

Abstract
Background
Reported odds ratios and population attributable fractions (PAF) for late-onset Alzheimer’s disease (LOAD) risk loci (BIN1, ABCA7, CR1, MS4A4E, CD2AP, PICALM, MS4A6A, CD33, and CLU) come from clinically ascertained samples. Little is known about the combined PAF for these LOAD risk alleles and the utility of these combined markers for case-control prediction. Here we evaluate these loci in a large population-based sample to estimate PAF and explore the effects of additive and non-additive interactions on LOAD status prediction performance.
Methods
2,419 samples from the Cache County Memory Study were genotyped for APOE and nine LOAD risk loci from AlzGene.org. We used logistic regression and ROC analysis to assess the LOAD status prediction performance of these loci using additive and non-additive models, and compared ORs and PAFs between AlzGene.org and Cache County.
Results
Odds ratios were comparable between Cache County and AlzGene.org when identical SNPs were genotyped. PAFs from AlzGene.org ranged from 2.25–37%; those from Cache County ranged from 0.05–20%. Including non-APOE alleles significantly improved LOAD status prediction performance (AUC = 0.80) over APOE alone (AUC = 0.78) when not constrained to an additive relationship (p < 0.03). We identified potential allelic interactions (p-values uncorrected): CD33-MS4A4E (Synergy Factor = 5.31; p < 0.003) and CLU-MS4A4E (SF = 3.81; p < 0.016).
Conclusions
While non-additive interactions between loci significantly improve diagnostic ability, the improvement does not reach the desired sensitivity or specificity for clinical use. Nevertheless, these results suggest that understanding gene-gene interactions may be important in resolving Alzheimer’s disease etiology.
Keywords: Alzheimer’s disease, epistasis, genetic interactions, population attributable fraction, odds ratio, risk
Introduction
Researchers have implicated several genes associated with late-onset Alzheimer’s disease (LOAD) including APOE. APOE ε4 increases LOAD risk and APOE ε2 reduces risk (1–4). According to AlzGene.org (5), nine additional genes significantly affect LOAD risk; BIN1 (rs744373), ABCA7 (rs3764650), CR1 (rs3818361), MS4A4E (rs670139), and CD2AP (rs9349407) are associated with increased risk for LOAD while PICALM (rs3851179), MS4A6A (rs610932), CD33 (rs3865444), and CLU (rs11136000) are associated with decreased risk (6–10). Only one study to date has examined the contribution of these nine risk alleles to LOAD status prediction (11). Verhaaren et al. calculated an additive genetic risk score and compared LOAD status prediction performance of age, gender, and APOE ε4 genotype using logistic regression with and without the additive genetic risk score. The genetic risk score did not improve prediction performance significantly, suggesting that the nine alleles may not be diagnostically useful when constrained to an additive relationship. The assumption of additive relationships between risk loci is common but is likely to be an oversimplification of the underlying biology for LOAD and other complex diseases (12–14). In fact, there may be underlying gene-gene interactions not examined in the Verhaaren et al. study or others that improve LOAD status prediction performance.
Some of the population attributable fractions for these nine loci have been reported individually and in different combinations (6, 8, 9); however, no study to date has reported the combined population attributable fraction for all nine risk alleles. Furthermore, previously reported odds ratios and population attributable fractions are from clinically ascertained samples rather than a population-based sample (6–10). The latter may provide a more reliable measure of population risk because clinically ascertained samples select for disease, enriching risk alleles in the sample.
In this study we estimated the allelic odds ratios and population attributable fractions for APOE ε2, APOE ε4, and the nine non-APOE LOAD risk alleles in a large population-based sample. We also extended the genetic risk score used by Verhaaren et al. by testing whether the nine non-APOE alleles contribute significantly to LOAD status prediction when interactions between loci are not constrained to additive relationships.
Methods and Materials
Sample Collection
The Cache County Study on Memory Health and Aging was initiated in 1994 (15). This cohort of 5,092 individuals represented approximately 90% of the Cache County population aged 65 and older. Specific details about data collection, obtaining consent, and phenotyping individuals in the Cache County population have been reported previously (15). Briefly, case-control status was determined in four triennial waves of data collection in a multi-stage dementia screening and assessment protocol. The first stage of screening consisted of administration of the Modified Mini-Mental State Exam-Revised (3MS-R) (16). Screen positive individuals and a randomly selected 19% designated subsample were invited to complete subsequent stages of evaluation consisting of an informant interview and the next stage, a clinical assessment including neuropsychological testing. The clinical assessment results were reviewed by a geropsychiatrist and neuropsychologist and preliminary diagnoses of dementia or other cognitive disorders were assigned. Those carrying a diagnosis of dementia or its prodrome were invited to complete standard laboratory tests for dementia, an MRI scan, and a geropsychiatrist examination. Final case-control status was determined by an expert panel of clinicians including study geropsychiatrists, neuropsychologists, a neurologist and cognitive neuroscientist. Diagnoses of AD followed NINCDS-ADRDA criteria (17), and cases included Possible or Probable AD. Controls were identified as those who were diagnosed with no dementia (per clinical assessment) or whose cognitive test result was negative at each preceding screening stage. Persons with incomplete screening results (i.e., those who were screen positive at one stage, but did not complete the subsequent stage), or missing genotype data were excluded from the analyses, leaving 2093 participants without dementia (controls) and 326 persons with LOAD (cases). All study procedures were approved by the Institutional Review Boards of Utah State, Duke and the Johns Hopkins University.
DNA from the 2,419 Cache County study participants was genotyped for the nine non-APOE LOAD risk alleles in the AlzGene.org “ALZGENE TOP RESULTS” list (18) using TaqMan Assays (Table 1). Genotyping failed for rs3764650 (ABCA7) and rs3818361 (CR1) so we selected rs3752246 and rs6656401 to represent the effects reported by ABCA7 and CR1 for AD risk, respectively. The CR1 SNPs are in high linkage disequilibrium (D’ = 0.995, R2 = 0.84) while both ABCA7 SNPs are within 10 kilobases of each other and rs3752246 was reported as significant by Naj et al. (9) APOE ε2 and APOE ε4 were previously genotyped as part of the Cache County study (15).
Table 1. Summary statistics for significant markers.
Minor allele frequencies, odds ratios, and population attributable fractions were calculated for all SNPs using both data from AlzGene.org and the Cache County population-based study. Population attributable fractions are reported as percentages. For better interpretation and comparison to previous studies, the risk allele for each locus (whether the major or the minor allele) was used to calculate population attributable fractions but the minor allele was used for odds ratios. Minor allele frequencies are comparable between AlzGene.org and the Cache County data. Odds ratios are generally similar except ABCA7 and CR1 differ in direction. Individual population attributable fractions in Cache County varied in magnitude when compared to those calculated for AlzGene.org. Combined population attributable fractions were also lower in Cache County. As expected APOE ε4 and APOE ε2 have strong population effects whereas the remaining alleles have minimal individual effect. Based on AlzGene.org data, combined population attributable fractions suggest the combined effect of the nine non-APOE alleles is approximately equal to APOE ε2 or APOE ε4 alone; however, the nine non-APOE alleles appear to have a larger effect than either APOE allele in the Cache County data.
| SNP | Nearest Gene | MAF
|
Odds Ratio
|
PAF
|
|||
|---|---|---|---|---|---|---|---|
| AlzGene | Cache Co. | AlzGene (95% CI) | Cache Co. (95% CI) | AlzGene | Cache Co. | ||
| rs3752246* | ABCA7 | 0.10 | 0.18 | 1.23 (1.18 – 1.28) | 0.94 (0.76 – 1.17) | 2.25 | 4.65 |
| rs7412 | APOE2 | 0.06 | 0.09 | 0.62 (0.46 – 0.85) | 0.89 (0.63 – 1.22) | 36 | 10 |
| rs429358 | APOE4 | 0.22 | 0.17 | 3.68 (3.30 – 4.11) | 2.51 (2.07 – 3.04) | 37 | 20 |
| rs744373 | BIN1 | 0.29 | 0.30 | 1.17 (1.13 – 1.20) | 1.02 (0.85 – 1.22) | 4.61 | 0.54 |
| rs9349407 | CD2AP | 0.29 | 0.28 | 1.12 (1.08 – 1.16) | 1.03 (0.85 –1.23) | 3.29 | 0.70 |
| rs3865444 | CD33 | 0.31 | 0.34 | 0.89 (0.86 – 0.92) | 1.00 (0.84 – 1.19) | 7.63 | 0.05 |
| rs11136000 | CLU | 0.38 | 0.39 | 0.88 (0.86 – 0.91) | 0.88 (0.74 – 1.04) | 7.85 | 7.98 |
| rs6656401 | CR1 | 0.19 | 0.19 | 1.19 (1.09 – 1.30) | 0.92 (0.74 –1.13) | 3.49 | 6.84 |
| rs670139 | MS4A4E | 0.41 | 0.41 | 1.08 (1.05 – 1.11) | 1.0 (0.84 – 1.18) | 3.14 | 0.05 |
| rs610932 | MS4A6A | 0.42 | 0.43 | 0.90 (0.88 – 0.93) | 0.89 (0.76 –1.06) | 5.81 | 6.33 |
| rs3851179 | PICALM | 0.35 | 0.38 | 0.88 (0.86 – 0.91) | 0.85 (0.72 – 1.01) | 8.19 | 9.69 |
|
| |||||||
| Combined PAF (All Alleles) | 75 | 51 | |||||
| Combined PAF (Excluding APOE) | 38 | 32 | |||||
The SNP for ABCA7 (rs3752246) was not reported on AlzGene.org, but was reported in Naj et al. as significant and was used in place of rs3764350
Statistical Analyses
All statistical analyses were performed in R (19). We used logistic regression and receiver operating characteristic (ROC) curve analysis to assess case-control predictive performance of the nine non-APOE alleles. Specifically, we tested whether the non-APOE alleles significantly improved LOAD status prediction performance over models excluding the non-APOE alleles. Two types of models were generated: additive risk profiles and genotype models to test potential additive and non-additive relationships, respectively. To assess efficacy of each model, we measured LOAD status prediction performance using the area under the curve (AUC) of the ROC curves. All models were adjusted for age and gender. A separate model using only age and gender was also generated to establish reference values.
We calculated three additive risk scores for participants in the Cache County Study to measure LOAD status prediction performance for the nine non-APOE LOAD risk alleles. Specifically, the following risk profiles were calculated: (1) APOE alone; (2) the nine LOAD risk alleles with APOE; and (3) the nine LOAD risk alleles without APOE. The risk allele (whether the major or the minor allele) and associated beta coefficient were used for each locus. We calculated additive risk scores as the sum of the risk across all alleles (equation 1), where β equals a previously calculated risk allele beta coefficient from odds ratios (β = ln(odds ratio)) reported by AlzGene.org (accessed February 2012), and N equals the subject’s number of risk alleles. APOE ε2 and APOE ε4 were coded jointly into a single class variable as 22, 23, 24, 33, 34, and 44.
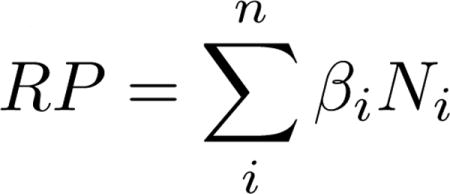 |
We also tested genotype models using genotype data in place of the risk profile score. We generated the following genotype models: (1) APOE alone; (2) the nine LOAD risk alleles with APOE; and (3) an optimized model. Using genotypes does not constrain the model to an additive relationship, allowing for other genetic models within each locus. The optimized model was generated using a stepwise regression method to test if interactions between loci contribute to LOAD status prediction and was selected using Akaike’s information criterion (AIC). To test for and avoid overfitting, we included three random variables while generating the optimized model. These variables were generated randomly with respect to all genotype and phenotype data in our study and were included to provide evidence that the selected variables provide meaningful information (20). While the absence of all random variables in the model does not guarantee the model was not overfit, it does suggest the included variables provide useful diagnostic information.
Synergy factors—a statistic that measures the strength of allelic interactions in case-controls studies (13, 21)—were calculated for any statistically significant allelic interactions using logistic regression. All synergy factors were adjusted for age, gender, and APOE ε4 by including only the main effects of the interacting alleles, the interaction term between the alleles, age, gender, and the number APOE ε4 alleles (Status = allele1*allele2 + age + gender + APOE4num). Synergy factor confidence intervals were calculated using the interaction term coefficient ±1.96 * standard error of the parameter estimate of the interaction term.
Odds ratios and population attributable fractions were also calculated. Odds ratios here estimate the relative risk of Alzheimer’s disease given allelic exposure while population attributable fractions estimate the proportional decrease in LOAD cases that would occur if the risk factor were removed from the population. Odds ratios were calculated only for the Cache County subjects but population attributable fractions were calculated for both Cache County subjects and the pooled AlzGene samples using published odds ratios and minor allele frequencies from AlzGene.org. We calculated population attributable fractions using equation 2 (9, 22), where p equals the allele frequency and OR is the odds ratio. A combined population attributable fraction was calculated for all risk factors and just the nine non-APOE risk factors using equation 3 (9, 22, 23) to estimate the proportional decrease in LOAD cases if all included risk factors were removed from the population. In this equation PAFj represents previously calculated PAFs from equation 2 and n is the number of loci included in the combined PAF. For better interpretation and comparison to previous studies, the risk allele for each locus (whether the major or the minor allele) was used to calculate population attributable fractions but the minor allele was used for odds ratios.
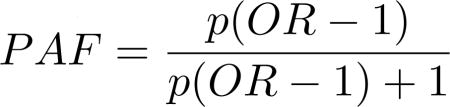 |
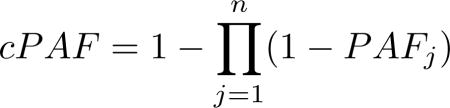 |
Results
Sample Demographics
The sample consisted of 1406 females and 1013 males. The mean age and standard deviation were 75.13 and 7.29 years, respectively. Mean age was significantly different between cases and controls (p < 2.2e-16), as were the proportion of males in each group (p < 0.04; Supplement: Table S1). Similarly, mean age was significantly different between participants included in the study and those excluded for reasons previously mentioned (p < 2.2e-16; Supplement: Table S2). The proportion of males, however, was not significantly different between included and excluded participants (p < 0.29).
Odds Ratios
Odds ratios calculated for the Cache County data were generally comparable in direction and magnitude to odds ratios from AlzGene.org when identical SNPs were genotyped. ABCA7 and CR1 varied, but a different SNP was genotyped for ABCA7 and the 95% confidence intervals for CR1 overlap between AlzGene.org and Cache County results (Table 1). Odds ratios from meta-analyses on AlzGene.org for ABCA7 and CR1 are 1.23 (95% CI 1.18 – 1.28) and 1.19 (95% CI 1.09 – 1.30), respectively, while from the Cache County data were 0.94 (95% CI 0.76 – 1.17) and 0.92 (95% CI 0.74 – 1.13), respectively. No alleles deviated significantly from Hardy Weinberg equilibrium.
Population Attributable Fraction
Population attributable fractions as calculated from AlzGene.org data ranged from 2.25% to 37% while those from Cache County ranged from 0.05% to 20% (Table 1). The highest risks were attributed to APOE ε4 (AlzGene = 37%; Cache = 20%) and lack of the APOE ε2 (AlzGene = 36%; Cache = 10%) whereas the next highest risk was attributed to PICALM (AlzGene = 8.19%; Cache = 9.69%). The smallest risk for AlzGene.org was from ABCA7 (2.2%) while the smallest for the Cache County data were CD33 and MS4A4E (0.05%). Combined population attributable fractions for all LOAD risk alleles (including APOE) were 75% and 51% for AlzGene.org and Cache County, respectively. Using only the nine non-APOE alleles were 38% and 32% for AlzGene.org and Cache County, respectively.
LOAD Status Prediction Performance
The non-APOE alleles combined with APOE (AUC = 0.782) did not improve LOAD status prediction performance over APOE alone (AUC = 0.783) when constrained to an additive model (Supplement: Figure S1), as previously reported (11); nor did the non-APOE alleles without APOE (AUC = 0.728) significantly improve LOAD status prediction performance over age and gender alone (AUC = 0.727; p < 0.2372). The model using all genotype data (full genotype model) when not constrained to an additive relationship (AUC = 0.796), however, did improve LOAD status prediction performance significantly over APOE alone (AUC = 0.783; p < 0.03; Figure 1). Moreover, the optimized model allowing for interactions between loci (AUC = 0.82) improves significantly over the full genotype model (p < 8.39e-07). All three genotype models improve prediction performance significantly over age and gender alone. None of the random variables previously mentioned were selected for the optimized model. Selected variables and interactions for the optimized model are as follows: rs3752246, rs6656401, rs11136000, rs610932, rs3865444, rs670139, Age, APOE.factor, rs3865444:rs670139, rs11136000:rs670139, rs3752246:APOE.factor, rs3752246:rs610932, and rs670139:Age.
Figure 1. Non-APOE LOAD risk loci contributions to LOAD status prediction performance.
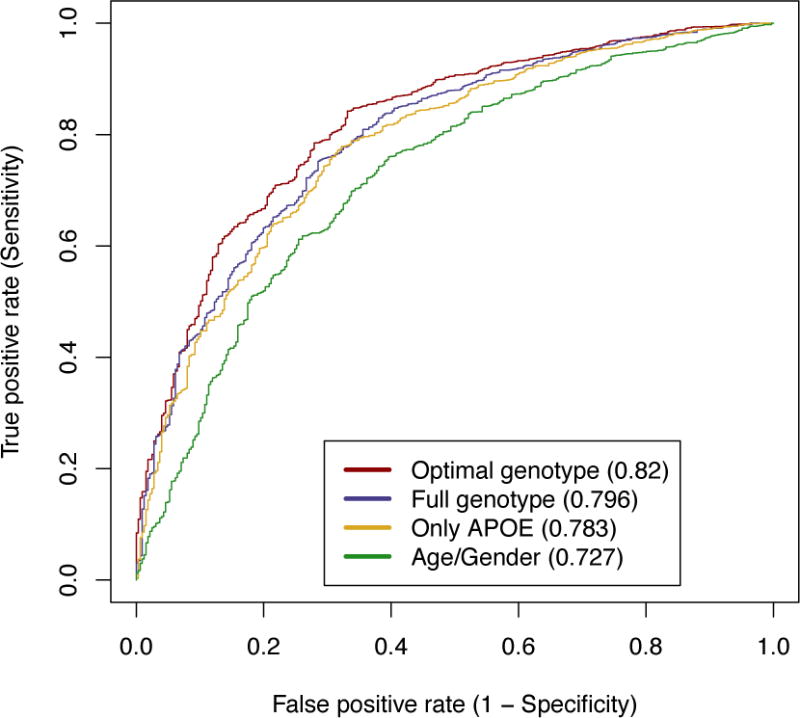
Three logistic regression models based on age, gender, and genetic information for APOE and the non-APOE LOAD risk loci illustrate the contribution of the non-APOE LOAD risk loci in LOAD status prediction performance. The models are as follows: APOE alone (Only APOE), all loci (Full genotype), and the optimized model (Optimal genotype). A fourth model using only age and gender (Age/Gender) was also generated as a baseline. The optimized model was optimized using Akaike’s Information Criterion (AIC). Comparing the full genotype model to APOE alone demonstrates that the LOAD risk loci contribute significantly to LOAD status prediction performance (p < 0.03) while the optimized model improves significantly over the full genotype model (p < 8.39e-07). Area under the curve (AUC) is listed in parentheses within the legend.
Locus Interactions
Investigating the optimized genotype model revealed two statistically significant alleles and two significant allelic interactions, though the p-values were not corrected for multiple testing. Genotypes A/G (p < 0.02) and G/G (p < 0.03) in rs6656401 (CR1) were significant individually. The significant interactions were between the rs3865444 C/C (CLU) genotype and the rs670139 G/G (MS4A4E) genotype (p < 0.016; SF 3.81, 95% CI 1.28 – 11.32) and the rs11136000 C/C (CD33) genotype and the rs670139 G/G (MS4A4E) genotype (p < 0.003; SF 5.31, 95% CI 1.79 – 15.77).
Discussion
Recent research has identified several alleles that may prove useful in resolving Alzheimer’s etiology (6–10), but until now there had not been an assessment of their population attributable fraction in a large, population-based sample. Similarly, deeper interrogation of the diagnostic utility of the Alzheimer’s disease candidate genes is needed. Verhaaren et al. explored the diagnostic utility based on an additive relationship, which we replicated in this work, but they did not test locus interactions—a major aim of this research. During this process we also estimated allelic odds ratios and population attributable fractions.
The data reported in this study are generalizable to other U.S. populations of northern European descent. The Cache County population has been included in the Centre d’Etude du Polymorphisme Humain (CEPH) families that are used to represent the European sample in the HapMap project (24, 25). Utah’s early pioneers were mostly unrelated and originated from various European locations (26–28), which is necessary for generalizability. The AlgGene.org data—a meta-analysis—varies between loci but is largely Caucasian-based as well. Many of the loci include populations of African, Asian, and Hispanic decent but the sample sizes for these populations are much smaller than the Caucasian populations.
Odds Ratios
We compared Cache County odds ratios to those reported in the meta-analyses on AlzGene.org and found them comparable. Minor differences were observed in ABCA7 and CR1 where we genotyped SNPs that are not listed on AlzGene.org. Specifically, minor alleles for both ABCA7 and CR1 were considered risk alleles (odds ratio > 1) according to data on AlzGene.org while odds ratios in the Cache County data suggest decreased risk, although the confidence intervals from both studies are broad and overlap each other so they may not be significantly different. Possible causes include: (1) differences in sample ascertainment between clinical and population studies (e.g. the cases in clinically ascertained samples are generally younger than those in the Cache County Sample; see AlzGene.org, Supplement: Table S1); and (2) allelic odds ratios are not adjusted for age, gender, and other loci—nor are they adjusted for undiscovered or uncharacterized allelic interactions (13, 29–31).
Clinical and population studies differ in sample ascertainment. Clinically ascertained cases and controls are selected to minimize confounding variables and maximize contrast between the true underlying causes by minimizing known differences between the two groups except for the phenotype of interest. Population-based studies, however, are designed to represent true population characteristics such as allele frequencies, odds ratios, and population attributable fractions, as reported here. While population-based studies are ideal for reporting population characteristics, small sample sizes for cases may reduce the accuracy of OR and PAF estimates. Because of the natural differences between clinically ascertained case-control and population-based studies, it is important to leverage the strengths of each of them.
The complex nature of Alzheimer’s disease inheritance, however, suggests that variations between studies may be exist because allelic odds ratios are not adjusted for age, gender, and other loci—nor are they adjusted for undiscovered and uncharacterized allelic interactions. Each of these factors plays a significant role in Alzheimer’s etiology and not adjusting for them introduces error into odds ratio estimates. Allelic interactions also likely contribute to the “missing heritability” in Alzheimer’s disease. No single genetic locus characterizes Alzheimer’s etiology. APOE alone is highly predictive, but the genetic loci included here also appear to influence Alzheimer’s susceptibility, as reported in this study and others (6–10). Furthermore the effects of APOE vary between ethnic groups (32–36). Failure to replicate established genome-wide association study findings in some populations (13, 37) further suggests the possible influence of environmental factors, gene-environment, and gene-gene interactions.
Population Attributable Fractions
Cache County population attributable fractions varied in magnitude when compared to those calculated from AlzGene.org data. Combined population attributable fractions were lower in Cache County. As expected APOE ε4 and APOE ε2 have strong population effects whereas the remaining alleles have minimal individual effects. Based on AlzGene.org data, combined population attributable fractions suggest the combined effect of the nine non-APOE alleles is approximately equal to APOE ε2 or APOE ε4 alone; however, the combined non-APOE alleles appear to have a larger effect than either APOE allele in the Cache County data. The Cache County values are of value because they are population-based and better represent risks within populations—the purpose of the PAF statistic. Despite being more conservative than other estimates (combined), however, the population attributable fractions reported in this study may still be inflated because they are based on the unadjusted allelic odds ratios and because the exposure frequency for the genotyped SNPs may vary from the functional variants they represent. They may also be biased as a consequence of potentially inaccurate odds ratios due to the small sample size for cases, as mentioned previously. Future estimates are also likely to change as allelic interactions are discovered and incorporated into the calculations.
Diagnostic Utility
Verhaaren et al. demonstrated that the nine non-APOE genes do not improve LOAD status prediction performance when constrained to an additive relationship, which we confirmed in this study. When unconstrained, however, the top nine alleles improved LOAD status prediction performance significantly, demonstrating these alleles may provide more information as we better understand their epistatic relationships. The optimized model further improved LOAD status prediction performance and revealed CLU-MS4A4E and CD33-MS4A4E interactions that may prove valuable in Alzheimer’s research. Synergy factors for both interactions suggest that being homozygous for both alleles in either interaction increases risk. Yet, although these data suggest the additional LOAD risk alleles significantly improve LOAD status prediction performance, the improvement is marginal and does not reach the desired sensitivity or specificity for clinical use.
The optimized model clearly improves LOAD status prediction performance over the full genotype model and over APOE alone, suggesting allelic interactions may be useful for diagnostic purposes; however, the p-values were not corrected for multiple testing. As such, these interactions need to be tested in an independent data set. It is also possible the optimized model is overfit; however, the random variables included in the model selection process were not selected for the final model, lending evidence that the final variables included provide non-random information. The revealed interactions also have strong synergy factors suggesting they may be important. Furthermore, the genotype model with all alleles improves LOAD status prediction performance over APOE alone, lending support for underlying relationships amongst the factors included in the model.
Implications and Future Directions
The results presented here offer evidence that gene-gene interactions play a role in Alzheimer’s susceptibility; however, the reported interactions, do not appear to improve LOAD status prediction performance by an amount that is relevant in a clinical diagnostic setting. These results do suggest that to fully understand the genetic basis of Alzheimer’s disease risk we must improve our efforts to characterize gene-gene and gene-environment interactions.
Additionally, environmental factors have not received as much attention as genetic factors in Alzheimer’s research and should be thoroughly investigated (12). Although the CLU-MS4A4E and CD33-MS4A4E interactions appear to have strong effects in the Cache County study, there may be unmeasured environmental factors that increase the effect of these interactions in the Cache County population. Other research has shown that only 30% of Alzheimer’s disease is explained by known genes, demonstrating that environmental effects and gene by environment interactions will be essential in future studies (38).
The CLU-MS4A4E and CD33-MS4A4E interactions have not been previously reported leaving the biological foundation in question. Using IPA (Ingenuity® Systems, www.ingenuity.com), we explored possible interactions between each pair and found that, while no information is available for MS4A4E specifically, both CLU and CD33 interact indirectly with MS4A2 (Supplement: Figure S2). According to IPA, both thioacetamide and TGFB1 act indirectly on both CLU and MS4A2 (Supplement: Figure S2A). CLU also binds to BCL2L1, which is acted upon by MS4A2. Likewise, CD33 acts on PTPN6, which binds to MS4A2 and CD33 binds to CBL, which then acts on MS4A2 (Supplement: Figure S2B). Both MS4A4E and MS4A2 are members of the membrane-spanning 4-domain gene family. A complete IPA legend is available in Ingenuity’s website (http://ingenuity.force.com/ipa/articles/Feature_Description/Legend).
Overall, the results presented in this paper suggest that gene-gene interactions (epistasis) may play an important role in Alzheimer’s etiology. While discovering and characterizing epistatic interactions is a non-trivial task, researchers and consortiums must heed the plentiful evidence that Alzheimer’s is driven by complex gene-gene and gene-environment interactions.
Supplementary Material
Acknowledgments
This work was supported by grants from NIH (R01AG11380, R01AG21136, R01AG31272, R01AG042611), the Alzheimer’s Association (MNIRG-11-205368) and the Utah Science, Technology, and Research initiative (USTAR), the Utah State University Agricultural Experiment Station, and the Brigham Young University Gerontology Program. The authors thank the participants and staff of the Dementia Progression Study, the Utah Population Database, and the Cache County Study on Memory Health and Aging for their important contributions to this work. Additionally, the authors acknowledge the assistance of Drs. David Ward and Ned Weinshenker. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Financial Disclosures
No authors report biomedical financial interests or potential conflicts of interest.
References
- 1.Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261:921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 2.Saunders AM, Strittmatter WJ, Schmechel D, George-Hyslop PH, Pericak-Vance MA, Joo SH, et al. Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer’s disease. Neurology. 1993;43:1467–1472. doi: 10.1212/wnl.43.8.1467. [DOI] [PubMed] [Google Scholar]
- 3.St Clair D, Rennie M, Slorach E, Norrman J, Yates C, Carothers A. Apolipoprotein E epsilon 4 allele is a risk factor for familial and sporadic presenile Alzheimer’s disease in both homozygote and heterozygote carriers. J Med Genet. 1995;32:642–644. doi: 10.1136/jmg.32.8.642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, Salvesen GS, et al. Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci U S A. 1993;90:1977–1981. doi: 10.1073/pnas.90.5.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bertram L, McQueen MB, Mullin K, Blacker D, Tanzi RE. Systematic meta-analyses of Alzheimer disease genetic association studies: the AlzGene database. Nat Genet. 2007;39:17–23. doi: 10.1038/ng1934. [DOI] [PubMed] [Google Scholar]
- 6.Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML, et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat Genet. 2009;41:1088–1093. doi: 10.1038/ng.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hollingworth P, Harold D, Sims R, Gerrish A, Lambert JC, Carrasquillo MM, et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat Genet. 2011;43:429–+. doi: 10.1038/ng.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lambert JC, Heath S, Even G, Campion D, Sleegers K, Hiltunen M, et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet. 2009;41:1094–1099. doi: 10.1038/ng.439. [DOI] [PubMed] [Google Scholar]
- 9.Naj AC, Jun G, Beecham GW, Wang LS, Vardarajan BN, Buros J, et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat Genet. 2011;43:436–+. doi: 10.1038/ng.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Seshadri S, Fitzpatrick AL, Ikram MA, DeStefano AL, Gudnason V, Boada M, et al. Genome-wide analysis of genetic loci associated with Alzheimer disease. JAMA. 2010;303:1832–1840. doi: 10.1001/jama.2010.574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Verhaaren BF, Vernooij MW, Koudstaal PJ, Uitterlinden AG, Duijn CM, Hofman A, et al. Alzheimer’s Disease Genes and Cognition in the Nondemented General Population. Biol Psychiatry. 2012 doi: 10.1016/j.biopsych.2012.04.009. [DOI] [PubMed] [Google Scholar]
- 12.Bullock JM, Medway C, Cortina-Borja M, Turton JC, Prince JA, Ibrahim-Verbaas CA, et al. Discovery by the Epistasis Project of an epistatic interaction between the GSTM3 gene and the HHEX/IDE/KIF11 locus in the risk of Alzheimer’s disease. Neurobiol Aging. doi: 10.1016/j.neurobiolaging.2012.08.010. [DOI] [PubMed] [Google Scholar]
- 13.Combarros O, Cortina-Borja M, Smith AD, Lehmann DJ. Epistasis in sporadic Alzheimer’s disease. Neurobiol Aging. 2009;30:1333–1349. doi: 10.1016/j.neurobiolaging.2007.11.027. [DOI] [PubMed] [Google Scholar]
- 14.Moore JH, Williams SM. Traversing the conceptual divide between biological and statistical epistasis: systems biology and a more modern synthesis. BioEssays. 2005;27:637–646. doi: 10.1002/bies.20236. [DOI] [PubMed] [Google Scholar]
- 15.Breitner JC, Wyse BW, Anthony JC, Welsh-Bohmer KA, Steffens DC, Norton MC, et al. APOE-epsilon4 count predicts age when prevalence of AD increases, then declines: the Cache County Study. Neurology. 1999;53:321–331. doi: 10.1212/wnl.53.2.321. [DOI] [PubMed] [Google Scholar]
- 16.Tschanz JT, Welsh-Bohmer KA, Plassman BL, Norton MC, Wyse BW, Breitner JC, et al. An adaptation of the modified mini-mental state examination: analysis of demographic influences and normative data: the cache county study. Neuropsychiatry Neuropsychol Behav Neurol. 2002;15:28–38. [PubMed] [Google Scholar]
- 17.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology. 1984;34:939–944. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- 18.Bertram LMM, Mullin K, Blacker D, Tanzi R. The AlzGene Database. Alzheimer Research Forum. 2007 doi: 10.1038/ng1934. [DOI] [PubMed] [Google Scholar]
- 19.R Core Team. R: A language and environment for statistical computing. 2.15.1. Vienna, Austria: R Foundation for Statistical Computing; 2012. [Google Scholar]
- 20.Gonzalez NA, Swain NR, Obregon O, Buehler BD, Williams GP, Nelson EJ, et al. Spatial and Temporal Statistical Analysis of Water Quality Patterns in a Small Temperate Supply Reservoir. American Society of Civil Engineers. 2012:1982–1992. [Google Scholar]
- 21.Cortina-Borja M, Smith AD, Combarros O, Lehmann D. The synergy factor: a statistic to measure interactions in complex diseases. BMC Research Notes. 2009:2. doi: 10.1186/1756-0500-2-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Imputation of sequence variants for identification of genetic risks for Parkinson’s disease: a meta-analysis of genome-wide association studies. The Lancet. 2011;377:641–649. doi: 10.1016/S0140-6736(10)62345-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Slatkin M. Exchangeable models of complex inherited diseases. Genetics. 2008;179:2253–2261. doi: 10.1534/genetics.107.077719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.International HapMap C The International HapMap Project. Nature. 2003;426:789–796. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- 25.Ridge PG, Maxwell TJ, Corcoran CD, Norton MC, Tschanz JT, O’Brien E, et al. Mitochondrial genomic analysis of late onset Alzheimer’s disease reveals protective haplogroups H6A1A/H6A1B: the Cache County Study on Memory in Aging. PLoS One. 2012;7:e45134. doi: 10.1371/journal.pone.0045134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.O’Brien E, Rogers AR, Beesley J, Jorde LB. Genetic structure of the Utah Mormons: comparison of results based on RFLPs, blood groups, migration matrices, isonymy, and pedigrees. Hum Biol. 1994;66:743–759. [PubMed] [Google Scholar]
- 27.Jorde LB, Morgan K. Genetic structure of the Utah Mormons: isonymy analysis. Am J Phys Anthropol. 1987;72:403–412. doi: 10.1002/ajpa.1330720313. [DOI] [PubMed] [Google Scholar]
- 28.Jorde LB. The genetic structure of the Utah Mormons: migration analysis. Hum Biol. 1982;54:583–597. [PubMed] [Google Scholar]
- 29.Moore JH, Williams SM. Traversing the conceptual divide between biological and statistical epistasis: systems biology and a more modern synthesis. BioEssays. 2005;27:637–646. doi: 10.1002/bies.20236. [DOI] [PubMed] [Google Scholar]
- 30.Cordell HJ. Epistasis: what it means, what it doesn’t mean, and statistical methods to detect it in humans. Hum Mol Genet. 2002;11:2463–2468. doi: 10.1093/hmg/11.20.2463. [DOI] [PubMed] [Google Scholar]
- 31.Moore JH. The ubiquitous nature of epistasis in determining susceptibility to common human diseases. Hum Hered. 2003;56:73–82. doi: 10.1159/000073735. [DOI] [PubMed] [Google Scholar]
- 32.Tang MX, Maestre G, Tsai WY, Liu XH, Feng L, Chung WY, et al. Relative risk of Alzheimer disease and age-at-onset distributions, based on APOE genotypes among elderly African Americans, Caucasians, and Hispanics in New York City. Am J Hum Genet. 1996;58:574–584. [PMC free article] [PubMed] [Google Scholar]
- 33.Murrell JR, Price B, Lane KA, Baiyewu O, Gureje O, Ogunniyi A, et al. Association of apolipoprotein E genotype and Alzheimer disease in African Americans. Arch Neurol. 2006;63:431–434. doi: 10.1001/archneur.63.3.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mayeux R. Apolipoprotein E, Alzheimer disease, and African Americans. Arch Neurol. 2003;60:161–163. doi: 10.1001/archneur.60.2.161. [DOI] [PubMed] [Google Scholar]
- 35.Desai PP, Hendrie HC, Evans RM, Murrell JR, DeKosky ST, Kamboh MI. Genetic variation in apolipoprotein D affects the risk of Alzheimer disease in African-Americans. Am J Med Genet B Neuropsychiatr Genet. 2003;116B:98–101. doi: 10.1002/ajmg.b.10798. [DOI] [PubMed] [Google Scholar]
- 36.Maestre G, Ottman R, Stern Y, Gurland B, Chun M, Tang MX, et al. Apolipoprotein E and Alzheimer’s disease: ethnic variation in genotypic risks. Ann Neurol. 1995;37:254–259. doi: 10.1002/ana.410370217. [DOI] [PubMed] [Google Scholar]
- 37.Healy DG. Case-control studies in the genomic era: a clinician’s guide. Lancet neurology. 2006;5:701–707. doi: 10.1016/S1474-4422(06)70524-5. [DOI] [PubMed] [Google Scholar]
- 38.Lee SH, Harold D, Nyholt DR, Consortium AN, International Endogene C, Genetic et al. Estimation and partitioning of polygenic variation captured by common SNPs for Alzheimer’s disease, multiple sclerosis and endometriosis. Hum Mol Genet. 2013;22:832–841. doi: 10.1093/hmg/dds491. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.