloch and LiVolsi, 2005). There are several histological subtypes of PTC, including classic PTC, follicular variant PTC (FVPTC), tall cell variant (TCV) and others. FVPTCs lack papillary structures but contain follicles and retain nuclear features of PTC; it is this latter attribute that separates them from pure FTCs. However, as will be described subsequently, the mutational profile of FVPTCs closely resembles that of FTCs. TCV PTCs contain cells that are at least twice as tall as wide. These carcinomas tend to be more aggressive, and the tall cell phenotype has a specific mutational correlate. Mutations or translocations in BRAF, RET, RAS, and NTRK1 – all participants in the same signaling cascade – are known in PTC (Fig. 2). In addition, translocations between PAX8 and PPARG occur in FVPTC.
Fig. 2.

Gene mutations and signaling pathways in thyroid carcinomas. Simplified schematic diagrams are shown of the RAS/MAPK and PI3K/AKT pathways. Proteins with reported mutations in thyroid carcinomas are in red. There are many protein targets of these pathways that contribute to oncogenesis, some of which are common to both pathways. A subset of these targets is shown. A question mark appears with the arrow from PAX8-PPARG because the mechanisms by which this fusion protein contributes to oncogenesis are not well understood. Similarly, the arrow from IDH1 has a question mark since the mechanisms by which IDH1 mutations contribute to thyroid cancer are poorly understood. RTK, receptor tyrosine kinase.
2.1. BRAF
BRAF is a member of a small family of RAF proteins that function as cytoplasmic serine/threonine protein kinases in the mitogen activated protein kinase (MAPK) signaling cascade. Members of the MAPK cascade modulate essential cellular processes such as proliferation, differentiation, and survival. BRAF is physiologically activated by RAS (Avruch et al., 2001). Once activated, BRAF can activate MEK by phosphorylating it and thereby transduce the mitogenic signals to downstream elements such as ERK. BRAF mutations found in cancer generally result in unregulated kinase activity and increased MEK activation; those mutations which do not exhibit increased kinase activity can nonetheless produce higher ERK activation downstream (Wan et al., 2004).
A wide variety of BRAF mutations are found in a wide range of cancers, but more than 95% of all cases are of a single variety denoted BRAFV600E. A missense thymine to adenine transversion at the 1799 nucleotide position leads to a valine to glutamate substitution at the 600 amino acid position. The mutation results in a constitutively active BRAF, no longer dependent on RAS, and hence causes constitutive MEK/ERK activation (Cantwell-Dorris et al., 2011; Wan et al., 2004). In addition to PTCs, BRAFV600E is found in the majority of malignant melanomas and hairy cell leukemias, as well as in significant fractions of other cancers such as astrocytomas and carcinomas of the colon, lung and ovary (reviewed in Pakneshan et al., 2013).
Mutant BRAF expression contributes to the transformation of cell lines and thyroid tumor formation in mice. BRAFV600E induces genomic instability in the rat PCCL3 thyrocyte cell line and increases focus formation in NIH3T3 cells (Mitsutake et al., 2005; Moretti et al., 2006). Expression of rare BRAF mutants, including fusion chimera generated by chromosomal rearrangements and truncated proteins generated by alternative splicing, shows that uninhibited BRAF kinase activity generally has pro-oncogenic effects (Baitei et al., 2009; Ciampi et al., 2005; Liu et al., 2007b). Early or late expression of BRAFV600E in transgenic mice rapidly produces enlarged and abnormal thyroids followed by invasive PTC within a year, confirming BRAF mutations as a causative event in PTCs (Charles et al., 2011; Knauf et al., 2005).
Due to differing techniques, cohorts, etc., the reported prevalence of BRAF mutations in PTC varies considerably, though it is clearly the most common mutation in PTC and therefore in thyroid carcinomas. The estimate for occurrences of BRAF mutations in general and BRAFV600E in particular range from 28% to 77% of all PTC cases; the average reported value is ~45% (Cohen et al., 2003; Fernandez et al., 2013; Frattini et al., 2004; Guerra et al., 2012; Lim et al., 2013; Namba et al., 2003; Nikiforova et al., 2003a; Nikiforov, 2011; Puxeddu et al., 2004). BRAF mutations are particularly common in TCV PTC, and are uncommon in FVPTC.
The presence of BRAF mutations correlates with more aggressive disease. Data from numerous groups and a meta-analysis associate BRAF mutations in PTC with large tumor size, extrathyroidal extension, higher stage at presentation, lymph node metastasis and increased risk of recurrence (Elisei et al., 2012; Fernandez et al., 2013; Jo et al., 2006; Kim et al., 2012; Lim et al., 2013; Namba et al., 2003; Nikiforova et al., 2003a; Xing et al., 2005). A recent retrospective analysis of 1849 PTC patients (Xing et al., 2013) found a mortality rate of 5.3% in BRAFV600E-positive patients versus 1.1% in mutation-negative patients. However, the association of BRAFV600E with mortality was lost after adjusting for standard clinical and histological prognostic features (e.g. the presence of extrathyroidal invasion or metastases). On the one hand, this suggests that BRAFV600E is a major driving force that underlies these traditional clinical and histological prognostic features. On the other hand, the data call into question the added value of determining the BRAF status of a given tumor, at least in terms of predicting mortality.
A clinically relevant feature of BRAF-mutated PTCs is their diminished responsiveness to radioactive iodine (RAI). Thyrocytes concentrate and organify iodine as part of their physiological function, making RAI useful to identify and ablate remnant and metastatic thyroid carcinoma. Loss of RAI avidity is a significant problem in progressive disease and severely limits the available treatment options. BRAF-mutated PTCs are associated with decreased expression of proteins involved in the uptake and organification of iodide, such as the sodium iodide symporter (NIS), resulting in an increased number of RAI courses required to achieve disease free status and increased incidence of RAI refractory recurrence (Barollo et al., 2010; Durante et al., 2007; Elisei et al., 2012; Gao et al., 2012; Riesco-Eizaguirre et al., 2006).
Therefore, re-establishment of RAI avidity is a major avenue of research in thyroid cancer. Small molecule inhibitors against BRAF or its downstream effector MEK are effective at increasing NIS expression in preclinical models (Durante et al., 2007; Liu et al., 2007a; Nucera et al., 2009). Selumentinib (AZD6244, ARRY-142886) is a MEK inhibitor that increases RAI uptake and effects partial responses in some BRAF or RAS -mutated, RAI-refractory patients (Ho et al., 2013).
Small molecule inhibitors of BRAF are potentially useful drugs in BRAF-mutated PTCs. Sorafenib (BAY 43-9006) was identified as part of a drug discovery program targeting BRAF (Lyons et al., 2001). It also blocks the actions of VEGF receptors, PDGF receptors, Kit, and RET, making it among the first multikinase inhibitors to be in clinical trials (Caronia et al., 2011; Murphy et al., 2006). Sorafenib exhibits broad but modest and ultimately transient anti-tumor effects in clinical trials on patients with advanced, metastatic thyroid cancer, though a positive response does not appear to depend upon the BRAF status of the tumor (Capdevila et al., 2012; Gupta-Abramson et al., 2008; Hoftijzer et al., 2009; Kloos et al., 2009; Marotta et al., 2013; Schneider et al., 2012; Shen et al., 2012). Vemurafenib is particularly interesting in that it is a specific inhibitor of BRAFV600E, not wild type BRAF. Vemurafenib is highly therapeutic in BRAFV600E melanomas, although resistance eventually develops. A phase I trial of vemurafenib in 3 BRAFV600E thyroid cancer patients resulted in one partial response (Kevin Kim et al., 2013).
2.2 RET
RET is a cell surface receptor tyrosine kinase that transmits environmental cues to RAS and the downstream MAPK pathway (Marotta et al., 2011). RET mutations in PTC are exclusively genetic rearrangements resulting in chimeric proteins denoted RET/PTC1, RET/PTC2, etc. RET/PTC is found in ~15% of PTCs. RET itself is not expressed in normal thyroid follicular cells, but the juxtaposition of the 3' kinase domain of RET to a 5' partner and its promoter leads to fusion oncoprotein expression and aberrant kinase activity, with downstream activation of the RAS/MAPK pathway and the phosphatidylinositide 3-kinase (PI3K)/AKT pathway (Bunone et al., 2000; Grieco et al., 1990; Melillo et al., 2005; Miyagi et al., 2004). Although many fusion partners of RET have been described, the most frequent are CCDC6 (resulting in RET/PTC1) and NCOA4 (resulting in RET/PTC3) (Bongarzone et al., 1994; Santoro et al., 1994). The common feature of the fusion partners is their ability to constitutively dimerize, which results in constitutive RET tyrosine kinase activity via autophosphorylation (Salvatore et al., 2000). There also is evidence that RET/PTC can heterodimerize with the EGFR, and that the EGFR kinase can then phosphorylate and activate the RET kinase domain (Croyle et al., 2008).
Exogenously induced expression of RET/PTC proteins results in PTC-like phenotypes in culture and mouse models. In non-malignant primary thyroid cells or cell lines, RET/PTC expression decreases thyroid specific gene expression, alters cell and colony morphology to resemble features of PTC, renders the cells insensitive to or independent of TSH signaling, and especially in the cases of RET/PTC3, markedly increases cell proliferation (Basolo et al., 2002; Bond et al., 1994; Santoro et al., 1993; Wang et al., 2003). Transgenic mice that express RET/PTC1 or RET/PTC3 in the thyroid acquire PTC-like thyroid carcinomas after long latency periods (Jhiang et al., 1996; Powell et al., 1998; Santoro et al., 1996).
Although RET/PTC rearrangements are substantially less common than BRAFV600E, there is a specific association of RET/PTC with radiation-induced thyroid cancer. For example, the Chernobyl accident which released nuclear materials including radioactive iodine was associated with increased numbers of RET/PTC thyroid cancers (Fugazzola et al., 1995; Rabes et al., 2000; Thomas et al., 1999). RET/PTC events also are prevalent in patients previously exposed to other forms of radiation, e.g. as part of medical treatment (Alipov et al., 1999; Bounacer et al., 1997; Collins et al., 2002; Nikiforov et al., 1997).
In contrast to the less common RET/PTC mutations, RET/PTC1 and RET/PTC3 involve fusion partners located on the same chromosome as RET. It is thought that chromosomal proximity of the fusion partners and fragile sites within those genes contribute to the high incidence of RET/PTC fusions in patients exposed to radiation (Gandhi et al., 2012, 2010a, 2010b; Nikiforova et al., 2000). In concordance with this hypothesis, irradiation of thyroid cell cultures generates RET/PTC fusions (Ito et al., 1993; Mizuno et al., 2000, 1997). Interestingly, several other thyroid cancer related mutations such as the AKAP9-BRAF, NTRK1-T1, and NTRK1-T2 fusions also involve intrachromosomal rearrangements (Santoro et al., 2006). AKAP9-BRAF in particular is overrepresented in PTCs attributable to radiation exposure, while BRAF point mutations are rare in the same population (Ciampi et al., 2005; Lima et al., 2004).
In general the prognosis for RET/PTC thyroid carcinomas is relatively good (Soares et al., 1998). Few correlations are found between RET/PTC mutations and clinical markers of increased mortality or morbidity, though increased lymph node metastases are sometimes noted (Adeniran et al., 2006; Santoro et al., 2002; Tallini et al., 1998). RET/PTC1 tumors are associated with classical PTC histology while RET/PTC3 are associated with more aggressive solid and TCV PTC (Basolo et al., 2002; Nikiforov et al., 1997; Thomas et al., 1999).
A number of small molecules targeting multiple tyrosine kinases inhibit RET and RET/PTC. Sunitinib targets RET among many other kinases and has modest efficacy in PTCs, although it has not been studied specifically in tumors expressing RET/PTC (Carr et al., 2010). Vandetanib and cabozantinib are inhibitors of multiple tyrosine kinases including RET and are approved for the treatment of medullary thyroid carcinoma (Thornton et al., 2012). Medullary thyroid carcinoma is a disease of calcitonin-producing thyroid C cells rather than thyroid follicular cells, and is frequently associated with activating point mutations in RET. Whether vandetanib and cabozantinib also may have specific efficacy in RET/PTC carcinomas is not known.
2.3. NTRK1
Chromosomal rearrangements involving the NTRK1 gene – another receptor tyrosine kinase - are found infrequently in PTC, probably in less than 5% of cases. Several fusion partners of NTRK1 have been described, and as with RET/PTC, the common feature is their ability to constitutively dimerize and thereby inappropriately activate the NTRK kinase (Bongarzone et al., 1989; Greco et al., 1995; Kaplan et al., 1991; Miozzo et al., 1990). Since NTRK1 rearrangements occur in a very small percentage of PTCs, data on their clinical significance is limited (Musholt et al., 2000; Rabes et al., 2000).
2.4. RAS and PAX8-PPARG
Activating RAS mutations and PAX8-PPARG translocations are the most common mutations found in FVPTCs. They also are the most common mutations in FTCs, and will be discussed under that heading.
3. Follicular thyroid carcinoma
FTCs account for the remaining differentiated thyroid carcinomas. Presently FTCs are identified by their thyroid follicular organization and by the lack of PTC nuclear features. The differences between FTCs and FVPTCs are not always clear. The diagnosis of FTC also depends on evidence of vascular or capsular invasion in the tissue sample, and therefore occasionally a minimally invasive FTC will mistakenly be classified as a benign follicular adenoma. Molecular markers that can reliably distinguish minimally invasive FTCs from adenomas have proved elusive but are continuously being sought.
3.1. RAS
The three RAS proteins H-RAS, N-RAS and K-RAS are small GTPases that regulate key cellular processes involved in growth, differentiation, survival, adhesion and migration. Downstream effectors of RAS include the RAF/MAPK pathway as noted above and the PI3K/AKT pathway. RAS exists in a GTP-bound active state or a GDP-bound inactive state, and RAS activation normally is induced by external signals such as from cell surface receptor tyrosine kinases. Constitutively activating RAS mutations probably are the most common mutations in all of cancer biology. In the thyroid, RAS mutations are present in ~40% of FTCs, 20–40% of benign adenomas, and 15–20% of PTCs – mostly FVPTC (Lemoine et al., 1988; Nikiforov, 2011; Suarez et al., 1990). Mutations in codon 61 of N-RAS are the most common, followed by mutations in codon 61 of H-RAS (Bamford et al., 2004; Namba et al., 1990; Volante et al., 2009).
Expression of mutated RAS oncoproteins transforms cells in vitro and in vivo. N-RASQ61 expression results in TSH independent proliferation and genomic instability in PCCL3 rat thyroid cell lines. Thyroid specific expression in mice results in the development of metastatic thyroid carcinoma with mixed follicular, papillary, and undifferentiated areas (Vitagliano et al., 2006). Expression of HRASV12 achieves similar results in cell lines and mice (Knauf et al., 2006; Rochefort et al., 1996; Saavedra et al., 2000).
The clinical significance of RAS mutations in thyroid carcinomas is unclear. A number of studies find correlation between RAS mutations and bone metastases, increased aggressiveness, or higher mortality across thyroid cancer types (Basolo et al., 2000; Garcia-Rostan et al., 2003; Hara et al., 1994; Karga et al., 1991; Manenti et al., 1994). On the other hand, RAS mutations also are prevalent in benign follicular adenomas, and are seen in unaggressive forms of FVPTC with few metastases (Gupta et al., 2013; Howitt et al., 2013; Zhu et al., 2003). A retrospective analysis found that RAS mutations are over-represented in differentiated thyroid carcinomas from patients with radioiodine-avid pulmonary metastases (Sabra et al., 2013), whereas BRAF mutations are over-represented in non radioiodine-avid metastatic disease. However, radioiodine-avid lung metastases were rarely cured by radioiodine therapy, and those with RAS mutations fared no better than radioiodine-avid metastases with BRAF mutations.
The broad scope of RAS actions presents many potential therapeutic targets. Farnesylthiosalicylic acid (FTS) blocks the association of RAS with the cell membrane, resulting in RAS degradation. FTS has efficacy against thyroid cancer cells in pre-clinical models (Biran et al., 2011; Levy et al., 2010; Marciano et al., 1995). FTS in combination with the multikinase inhibitor sorafenib may have some activity in patients with metastatic thyroid carcinoma (Hong et al., 2011). Other drugs may disrupt RAS actions by inhibiting downstream targets in the MAPK and/or PI3K pathways (Chan et al., 2012; Jin et al., 2011; Liu et al., 2012). As noted previously, the MEK inhibitor selumentinib increased RAI uptake in iodide-refractory patients, particularly those with RAS mutations (Ho et al., 2013).
3.2. PAX8-PPARG
PAX8-PPARG is the other major mutation found in FTC, accounting for ~35% of cases. It also is found in FVPTC and occasionally in benign follicular adenomas (Eberhardt et al., 2010). A t(2;3)(q13;p25) chromosomal translocation fuses the promoter and most of the PAX8 gene to the coding exons of the PPARG gene. Thus the PAX8-PPARG fusion protein (PPFP) is expressed under control of the PAX8 promoter, which is highly active in the thyroid (Kroll et al., 2000). PAX8 is a transcription factor that is important for thyroid development, and in the mature gland it drives the expression of many thyroid-specific genes such as those encoding thyroglobulin, thyroid peroxidase and the sodium iodide symporter. PPARG is a nuclear receptor transcription factor that is essential for adipogenesis, but is expressed at very low levels in the normal thyroid and has no known function in that organ. A separate fusion protein between CREB3L2 and PPARG has been reported in two cases of FTC (Lui et al., 2008). That two distinct fusion proteins involving PPARG have been associated with FTC suggests that modulation of PPARG-regulated pathways is important for PPFP-mediated carcinogenesis. However, the oncogenic mechanism of PPFP is poorly understood and its functional relationship to PPARG is complex.
PPFP has been shown to be a dominant negative inhibitor of PPARG-mediated gene activation in numerous transfection systems (Kroll et al., 2000; Powell et al., 2004; Yin et al., 2009, 2006). Additionally PPARG often is downregulated in other types of thyroid carcinomas, PPARG agonists have therapeutic effects in cell and mouse models of various cancers, and heterozygous PPARG deletion enhances tumorigenesis in a mouse model of non-PPFP thyroid carcinoma (Aldred et al., 2003; Kato et al., 2006; Marques et al., 2004; Park et al., 2005). Overall these studies suggest that PPARG can have tumor suppressive function, and that PPFP may contribute to thyroid carcinogenesis by impeding this activity of PPARG.
On the other hand, there also is evidence that PPFP can transactivate at least some PPARG target genes. Significant numbers of PPARG target genes are upregulated in PPFP tumor samples compared to non-PPFP FTC or normal thyroid (Giordano et al., 2006; Lacroix et al., 2005). In vitro, exogenously induced PPFP expression stimulates the promoters of some PPARG genes while repressing others or sometimes both depending on the cellular context (Au et al., 2006; Giordano et al., 2006; Powell et al., 2004). Similarly, some PPARG target genes are induced and others are repressed in a transgenic mouse model of PPFP FTC (Dobson et al., 2011). However, treatment of these mice with the PPARG agonist pioglitazone caused broad upregulation of PPARG target genes and induced adipocyte transdifferentiation in the PPFP thyroid cancer cells. PPFP functional domains therefore necessarily retain their capacity to act in a strongly PPARG-like manner.
Stable transfection of PPFP in thyroid cell lines results in increased cell division, decreased apoptosis, and increased colony growth in soft agar (Au et al., 2006; Espadinha et al., 2007; Powell et al., 2004). Thyroid specific expression of PPFP in transgenic mice results in mild thyroid hyperplasia, but PPFP expression and thyroid-specific deletion of the tumor suppressor PTEN synergize to induce metastatic FTC (Diallo-Krou et al., 2009; Dobson et al., 2011). The mouse data are consistent with the biology of human PPFP FTCs, since these cancers have increased activated AKT (Diallo-Krou et al., 2009) and PTEN is a negative regulator of AKT. However, the role of PPFP in thyroid carcinogenesis may be complex because data suggest that PPFP also may inhibit tumor growth by suppressing neovascularization (Reddi et al., 2011, 2010).
The prognostic significance of PPFP expression in FTC or FVPTC has not been extensively studied. PPFP is associated with a younger age at presentation and increased vascular invasion (French et al., 2003; Marques et al., 2004; Nikiforova et al., 2003b). However, others report that PPFP is associated with indicators of good prognosis including markers of differentiation and few metastases (Sahin et al., 2005).
PPARG agonists have been disappointing as therapeutic agents in human cancers, despite the evidence that PPARG may be a tumor suppressor. However, in the above-mentioned mouse model of PPFP FTC, pioglitazone was strongly therapeutic, eliminating metastatic disease and greatly shrinking the size of the primary tumors. In addition, the remaining thyroid cells became adipocyte-like cells, accumulating large amounts of lipid (Dobson et al., 2011). It is hypothesized that this unique transdifferentiation event underlies the anti-tumor efficacy, which reflects the fact that the drug target is PPFP, not PPARG. The results suggest pioglitazone may be an effective treatment for PPFP thyroid carcinomas in patients.
3.3. PTEN/PI3K/AKT
PTEN is a phosphatidylinositol-3,4,5-trisphosphate 3-phosphatase that functions as a negative regulator of PI3K (Fig. 2). Activated PI3K phosphorylates AKT, which then regulates a variety of cellular processes such as protein synthesis, survival and proliferation via the phosphorylation of many intermediates such as mTOR and FOXO. AKT is inappropriately active in many cancers, and as a negative regulator of AKT signaling, PTEN is an important tumor suppressor. Germline PTEN mutations cause Cowden syndrome, which is associated with increased risk of thyroid cancer, but which will not be discussed here since the focus of this review is sporadic thyroid cancers.
However, increased AKT activation also can be seen in sporadic thyroid cancers due to mutations or epigenetic changes (discussed subsequently) in PTEN or downstream components of this pathway (Orloff et al., 2013; Paes and Ringel, 2008). The overall incidence of PTEN mutation in sporadic thyroid carcinoma is ~8% in FTC, 2% in PTC, and 11% in anaplastic thyroid carcinoma (ATC) (Bamford et al., 2004; Dahia et al., 1997; Halachmi et al., 1998). Activating mutations of PIK3CA, the catalytic subunit of PI3K, are roughly as prevalent as PTEN mutations, although they occur in a mutually exclusive manner. In addition, PIK3CA gene copy number gain is found in 28% of FTCs and ~40% of ATCs, and this also might lead to increased AKT activation (Hou et al., 2007; Liu et al., 2008; Wang et al., 2007). Although inhibitors of the PI3K/AKT/mTOR pathway have shown efficacy against thyroid cancer in pre-clinical models, their success in clinical trials remains to be seen (Lin et al., 2012; Liu et al., 2011; Mandal et al., 2005; Papewalis et al., 2009).
3.4 IDH1
Isocitrate dehydrogenases 1 and 2 are enzymes of intermediary metabolism that interconvert isocitrate and 2-oxoglutarate (alpha ketoglutarate). IDH1 is cytoplasmic, whereas IDH2 is mitochondrial. Mutations in both enzymes have been described in a number of cancers, most prominently in glioblastoma multiforme and chondrosarcoma (reviewed in Losman and Kaelin, Jr., 2013). The cancer-associated mutations result in altered enzymatic activity such that 2-oxoglutarate is converted to 2-hydroxyglutarate rather than isocitrate. Evidence indicates that accumulation of 2-hydroxyglutarate is important to the role of IDH mutations in cancer, but the mechanisms by which this happens are poorly understood. Recently, IDH1 mutations have been identified in thyroid carcinomas (Hemerly et al., 2010; Murugan et al., 2010). In the study by Hemerly and colleagues, IDH1 mutations were found in 24% of follicular carcinomas, 18% of follicular variant of papillary carcinomas, and 8% of papillary carcinomas, but these differences in frequency were not significant due to small numbers of cases. IDH1 mutations also have been found in anaplastic and Hurthle cell carcinomas. The clinical significance of these mutations is not known. IDH2 mutations have yet to be identified in thyroid carcinomas.
3.5. THRB
The thyroid hormone receptor beta (THRB) mutation PV was initially knocked into mice to create a mouse model of resistance to thyroid hormone (Kaneshige et al., 2000). Homozygous or heterozygous THRB(PV) mice (in conjunction with markedly increased TSH stimulation) develop metastatic FTC, which was unforeseen (Kato et al., 2004; Suzuki et al., 2002). Mutations in THRB are generally thought to be very rare to non-existent in human PTC and FTC (Rocha et al., 2007; Takano et al., 2003), although one publication reported that nearly all PTCs contain THRB mutations (Puzianowska-Kuznicka et al., 2002).
However the mouse model remains useful to tease out how other clinically relevant pathways interact to cause human FTC (Guigon and Cheng, 2009). The important role of the PI3K/AKT pathway in human FTC is recapitulated in these mice, along with the discovery of a novel interaction between THRB and PI3K regulatory subunit p85 (Furuya et al., 2007, 2006; Guigon et al., 2009; Kim et al., 2005; Saji et al., 2011). Downstream of AKT, beta-catenin is protected from degradation by THRB(PV) and unliganded wildtype THRB, but not liganded THRB, suggesting a role for thyroid hormone in beta-catenin regulation (Guigon et al., 2008).
4. Anaplastic thyroid carcinoma
Anaplastic thyroid carcinoma is the deadliest subtype of thyroid cancer. It is rare, accounting for at most 2% of cases but causes up to 50% of thyroid cancer deaths (Nagaiah et al., 2011). Mean survival time is ~5 months from diagnosis. ATCs are completely undifferentiated, aggressive, and therapy resistant. Treatment strategies are mostly palliative or designed to prevent airway compromise. Many patients present with widespread metastases but there are no effective systemic therapies (Smallridge et al., 2012). Targeted therapies may provide much needed options.
4.1. RAS, BRAF, PTEN, PI3K
A number of mutations described previously in differentiated thyroid carcinomas also are found in ATC. RAS mutations are found in 27% of cases, BRAF in 25%, PTEN in 11%, and PI3K catalytic subunit PIK3CA in 12% (Bamford et al., 2004; Smallridge et al., 2009). RET gene amplifications are reported, but not RET/PTC (Nakashima et al., 2007). The presence of these genetic changes alongside those more exclusively associated with ATC suggests a stepwise accumulation of mutations and dedifferentiated phenotypes (Nikiforov, 2004). Promising preclinical data indicate that these mutations remain relevant and are viable targets of therapy (Jin et al., 2011; Kim et al., 2007; Lin et al., 2012; Nehs et al., 2012; Yeung et al., 2007)..
4.2. TP53
TP53 is a tumor suppressor known to be mutated in a large variety of cancers. TP53 mutations are associated with 55% of ATCs but not differentiated thyroid cancers (Donghi et al., 1993; Fagin et al., 1993; Nakamura et al., 1992). Acquisition of TP53 mutations is thought to drive disease progression towards anaplasia (La Perle et al., 2000; Matias-Guiu et al., 1994). TP53 is a transcription factor that modulates the expression of a large number of genes in response to cellular stresses. TP53 regulates apoptosis, cell cycle progression, DNA repair, senescence, and stem cell homeostastasis, among many other processes. Loss of TP53 function abrogates the cell's ability to enter senescence or cell death appropriately (Stegh, 2012).
Loss of TP53 in transgenic mice leads to tumor formation. TP53 null mice crossed with RET/PTC1 transgenic mice generate larger, more anaplastic and invasive thyroid tumors (La Perle et al., 2000). Thyroid-specific loss of TP53 and PTEN results in the development of undifferentiated, aggressive thyroid carcinomas with substantial gene expression overlap with human ATC (Antico Arciuch et al., 2011).
Unfortunately, TP53 loss of function through mutation or deletion is difficult to restore. A number of small molecules exist which inhibit Mdm2, a negative regulator of TP53. However, a wild-type TP53 still needs to exist in the tumor population for this strategy to be useful (Shangary et al., 2008; Vassilev et al., 2004). Those patients with genomic loss of TP53 need another option. Re-introduction of the gene via an adenovirus vector achieved expression of TP53, but only near the injection site (Lang et al., 2003).
5. Epigenetic changes in thyroid cancer
Epigenetics refers to chromosomal changes that do not involve the nucleotide sequence and that have the potential to alter gene expression and to be heritable. DNA methylation and histone modification are common examples of epigenetic changes. The effects of epigenetic modifications are powerful, yet theoretically reversible by small molecules and endogenous enzymes, making them attractive targets to turn the course of malignant transformation.
5.1. DNA methylation
DNA methylation – the addition of a methyl group to the 5 position of cytosine – can lead to transcriptional silencing of a gene. The mechanism was first proposed for X chromosome silencing, and later shown to be at work much more widely (Li et al., 1993; Riggs, 1975). DNA methylation can be permanent and heritable, a mechanism to prevent reversion to less differentiated states. However, methylation patterns in many cancers including thyroid cancer are unstable, which may contribute to dedifferentiation as well as to the generation tumor heterogeneity (Hansen et al., 2011).
Methylation of tumor suppressor genes and markers of differentiation is common during oncogenesis. P16INK4A, RASSF members 1A, 2, and 10, and PTEN are downregulated in thyroid cancer by promoter methylation (Elisei et al., 1998; Hou et al., 2008; Schagdarsurengin et al., 2010, 2009, 2002). PTEN often is methylated in FTC and exhibits a pattern of increasing methylation with decreasing differentiation. Genes associated with differentiated function such as SLC5A5 (NIS) and NKX2-1 also are hypermethylated in undifferentiated thyroid cancer (Kondo et al., 2009; Venkataraman et al., 1999).
While methylation pattern changes are likely part of the transcriptional programs driving oncogenesis, specific mutations in the methylation pathway also can contribute to the disease state. Mutations in methylenetetrahydrofolate reductase (MTHFR), an enzyme in the folic acid metabolism pathway, can lead to defects in DNA methylation (Goyette et al., 1994; Paz et al., 2002). MTHFR mutations are associated with increased risk of thyroid carcinoma (Fard-Esfahani et al., 2011; Ozdemir et al., 2012; Prasad and Wilkhoo, 2011).
Treatment with 5-azacytidine or other DNA methyltransferase inhibitors can reverse the transcriptional repression of DNA methylation. THRB and RAP1GAP are frequently hypermethylated; 5 azacytidine restored their expression (W. G. Kim et al., 2013; Zuo et al., 2010). However, the efficacy of 5 azacytidine or similar drugs as tools for redifferentiation is not well established (Dom et al., 2013; Vivaldi et al., 2009), and these agents would likely seem to be too non-specific in their effects.
5.2. Histone modifications
Histones are the main protein component of chromosome structure, being the spool around which short segments of DNA are bound. Post-translational modifications of histones are numerous and include ADP ribosylation, acetylation, methylation, phosphorylation, ubiquitination and SUMOylation. These modifications affect transcriptional access to genes by changing DNA conformation. For example, histone acetylation can cause activation of gene transcription, deacetylation its arrest.
Global dysregulation of histone modifications is a feature and contributing factor of cancer (Chi et al., 2010). Acetylation at lysines 9–14 of histone H3 is higher in thyroid cancers compared to normal, and also higher in response to oncogene expression (Puppin et al., 2011).
It is not well known how histone modifications at specific sites in the genome contribute to thyroid carcinoma. However, treatment of thyroid cancer cell lines with histone deacetylase (HDAC) inhibitors changes the expression levels of key genes. HDAC inhibitors increase the transcript levels of markers of thyroid differentiation such as NIS, TPO, thyroglobulin and PAX8, making these possible reagents for restoring RAI avidity or effecting re-differentiation (Furuya et al., 2004; Kitazono et al., 2001; Pugliese et al., 2013; Puppin et al., 2005; Zarnegar et al., 2002). The HDAC inhibitor romidepsin restored some degree of RAI uptake in 2 of 16 patients in a clinical trial, although subsequent therapy with RAI did not result in improvement in the overall disease status of these two patients (Sherman et al., 2012).
The broad actions of HDAC inhibitors also can increase tumor sensitivity to other treatments or be cytotoxic themselves. Valproic acid increases ATC sensitivity to doxorubicin and imatinib (Catalano et al., 2009, 2006). HDAC inhibitors alone induce ATC cell death in cell culture and xenograft models (Borbone et al., 2010; Catalano et al., 2012). A patient was apparently cured of ATC with multimodality therapy consisting of valproic acid, cisplatin, doxorubicin, radiation therapy and surgery (Noguchi et al., 2009).
Though not a direct epigenetic effect, inhibition of aurora kinases is another promising approach. Aurora kinases phosphorylate histones during mitosis to ensure correct chromosome segregation. Aurora kinases are overexpressed in thyroid carcinomas (Sorrentino et al., 2005). Multiple aurora kinase inhibitors show antitumor activity in ATC cell lines and mouse models by inducing mitotic catastrophe (Arlot-Bonnemains et al., 2008; Baldini et al., 2012; Libertini et al., 2011; Wunderlich et al., 2011).
5.3. Non-coding RNA
The mammalian transcriptome includes many RNA molecules that do not encode proteins. In fact, it is estimated that non-coding RNA transcripts outnumber mRNAs 10:1. A number of these play a role in epigenetic processes (Lee, 2012).
Long non-coding RNAs (lncRNAs) can bind to specific genomic DNA regions and direct epigenetic changes to chromosome structure. For example, HOTAIR is a lncRNA that binds to Polycomb Repressive Complex 2 and mislocalizes it on the genome, resulting in inappropriate gene silencing via histone H3 lysine 27 methylation. HOTAIR is overexpressed in a number of cancers and is predictive of aggressive disease and death. Loss of HOTAIR conversely inhibits metastatic potential (Gupta et al., 2010; K Kim et al., 2013; Kogo et al., 2011; Niinuma et al., 2012; Yang et al., 2011). PTEN is reportedly a target of HOTAIR (Li et al., 2013). However, HOTAIR expression has not been studied in thyroid carcinoma.
PTC susceptibility candidate 3 (PTCSC3) is a thyroid-specific lncRNA that is dramatically downregulated in PTC and appears to function as a tumor suppressor, possibly by regulating the activities of miRNAs (Fan et al., 2013; Jendrzejewski et al., 2012). NAMA is a lncRNA whose genomic locus exhibits loss of heterozygosity in thyroid carcinoma and whose expression is downregulated by BRAF activation (BRAFV600E) (Yoon et al., 2007). However, the function of NAMA has not been characterized.
6. Conclusion
Cancer is a disease of accumulated DNA damage whose progression is thought to be driven by the acquisition of additional genetic abnormalities. Understanding how these changes contribute to the disease process is essential to the development of novel prognostic and therapeutic strategies. The association of recurrent mutations with specific histological subtypes of thyroid carcinoma corroborates the visual acumen of past pathologists while offering new ways to confront ambiguous subtypes such as FVPTC. Targeting recurrent mutations singly can result in transient relief from disease progression. Combination therapy addressing a more comprehensive catalogue of driving mutations may achieve more durable responses and perhaps cures. Finally, epigenetic control of the tumor genome also is an attractive target to reverse disease progression.
Highlights
Most thyroid carcinomas contain one of a small number of recurring gene mutations.
Specific mutations correlate with histological subtypes of thyroid carcinoma.
Gene mutations provide insight into thyroid carcinoma pathogenesis.
Thyroid carcinoma gene mutations are potential drug targets.
Acknowledgements
This work was supported by NIH grants R01CA151842 and R01CA166033.
Abbreviations
- ATC
anaplastic thyroid carcinoma
- DTC
differentiated thyroid carcinomas
- FTC
follicular thyroid carcinoma
- FTS
farnesylthiosalicylic acid
- FVPTC
follicular variant papillary thyroid carcinoma
- HDAC
histone deacetylase
- IDH
isocitrate dehydrogenase
- lncRNA
long non-coding RNA
- MAPK
mitogen activated protein kinase
- MTHFR
methylenetetrahydrofolate reductase
- NIS
sodium iodide symporter
- PI3K
phosphatidylinositide 3-kinase
- PPFP
PAX8-PPARG fusion protein
- PTC
papillary thyroid carcinoma
- RAI
radioactive iodine
- TCV
tall cell variant
Footnotes
Publisher's Disclaimer: This is a PDF le of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its nal citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adeniran AJ, Zhu Z, Gandhi M, Steward DL, Fidler JP, Giordano TJ, Biddinger PW, Nikiforov YE. Correlation between genetic alterations and microscopic features, clinical manifestations, and prognostic characteristics of thyroid papillary carcinomas. Am. J. Surg. Pathol. 2006;30:216–222. doi: 10.1097/01.pas.0000176432.73455.1b. [DOI] [PubMed] [Google Scholar]
- Aldred MA, Morrison C, Gimm O, Hoang-Vu C, Krause U, Dralle H, Jhiang S, Eng C. Peroxisome proliferator-activated receptor gamma is frequently downregulated in a diversity of sporadic nonmedullary thyroid carcinomas. Oncogene. 2003;22:3412–3416. doi: 10.1038/sj.onc.1206400. [DOI] [PubMed] [Google Scholar]
- Alipov G, Ito M, Prouglo Y, Takamura N, Yamashita S. Ret proto-oncogene rearrangement in thyroid cancer around Semipalatinsk nuclear testing site. Lancet. 1999;354:1528–1529. doi: 10.1016/S0140-6736(99)03548-5. [DOI] [PubMed] [Google Scholar]
- Antico Arciuch VG, Russo MA, Dima M, Kang KS, Dasrath F, Liao XH, Refetoff S, Montagna C, Di Cristofano A. Thyrocyte-specific inactivation of p53 and Pten results in anaplastic thyroid carcinomas faithfully recapitulating human tumors. Oncotarget. 2011;2:1109–26. doi: 10.18632/oncotarget.380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arlot-Bonnemains Y, Baldini E, Martin B, Delcros J-G, Toller M, Curcio F, Ambesi-Impiombato FS, D'Armiento M, Ulisse S. Effects of the Aurora kinase inhibitor VX-680 on anaplastic thyroid cancer-derived cell lines. Endocr. Relat. Cancer. 2008;15:559–568. doi: 10.1677/ERC-08-0021. [DOI] [PubMed] [Google Scholar]
- Au AYM, McBride C, Wilhelm KG, Koenig RJ, Speller B, Cheung L, Messina M, Wentworth J, Tasevski V, Learoyd D, Robinson BG, Clifton-Bligh RJ. PAX8-Peroxisome Proliferator-Activated Receptor γ (PPARγ) Disrupts Normal PAX8 or PPARγ Transcriptional Function and Stimulates Follicular Thyroid Cell Growth. Endocrinology. 2006;147:367–376. doi: 10.1210/en.2005-0147. [DOI] [PubMed] [Google Scholar]
- Avruch J, Khokhlatchev A, Kyriakis JM, Luo Z, Tzivion G, Vavvas D, Zhang X-F. Ras Activation of the Raf Kinase: Tyrosine Kinase Recruitment of the MAP Kinase Cascade. Recent Prog Horm Res. 2001;56:127–156. doi: 10.1210/rp.56.1.127. [DOI] [PubMed] [Google Scholar]
- Baitei EY, Zou M, Al-Mohanna F, Collison K, Alzahrani AS, Farid NR, Meyer B, Shi Y. Aberrant BRAF splicing as an alternative mechanism for oncogenic B-Raf activation in thyroid carcinoma. J. Pathol. 2009;217:707–715. doi: 10.1002/path.2496. [DOI] [PubMed] [Google Scholar]
- Baldini E, Sorrenti S, D'Armiento E, Guaitoli E, Morrone S, D'Andrea V, Gnessi L, Moretti C, Antonelli A, Catania A, De Antoni E, Ulisse S. Effects of the Aurora kinases pan-inhibitor SNS-314 mesylate on anaplastic thyroid cancer derived cell lines. Clin Ter. 2012;163:e307–313. [PubMed] [Google Scholar]
- Baloch ZW, LiVolsi VA. Pathologic diagnosis of papillary thyroid carcinoma: today and tomorrow. Expert Review of Molecular Diagnostics. 2005;5:573. doi: 10.1586/14737159.5.4.573. [DOI] [PubMed] [Google Scholar]
- Bamford S, Dawson E, Forbes S, Clements J, Pettett R, Dogan A, Flanagan A, Teague J, Futreal PA, Stratton MR, Wooster R. The COSMIC (Catalogue of Somatic Mutations in Cancer) database and website. Br. J. Cancer. 2004;91:355–358. doi: 10.1038/sj.bjc.6601894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barollo S, Pennelli G, Vianello F, Watutantrige Fernando S, Negro I, Merante Boschin I, Pelizzo MR, Rugge M, Mantero F, Nacamulli D, Girelli ME, Busnardo B, Mian C. BRAF in primary and recurrent papillary thyroid cancers: the relationship with (131)I and 2-[(18)F]fluoro-2-deoxy-D-glucose uptake ability. Eur. J. Endocrinol. 2010;163:659–663. doi: 10.1530/EJE-10-0290. [DOI] [PubMed] [Google Scholar]
- Basolo F, Giannini R, Monaco C, Melillo RM, Carlomagno F, Pancrazi M, Salvatore G, Chiappetta G, Pacini F, Elisei R, Miccoli P, Pinchera A, Fusco A, Santoro M. Potent Mitogenicity of the RET/PTC3 Oncogene Correlates with Its Prevalence in Tall-Cell Variant of Papillary Thyroid Carcinoma. Am J Pathol. 2002;160:247–254. doi: 10.1016/S0002-9440(10)64368-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basolo F, Pisaturo F, Pollina LE, Fontanini G, Elisei R, Molinaro E, Iacconi P, Miccoli P, Pacini F. N-ras mutation in poorly differentiated thyroid carcinomas: correlation with bone metastases and inverse correlation to thyroglobulin expression. Thyroid. 2000;10:19–23. doi: 10.1089/thy.2000.10.19. [DOI] [PubMed] [Google Scholar]
- Biran A, Brownstein M, Haklai R, Kloog Y. Downregulation of survivin and aurora A by histone deacetylase and RAS inhibitors: a new drug combination for cancer therapy. Int. J. Cancer. 2011;128:691–701. doi: 10.1002/ijc.25367. [DOI] [PubMed] [Google Scholar]
- Bond JA, Wyllie FS, Rowson J, Radulescu A, Wynford-Thomas D. In vitro reconstruction of tumour initiation in a human epithelium. Oncogene. 1994;9:281–290. [PubMed] [Google Scholar]
- Bongarzone I, Butti MG, Coronelli S, Borrello MG, Santoro M, Mondellini P, Pilotti S, Fusco A, Della Porta G, Pierotti MA. Frequent activation of ret protooncogene by fusion with a new activating gene in papillary thyroid carcinomas. Cancer Res. 1994;54:2979–2985. [PubMed] [Google Scholar]
- Bongarzone I, Pierotti MA, Monzini N, Mondellini P, Manenti G, Donghi R, Pilotti S, Grieco M, Santoro M, Fusco A. High frequency of activation of tyrosine kinase oncogenes in human papillary thyroid carcinoma. Oncogene. 1989;4:1457–1462. [PubMed] [Google Scholar]
- Borbone E, Berlingieri MT, De Bellis F, Nebbioso A, Chiappetta G, Mai A, Altucci L, Fusco A. Histone deacetylase inhibitors induce thyroid cancer-specific apoptosis through proteasome-dependent inhibition of TRAIL degradation. Oncogene. 2010;29:105–116. doi: 10.1038/onc.2009.306. [DOI] [PubMed] [Google Scholar]
- Bounacer A, Wicker R, Caillou B, Cailleux AF, Sarasin A, Schlumberger M, Suárez HG. High prevalence of activating ret proto-oncogene rearrangements, in thyroid tumors from patients who had received external radiation. Oncogene. 1997;15:1263–1273. doi: 10.1038/sj.onc.1200206. [DOI] [PubMed] [Google Scholar]
- Bunone G, Uggeri M, Mondellini P, Pierotti MA, Bongarzone I. RET Receptor Expression in Thyroid Follicular Epithelial Cell-derived Tumors. Cancer Res. 2000;60:2845–2849. [PubMed] [Google Scholar]
- [accessed 10.4.12];Cancer of the Thyroid - SEER Stat Fact Sheets [WWW Document] n.d. URL http://seer.cancer.gov/statfacts/html/thyro.html.
- Cantwell-Dorris ER, O'Leary JJ, Sheils OM. BRAFV600E: Implications for Carcinogenesis and Molecular Therapy. Mol Cancer Ther. 2011;10:385–394. doi: 10.1158/1535-7163.MCT-10-0799. [DOI] [PubMed] [Google Scholar]
- Capdevila J, Iglesias L, Halperin I, Segura A, Martínez-Trufero J, Vaz MÁ, Corral J, Obiols G, Grande E, Grau JJ, Tabernero J. Sorafenib in metastatic thyroid cancer. Endocr. Relat. Cancer. 2012;19:209–216. doi: 10.1530/ERC-11-0351. [DOI] [PubMed] [Google Scholar]
- Caronia LM, Phay JE, Shah MH. Role of BRAF in Thyroid Oncogenesis. Clin Cancer Res. 2011;17:7511–7517. doi: 10.1158/1078-0432.CCR-11-1155. [DOI] [PubMed] [Google Scholar]
- Carr LL, Mankoff DA, Goulart BH, Eaton KD, Capell PT, Kell EM, Bauman JE, Martins RG. Phase II study of daily sunitinib in FDG-PET-positive, iodine-refractory differentiated thyroid cancer and metastatic medullary carcinoma of the thyroid with functional imaging correlation. Clin. Cancer Res. 2010;16:5260–5268. doi: 10.1158/1078-0432.CCR-10-0994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catalano MG, Fortunati N, Pugliese M, Poli R, Bosco O, Mastrocola R, Aragno M, Boccuzzi G. Valproic acid, a histone deacetylase inhibitor, enhances sensitivity to doxorubicin in anaplastic thyroid cancer cells. J. Endocrinol. 2006;191:465–472. doi: 10.1677/joe.1.06970. [DOI] [PubMed] [Google Scholar]
- Catalano MG, Pugliese M, Gargantini E, Grange C, Bussolati B, Asioli S, Bosco O, Poli R, Compagnone A, Bandino A, Mainini F, Fortunati N, Boccuzzi G. Cytotoxic activity of the histone deacetylase inhibitor panobinostat (LBH589) in anaplastic thyroid cancer in vitro and in vivo. Int. J. Cancer. 2012;130:694–704. doi: 10.1002/ijc.26057. [DOI] [PubMed] [Google Scholar]
- Catalano MG, Pugliese M, Poli R, Bosco O, Bertieri R, Fortunati N, Boccuzzi G. Effects of the histone deacetylase inhibitor valproic acid on the sensitivity of anaplastic thyroid cancer cell lines to imatinib. Oncol. Rep. 2009;21:515–521. [PubMed] [Google Scholar]
- Chan D, Tyner JW, Chng WJ, Bi C, Okamoto R, Said J, Ngan BD, Braunstein GD, Koeffler HP. Effect of dasatinib against thyroid cancer cell lines in vitro and a xenograft model in vivo. Oncol Lett. 2012;3:807–815. doi: 10.3892/ol.2012.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charles R-P, Iezza G, Amendola E, Dankort D, McMahon M. Mutationally activated BRAF(V600E) elicits papillary thyroid cancer in the adult mouse. Cancer Res. 2011;71:3863–3871. doi: 10.1158/0008-5472.CAN-10-4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi P, Allis CD, Wang GG. Covalent histone modifications--miswritten, misinterpreted and mis-erased in human cancers. Nat. Rev. Cancer. 2010;10:457–469. doi: 10.1038/nrc2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciampi R, Knauf JA, Kerler R, Gandhi M, Zhu Z, Nikiforova MN, Rabes HM, Fagin JA, Nikiforov YE. Oncogenic AKAP9-BRAF fusion is a novel mechanism of MAPK pathway activation in thyroid cancer. J. Clin. Invest. 2005;115:94–101. doi: 10.1172/JCI23237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen Y, Xing M, Mambo E, Guo Z, Wu G, Trink B, Beller U, Westra WH, Ladenson PW, Sidransky D. BRAF mutation in papillary thyroid carcinoma. J. Natl. Cancer Inst. 2003;95:625–627. doi: 10.1093/jnci/95.8.625. [DOI] [PubMed] [Google Scholar]
- Collins BJ, Chiappetta G, Schneider AB, Santoro M, Pentimalli F, Fogelfeld L, Gierlowski T, Shore-Freedman E, Jaffe G, Fusco A. RET Expression in Papillary Thyroid Cancer from Patients Irradiated in Childhood for Benign Conditions. J. Clin. Endocrinol. Metab. 2002;87:3941–3946. doi: 10.1210/jcem.87.8.8748. [DOI] [PubMed] [Google Scholar]
- Cooper DS, Doherty GM, Haugen BR, Kloos RT, Lee SL, Mandel SJ, Mazzaferri EL, McIver B, Pacini F, Schlumberger M, Sherman SI, Steward DL, Tuttle RM. Revised American Thyroid Association Management Guidelines for Patients with Thyroid Nodules and Differentiated Thyroid Cancer. Thyroid. 2009;19:1167–1214. doi: 10.1089/thy.2009.0110. [DOI] [PubMed] [Google Scholar]
- Croyle M, Akeno N, Knauf JA, Fabbro D, Chen X, Baumgartner JE, Lane HA, Fagin JA. RET/PTC-Induced Cell Growth Is Mediated in Part by Epidermal Growth Factor Receptor (EGFR) Activation: Evidence for Molecular and Functional Interactions Between RET and EGFR. Cancer Res. 2008;68:4183–4191. doi: 10.1158/0008-5472.CAN-08-0413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahia PL, Marsh DJ, Zheng Z, Zedenius J, Komminoth P, Frisk T, Wallin G, Parsons R, Longy M, Larsson C, Eng C. Somatic deletions and mutations in the Cowden disease gene, PTEN, in sporadic thyroid tumors. Cancer Res. 1997;57:4710–4713. [PubMed] [Google Scholar]
- Diallo-Krou E, Yu J, Colby LA, Inoki K, Wilkinson JE, Thomas DG, Giordano TJ, Koenig RJ. Paired Box Gene 8-Peroxisome Proliferator-Activated Receptor-γ Fusion Protein and Loss of Phosphatase and Tensin Homolog Synergistically Cause Thyroid Hyperplasia in Transgenic Mice. Endocrinology. 2009;150:5181–5190. doi: 10.1210/en.2009-0701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobson ME, Diallo-Krou E, Grachtchouk V, Yu J, Colby LA, Wilkinson JE, Giordano TJ, Koenig RJ. Pioglitazone Induces a Proadipogenic Antitumor Response in Mice with PAX8-PPARγ Fusion Protein Thyroid Carcinoma. Endocrinology. 2011;152:4455–4465. doi: 10.1210/en.2011-1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dom G, Galdo VC, Tarabichi M, Tomás G, Hébrant A, Andry G, De Martelar V, Libert F, Leteurtre E, Dumont JE, Maenhaut C, van Staveren WCG. 5-aza-2'-deoxycytidine has minor effects on differentiation in human thyroid cancer cell lines, but modulates genes that are involved in adaptation in vitro. Thyroid. 2013;23:317–328. doi: 10.1089/thy.2012.0388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donghi R, Longoni A, Pilotti S, Michieli P, Della Porta G, Pierotti MA. Gene p53 mutations are restricted to poorly differentiated and undifferentiated carcinomas of the thyroid gland. J. Clin. Invest. 1993;91:1753–1760. doi: 10.1172/JCI116385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durante C, Puxeddu E, Ferretti E, Morisi R, Moretti S, Bruno R, Barbi F, Avenia N, Scipioni A, Verrienti A, Tosi E, Cavaliere A, Gulino A, Filetti S, Russo D. BRAF Mutations in Papillary Thyroid Carcinomas Inhibit Genes Involved in Iodine Metabolism. J. Clin. Endocrinol. Metab. 2007;92:2840–2843. doi: 10.1210/jc.2006-2707. [DOI] [PubMed] [Google Scholar]
- Eberhardt NL, Grebe SKG, McIver B, Reddi HV. The Role of the PAX8/PPARγ Fusion Oncogene in the Pathogenesis of Follicular Thyroid Cancer. Mol Cell Endocrinol. 2010;321:50–56. doi: 10.1016/j.mce.2009.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elisei R, Shiohara M, Koeffler HP, Fagin JA. Genetic and epigenetic alterations of the cyclin-dependent kinase inhibitors p15INK4b and p16INK4a in human thyroid carcinoma cell lines and primary thyroid carcinomas. Cancer. 1998;83:2185–2193. [PubMed] [Google Scholar]
- Elisei R, Viola D, Torregrossa L, Giannini R, Romei C, Ugolini C, Molinaro E, Agate L, Biagini A, Lupi C, Valerio L, Materazzi G, Miccoli P, Piaggi P, Pinchera A, Vitti P, Basolo F. The BRAFV600E Mutation Is an Independent, Poor Prognostic Factor for the Outcome of Patients with Low-Risk Intrathyroid Papillary Thyroid Carcinoma: Single-Institution Results from a Large Cohort Study. J. Clin. Endocrinol. Metab. 2012;97:4390–4398. doi: 10.1210/jc.2012-1775. [DOI] [PubMed] [Google Scholar]
- Espadinha C, Cavaco BM, Leite V. PAX8PPARgamma stimulates cell viability and modulates expression of thyroid-specific genes in a human thyroid cell line. Thyroid. 2007;17:497–509. doi: 10.1089/thy.2006.0263. [DOI] [PubMed] [Google Scholar]
- Fagin JA, Matsuo K, Karmakar A, Chen DL, Tang SH, Koeffler HP. High prevalence of mutations of the p53 gene in poorly differentiated human thyroid carcinomas. J. Clin. Invest. 1993;91:179–184. doi: 10.1172/JCI116168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan M, Li X, Jiang W, Huang Y, Li J, Wang Z. A long non-coding RNA, PTCSC3, as a tumor suppressor and a target of miRNAs in thyroid cancer cells. Exp Ther Med. 2013;5:1143–1146. doi: 10.3892/etm.2013.933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fard-Esfahani P, Fard-Esfahani A, Saidi P, Fayaz S, Mohabati R, Majdi M. An increased risk of differentiated thyroid carcinoma in Iran with the 677C→T homozygous polymorphism in the MTHFR Gene. Cancer Epidemiol. 2011;35:56–58. doi: 10.1016/j.canep.2010.10.001. [DOI] [PubMed] [Google Scholar]
- Fernandez IJ, Piccin O, Sciascia S, Cavicchi O, Repaci A, Vicennati V, Fiorentino M. Clinical Significance of BRAF Mutation in Thyroid Papillary Cancer. Otolaryngol. Head Neck Surg. 2013;148:919–25. doi: 10.1177/0194599813481942. [DOI] [PubMed] [Google Scholar]
- Frattini M, Ferrario C, Bressan P, Balestra D, De Cecco L, Mondellini P, Bongarzone I, Collini P, Gariboldi M, Pilotti S, Pierotti MA, Greco A. Alternative mutations of BRAF, RET and NTRK1 are associated with similar but distinct gene expression patterns in papillary thyroid cancer. Oncogene. 2004;23:7436–7440. doi: 10.1038/sj.onc.1207980. [DOI] [PubMed] [Google Scholar]
- French CA, Alexander EK, Cibas ES, Nose V, Laguette J, Faquin W, Garber J, Moore F, Fletcher JA, Larsen PR, Kroll TG. Genetic and Biological Subgroups of Low-Stage Follicular Thyroid Cancer. Am J Pathol. 2003;162:1053–1060. doi: 10.1016/S0002-9440(10)63902-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fugazzola L, Pilotti S, Pinchera A, Vorontsova TV, Mondellini P, Bongarzone I, Greco A, Astakhova L, Butti MG, Demidchik EP. Oncogenic rearrangements of the RET proto-oncogene in papillary thyroid carcinomas from children exposed to the Chernobyl nuclear accident. Cancer Res. 1995;55:5617–5620. [PubMed] [Google Scholar]
- Furuya F, Hanover JA, Cheng S. Activation of phosphatidylinositol 3-kinase signaling by a mutant thyroid hormone beta receptor. Proc. Natl. Acad. Sci. U.S.A. 2006;103:1780–1785. doi: 10.1073/pnas.0510849103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuya F, Lu C, Willingham MC, Cheng S-Y. Inhibition of phosphatidylinositol 3-kinase delays tumor progression and blocks metastatic spread in a mouse model of thyroid cancer. Carcinogenesis. 2007;28:2451–2458. doi: 10.1093/carcin/bgm174. [DOI] [PubMed] [Google Scholar]
- Furuya F, Shimura H, Suzuki H, Taki K, Ohta K, Haraguchi K, Onaya T, Endo T, Kobayashi T. Histone deacetylase inhibitors restore radioiodide uptake and retention in poorly differentiated and anaplastic thyroid cancer cells by expression of the sodium/iodide symporter thyroperoxidase and thyroglobulin. Endocrinology. 2004;145:2865–2875. doi: 10.1210/en.2003-1258. [DOI] [PubMed] [Google Scholar]
- Gandhi M, Dillon LW, Pramanik S, Nikiforov YE, Wang Y-H. DNA Breaks at Fragile Sites Generate Oncogenic RET/PTC Rearrangements in Human Thyroid Cells. Oncogene. 2010a;29:2272–2280. doi: 10.1038/onc.2009.502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi M, Evdokimova V, Nikiforov YE. Mechanisms of chromosomal rearrangements in solid tumors: the model of papillary thyroid carcinoma. Mol Cell Endocrinol. 2010b;321:36–43. doi: 10.1016/j.mce.2009.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi M, Evdokimova V, Nikiforov YE. Frequency of close positioning of chromosomal loci detected by FRET correlates with their participation in carcinogenic rearrangements in human cells. Genes Chromosomes Cancer. 2012;51:1037–1044. doi: 10.1002/gcc.21988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao WL, Wie LL, Chao YG, Wie L, Song TL. Prognostic prediction of BRAF(V600E) and its relationship with sodium iodide symporter in classic variant of papillary thyroid carcinomas. Clin. Lab. 2012;58:919–926. [PubMed] [Google Scholar]
- Garcia-Rostan G, Zhao H, Camp RL, Pollan M, Herrero A, Pardo J, Wu R, Carcangiu ML, Costa J, Tallini G. Ras mutations are associated with aggressive tumor phenotypes and poor prognosis in thyroid cancer. J. Clin. Oncol. 2003;21:3226–3235. doi: 10.1200/JCO.2003.10.130. [DOI] [PubMed] [Google Scholar]
- Giordano TJ, Au AYM, Kuick R, Thomas DG, Rhodes DR, Wilhelm KG, Vinco M, Misek DE, Sanders D, Zhu Z, Ciampi R, Hanash S, Chinnaiyan A, Clifton-Bligh RJ, Robinson BG, Nikiforov YE, Koenig RJ. Delineation, Functional Validation, and Bioinformatic Evaluation of Gene Expression in Thyroid Follicular Carcinomas with the PAX8-PPARG Translocation. Clin. Cancer Res. 2006;12:1983–1993. doi: 10.1158/1078-0432.CCR-05-2039. [DOI] [PubMed] [Google Scholar]
- Goyette P, Sumner JS, Milos R, Duncan AM, Rosenblatt DS, Matthews RG, Rozen R. Human methylenetetrahydrofolate reductase: isolation of cDNA, mapping and mutation identification. Nat. Genet. 1994;7:195–200. doi: 10.1038/ng0694-195. [DOI] [PubMed] [Google Scholar]
- Greco A, Mariani C, Miranda C, Lupas A, Pagliardini S, Pomati M, Pierotti MA. The DNA rearrangement that generates the TRK-T3 oncogene involves a novel gene on chromosome 3 whose product has a potential coiled-coil domain. Mol. Cell. Biol. 1995;15:6118–6127. doi: 10.1128/mcb.15.11.6118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grieco M, Santoro M, Berlingieri MT, Melillo RM, Donghi R, Bongarzone I, Pierotti MA, Della Porta G, Fusco A, Vecchio G. PTC is a novel rearranged form of the ret proto-oncogene and is frequently detected in vivo in human thyroid papillary carcinomas. Cell. 1990;60:557–563. doi: 10.1016/0092-8674(90)90659-3. [DOI] [PubMed] [Google Scholar]
- Guerra A, Sapio MR, Marotta V, Campanile E, Rossi S, Forno I, Fugazzola L, Budillon A, Moccia T, Fenzi G, Vitale M. The Primary Occurrence of BRAFV600E Is a Rare Clonal Event in Papillary Thyroid Carcinoma. J. Clin. Endocrinol. Metab. 2012;97:517–524. doi: 10.1210/jc.2011-0618. [DOI] [PubMed] [Google Scholar]
- Guigon CJ, Cheng S. Novel Nongenomic Signaling of Thyroid Hormone Receptors in Thyroid Carcinogenesis. Mol. Cell. Endocrinol. 2009;308:63–69. doi: 10.1016/j.mce.2009.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guigon CJ, Zhao L, Lu C, Willingham MC, Cheng S. Regulation of β-Catenin by a Novel Nongenomic Action of Thyroid Hormone β Receptor. Mol. Cell. Biol. 2008;28:4598–4608. doi: 10.1128/MCB.02192-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guigon CJ, Zhao L, Willingham MC, Cheng S-Y. PTEN deficiency accelerates tumour progression in a mouse model of thyroid cancer. Oncogene. 2009;28:509–517. doi: 10.1038/onc.2008.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta-Abramson V, Troxel AB, Nellore A, Puttaswamy K, Redlinger M, Ransone K, Mandel SJ, Flaherty KT, Loevner LA, O'Dwyer PJ, Brose MS. Phase II Trial of Sorafenib in Advanced Thyroid Cancer. J. Clin. Oncol. 2008;26:4714–4719. doi: 10.1200/JCO.2008.16.3279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta N, Dasyam AK, Carty SE, Nikiforova MN, Ohori NP, Armstrong M, Yip L, Lebeau SO, McCoy KL, Coyne C, Stang MT, Johnson J, Ferris RL, Seethala R, Nikiforov YE, Hodak SP. RAS Mutations in Thyroid FNA Specimens Are Highly Predictive of Predominantly Low-Risk Follicular-Pattern Cancers. J. Clin. Endocrinol. Metab. 2013;98:E914–922. doi: 10.1210/jc.2012-3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta RA, Shah N, Wang KC, Kim J, Horlings HM, Wong DJ, Tsai M-C, Hung T, Argani P, Rinn JL, Wang Y, Brzoska P, Kong B, Li R, West RB, van de Vijver MJ, Sukumar S, Chang HY. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature. 2010;464:1071–1076. doi: 10.1038/nature08975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halachmi N, Halachmi S, Evron E, Cairns P, Okami K, Saji M, Westra WH, Zeiger MA, Jen J, Sidransky D. Somatic mutations of the PTEN tumor suppressor gene in sporadic follicular thyroid tumors. Genes Chromosomes Cancer. 1998;23:239–243. doi: 10.1002/(sici)1098-2264(199811)23:3<239::aid-gcc5>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- Hansen KD, Timp W, Bravo HC, Sabunciyan S, Langmead B, McDonald OG, Wen B, Wu H, Liu Y, Diep D, Briem E, Zhang K, Irizarry RA, Feinberg AP. Increased methylation variation in epigenetic domains across cancer types. Nat. Genet. 2011;43:768–775. doi: 10.1038/ng.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara H, Fulton N, Yashiro T, Ito K, DeGroot LJ, Kaplan EL. N-ras mutation: an independent prognostic factor for aggressiveness of papillary thyroid carcinoma. Surgery. 1994;116:1010–1016. [PubMed] [Google Scholar]
- Hemerly JP, Bastos AU, Cerutti JM. Identification of several novel non-p.R132 IDH1 variants in thyroid carcinomas. Eur J Endocrinol. 2010;163:747–755. doi: 10.1530/EJE-10-0473. [DOI] [PubMed] [Google Scholar]
- Ho AL, Grewal RK, Leboeuf R, Sherman EJ, Pfister DG, Deandreis D, Pentlow KS, Zanzonico PB, Haque S, Gavane S, Ghossein RA, Ricarte-Filho JC, Domínguez JM, Shen R, Tuttle RM, Larson SM, Fagin JA. Selumetinib-enhanced radioiodine uptake in advanced thyroid cancer. N. Engl. J. Med. 2013;368:623–632. doi: 10.1056/NEJMoa1209288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoftijzer H, Heemstra KA, Morreau H, Stokkel MP, Corssmit EP, Gelderblom H, Weijers K, Pereira AM, Huijberts M, Kapiteijn E, Romijn JA, Smit JW. Beneficial effects of sorafenib on tumor progression, but not on radioiodine uptake, in patients with differentiated thyroid carcinoma. Eur J Endocrinol. 2009;161:923–931. doi: 10.1530/EJE-09-0702. [DOI] [PubMed] [Google Scholar]
- Hong DS, Cabanillas ME, Wheler J, Naing A, Tsimberidou AM, Ye L, Waguespack SG, Hernandez M, Naggar AKE, Bidyasar S, Wright J, Sherman SI, Kurzrock R. Inhibition of the Ras/Raf/MEK/ERK and RET Kinase Pathways with the Combination of the Multikinase Inhibitor Sorafenib and the Farnesyltransferase Inhibitor Tipifarnib in Medullary and Differentiated Thyroid Malignancies. J. Clin. Endocrinol. Metab. 2011;96:997–1005. doi: 10.1210/jc.2010-1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou P, Ji M, Xing M. Association of PTEN gene methylation with genetic alterations in the phosphatidylinositol 3-kinase/AKT signaling pathway in thyroid tumors. Cancer. 2008;113:2440–2447. doi: 10.1002/cncr.23869. [DOI] [PubMed] [Google Scholar]
- Hou P, Liu D, Shan Y, Hu S, Studeman K, Condouris S, Wang Y, Trink A, El-Naggar AK, Tallini G, Vasko V, Xing M. Genetic alterations and their relationship in the phosphatidylinositol 3-kinase/Akt pathway in thyroid cancer. Clin. Cancer Res. 2007;13:1161–1170. doi: 10.1158/1078-0432.CCR-06-1125. [DOI] [PubMed] [Google Scholar]
- Howitt BE, Jia Y, Sholl LM, Barletta J. Molecular alterations in partially-encapsulated/well-circumscribed follicular variant of papillary thyroid carcinoma. Thyroid. 2013 doi: 10.1089/thy.2013.0018. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Ito T, Seyama T, Iwamoto KS, Hayashi T, Mizuno T, Tsuyama N, Dohi K, Nakamura N, Akiyama M. In Vitro Irradiation Is Able to Cause RET Oncogene Rearrangement. Cancer Res. 1993;53:2940–2943. [PubMed] [Google Scholar]
- Jendrzejewski J, He H, Radomska HS, Li W, Tomsic J, Liyanarachchi S, Davuluri RV, Nagy R, de la Chapelle A. The polymorphism rs944289 predisposes to papillary thyroid carcinoma through a large intergenic noncoding RNA gene of tumor suppressor type. Proc. Natl. Acad. Sci. U.S.A. 2012;109:8646–8651. doi: 10.1073/pnas.1205654109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jhiang SM, Sagartz JE, Tong Q, Parker-Thornburg J, Capen CC, Cho JY, Xing S, Ledent C. Targeted expression of the ret/PTC1 oncogene induces papillary thyroid carcinomas. Endocrinology. 1996;137:375–378. doi: 10.1210/endo.137.1.8536638. [DOI] [PubMed] [Google Scholar]
- Jin N, Jiang T, Rosen DM, Nelkin BD, Ball DW. Synergistic action of a RAF inhibitor and a dual PI3K/mTOR inhibitor in thyroid cancer. Clin. Cancer Res. 2011;17:6482–6489. doi: 10.1158/1078-0432.CCR-11-0933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo YS, Li S, Song JH, Kwon KH, Lee JC, Rha SY, Lee HJ, Sul JY, Kweon GR, Ro H-K, Kim J-M, Shong M. Influence of the BRAF V600E mutation on expression of vascular endothelial growth factor in papillary thyroid cancer. J. Clin. Endocrinol. Metab. 2006;91:3667–3670. doi: 10.1210/jc.2005-2836. [DOI] [PubMed] [Google Scholar]
- Kaneshige M, Kaneshige K, Zhu X, Dace A, Garrett L, Carter TA, Kazlauskaite R, Pankratz DG, Wynshaw-Boris A, Refetoff S, Weintraub B, Willingham MC, Barlow C, Cheng S. Mice with a targeted mutation in the thyroid hormone beta receptor gene exhibit impaired growth and resistance to thyroid hormone. Proc. Natl. Acad. Sci. U.S.A. 2000;97:13209–13214. doi: 10.1073/pnas.230285997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan DR, Hempstead BL, Martin-Zanca D, Chao MV, Parada LF. The trk protooncogene product: a signal transducing receptor for nerve growth factor. Science. 1991;252:554–558. doi: 10.1126/science.1850549. [DOI] [PubMed] [Google Scholar]
- Karga H, Lee JK, Vickery AL, Jr, Thor A, Gaz RD, Jameson JL. Ras oncogene mutations in benign and malignant thyroid neoplasms. J. Clin. Endocrinol. Metab. 1991;73:832–836. doi: 10.1210/jcem-73-4-832. [DOI] [PubMed] [Google Scholar]
- Kato Y, Ying H, Willingham MC, Cheng S-Y. A Tumor Suppressor Role for Thyroid Hormone β Receptor in a Mouse Model of Thyroid Carcinogenesis. Endocrinology. 2004;145:4430–4438. doi: 10.1210/en.2004-0612. [DOI] [PubMed] [Google Scholar]
- Kato Y, Ying H, Zhao L, Furuya F, Araki O, Willingham MC, Cheng S-Y. PPARgamma insufficiency promotes follicular thyroid carcinogenesis via activation of the nuclear factor-kappaB signaling pathway. Oncogene. 2006;25:2736–2747. doi: 10.1038/sj.onc.1209299. [DOI] [PubMed] [Google Scholar]
- Kim CS, Vasko VV, Kato Y, Kruhlak M, Saji M, Cheng S-Y, Ringel MD. AKT activation promotes metastasis in a mouse model of follicular thyroid carcinoma. Endocrinology. 2005;146:4456–4463. doi: 10.1210/en.2005-0172. [DOI] [PubMed] [Google Scholar]
- Kim K, Cabanillas M, Lazar AJ, Williams MD, Sanders DL, Ilagan JL, Nolop K, Lee RJ, Sherman SI. Clinical Responses to Vemurafenib in Patients with Metastatic Papillary Thyroid Cancer Harboring V600EBRAF Mutation. Thyroid. 2013 doi: 10.1089/thy.2013.0057. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K, Jutooru I, Chadalapaka G, Johnson G, Frank J, Burghardt R, Kim S, Safe S. HOTAIR is a negative prognostic factor and exhibits pro-oncogenic activity in pancreatic cancer. Oncogene. 2013;32:1616–1625. doi: 10.1038/onc.2012.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Yazici YD, Calzada G, Wang Z-Y, Younes MN, Jasser SA, El-Naggar AK, Myers JN. Sorafenib inhibits the angiogenesis and growth of orthotopic anaplastic thyroid carcinoma xenografts in nude mice. Mol. Cancer Ther. 2007;6:1785–1792. doi: 10.1158/1535-7163.MCT-06-0595. [DOI] [PubMed] [Google Scholar]
- Kim TH, Park YJ, Lim JA, Ahn HY, Lee EK, Lee YJ, Kim KW, Hahn SK, Youn YK, Kim KH, Cho BY, Park DJ. The association of the BRAF(V600E) mutation with prognostic factors and poor clinical outcome in papillary thyroid cancer: a meta-analysis. Cancer. 2012;118:1764–1773. doi: 10.1002/cncr.26500. [DOI] [PubMed] [Google Scholar]
- Kim WG, Zhu X, Kim DW, Zhang L, Kebebew E, Cheng S-Y. Reactivation of the silenced thyroid hormone receptor β gene expression delays thyroid tumor progression. Endocrinology. 2013;154:25–35. doi: 10.1210/en.2012-1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitazono M, Robey R, Zhan Z, Sarlis NJ, Skarulis MC, Aikou T, Bates S, Fojo T. Low concentrations of the histone deacetylase inhibitor, depsipeptide (FR901228), increase expression of the Na(+)/I(−) symporter and iodine accumulation in poorly differentiated thyroid carcinoma cells. J. Clin. Endocrinol. Metab. 2001;86:3430–3435. doi: 10.1210/jcem.86.7.7621. [DOI] [PubMed] [Google Scholar]
- Kloos RT, Ringel MD, Knopp MV, Hall NC, King M, Stevens R, Liang J, Wakely PE, Vasko VV, Saji M, Rittenberry J, Wei L, Arbogast D, Collamore M, Wright JJ, Grever M, Shah MH. Phase II Trial of Sorafenib in Metastatic Thyroid Cancer. J. Clin. Oncol. 2009;27:1675–1684. doi: 10.1200/JCO.2008.18.2717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knauf JA, Ma X, Smith EP, Zhang L, Mitsutake N, Liao X-H, Refetoff S, Nikiforov YE, Fagin JA. Targeted Expression of BRAFV600E in Thyroid Cells of Transgenic Mice Results in Papillary Thyroid Cancers that Undergo Dedifferentiation. Cancer Res. 2005;65:4238–4245. doi: 10.1158/0008-5472.CAN-05-0047. [DOI] [PubMed] [Google Scholar]
- Knauf JA, Ouyang B, Knudsen ES, Fukasawa K, Babcock G, Fagin JA. Oncogenic RAS induces accelerated transition through G2/M and promotes defects in the G2 DNA damage and mitotic spindle checkpoints. J. Biol. Chem. 2006;281:3800–3809. doi: 10.1074/jbc.M511690200. [DOI] [PubMed] [Google Scholar]
- Kogo R, Shimamura T, Mimori K, Kawahara K, Imoto S, Sudo T, Tanaka F, Shibata K, Suzuki A, Komune S, Miyano S, Mori M. Long noncoding RNA HOTAIR regulates polycomb-dependent chromatin modification and is associated with poor prognosis in colorectal cancers. Cancer Res. 2011;71:6320–6326. doi: 10.1158/0008-5472.CAN-11-1021. [DOI] [PubMed] [Google Scholar]
- Kondo T, Nakazawa T, Ma D, Niu D, Mochizuki K, Kawasaki T, Nakamura N, Yamane T, Kobayashi M, Katoh R. Epigenetic silencing of TTF-1/NKX2-1 through DNA hypermethylation and histone H3 modulation in thyroid carcinomas. Lab. Invest. 2009;89:791–799. doi: 10.1038/labinvest.2009.50. [DOI] [PubMed] [Google Scholar]
- Kroll TG, Sarraf P, Pecciarini L, Chen C-J, Mueller E, Spiegelman BM, Fletcher JA. PAX8-PPARγ1 Fusion in Oncogene Human Thyroid Carcinoma. Science. 2000;289:1357–1360. doi: 10.1126/science.289.5483.1357. [DOI] [PubMed] [Google Scholar]
- Lacroix L, Lazar V, Michiels S, Ripoche H, Dessen P, Talbot M, Caillou B, Levillain J-P, Schlumberger M, Bidart J-M. Follicular Thyroid Tumors with the PAX8-PPARγ1 Rearrangement Display Characteristic Genetic Alterations. Am. J. Pathol. 2005;167:223–231. doi: 10.1016/s0002-9440(10)62967-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang FF, Bruner JM, Fuller GN, Aldape K, Prados MD, Chang S, Berger MS, McDermott MW, Kunwar SM, Junck LR, Chandler W, Zwiebel JA, Kaplan RS, Yung WKA. Phase I trial of adenovirus-mediated p53 gene therapy for recurrent glioma: biological and clinical results. J. Clin. Oncol. 2003;21:2508–2518. doi: 10.1200/JCO.2003.21.13.2508. [DOI] [PubMed] [Google Scholar]
- La Perle KM, Jhiang SM, Capen CC. Loss of p53 promotes anaplasia and local invasion in ret/PTC1-induced thyroid carcinomas. Am. J. Pathol. 2000;157:671–677. doi: 10.1016/S0002-9440(10)64577-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JT. Epigenetic regulation by long noncoding RNAs. Science. 2012;338:1435–1439. doi: 10.1126/science.1231776. [DOI] [PubMed] [Google Scholar]
- Lemoine NR, Mayall ES, Wyllie FS, Farr CJ, Hughes D, Padua RA, Thurston V, Williams ED, Wynford-Thomas D. Activated ras oncogenes in human thyroid cancers. Cancer Res. 1988;48:4459–4463. [PubMed] [Google Scholar]
- Levy R, Grafi-Cohen M, Kraiem Z, Kloog Y. Galectin-3 promotes chronic activation of K-Ras and differentiation block in malignant thyroid carcinomas. Mol. Cancer Ther. 2010;9:2208–2219. doi: 10.1158/1535-7163.MCT-10-0262. [DOI] [PubMed] [Google Scholar]
- Libertini S, Abagnale A, Passaro C, Botta G, Barbato S, Chieffi P, Portella G. AZD1152 negatively affects the growth of anaplastic thyroid carcinoma cells and enhances the effects of oncolytic virus dl922-947. Endocr. Relat. Cancer. 2011;18:129–141. doi: 10.1677/ERC-10-0234. [DOI] [PubMed] [Google Scholar]
- Li D, Feng J, Wu T, Wang Y, Sun Y, Ren J, Liu M. Long intergenic noncoding RNA HOTAIR is overexpressed and regulates PTEN methylation in laryngeal squamous cell carcinoma. Am. J. Pathol. 2013;182:64–70. doi: 10.1016/j.ajpath.2012.08.042. [DOI] [PubMed] [Google Scholar]
- Li E, Beard C, Jaenisch R. Role for DNA methylation in genomic imprinting. Nature. 1993;366:362–365. doi: 10.1038/366362a0. [DOI] [PubMed] [Google Scholar]
- Lima J, Trovisco V, Soares P, Máximo V, Magalhães J, Salvatore G, Santoro M, Bogdanova T, Tronko M, Abrosimov A, Jeremiah S, Thomas G, Williams D, Sobrinho-Simões M. BRAF Mutations Are Not a Major Event in Post-Chernobyl Childhood Thyroid Carcinomas. J. Clin. Endocrinol. Metab. 2004;89:4267–4271. doi: 10.1210/jc.2003-032224. [DOI] [PubMed] [Google Scholar]
- Lim JY, Hong SW, Lee YS, Kim B-W, Park CS, Chang H-S, Cho JY. Clinicopathologic implications of the BRAFV600E mutation in papillary thyroid cancer; a subgroup analysis of 3130 cases in a single center. Thyroid. 2013 doi: 10.1089/thy.2013.0036. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Lin S-F, Huang Y-Y, Lin J-D, Chou T-C, Hsueh C, Wong RJ. Utility of a PI3K/mTOR Inhibitor (NVP-BEZ235) for Thyroid Cancer Therapy. PLoS One. 2012;7:e46726. doi: 10.1371/journal.pone.0046726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Hu S, Hou P, Jiang D, Condouris S, Xing M. Suppression of BRAF/MEK/MAP Kinase Pathway Restores Expression of Iodide-Metabolizing Genes in Thyroid Cells Expressing the V600E BRAF Mutant. Clin. Cancer Res. 2007a;13:1341–1349. doi: 10.1158/1078-0432.CCR-06-1753. [DOI] [PubMed] [Google Scholar]
- Liu D, Liu Z, Condouris S, Xing M. BRAF V600E maintains proliferation, transformation, and tumorigenicity of BRAF-mutant papillary thyroid cancer cells. J. Clin. Endocrinol. Metab. 2007b;92:2264–2271. doi: 10.1210/jc.2006-1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R, Liu D, Trink E, Bojdani E, Ning G, Xing M. The Akt-specific inhibitor MK2206 selectively inhibits thyroid cancer cells harboring mutations that can activate the PI3K/Akt pathway. J. Clin. Endocrinol. Metab. 2011;96:E577–585. doi: 10.1210/jc.2010-2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R, Liu D, Xing M. The Akt inhibitor MK2206 synergizes, but perifosine antagonizes, the BRAF(V600E) inhibitor PLX4032 and the MEK1/2 inhibitor AZD6244 in the inhibition of thyroid cancer cells. J. Clin. Endocrinol. Metab. 2012;97:E173–182. doi: 10.1210/jc.2011-1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Hou P, Ji M, Guan H, Studeman K, Jensen K, Vasko V, El-Naggar AK, Xing M. Highly prevalent genetic alterations in receptor tyrosine kinases and phosphatidylinositol 3-kinase/akt and mitogen-activated protein kinase pathways in anaplastic and follicular thyroid cancers. J. Clin. Endocrinol. Metab. 2008;93:3106–3116. doi: 10.1210/jc.2008-0273. [DOI] [PubMed] [Google Scholar]
- Losman JA, Kaelin WG., Jr. What a difference a hydroxyl makes: mutant IDH, (R)-2-hydroxyglutarate, and cancer. Genes Dev. 2013;27:836–852. doi: 10.1101/gad.217406.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lui W-O, Zeng L, Rehrmann V, Deshpande S, Tretiakova M, Kaplan EL, Leibiger I, Leibiger B, Enberg U, Höög A, Larsson C, Kroll TG. CREB3L2-PPARγ Fusion Mutation Identifies a Thyroid Signaling Pathway Regulated by Intramembrane Proteolysis. Cancer Research. 2008;68:7156–7164. doi: 10.1158/0008-5472.CAN-08-1085. [DOI] [PubMed] [Google Scholar]
- Lyons JF, Wilhelm S, Hibner B, Bollag G. Discovery of a novel Raf kinase inhibitor. Endocr Relat Cancer. 2001;8:219–225. doi: 10.1677/erc.0.0080219. [DOI] [PubMed] [Google Scholar]
- Mandal M, Kim S, Younes MN, Jasser SA, El-Naggar AK, Mills GB, Myers JN. The Akt inhibitor KP372-1 suppresses Akt activity and cell proliferation and induces apoptosis in thyroid cancer cells. Br. J. Cancer. 2005;92:1899–1905. doi: 10.1038/sj.bjc.6602595. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Manenti G, Pilotti S, Re FC, Della Porta G, Pierotti MA. Selective activation of ras oncogenes in follicular and undifferentiated thyroid carcinomas. Eur. J. Cancer. 1994;30A:987–993. doi: 10.1016/0959-8049(94)90130-9. [DOI] [PubMed] [Google Scholar]
- Marciano D, Ben-Baruch G, Marom M, Egozi Y, Haklai R, Kloog Y. Farnesyl derivatives of rigid carboxylic acids-inhibitors of ras-dependent cell growth. J. Med. Chem. 1995;38:1267–1272. doi: 10.1021/jm00008a004. [DOI] [PubMed] [Google Scholar]
- Marotta V, Guerra A, Sapio MR, Vitale M. RET/PTC rearrangement in benign and malignant thyroid diseases: a clinical standpoint. Eur J Endocrinol. 2011;165:499–507. doi: 10.1530/EJE-11-0499. [DOI] [PubMed] [Google Scholar]
- Marotta V, Ramundo V, Camera L, Prete MD, Fonti R, Esposito R, Palmieri G, Salvatore M, Vitale M, Colao A, Faggiano A. Sorafenib in advanced iodine-refractory differentiated thyroid cancer: efficacy, safety and exploratory analysis of role of serum thyroglobulin and FDG-PET. Clin. Endocrinol. (Oxf) 2013;78:760–767. doi: 10.1111/cen.12057. [DOI] [PubMed] [Google Scholar]
- Marques AR, Espadinha C, Frias MJ, Roque L, Catarino AL, Sobrinho LG, Leite V. Underexpression of peroxisome proliferator-activated receptor (PPAR)γ in PAX8/PPARγ-negative thyroid tumours. Br. J. Cancer. 2004;91:732–738. doi: 10.1038/sj.bjc.6601989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matias-Guiu X, Cuatrecasas M, Musulen E, Prat J. p53 expression in anaplastic carcinomas arising from thyroid papillary carcinomas. J. Clin. Pathol. 1994;47:337–339. doi: 10.1136/jcp.47.4.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melillo RM, Castellone MD, Guarino V, De Falco V, Cirafici AM, Salvatore G, Caiazzo F, Basolo F, Giannini R, Kruhoffer M, Orntoft T, Fusco A, Santoro M. The RET/PTC-RAS-BRAF linear signaling cascade mediates the motile and mitogenic phenotype of thyroid cancer cells. J. Clin. Invest. 2005;115:1068–1081. doi: 10.1172/JCI22758. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Miozzo M, Pierotti MA, Sozzi G, Radice P, Bongarzone I, Spurr NK, Della Porta G. Human TRK proto-oncogene maps to chromosome 1q32-q41. Oncogene. 1990;5:1411–1414. [PubMed] [Google Scholar]
- Mitsutake N, Knauf JA, Mitsutake S, Mesa C, Jr, Zhang L, Fagin JA. Conditional BRAFV600E expression induces DNA synthesis, apoptosis, dedifferentiation, and chromosomal instability in thyroid PCCL3 cells. Cancer Res. 2005;65:2465–2473. doi: 10.1158/0008-5472.CAN-04-3314. [DOI] [PubMed] [Google Scholar]
- Miyagi E, Braga-Basaria M, Hardy E, Vasko V, Burman KD, Jhiang S, Saji M, Ringel MD. Chronic expression of RET/PTC 3 enhances basal and insulin-stimulated PI3 kinase/AKT signaling and increases IRS-2 expression in FRTL-5 thyroid cells. Mol. Carcinog. 2004;41:98–107. doi: 10.1002/mc.20042. [DOI] [PubMed] [Google Scholar]
- Mizuno T, Iwamoto KS, Kyoizumi S, Nagamura H, Shinohara T, Koyama K, Seyama T, Hamatani K. Preferential induction of RET/PTC1 rearrangement by X-ray irradiation. Oncogene. 2000;19:438–443. doi: 10.1038/sj.onc.1203343. [DOI] [PubMed] [Google Scholar]
- Mizuno T, Kyoizumi S, Suzuki T, Iwamoto KS, Seyama T. Continued expression of a tissue specific activated oncogene in the early steps of radiation-induced human thyroid carcinogenesis. Oncogene. 1997;15:1455–1460. doi: 10.1038/sj.onc.1201313. [DOI] [PubMed] [Google Scholar]
- Moretti S, Macchiarulo A, De Falco V, Avenia N, Barbi F, Carta C, Cavaliere A, Melillo RM, Passeri L, Santeusanio F, Tartaglia M, Santoro M, Puxeddu E. Biochemical and molecular characterization of the novel BRAF(V599Ins) mutation detected in a classic papillary thyroid carcinoma. Oncogene. 2006;25:4235–4240. doi: 10.1038/sj.onc.1209448. [DOI] [PubMed] [Google Scholar]
- Murphy DA, Makonnen S, Lassoued W, Feldman MD, Carter C, Lee WMF. Inhibition of tumor endothelial ERK activation, angiogenesis, and tumor growth by sorafenib (BAY43-9006) Am. J. Pathol. 2006;169:1875–1885. doi: 10.2353/ajpath.2006.050711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murugan AK, Bojdani E, Xing M. Identification and functional characterization of isocitrate dehydrogenase 1 (IDH1) mutations in thyroid cancer. Biochem Biophys Res Commun. 2010;393:555–559. doi: 10.1016/j.bbrc.2010.02.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musholt TJ, Musholt PB, Khaladj N, Schulz D, Scheumann GF, Klempnauer J. Prognostic significance of RET and NTRK1 rearrangements in sporadic papillary thyroid carcinoma. Surgery. 2000;128:984–993. doi: 10.1067/msy.2000.110845. [DOI] [PubMed] [Google Scholar]
- Nagaiah G, Hossain A, Mooney CJ, Parmentier J, Remick SC. Anaplastic thyroid cancer: a review of epidemiology, pathogenesis, and treatment. J. Oncol. 2011;2011:542358. doi: 10.1155/2011/542358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Yana I, Kobayashi T, Shin E, Karakawa K, Fujita S, Miya A, Mori T, Nishisho I, Takai S. p53 gene mutations associated with anaplastic transformation of human thyroid carcinomas. Jpn. J. Cancer Res. 1992;83:1293–1298. doi: 10.1111/j.1349-7006.1992.tb02761.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakashima M, Takamura N, Namba H, Saenko V, Meirmanov S, Matsumoto N, Hayashi T, Maeda S, Sekine I. RET oncogene amplification in thyroid cancer: correlations with radiation-associated and high-grade malignancy. Hum. Pathol. 2007;38:621–628. doi: 10.1016/j.humpath.2006.10.013. [DOI] [PubMed] [Google Scholar]
- Namba H, Nakashima M, Hayashi T, Hayashida N, Maeda S, Rogounovitch TI, Ohtsuru A, Saenko VA, Kanematsu T, Yamashita S. Clinical Implication of Hot Spot BRAF Mutation, V599E, in Papillary Thyroid Cancers. J. Clin. Endocrinol. Metab. 2003;88:4393–4397. doi: 10.1210/jc.2003-030305. [DOI] [PubMed] [Google Scholar]
- Namba H, Rubin SA, Fagin JA. Point mutations of ras oncogenes are an early event in thyroid tumorigenesis. Mol. Endocrinol. 1990;4:1474–1479. doi: 10.1210/mend-4-10-1474. [DOI] [PubMed] [Google Scholar]
- Nehs MA, Nucera C, Nagarkatti SS, Sadow PM, Morales-Garcia D, Hodin RA, Parangi S. Late intervention with anti-BRAF(V600E) therapy induces tumor regression in an orthotopic mouse model of human anaplastic thyroid cancer. Endocrinology. 2012;153:985–994. doi: 10.1210/en.2011-1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niinuma T, Suzuki H, Nojima M, Nosho K, Yamamoto H, Takamaru H, Yamamoto E, Maruyama R, Nobuoka T, Miyazaki Y, Nishida T, Bamba T, Kanda T, Ajioka Y, Taguchi T, Okahara S, Takahashi H, Nishida Y, Hosokawa M, Hasegawa T, Tokino T, Hirata K, Imai K, Toyota M, Shinomura Y. Upregulation of miR-196a and HOTAIR drive malignant character in gastrointestinal stromal tumors. Cancer Res. 2012;72:1126–1136. doi: 10.1158/0008-5472.CAN-11-1803. [DOI] [PubMed] [Google Scholar]
- Nikiforova MN, Kimura ET, Gandhi M, Biddinger PW, Knauf JA, Basolo F, Zhu Z, Giannini R, Salvatore G, Fusco A, Santoro M, Fagin JA, Nikiforov YE. BRAF Mutations in Thyroid Tumors Are Restricted to Papillary Carcinomas and Anaplastic or Poorly Differentiated Carcinomas Arising from Papillary Carcinomas. J. Clin. Endocrinol. Metab. 2003a;88:5399–5404. doi: 10.1210/jc.2003-030838. [DOI] [PubMed] [Google Scholar]
- Nikiforova MN, Lynch RA, Biddinger PW, Alexander EK, Dorn GW, Tallini G, Kroll TG, Nikiforov YE. RAS Point Mutations and PAX8-PPARγ Rearrangement in Thyroid Tumors: Evidence for Distinct Molecular Pathways in Thyroid Follicular Carcinoma. J. Clin. Endocrinol. Metab. 2003b;88:2318–2326. doi: 10.1210/jc.2002-021907. [DOI] [PubMed] [Google Scholar]
- Nikiforova MN, Stringer JR, Blough R, Medvedovic M, Fagin JA, Nikiforov YE. Proximity of chromosomal loci that participate in radiation-induced rearrangements in human cells. Science. 2000;290:138–141. doi: 10.1126/science.290.5489.138. [DOI] [PubMed] [Google Scholar]
- Nikiforov YE. Genetic alterations involved in the transition from well-differentiated to poorly differentiated and anaplastic thyroid carcinomas. Endocr. Pathol. 2004;15:319–327. doi: 10.1385/ep:15:4:319. [DOI] [PubMed] [Google Scholar]
- Nikiforov YE. Molecular analysis of thyroid tumors. Mod Pathol. 2011;24:S34–S43. doi: 10.1038/modpathol.2010.167. [DOI] [PubMed] [Google Scholar]
- Nikiforov YE, Rowland JM, Bove KE, Monforte-Munoz H, Fagin JA. Distinct pattern of ret oncogene rearrangements in morphological variants of radiation-induced and sporadic thyroid papillary carcinomas in children. Cancer Res. 1997;57:1690–1694. [PubMed] [Google Scholar]
- Noguchi H, Yamashita H, Murakami T, Hirai K, Noguchi Y, Maruta J, Yokoi T, Noguchi S. Successful treatment of anaplastic thyroid carcinoma with a combination of oral valproic acid, chemotherapy, radiation and surgery. Endocr. J. 2009;56:245–249. doi: 10.1507/endocrj.k08e-016. [DOI] [PubMed] [Google Scholar]
- Nucera C, Goldfarb M, Hodin R, Parangi S. Role of B-Raf(V600E) in differentiated thyroid cancer and preclinical validation of compounds against B-Raf(V600E) Biochim. Biophys. Acta. 2009;1795:152–161. doi: 10.1016/j.bbcan.2009.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orloff MS, He X, Peterson C, Chen F, Chen J-L, Mester JL, Eng C. Germline PIK3CA and AKT1 mutations in Cowden and Cowden-like syndromes. Am. J. Hum. Genet. 2013;92:76–80. doi: 10.1016/j.ajhg.2012.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozdemir S, Silan F, Hasbek Z, Uludag A, Atik S, Erselcan T, Ozdemir O. Increased T-allele frequency of 677 C>T polymorphism in the methylenetetrahydrofolate reductase gene in differentiated thyroid carcinoma. Genet Test Mol Biomarkers. 2012;16:780–784. doi: 10.1089/gtmb.2011.0347. [DOI] [PubMed] [Google Scholar]
- Paes JE, Ringel MD. Dysregulation of the Phosphatidylinositol 3-kinase Pathway in Thyroid Neoplasia. Endocrinol Metab Clin North Am. 2008;37:375–387. doi: 10.1016/j.ecl.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pakneshan S, Salajegheh A, Smith RA, Lam AK-Y. Clinicopathological relevance of BRAF mutations in human cancer. Pathology. 2013;45:346–356. doi: 10.1097/PAT.0b013e328360b61d. [DOI] [PubMed] [Google Scholar]
- Papewalis C, Wuttke M, Schinner S, Willenberg HS, Baran AM, Scherbaum WA, Schott M. Role of the novel mTOR inhibitor RAD001 (everolimus) in anaplastic thyroid cancer. Horm. Metab. Res. 2009;41:752–756. doi: 10.1055/s-0029-1224116. [DOI] [PubMed] [Google Scholar]
- Park J-W, Zarnegar R, Kanauchi H, Wong MG, Hyun WC, Ginzinger DG, Lobo M, Cotter P, Duh Q-Y, Clark OH. Troglitazone, the Peroxisome Proliferator-Activated Receptor-γ Agonist, Induces Antiproliferation and Redifferentiation in Human Thyroid Cancer Cell Lines. Thyroid. 2005;15:222–231. doi: 10.1089/thy.2005.15.222. [DOI] [PubMed] [Google Scholar]
- Paz MF, Avila S, Fraga MF, Pollan M, Capella G, Peinado MA, Sanchez-Cespedes M, Herman JG, Esteller M. Germ-line variants in methyl-group metabolism genes and susceptibility to DNA methylation in normal tissues and human primary tumors. Cancer Res. 2002;62:4519–4524. [PubMed] [Google Scholar]
- Powell DJ, Russell J, Nibu K, Li G, Rhee E, Liao M, Goldstein M, Keane WM, Santoro M, Fusco A, Rothstein JL. The RET/PTC3 Oncogene: Metastatic Solid-Type Papillary Carcinomas in Murine Thyroids. Cancer Res. 1998;58:5523–5528. [PubMed] [Google Scholar]
- Powell JG, Wang X, Allard BL, Sahin M, Wang X-L, Hay ID, Hiddinga HJ, Deshpande SS, Kroll TG, Grebe SK, Eberhardt NL, McIver B. The PAX8/PPARγ fusion oncoprotein transforms immortalized human thyrocytes through a mechanism probably involving wild-type PPARγ inhibition. Oncogene. 2004;23:3634–3641. doi: 10.1038/sj.onc.1207399. [DOI] [PubMed] [Google Scholar]
- Prasad VVTS, Wilkhoo H. Association of the functional polymorphism C677T in the methylenetetrahydrofolate reductase gene with colorectal, thyroid, breast, ovarian, and cervical cancers. Onkologie. 2011;34:422–426. doi: 10.1159/000331131. [DOI] [PubMed] [Google Scholar]
- Pugliese M, Fortunati N, Germano A, Asioli S, Marano F, Palestini N, Frairia R, Boccuzzi G, Catalano MG. Histone deacetylase inhibition affects sodium iodide symporter (NIS) expression and induces 131I cytotoxicity in anaplastic thyroid cancer cells. Thyroid. 2013 doi: 10.1089/thy.2012.0359. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Puppin C, D'Aurizio F, D'Elia AV, Cesaratto L, Tell G, Russo D, Filetti S, Ferretti E, Tosi E, Mattei T, Pianta A, Pellizzari L, Damante G. Effects of histone acetylation on sodium iodide symporter promoter and expression of thyroid-specific transcription factors. Endocrinology. 2005;146:3967–3974. doi: 10.1210/en.2005-0128. [DOI] [PubMed] [Google Scholar]
- Puppin C, Passon N, Lavarone E, Di Loreto C, Frasca F, Vella V, Vigneri R, Damante G. Levels of histone acetylation in thyroid tumors. Biochem. Biophys. Res. Commun. 2011;411:679–683. doi: 10.1016/j.bbrc.2011.06.182. [DOI] [PubMed] [Google Scholar]
- Puxeddu E, Moretti S, Elisei R, Romei C, Pascucci R, Martinelli M, Marino C, Avenia N, Rossi ED, Fadda G, Cavaliere A, Ribacchi R, Falorni A, Pontecorvi A, Pacini F, Pinchera A, Santeusanio F. BRAFV599E Mutation Is the Leading Genetic Event in Adult Sporadic Papillary Thyroid Carcinomas. J. Clin. Endocrinol. Metab. 2004;89:2414–2420. doi: 10.1210/jc.2003-031425. [DOI] [PubMed] [Google Scholar]
- Puzianowska-Kuznicka M, Krystyniak A, Madej A, Cheng S-Y, Nauman J. Functionally impaired TR mutants are present in thyroid papillary cancer. J. Clin. Endocrinol. Metab. 2002;87:1120–1128. doi: 10.1210/jcem.87.3.8296. [DOI] [PubMed] [Google Scholar]
- Rabes HM, Demidchik EP, Sidorow JD, Lengfelder E, Beimfohr C, Hoelzel D, Klugbauer S. Pattern of radiation-induced RET and NTRK1 rearrangements in 191 post-chernobyl papillary thyroid carcinomas: biological, phenotypic, and clinical implications. Clin. Cancer Res. 2000;6:1093–1103. [PubMed] [Google Scholar]
- Reddi HV, Madde P, Marlow LA, Copland JA, McIver B, Grebe SKG, Eberhardt NL. Expression of the PAX8/PPARγ fusion protein is associated with decreased neovascularization in vivo: Impact on tumorigenesis and disease prognosis. Genes Cancer. 2010;1:480–492. doi: 10.1177/1947601910373545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddi HV, Madde P, Milosevic D, Hackbarth JS, Algeciras-Schimnich A, McIver B, Grebe SKG, Eberhardt NL. The putative PAX8/PPARγ fusion oncoprotein exhibits partial tumor suppressor activity through up-regulation of micro-RNA-122 and dominant-negative PPARγ activity. Genes Cancer. 2011;2:46–55. doi: 10.1177/1947601911405045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riesco-Eizaguirre G, Gutiérrez-Martínez P, García-Cabezas MA, Nistal M, Santisteban P. The oncogene BRAF V600E is associated with a high risk of recurrence and less differentiated papillary thyroid carcinoma due to the impairment of Na+/I− targeting to the membrane. Endocr. Relat. Cancer. 2006;13:257–269. doi: 10.1677/erc.1.01119. [DOI] [PubMed] [Google Scholar]
- Riggs AD. X inactivation, differentiation, and DNA methylation. Cytogenet. Cell Genet. 1975;14:9–25. doi: 10.1159/000130315. [DOI] [PubMed] [Google Scholar]
- Rocha AS, Marques R, Bento I, Soares R, Magalhães J, de Castro IV, Soares P. Thyroid hormone receptor beta mutations in the “hot-spot region” are rare events in thyroid carcinomas. J. Endocrinol. 2007;192:83–86. doi: 10.1677/JOE-06-0009. [DOI] [PubMed] [Google Scholar]
- Rochefort P, Caillou B, Michiels FM, Ledent C, Talbot M, Schlumberger M, Lavelle F, Monier R, Feunteun J. Thyroid pathologies in transgenic mice expressing a human activated Ras gene driven by a thyroglobulin promoter. Oncogene. 1996;12:111–118. [PubMed] [Google Scholar]
- Saavedra HI, Knauf JA, Shirokawa JM, Wang J, Ouyang B, Elisei R, Stambrook PJ, Fagin JA. The RAS oncogene induces genomic instability in thyroid PCCL3 cells via the MAPK pathway. Oncogene. 2000;19:3948–3954. doi: 10.1038/sj.onc.1203723. [DOI] [PubMed] [Google Scholar]
- Sabra M, Dominguez J, Grewal R, Larson S, Ghossein R, Tuttle R, Fagin J. Clinical outcomes and molecular profile of differentiated thyroid cancers with radioiodine-avid distant metastases. J. Clin. Endocrinol. Metab. 2013;98:E829–36. doi: 10.1210/jc.2012-3933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahin M, Allard BL, Yates M, Powell JG, Wang X-L, Hay ID, Zhao Y, Goellner JR, Sebo TJ, Grebe SKG, Eberhardt NL, McIver B. PPARγ Staining as a Surrogate for PAX8/PPARγ Fusion Oncogene Expression in Follicular Neoplasms: Clinicopathological Correlation and Histopathological Diagnostic Value. J. Clin. Endocrinol. Metab. 2005;90:463–468. doi: 10.1210/jc.2004-1203. [DOI] [PubMed] [Google Scholar]
- Saji M, Narahara K, McCarty SK, Vasko VV, La Perle KM, Porter K, Jarjoura D, Lu C, Cheng S-Y, Ringel MD. Akt1 deficiency delays tumor progression, vascular invasion, and distant metastasis in a murine model of thyroid cancer. Oncogene. 2011;30:4307–4315. doi: 10.1038/onc.2011.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvatore D, Barone MV, Salvatore G, Melillo RM, Chiappetta G, Mineo A, Fenzi G, Vecchio G, Fusco A, Santoro M. Tyrosines 1015 and 1062 are in vivo autophosphorylation sites in ret and ret-derived oncoproteins. J. Clin. Endocrinol. Metab. 2000;85:3898–3907. doi: 10.1210/jcem.85.10.6882. [DOI] [PubMed] [Google Scholar]
- Santoro M, Chiappetta G, Cerrato A, Salvatore D, Zhang L, Manzo G, Picone A, Portella G, Santelli G, Vecchio G, Fusco A. Development of thyroid papillary carcinomas secondary to tissue-specific expression of the RET/PTC1 oncogene in transgenic mice. Oncogene. 1996;12:1821–1826. [PubMed] [Google Scholar]
- Santoro M, Dathan NA, Berlingieri MT, Bongarzone I, Paulin C, Grieco M, Pierotti MA, Vecchio G, Fusco A. Molecular characterization of RET/PTC3; a novel rearranged version of the RETproto-oncogene in a human thyroid papillary carcinoma. Oncogene. 1994;9:509–516. [PubMed] [Google Scholar]
- Santoro M, Melillo R, Grieco M, Berlingieri M, Vecchio G, Fusco A. The TRK and RET tyrosine kinase oncogenes cooperate with ras in the neoplastic transformation of a rat thyroid epithelial cell line. Cell Growth Differ. 1993;4:77–84. [PubMed] [Google Scholar]
- Santoro M, Melillo RM, Fusco A. RET/PTC activation in papillary thyroid carcinoma: European Journal of Endocrinology Prize Lecture. Eur J Endocrinol. 2006;155:645–653. doi: 10.1530/eje.1.02289. [DOI] [PubMed] [Google Scholar]
- Santoro M, Papotti M, Chiappetta G, Garcia-Rostan G, Volante M, Johnson C, Camp RL, Pentimalli F, Monaco C, Herrero A, Carcangiu ML, Fusco A, Tallini G. RET Activation and Clinicopathologic Features in Poorly Differentiated Thyroid Tumors. J. Clin. Endocrinol. Metab. 2002;87:370–379. doi: 10.1210/jcem.87.1.8174. [DOI] [PubMed] [Google Scholar]
- Schagdarsurengin U, Gimm O, Hoang-Vu C, Dralle H, Pfeifer GP, Dammann R. Frequent epigenetic silencing of the CpG island promoter of RASSF1A in thyroid carcinoma. Cancer Res. 2002;62:3698–3701. [PubMed] [Google Scholar]
- Schagdarsurengin U, Richter AM, Hornung J, Lange C, Steinmann K, Dammann RH. Frequent epigenetic inactivation of RASSF2 in thyroid cancer and functional consequences. Mol. Cancer. 2010;9:264. doi: 10.1186/1476-4598-9-264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schagdarsurengin U, Richter AM, Wöhler C, Dammann RH. Frequent epigenetic inactivation of RASSF10 in thyroid cancer. Epigenetics. 2009;4:571–576. doi: 10.4161/epi.4.8.10056. [DOI] [PubMed] [Google Scholar]
- Schneider TC, Abdulrahman RM, Corssmit EP, Morreau H, Smit JWA, Kapiteijn E. Long-term analysis of the efficacy and tolerability of sorafenib in advanced radio-iodine refractory differentiated thyroid carcinoma: final results of a phase II trial. Eur. J. Endocrinol. 2012;167:643–650. doi: 10.1530/EJE-12-0405. [DOI] [PubMed] [Google Scholar]
- Shangary S, Ding K, Qiu S, Nikolovska-Coleska Z, Bauer JA, Liu M, Wang G, Lu Y, McEachern D, Bernard D, Bradford CR, Carey TE, Wang S. Reactivation of p53 by a specific MDM2 antagonist (MI-43) leads to p21-mediated cell cycle arrest and selective cell death in colon cancer. Mol. Cancer Ther. 2008;7:1533–1542. doi: 10.1158/1535-7163.MCT-08-0140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Ruan M, Luo Q, Yu Y, Lu H, Zhu R, Chen L. Brain metastasis from follicular thyroid carcinoma: treatment with sorafenib. Thyroid. 2012;22:856–860. doi: 10.1089/thy.2011.0419. [DOI] [PubMed] [Google Scholar]
- Sherman EJ, Su YB, Lyall A, Schoder H, Fury MG, Ghossein RA, Haque S, Lisa D, Shaha AR, Tuttle RM, Pfister DG. Evaluation of romidepsin for clinical activity and radioactive iodine reuptake in radioactive iodine-refractory thyroid carcinoma. Thyroid. 2012;23:593–9. doi: 10.1089/thy.2012.0393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smallridge RC, Ain KB, Asa SL, Bible KC, Brierley JD, Burman KD, Kebebew E, Lee NY, Nikiforov YE, Rosenthal MS, Shah MH, Shaha AR, Tuttle RM. American Thyroid Association guidelines for management of patients with anaplastic thyroid cancer. Thyroid. 2012;22:1104–1139. doi: 10.1089/thy.2012.0302. [DOI] [PubMed] [Google Scholar]
- Smallridge RC, Marlow LA, Copland JA. Anaplastic thyroid cancer: molecular pathogenesis and emerging therapies. Endocr Relat Cancer. 2009;16:17–44. doi: 10.1677/ERC-08-0154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soares P, Fonseca E, Wynford-Thomas D, Sobrinho-Simões M. Sporadic ret-rearranged papillary carcinoma of the thyroid: a subset of slow growing, less aggressive thyroid neoplasms? J. Pathol. 1998;185:71–78. doi: 10.1002/(SICI)1096-9896(199805)185:1<71::AID-PATH42>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Sorrentino R, Libertini S, Pallante PL, Troncone G, Palombini L, Bavetsias V, Spalletti-Cernia D, Laccetti P, Linardopoulos S, Chieffi P, Fusco A, Portella G. Aurora B overexpression associates with the thyroid carcinoma undifferentiated phenotype and is required for thyroid carcinoma cell proliferation. J. Clin. Endocrinol. Metab. 2005;90:928–935. doi: 10.1210/jc.2004-1518. [DOI] [PubMed] [Google Scholar]
- Stegh AH. Targeting the p53 signaling pathway in cancer therapy - The promises, challenges, and perils. Expert Opin Ther Targets. 2012;16:67–83. doi: 10.1517/14728222.2011.643299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suarez HG, du Villard JA, Severino M, Caillou B, Schlumberger M, Tubiana M, Parmentier C, Monier R. Presence of mutations in all three ras genes in human thyroid tumors. Oncogene. 1990;5:565–570. [PubMed] [Google Scholar]
- Suzuki H, Willingham MC, Cheng S-Y. Mice with a mutation in the thyroid hormone receptor beta gene spontaneously develop thyroid carcinoma: a mouse model of thyroid carcinogenesis. Thyroid. 2002;12:963–969. doi: 10.1089/105072502320908295. [DOI] [PubMed] [Google Scholar]
- Takano T, Miyauchi A, Yoshida H, Nakata Y, Kuma K, Amino N. Expression of TRbeta1 mRNAs with functionally impaired mutations is rare in thyroid papillary carcinoma. J. Clin. Endocrinol. Metab. 2003;88:3447–3449. doi: 10.1210/jc.2003-030012. [DOI] [PubMed] [Google Scholar]
- Tallini G, Santoro M, Helie M, Carlomagno F, Salvatore G, Chiappetta G, Carcangiu ML, Fusco A. RET/PTC oncogene activation defines a subset of papillary thyroid carcinomas lacking evidence of progression to poorly differentiated or undifferentiated tumor phenotypes. Clin. Cancer Res. 1998;4:287–294. [PubMed] [Google Scholar]
- Thomas GA, Bunnell H, Cook HA, Williams ED, Nerovnya A, Cherstvoy ED, Tronko ND, Bogdanova TI, Chiappetta G, Viglietto G, Pentimalli F, Salvatore G, Fusco A, Santoro M, Vecchio G. High prevalence of RET/PTC rearrangements in Ukrainian and Belarussian post-Chernobyl thyroid papillary carcinomas: a strong correlation between RET/PTC3 and the solid-follicular variant. J. Clin. Endocrinol. Metab. 1999;84:4232–4238. doi: 10.1210/jcem.84.11.6129. [DOI] [PubMed] [Google Scholar]
- Thornton K, Kim G, Maher VE, Chattopadhyay S, Tang S, Moon YJ, Song P, Marathe A, Balakrishnan S, Zhu H, Garnett C, Liu Q, Booth B, Gehrke B, Dorsam R, Verbois L, Ghosh D, Wilson W, Duan J, Sarker H, Miksinski SP, Skarupa L, Ibrahim A, Justice R, Murgo A, Pazdur R. Vandetanib for the treatment of symptomatic or progressive medullary thyroid cancer in patients with unresectable locally advanced or metastatic disease: U.S. Food and Drug Administration drug approval summary. Clin. Cancer Res. 2012;18:3722–3730. doi: 10.1158/1078-0432.CCR-12-0411. [DOI] [PubMed] [Google Scholar]
- Tuttle RM, Ball DW, Byrd D, Dilawari RA, Doherty GM, Duh Q-Y, Ehya H, Farrar WB, Haddad RI, Kandeel F, Kloos RT, Kopp P, Lamonica DM, Loree TR, Lydiatt WM, McCaffrey JC, Olson JA, Parks L, Ridge JA, Shah JP, Sherman SI, Sturgeon C, Waguespack SG, Wang TN, Wirth LJ. Thyroid Carcinoma. J Natl Compr Canc Netw. 2010;8:1228–1274. doi: 10.6004/jnccn.2010.0093. [DOI] [PubMed] [Google Scholar]
- Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, Fotouhi N, Liu EA. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- Venkataraman GM, Yatin M, Marcinek R, Ain KB. Restoration of iodide uptake in dedifferentiated thyroid carcinoma: relationship to human Na+/I-symporter gene methylation status. J. Clin. Endocrinol. Metab. 1999;84:2449–2457. doi: 10.1210/jcem.84.7.5815. [DOI] [PubMed] [Google Scholar]
- Vitagliano D, Portella G, Troncone G, Francione A, Rossi C, Bruno A, Giorgini A, Coluzzi S, Nappi TC, Rothstein JL, Pasquinelli R, Chiappetta G, Terracciano D, Macchia V, Melillo RM, Fusco A, Santoro M. Thyroid targeting of the N-ras(Gln61Lys) oncogene in transgenic mice results in follicular tumors that progress to poorly differentiated carcinomas. Oncogene. 2006;25:5467–5474. doi: 10.1038/sj.onc.1209527. [DOI] [PubMed] [Google Scholar]
- Vivaldi A, Miasaki FY, Ciampi R, Agate L, Collecchi P, Capodanno A, Pinchera A, Elisei R. Re-differentiation of thyroid carcinoma cell lines treated with 5-Aza-2'-deoxycytidine and retinoic acid. Mol. Cell. Endocrinol. 2009;307:142–148. doi: 10.1016/j.mce.2009.03.020. [DOI] [PubMed] [Google Scholar]
- Volante M, Rapa I, Gandhi M, Bussolati G, Giachino D, Papotti M, Nikiforov YE. RAS mutations are the predominant molecular alteration in poorly differentiated thyroid carcinomas and bear prognostic impact. J. Clin. Endocrinol. Metab. 2009;94:4735–4741. doi: 10.1210/jc.2009-1233. [DOI] [PubMed] [Google Scholar]
- Wang J, Knauf JA, Basu S, Puxeddu E, Kuroda H, Santoro M, Fusco A, Fagin JA. Conditional Expression of RET/PTC Induces a Weak Oncogenic Drive in Thyroid PCCL3 Cells and Inhibits Thyrotropin Action at Multiple Levels. Molecular Endocrinology. 2003;17:1425–1436. doi: 10.1210/me.2003-0041. [DOI] [PubMed] [Google Scholar]
- Wang Y, Hou P, Yu H, Wang W, Ji M, Zhao S, Yan S, Sun X, Liu D, Shi B, Zhu G, Condouris S, Xing M. High Prevalence and Mutual Exclusivity of Genetic Alterations in the Phosphatidylinositol-3-Kinase/Akt Pathway in Thyroid Tumors. J. Clin. Endocrinol. Metab. 2007;92:2387–2390. doi: 10.1210/jc.2006-2019. [DOI] [PubMed] [Google Scholar]
- Wan PTC, Garnett MJ, Roe SM, Lee S, Niculescu-Duvaz D, Good VM, Jones CM, Marshall CJ, Springer CJ, Barford D, Marais R. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell. 2004;116:855–867. doi: 10.1016/s0092-8674(04)00215-6. [DOI] [PubMed] [Google Scholar]
- Wunderlich A, Fischer M, Schlosshauer T, Ramaswamy A, Greene BH, Brendel C, Doll D, Bartsch D, Hoffmann S. Evaluation of Aurora kinase inhibition as a new therapeutic strategy in anaplastic and poorly differentiated follicular thyroid cancer. Cancer Sci. 2011;102:762–768. doi: 10.1111/j.1349-7006.2011.01853.x. [DOI] [PubMed] [Google Scholar]
- Xing M, Alzahrani AS, Carson KA, Viola D, Elisei R, Bendlova B, Yip L, Mian C, Vianello F, Tuttle RM, Robenshtok E, Fagin JA, Puxeddu E, Fugazzola L, Czarniecka A, Jarzab B, O'Neill CJ, Sywak MS, Lam AK, Riesco-Eizaguirre G, Santisteban P, Nakayama H, Tufano RP, Pai SI, Zeiger MA, Westra WH, Clark DP, Clifton-Bligh R, Sidransky D, Ladenson PW, Sykorova V. Association between BRAF V600E mutation and mortality in patients with papillary thyroid cancer. JAMA. 2013;309:1493–1501. doi: 10.1001/jama.2013.3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing M, Westra WH, Tufano RP, Cohen Y, Rosenbaum E, Rhoden KJ, Carson KA, Vasko V, Larin A, Tallini G, Tolaney S, Holt EH, Hui P, Umbricht CB, Basaria S, Ewertz M, Tufaro AP, Califano JA, Ringel MD, Zeiger MA, Sidransky D, Ladenson PW. BRAF Mutation Predicts a Poorer Clinical Prognosis for Papillary Thyroid Cancer. J. Clin. Endocrinol. Metab. 2005;90:6373–6379. doi: 10.1210/jc.2005-0987. [DOI] [PubMed] [Google Scholar]
- Yang Z, Zhou L, Wu L-M, Lai M-C, Xie H-Y, Zhang F, Zheng S-S. Overexpression of long non-coding RNA HOTAIR predicts tumor recurrence in hepatocellular carcinoma patients following liver transplantation. Ann. Surg. Oncol. 2011;18:1243–1250. doi: 10.1245/s10434-011-1581-y. [DOI] [PubMed] [Google Scholar]
- Yeung S-CJ, She M, Yang H, Pan J, Sun L, Chaplin D. Combination chemotherapy including combretastatin A4 phosphate and paclitaxel is effective against anaplastic thyroid cancer in a nude mouse xenograft model. J. Clin. Endocrinol. Metab. 2007;92:2902–2909. doi: 10.1210/jc.2007-0027. [DOI] [PubMed] [Google Scholar]
- Yin Y, Yuan H, Wang C, Pattabiraman N, Rao M, Pestell RG, Glazer RI. 3-Phosphoinositide-Dependent Protein Kinase-1 Activates the Peroxisome Proliferator-Activated Receptor-{gamma} and Promotes Adipocyte Differentiation. Mol Endocrinol. 2006;20:268–278. doi: 10.1210/me.2005-0197. [DOI] [PubMed] [Google Scholar]
- Yin Y, Yuan H, Zeng X, Kopelovich L, Glazer RI. Inhibition of Peroxisome Proliferator-Activated Receptor γ Increases Estrogen Receptor–Dependent Tumor Specification. Cancer Res. 2009;69:687–694. doi: 10.1158/0008-5472.CAN-08-2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon H, He H, Nagy R, Davuluri R, Suster S, Schoenberg D, Pellegata N, de la Chapelle A. Identification of a novel noncoding RNA gene, NAMA, that is downregulated in papillary thyroid carcinoma with BRAF mutation and associated with growth arrest. Int. J. Cancer. 2007;121:767–775. doi: 10.1002/ijc.22701. [DOI] [PubMed] [Google Scholar]
- Zarnegar R, Brunaud L, Kanauchi H, Wong M, Fung M, Ginzinger D, Duh Q-Y, Clark OH. Increasing the effectiveness of radioactive iodine therapy in the treatment of thyroid cancer using Trichostatin A, a histone deacetylase inhibitor. Surgery. 2002;132:984–990. doi: 10.1067/msy.2002.128690. discussion 990. [DOI] [PubMed] [Google Scholar]
- Zhu Z, Gandhi M, Nikiforova MN, Fischer AH, Nikiforov YE. Molecular profile and clinical-pathologic features of the follicular variant of papillary thyroid carcinoma. An unusually high prevalence of ras mutations. Am. J. Clin. Pathol. 2003;120:71–77. doi: 10.1309/ND8D-9LAJ-TRCT-G6QD. [DOI] [PubMed] [Google Scholar]
- Zuo H, Gandhi M, Edreira MM, Hochbaum D, Nimgaonkar VL, Zhang P, Dipaola J, Evdokimova V, Altschuler DL, Nikiforov YE. Downregulation of Rap1GAP through epigenetic silencing and loss of heterozygosity promotes invasion and progression of thyroid tumors. Cancer Res. 2010;70:1389–1397. doi: 10.1158/0008-5472.CAN-09-2812. [DOI] [PMC free article] [PubMed] [Google Scholar]