

Abstract
Embryonal tumor with abundant neuropil and true rosettes (ETANTR) is a recently described embryonal neoplasm of the central nervous system, consisting of a well‐circumscribed embryonal tumor of infancy with mixed features of ependymoblastoma (multilayer ependymoblastic rosettes and pseudorosettes) and neuroblastoma (neuroblastic rosettes) in the presence of neuropil‐like islands. We present the case of a young child with a very aggressive tumor that rapidly recurred after gross total resection, chemotherapy and radiation. Prominent vascular sclerosis and circumscribed tumor led to the diagnosis of malignant astroblastoma; however, rapid recurrence and progression of this large tumor after gross total resection prompted review of the original pathology. ETANTR is histologically distinct with focal glial fibrillary acid protein (GFAP) and synaptophysin expression in the presence of neuronal and ependymoblastic rosettes with focal neuropil islands. These architectural features, combined with unique chromosome 19q13.42 amplification, confirmed the diagnosis. In this report, we describe tumor stem cell (TSC) marker CD133, CD15 and nestin alterations in ETANTR before and after chemotherapy. We found that TSC marker CD133 was richly expressed after chemotherapy in recurrent ETANTR, while CD15 is depleted compared with that expressed in the original tumor, suggesting that CD133+ cells likely survived initial treatment, further contributing to formation of the recurrent tumor.
Keywords: embryonal tumor with abundant neuropil and true rosettes, ETANTR, microRNA, tumor stem cell
Introduction
Embryonal tumors are malignant neoplasms that predominantly affect children. According to the 2007 World Health Organization (WHO) classification of central nervous system (CNS) tumors, there are three types of embryonal tumors: medulloblastoma, atypical teratoid/rhabdoid tumor (AT/RT) and primitive neuroectodermal tumor (PNET) 23, 24. PNETs are further subdivided into neuroblastoma, ganglioneuroblastoma, medulloepithelioma, ependymoblastoma and PNET‐not otherwise specified (NOS). Embryonal tumor with abundant neuropil and true rosettes (ETANTR) is considered a variant within the PNET‐NOS category of the WHO classification. Because of its unique clinical and histological features, it has been suggested that this tumor may eventually be considered a separate entity 1.
ETANTR was first described in 2000 by Eberhart et al 8. They reported on nine children with tumors that had hybrid features of both cerebral neuroblastoma and ependymoblastoma along with unique histological, immunohistochemical and ultrastructural characteristics. The composite features of ETANTR as initially observed by Eberhart et al 8 included fine and abundant fibrillary neuropil, undifferentiated neuroepithelial cells arranged in clusters and several types of rosettes, such as Homer‐Wright, Flexner–Wintersteiner and ependymoblastic like. Recently a specific amplification of 19q13.42 region containing a cluster of microRNA (miR) coding genes was found in embryonal tumors with multilayered rosettes, which can be helpful in narrowing the diagnosis of these primitive tumors 15, 21, 30. To date, 30 cases of ETANTR have been reported in the literature 8, 16, 17, 26, 35. Age at diagnosis ranged from 6 months to 4 years. ETANTR is associated with high potential for malignancy and poor prognosis 19. Mean survival is approximately 16 months, although one long‐term survivor after 7 years of follow‐up has been reported 7, 16, 26, 35.
The tumor stem cell (TSC) theory suggests that the tumorigenic potential of CNS tumors is limited to a subpopulation of cells with stem‐like properties (CD133, CD15) 2, 33. It has been further suggested that these subpopulations confer chemotherapy and radiation resistance by their slow growth and ability to remain dormant, evading conventional therapeutic strategies 31. ETANTR remains poorly characterized clinically as well as pathologically. The characterization of tumor stem‐like cells in ETANTR will improve our understanding of this disease, leading to a better understanding of tumor biology and treatment design. In this study, we investigate the presence of stem‐like cell tumor markers CD133, CD15 and nestin from initial diagnosis, tumor recurrence and within different regions of a single ETANTR tumor.
Clinical History
A 3‐year‐old girl presented with a 4‐day history of worsening headache, nausea, vomiting and ataxia. After presentation, she became somnolent and her Glasgow Coma Score (GCS) dropped from 15 on presentation to 10. She was intubated and transferred to our hospital for further management.
T1‐weighted head magnetic resonance imaging (MRI; Figure 1A) showed a heterogeneous mass in the left frontoparietal region measuring 6.5 × 5.5 cm with areas of calcification and hydrocephalus. The patient underwent a left parietal occipital craniotomy for tumor resection. Gross total resection was achieved (Figure 1B). Tumor pathology was initially interpreted as malignant astroblastoma. The patient was treated under Children's Cancer Group Protocol 99703 and received three cycles of multiagent chemotherapy, including vincristine, etoposide and Cytoxan. This was followed by three cycles of non‐ablative stem cell chemotherapy with carboplatin and thiotepa. Follow‐up MRI of the brain 6 months after initial craniotomy revealed no evidence of recurrence. Routine 8‐month surveillance MRI demonstrated an enhancing mass in the left parietal occipital surgical resection cavity measuring 3.8 cm in maximal diameter (Figure 1C). A second tumor resection was performed, after which postoperative MRI scan demonstrated gross total resection. Following the second resection, the tumor was reclassified as an ETANTR rather than malignant astroblastoma.
Figure 1.
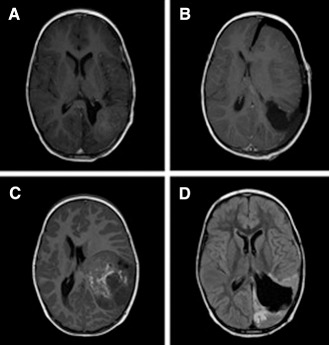
Axial contrast‐enhanced T1‐weighted magnetic resonance images (MRIs). A. Preoperative image demonstrating a 6.5 × 5.5 cm heterogeneous mass in left parietal region. B. Postoperative MRI following gross total resection. C. Axial MRI 8 months postoperatively reveals recurrent tumor within the resection cavity of the initial tumor. D. MRI 4 months after a second craniotomy reveals progression of disease with numerous new small lesions along brain surface and cranial nerves, most notably in posterior fossa, periventricular region and along the surgical cavity.
The patient was treated with craniospinal proton beam radiation. Four months following the second surgery, she became progressively somnolent with worsening headaches and vomiting. MRI revealed infiltrative tumor with diffuse leptomeningeal spread to intraventricular, supratentorial and infratentorial locations, along with coating of multiple cranial nerves and nodular disease in the entire spine (Figure 1D). Treatment with temozolomide, irinotecan and Avastin was initiated. The patient died of disease progression 13 months after initial diagnosis and 5 months following initial recurrence.
Methods
DNA, RNA and total protein were extracted from tumor samples at the time of initial resection and recurrence. Regional tumor heterogeneity was examined in the recurrent tumor by taking specimens from the tumor core and periventricular regions closest to the subventricular zone as evident at the time of surgery via the use of stereotactic navigation. Tissue protein, total RNA and DNA were isolated using previously established protocols 10.
Immunohistochemistry
Immunohistochemical analysis was performed on paraffin‐embedded and frozen sections according to established protocols 10. Primary antibodies included GFAP (polyclonal, 1:3200; DAKO Corp., Carpinteria, CA, USA), synaptophysin (prediluted, clone SP11; Ventana Medical Systems, Inc., Oro Valley, AZ, USA), INI‐1 (prediluted clone MRQ‐17; Cell Marque Corp., Rocklin, CA, USA) and Ki‐67 (prediluted, clone 30‐9; Ventana Medical Systems), nestin (AB5922 rabbit polyclonal, 1:1000; Millipore, Billerica, MA, USA), CD133 [3663 rabbit monoclonal immunoglobulin G (IgG), 1:100; Cell Signaling, Boston, MA, USA)] and CD15 (4744 mouse monoclonal IgM, 1:100; Cell Signaling). The percentage of cells that displayed positive immunoreactivity for CD15 and CD133 were calculated by analyzing five images taken in 10 random sections at 40× magnification based on a standard grid system. Positive control staining was performed using cultured human glioblastoma cell line, and negative control staining was performed using sections incubated in primary antibody dilution buffer without the primary antibody. Counts were performed by two masked independent investigators (DBA and ACW). Mean cell count for positive immunoreactivity was recorded, and a one‐way analysis of variance (ANOVA) was performed to analyze the differences between CD133+ and CD15+ populations in the tumor before chemotherapy and after chemotherapy in the core and periventricular regions. All statistical analysis was performed using PASW Statistics 18 software (SPSS Inc., Chicago, IL, USA).
Real‐time reverse transcription polymerase chain reaction (PCR)
The protocol followed for quantitative reverse transcription‐PCR (RT‐PCR) for CD133, CD15 and chromosome 19q13.42 has previously been described by Fan et al 9. PCR primers used for the analysis of chromosome 19q13.42 amplification were designed to amplify the coding region for miR‐512. PCR primers used for the analysis of miR‐512 were miR‐512‐QF (forward primer: 5′‐GAGCCACTGCAGGTAAAAGC‐3′) and miR‐512‐QR (reverse primer: 5′‐CAGGCTATCCCTGAATCCAA‐3′). Amplification of miR‐512 was compared with amplification of the coding region for KIR3DL3, a natural killer cell receptor that is located on 19q13.42 and has been shown to remain unamplified in PNETs 21. The primer sequences used for KIR3DL3 were 19q13.42 forward (forward primer: 5′‐CACCTTCTAAACTCACAACCT‐3′) and 19q13.42 reverse (reverse primer: 5′‐CTGTCTCTTCTGATTTCACCA‐3′). The reactions for CD15 and CD133 were normalized to β‐actin, while the reactions for miR‐512 amplification were normalized to that of three non‐pooled samples of normal human brain tissue. All samples were analyzed in triplicate and confirmed via two separate trials.
Protein analysis
Western blot analysis was performed according to established protocols 10 to compare stem cell marker expression levels in the pre‐chemotherapy tumor sample with those in the core and periventricular regions of the recurrent tumor following chemotherapy. Primary antibodies were directed against CD133 (3663, 1:1000; Cell Signaling).
Results
Anatomic pathology and analysis of 19q13.42 amplification
Histology sections of the initial specimen showed a highly necrotic tumor, composed of small round blue cells with high nuclear‐to‐cytoplasmic ratio, numerous mitoses and apoptotic bodies. Neoplastic cells arranged around sclerotic blood vessels formed pseudorosettes. True rosette formations were also present, some of which were multilayered. Focally the tumor had prominent neuropil islands when highlighted by synaptophysin. The tumor showed only focal GFAP immunoreactivity, with normal preservation of INI‐1 expression and markedly elevated proliferation index (Ki‐67). Trichrome highlighted prominent hyalinization of some vessels surrounded by tumor (Figure 2). The latter finding, in association with only focal GFAP expression in the presence of a highly malignant neoplasm, was originally interpreted as malignant astroblastoma.
Figure 2.
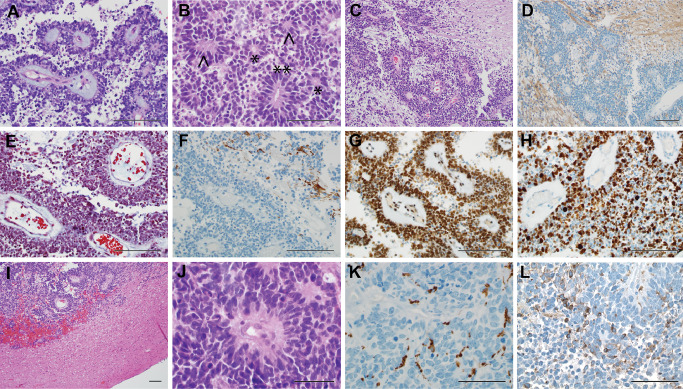
Tumor histopathology [embryonal tumor with abundant neuropil and true rosettes (ETANTR) ]. First resection (A–H): A. Largely necrotic tumor with viable cells predominately present around hyalinized blood vessels. B. The tumor is composed of small round blue cells, some of them forming ependymal (*), ependymoblastic (**) and neuronal rosettes (∧). C. Hematoxylin and eosin (H&E) staining shows neuropil islands around the tumor. D. Synaptophysin highlights neuropil. E. Trichrome demonstrates prominent perivascular hyalinization. F. GFAP shows only focal immunoreactivity of tumor cells. G. INI‐1 is preserved. H. Proliferation index (Ki‐67) is highly elevated. Second resection (I–L): I. Second resected specimen demonstrates a well‐circumscribed tumor with surrounding gliotic and mildly ischemic brain parenchyma. J. Small round blue cells, similar to original resection, with pseudorosettes and true rosette formation. K. GFAP demonstrates similar focal immunoreactivity of tumor cells. L. In addition, some tumor cells are also focally immunoreactive for synaptophysin. Scale bars represent 100 μm.
Histopathology of the recurrent tumor 8 months after gross total resection showed a well‐circumscribed tumor with morphology similar to the prior specimen: small round blue cells with hyperchromatic nuclei and high nuclear‐to‐cytoplasmic ratio. Cells were arranged in sheets palisading around vessels forming pseudorosettes. Mitoses and small cell necrosis were prominent. There was focal parenchymal sclerosis within the tumor and some vessels still displayed hyalinized walls. Immunohistochemical studies showed focal immunoreactivity for GFAP and synaptophysin in tumor cells (Figure 2). The tumor still had preservation of INI‐1, and the proliferation index was similar to the original specimen. In view of the tumor's aggressive behavior and rapid recurrence after gross total resection and chemotherapy, the pathology specimens were rereviewed, and the histological features were considered to be more consistent with ETANTR than malignant astroblastoma. In addition, molecular studies in the recurrent tumor demonstrated focal amplification of chromosome 19q13.42 (Figure 3). A combination of clinicopathological and molecular features in addition to the presence of this focal amplification confirmed the diagnosis of ETANTR.
Figure 3.
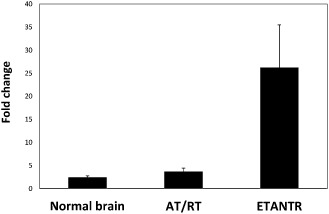
Bar graph comparison of focal amplification of microRNA (miR)‐512 in the index case [embryonal tumor with abundant neuropil and true rosettes (ETANTR) ], an embryonal tumor [atypical teratoid/rhabdoid tumor (AT/RT)] and three normal brains. The genomic copy number of miR‐512 is compared with that of KIRL3DL3 within each sample by reverse transcription‐polymerase chain reaction (RT‐PCR). Both loci are located on chromosome 19q13.42; there is focal amplification of distant regions within band 13, and sub‐band 42 is observed frequently in ependymoblastoma and ETANTR. miR‐512 amplification is significantly increased in ETANTR, confirming the diagnosis of ETANTR.
Analysis of stem cell markers
Stem cell markers CD133, CD15 and nestin were analyzed at the time of initial resection and at recurrence following chemotherapy. Analysis by Western blot demonstrated a marked increase in CD133 expression in the recurrent tumor in both the core and periventricular regions compared with tumor at the time of initial resection (Figure 4). RT‐PCR quantification of CD133 transcript levels showed a greater than 40‐ and 60‐fold increase in CD133 in the core and periventricular region of the recurrent tumor, respectively, compared with the initial tumor (P < 0.05; Figure 4). There was no difference in CD133 expression between the core and rim of the recurrent tumor. Fluorescently labeled tissue sections confirmed increased density and distribution of CD133 immunoreactivity in the core and periventricular regions of the recurrent tumor compared with the initial tumor, with 50–55 CD133+ cells per high‐powered field (HPF) compared with 20 cells per HPF in the initial tumor (although this number did not reach statistical significance given the sample size; P = 0.1; Figure 4). CD133 immunostaining showed a typical cell membrane distribution in the small round cells of this tumor.
Figure 4.
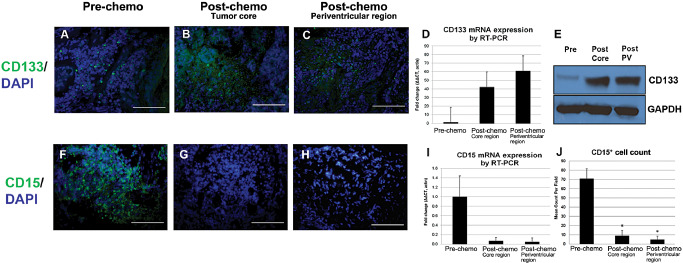
Stem cell marker expression pre‐ and post‐chemotherapy. A–E. CD133 expression. A–C. Immunofluorescence studies demonstrate CD133 increases (P < 0.05) after chemotherapy, predominantly in the tumor core area. D. Relative reverse transcription‐polymerase chain reaction (RT‐PCR) demonstrates a 40‐ and 60‐fold increase in CD133 expression in core and periventricular regions, respectively, after therapy (P < 0.05). E. Western blot analysis reveals increased CD133 in both the core and periventricular regions of the recurrent tumor compared with tumor at time of initial resection. F–J: CD15 expression. There is robust expression of CD15 on initial presentation, dropping significantly after therapy, as seen on immunofluorescence studies (P < 0.001) (F–H), RT‐PCR (I) and total cell count (J). All images at 40× magnification; scale bar represents 100 μm. DAPI, 4',6‐diaminodino‐2‐phenylindole; GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; mRNA, messenger RNA; PV, periventricular region.
CD15 expression decreased precipitously in both the core and periventricular region of the recurrent tumor compared with the initial tumor (Figure 4). In the initial tumor, 87% of cells were CD15+, but fewer than 1% of cells in the core and periventricular region of the recurrent tumor were CD15+ (P < 0.001). Quantification of CD15 transcript levels followed a similar pattern (Figure 4). CD15 transcript levels were decreased 14‐ and 20‐fold in the core and periventricular regions of the recurrent tumor, respectively, compared with the initial tumor.
Nestin expression was uniform and ubiquitous in sections of the primary and recurrent tumor when examined by immunofluorescence. Cytoplasmic expression of nestin was observed in nearly all cells of the tumor specimens. Blood vessels did not show nestin immunoreactivity. There were no differences in nestin expression seen by Western blot or RT‐PCR analysis between pre‐ and post‐chemotherapy ETANTR specimens.
In summary, the recurrent tumor shows a significant increase in CD133+ cells by Western blot and RT‐PCR analysis. In contrast, there was a significant decline in the subpopulation of CD15+ cells post‐chemotherapy.
Discussion
ETANTR is a recently described embryonal neoplasm of the CNS. It is histologically distinct and associated with a poor prognosis 8, 11, 19, 29. In this report, we describe TSC marker alterations in ETANTR before and after chemotherapy.
According to the current WHO classification of CNS tumors 24, ETANTR is an embryonal tumor that falls into the subgroup of CNS PNET‐NOS, which are composed of poorly differentiated neuroepithelial cells. CNS PNETs that lack neural tube formation (medulloepithelioma), true ependymoblastic rosettes (ependymoblastoma) or neuronal differentiation, with or without ganglion cells (neuroblastoma or ganglioneuroblastoma), are classified as CNS PNET‐NOS. ETANTR has been described as a well‐circumscribed embryonal tumor of infancy with mixed features of ependymoblastoma (multilayered ependymoblastic rosettes and pseudorosettes) and neuroblastoma (neuroblastic rosettes) in the presence of neuropil‐like islands.
We present the case of a young child with a very aggressive tumor that rapidly recurred after gross total resection, chemotherapy and radiation. Histology sections of the first resected specimen showed a highly malignant neoplasm with prominent necrosis and small round blue cells frequently arranged around sclerotic blood vessels. Grossly the tumor appeared well circumscribed. The prominent vascular sclerosis and well‐circumscribed appearance of the tumor led to a diagnosis of malignant astroblastoma. Astroblastomas are considered to be circumscribed vasocentric glial neoplasms of children, adolescents and young adults. Histologically, astroblastomas are described as unipolar cytoplasmic processes that anchor the tumor cell to the stroma of a blood vessel, giving the appearance of pseudorosette or papillary formation. Vascular and tissue sclerosis or hyalinization is often seen. The tumor is thought to originate from the tanycyte, a cell with intermediate features between astrocyte and ependymal cells. Astroblastomas are rare tumors that have been divided into well‐differentiated and anaplastic (malignant) subtypes. The latter is even rarer and has prominent mitotic activity, cytologic atypia and architectural disorganization. It is thought that gross total resection, even in high‐grade lesions, usually results in a favorable outcome 3. The fact that the tumor recurred in such a short period of time after gross total resection and aggressive therapy prompted a review of the original pathology specimens.
As described earlier, this is a poorly differentiated tumor, with only focal GFAP and synaptophysin expression seen in the specimen from the second resection. Both of these findings were seen in a minority of the tumor cells, but are not uncommon in undifferentiated tumors including malignant astroblastomas 4. The presence of architectural features such as ependymoblastic and neuronal rosettes with focal neuropil islands suggested that the tumor was more consistent with the diagnosis of ETANTR. The presence of a unique chromosome 19q13.42 amplification in this case added clarity to the diagnosis.
A diverse set of molecular and genetic alterations in PNETs, along with unique histological characteristics, validates the subclassification of embryonal tumors into five distinct entities. Medulloblastomas are commonly known by their i(17)q abnormalities. Similarly, AT/RT is characterized by mutations or loss of the INI1 locus at 22q11.2. These changes are almost never found in supratentorial PNET and therefore are considered to be a different entity. Within PNETs, neuroblastomas show 13q telomeric deletion, 14q deletion and homozygous deletion of 9p21.3 spanning the CDKN2A and CDKN2B loci 20, 32. In contrast, chromosome 2 gain is frequently observed in ependymoblastoma and ETANTR 6. More recently, a highly specific cluster of focal amplifications at chromosome region 19q13.42 were found in both ependymoblastoma and ETANTR 15, 35. Focal amplification of 19q13.42 has previously been demonstrated in 11/45 PNETs that were not further subclassified 21. miR‐512 is one of 50 miRs within chromosome 19q13.42. miR‐512 was chosen for this experiment because it has been shown to be one of seven focally amplified miRs within this cluster and has a potentially oncogenic role 21. This aberration is unique to ETANTR as it has not been detected in any other pediatric brain tumors studied including 150 medulloblastomas, 122 ependymomas, 12 supratentorial PNETs and 12 AT/RTs 15, 17, 29, 30. The functional consequence of this chromosomal amplification in ETANTR is unknown; however, miR posttranscriptional silencing is a potential mechanism. Identification of amplification in this region and downstream effectors may not only serve as diagnostic markers, but also as important therapeutic targets for the development of novel treatment strategies for these rare pediatric tumors.
The brain tumor initiating cell (BTIC) or TSC theory has provided a new pattern for understanding tumor cell biology. It suggests that tumorigenic potential is limited to a subpopulation of cells with stem‐like properties with the ability to proliferate, self‐renew and differentiate into different lineages. Subpopulations of human brain tumor cells expressing the surface markers CD133, CD15 or nestin have been thought to behave in a “stem‐like fashion” and have therefore been cautiously identified as markers for BTICs in select tumor types 12, 18, 28.
CD133 is a stem cell marker expressed on neural, hepatic and hematopoietic progenitor cells 27. Enhanced CD133 expression has been correlated with poor prognosis and decreased survival in patients with medulloblastoma and neuroblastoma 12, 34. A higher proportion of CD133+ cells in medulloblastoma, neuroblastoma and ganglioneuroblastoma correlated with shorter survival and unfavorable histology 12, 34. Gowda et al 12 analyzed the prevalence and prognostic significance of TSC markers CD133 and CD15 in 60 cases of medulloblastoma. They found 70% of medulloblastomas are CD133+, with positivity as high as 17% of cells 12. CD133+ cells have also been shown to be resistant to chemotherapy and radiation treatment 25. Radioresistant and chemoresistant tumors display an enriched population of CD133+ cells compared with the initial tumor population. Our analysis of this patient with ETANTR is congruent with these observations. Western blot analysis and RT‐PCR demonstrated that CD133 protein and transcript level is increased in the recurrent form of the tumor compared with the tumor at the time of initial resection. Although we have no functional cellular data for further investigation, our data suggest that the small number of CD133+ cells in the initial tumor may have been spared by the initial chemotherapy regimen given to this patient. The recurrent tumor's enriched population of CD133+ cells likely survived initial treatment, further contributing to the formation of the recurrent tumor.
CD15, also known as stage‐specific embryonic antigen 1 (SSEA‐1/LeX), is a potential surface marker for BTICs first introduced by Son et al 33. A CD15+ subpopulation has been identified in medulloblastoma and glioblastoma 12, 33. Sixty‐seven percent of medulloblastomas are CD15+ (with positivity as high as 17.1% of the population) 12. CD15+ cells have been shown to be highly tumorigenic in vivo, and these cells also have self‐renewal and multi‐lineage differentiation potential 33. Our results showed a decrease in CD15+ cells following chemotherapy in ETANTR. The depleted population of CD15+ cells after chemotherapy in ETANTR may be caused by effective treatment against this subpopulation of cells by the therapeutic protocol given or the failure of CD15+ cells to adequately label TSCs in ETANTR.
The immature intermediate filament, nestin, is commonly expressed in CNS progenitor and neural stem cells. It is considered a marker of poor prognosis and is also considered to be a brain TSC marker for tumors originating from neuroectoderm 5, 18. In a study of 22 pediatric brain tumors including gliomas and medulloblastomas, Hemmati et al 13 showed that the most common characteristic between tumor‐derived neurospheres and normal neural stem cells was the expression of CD133 and nestin. Our study found no difference in the expression of nestin before or after chemoradiation.
The subventricular zone adjacent to the lateral wall of the lateral ventricle in the human CNS contains adult neural stem cells 14. Some have suggested that tumors with a relationship to neural stem cell regions display a more aggressive phenotype 22. For this reason, recurrent ETANTR tumor specimens were taken from both within the tumor core as well as from the region closest to the lateral ventricular wall. Our results showed no difference in CD133, CD15 or nestin positivity from these regions within the tumor.
The rarity of this tumor limits robust experimental analysis. Thus far, fewer than 30 cases have been described in the literature. We present this observational data of TSC markers with caution, noting the absence of validation in this paper or in the literature to support proliferative, self‐renewal or multi‐lineage capacity of CD133, CD15 or nestin‐positive cells in ETANTR.
Conclusions
We observed an increase in protein level and transcript number of the BTIC marker CD133 after chemotherapy and radiation in this case of ETANTR. We also found a decrease in CD15 positivity. While suggestive, this observational data must be further validated with functional studies to prove whether or not this subpopulation offers chemotherapy and radiation resistance in ETANTR. It may be necessary to develop multifaceted and targeted therapeutic strategies including those that target TSCs.
Acknowledgments and Disclosures
The authors would like to thank the Neuropathology Division at Massachusetts General Hospital, Boston, MA, particularly Dr. E. Tessa Hedley‐Whyte for important discussion and input regarding best histopathologic classification of the tumor. Thanks also to Holly Wagner for providing editorial assistance.
This study was supported by a Neurosurgery Research and Education Foundation (NREF) fellowship (SHJ), NIH/NCI Grant No. T32 CA009676‐18 (SHJ). National Cancer Institute Grant No. R01CA148621 (X.F.) and R01CA163737 (X.F.), Accelerate Brain Cancer Cure Project Award, American Brain Tumor Association Translational Grant, and Voices Against Brain Cancer Research Grant.
References
- 1. Al‐Hussain TO, Dababo MA (2009) Posterior fossa tumor in a 2 year‐old girl. Brain Pathol 19:343–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB et al (2006) Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 444:756–760. [DOI] [PubMed] [Google Scholar]
- 3. Bonnin JM, Rubinstein LJ (1989) Astroblastomas: a pathological study of 23 tumors, with a postoperative follow‐up in 13 patients. Neurosurgery 25:6–13. [PubMed] [Google Scholar]
- 4. Cabello A, Madero S, Castresana A, Diaz‐Lobato R (1991) Astroblastoma: electron microscopy and immunohistochemical findings: case report. Surg Neurol 35:116–121. [DOI] [PubMed] [Google Scholar]
- 5. Dahlstrand J, Collins VP, Lendahl U (1992) Expression of the class VI intermediate filament nestin in human central nervous system tumors. Cancer Res 52:5334–5341. [PubMed] [Google Scholar]
- 6. Dunham C, Sugo E, Tobias V, Wills E, Perry A (2007) Embryonal tumor with abundant neuropil and true rosettes (ETANTR): report of a case with prominent neurocytic differentiation. J Neurooncol 84:91–98. [DOI] [PubMed] [Google Scholar]
- 7. Eberhart CG (2011) Molecular diagnostics in embryonal brain tumors. Brain Pathol 21:96–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Eberhart CG, Brat DJ, Cohen KJ, Burger PC (2000) Pediatric neuroblastic brain tumors containing abundant neuropil and true rosettes. Pediatr Dev Pathol 3:346–352. [DOI] [PubMed] [Google Scholar]
- 9. Fan X, Mikolaenko I, Elhassan I, Ni X, Wang Y, Ball D et al (2004) Notch1 and notch2 have opposite effects on embryonal brain tumor growth. Cancer Res 64:7787–7793. [DOI] [PubMed] [Google Scholar]
- 10. Fan X, Khaki L, Zhu TS, Soules ME, Talsma CE, Gul N et al (2010) NOTCH pathway blockade depletes CD133‐positive glioblastoma cells and inhibits growth of tumor neurospheres and xenografts. Stem Cells 28:5–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ferri Niguez B, Martinez‐Lage JF, Almagro MJ, Fuster JL, Serrano C, Torroba MA et al (2010) Embryonal tumor with abundant neuropil and true rosettes (ETANTR): a new distinctive variety of pediatric PNET: a case‐based update. Childs Nerv Syst 26:1003–1008. [DOI] [PubMed] [Google Scholar]
- 12. Gowda KK, Gupta K, Kapoor R, Vasishta RK (2012) Nuclear expression of beta‐catenin and stem cell markers as potential prognostic indicators in medulloblastoma. Neurol India 60:487–494. [DOI] [PubMed] [Google Scholar]
- 13. Hemmati HD, Nakano I, Lazareff JA, Masterman‐Smith M, Geschwind DH, Bronner‐Fraser M et al (2003) Cancerous stem cells can arise from pediatric brain tumors. Proc Natl Acad Sci U S A 100:15178–15183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jackson EL, Garcia‐Verdugo JM, Gil‐Perotin S, Roy M, Quinones‐Hinojosa A, VandenBerg S et al (2006) PDGFR alpha‐positive B cells are neural stem cells in the adult SVZ that form glioma‐like growths in response to increased PDGF signaling. Neuron 51:187–199. [DOI] [PubMed] [Google Scholar]
- 15. Korshunov A, Remke M, Gessi M, Ryzhova M, Hielscher T, Witt H et al (2010) Focal genomic amplification at 19q13.42 comprises a powerful diagnostic marker for embryonal tumors with ependymoblastic rosettes. Acta Neuropathol 120:253–260. [DOI] [PubMed] [Google Scholar]
- 16. Korshunov A, Remke M, Werft W, Benner A, Ryzhova M, Witt H et al (2010) Adult and pediatric medulloblastomas are genetically distinct and require different algorithms for molecular risk stratification. J Clin Oncol 28:3054–3060. [DOI] [PubMed] [Google Scholar]
- 17. Korshunov A, Witt H, Hielscher T, Benner A, Remke M, Ryzhova M et al (2010) Molecular staging of intracranial ependymoma in children and adults. J Clin Oncol 28:3182–3190. [DOI] [PubMed] [Google Scholar]
- 18. Krupkova O, Jr , Loja T, Redova M, Neradil J, Zitterbart K, Sterba J et al (2011) Analysis of nuclear nestin localization in cell lines derived from neurogenic tumors. Tumour Biol 32:631–639. [DOI] [PubMed] [Google Scholar]
- 19. La Spina M, Pizzolitto S, Skrap M, Nocerino A, Russo G, Di Cataldo A et al (2006) Embryonal tumor with abundant neuropil and true rosettes. A new entity or only variations of a parent neoplasms (PNETs)? This is the dilemma. J Neurooncol 78:317–320. [DOI] [PubMed] [Google Scholar]
- 20. Lamont JM, McManamy CS, Pearson AD, Clifford SC, Ellison DW (2004) Combined histopathological and molecular cytogenetic stratification of medulloblastoma patients. Clin Cancer Res 10:5482–5493. [DOI] [PubMed] [Google Scholar]
- 21. Li M, Lee KF, Lu Y, Clarke I, Shih D, Eberhart C et al (2009) Frequent amplification of a chr19q13.41 microRNA polycistron in aggressive primitive neuroectodermal brain tumors. Cancer Cell 16:533–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lim DA, Cha S, Mayo MC, Chen MH, Keles E, VandenBerg S et al (2007) Relationship of glioblastoma multiforme to neural stem cell regions predicts invasive and multifocal tumor phenotype. Neuro Oncol 9:424–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A et al (2007) The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol 114:97–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Louis DN, Ohgaki H, Wiestler OD, Webster KC (eds) (2007) WHO Classification of Tumours of the Central Nervous System, 4th edn. International Agency for Research on Cancer: Lyon. [Google Scholar]
- 25. Ma HI, Chiou SH, Hueng DY, Tai LK, Huang PI, Kao CL et al (2011) Celecoxib and radioresistant glioblastoma‐derived CD133+ cells: improvement in radiotherapeutic effects. Laboratory investigation. J Neurosurg 114:651–662. [DOI] [PubMed] [Google Scholar]
- 26. Manjila S, Ray A, Hu Y, Cai DX, Cohen ML, Cohen AR (2011) Embryonal tumors with abundant neuropil and true rosettes: 2 illustrative cases and a review of the literature. Neurosurg Focus 30:E2. [DOI] [PubMed] [Google Scholar]
- 27. Mizrak D, Brittan M, Alison M (2008) CD133: molecule of the moment. J Pathol 214:3–9. [DOI] [PubMed] [Google Scholar]
- 28. Panagiotakos G, Tabar V (2007) Brain tumor stem cells. Curr Neurol Neurosci Rep 7:215–220. [DOI] [PubMed] [Google Scholar]
- 29. Pfister S, Remke M, Toedt G, Werft W, Benner A, Mendrzyk F et al (2007) Supratentorial primitive neuroectodermal tumors of the central nervous system frequently harbor deletions of the CDKN2A locus and other genomic aberrations distinct from medulloblastomas. Genes Chromosomes Cancer 46:839–851. [DOI] [PubMed] [Google Scholar]
- 30. Pfister S, Remke M, Castoldi M, Bai AH, Muckenthaler MU, Kulozik A et al (2009) Novel genomic amplification targeting the microRNA cluster at 19q13.42 in a pediatric embryonal tumor with abundant neuropil and true rosettes. Acta Neuropathol 117:457–464. [DOI] [PubMed] [Google Scholar]
- 31. Reya T, Morrison SJ, Clarke MF, Weissman IL (2001) Stem cells, cancer, and cancer stem cells. Nature 414:105–111. [DOI] [PubMed] [Google Scholar]
- 32. Russo C, Pellarin M, Tingby O, Bollen AW, Lamborn KR, Mohapatra G et al (1999) Comparative genomic hybridization in patients with supratentorial and infratentorial primitive neuroectodermal tumors. Cancer 86:331–339. [DOI] [PubMed] [Google Scholar]
- 33. Son MJ, Woolard K, Nam DH, Lee J, Fine HA (2009) SSEA‐1 is an enrichment marker for tumor‐initiating cells in human glioblastoma. Cell Stem Cell 4:440–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tong QS, Zheng LD, Tang ST, Ruan QL, Liu Y, Li SW et al (2008) Expression and clinical significance of stem cell marker CD133 in human neuroblastoma. World J Pediatr 4:58–62. [DOI] [PubMed] [Google Scholar]
- 35. Wang Y, Chu SG, Xiong J, Cheng HX, Chen H, Yao XH (2011) Embryonal tumor with abundant neuropil and true rosettes (ETANTR) with a focal amplification at chromosome 19q13.42 locus: further evidence of two new instances in China. Neuropathology 31:639–647. [DOI] [PubMed] [Google Scholar]