

Abstract
Point-of-care diagnostics show promise in removing reliance on centralized lab testing facilities, and may help increase both the survival rate for infectious diseases as well as monitoring of chronic illnesses. CMOS compatible diagnostic platforms are currently being considered possible solution as they can be easily miniaturized and can be cost-effective. Top-down fabricated silicon nanowires are a CMOS-compatible technology which have demonstrated high sensitivities in detecting biological analytes, such as proteins, DNA, and RNA. However, the reported response of nanowires to these analytes has varied widely since several different functionalization protocols have been attempted with little characterization and comparison. Here we report protocols for fabrication and functionalization of silicon nanowires which yield highly stable nanowires in aqueous solutions, and limits of detection to ~1pg/mL of the model protein used in the study. A thorough characterization was done into optimizing the release of the silicon nanowires using combined dry and wet etch techniques, which yielded nanowires that could be directly compared to increase output statistics. Moreover, a range of different linker chemistries were tried for reacting the primary antibody, and its response to target and non-specific antigens, with polyethylene glycol based linker BS(PEG)5 providing the best response. Consequently, this chemistry was used to characterize different oxide thicknesses and their responses to the mouse IgG antigen, which with the smallest oxide thickness yielded 0.1–1pg/mL limits of detection and a dynamic range over 3 orders of magnitude.
Introduction
The electronics technology revolution which has occurred over the past decade, in large part due to the aggressive scaling of semiconductors dictated by Moore’s Law1, has allowed for Complementary Metal-Oxide Semiconductor (CMOS) technology to become a plausible platform to meet many of the requirements for portable biosensors, especially when it comes to cost and miniaturization.2 Metal oxide semiconductor field-effect transistors (MOSFET’s), the workhorse of CMOS technology, can be configured as a biosensor by modifying the gate with biological entities specific to the analyte of interest. Attachment of chemical and biological species to the device surfaces (with or without a metal gate) has allowed for a wide variety of analytes to be detected such as metal ions3–10, small molecules11–20, proteins21–27, and DNA28–32. Silicon nanowire FET’s have proven to sense biomarkers in clinically relevant levels33–40, and more recently demonstrated using CMOS compatible processing techniques41–43. The high sensitivities of nanowires have often been attributed to their high surface area to volume ratio, as well as their widths being similar in dimension to biological species such as proteins and DNA.44,45 Even though nanowires promise incredible sensitivity, the variety of device configurations (floating gates, with and without reference electrode, enhancement or depletion mode) in conjunction with the different functionalization and sensing protocols have led to large discrepancies in the magnitude of signal output.46 Surface functionalization protocols for analyte detection using optical methods has been well established47–52, with a multitude of protocols which yield detection limits in the pg-ng/mL range of analytes53,54. However, very little has been done in regards to understanding sensing protocols for electronic-based, label-free sensors.
In this work we characterize and provide possible solutions for two important problems in silicon nanowire sensing: the fabrication and device release of silicon on insulator (SOI) based nanowire FET’s, and the surface functionalization of nanowire FET’s. Silicon nanowire FET’s of different gate oxide thicknesses were fabricated and released using combined dry and wet etch techniques, yielding devices with threshold stabilities in the single mV range in aqueous solution. Previously we showed that monofunctional silanes could be used for high density, sub-nanometer interfacing to oxide surfaces, providing attractive qualities for interface dependent sensors.55 Here we use these monofunctional silanes with different linkers to elucidate protocols for attaching primary antibodies to surfaces which yield high specificity and sensitivity, while adhering to mainstream functionalization techniques. Using mouse immunoglobulins as the model antigen, goat-antimouse IgG’s were functionalized to the surfaces using an optimized protocol, which yielded sensitivities between 0.1–1 pg/mL for a 50A˚́ gate oxide thickness. Moreover, sensitivities achieved against other similar IgG’s from rabbits and different isotypes yielded minimal signal change. Current work involves using these protocols on foundry-grade CMOS chips to sense a wide variety of cancer biomarkers, in hope to improve the understanding of how to generate repeatable results on electronic-based biosensor platforms.
Experimental Section
The detailed fabrication outline of the SiO2 nanowire process and materials, as well the formation of the 3-aminopropyldimethylethoxysilane (APDMS) monolayer, can be found in the supporting information.
Materials
Dissucinimidyl Carbonate (DSC), glutaraldehyde (grade I, 50% in H2O), 1x PBS (molecular biology grade), Tween-20, and sodium cyanoborohyrdide were purchased from Sigma-Aldrich. The linker BS(PEG)5 was acquired from Pierce Scientific and a septum applied to the vial for air-free extraction using a syringe. The molecule was stored at −20°C until use.
The linker chemistries were then reacted onto the chips before primary antibody attachment. The DSC, BS(PEG)5, and glutaraldehyde linker chemistries were reacted with the APDMS monolayer at 2% (w/v) in dry DMF for 2 hours. The glutaraldehyde layer was then reduced with a 1% sodium cyanoborohydride solution to remove Schiff bases. All chips were finally rinsed in acetone and methanol, then blown dry with N2. The primary antibody was then reacted on the chips in 150 mM sodium bicarbonate buffer, pH 8.5 and rinsed with 1x PBS/0.05% Tween-20 for 1min, then 0.1x PBS for 1min. Devices were blocked with a 1x solution of BioFx Casein blocker for 30 mins, rinsed in 1xPBS/0.05% Tween-20 for 1min, then 0.1x PBS for 1min.
Measurement Details
Measurements were carried out using a Keithley SCS-4200 semiconductor characterization system and Agilent multiplexer. Devices were anchored to ceramic packages and wire bonded to leads on the outside of the package, and backfilled with epoxy for isolation. For fluidic measurements, the device was biased using an Ag/AgCl reference electrode in an aqueous electrolyte solution, and a PDMS fluidic well (volume ~10 uL) was bonded on top of the chip (Supplementary Fig. 1). The platinum window was not utilized as an electrode for measurements since its stability in solution was worse than the Ag/AgCl electrode and led to larger drifts.56 A Harvard Apparatus syringe pump was used at a flow rate of 20 uL/min to deliver analytes through microbore PTFE tubing (Upchurch Scientific) attached to micromanipulators, while a dry vacuum pump (Chemglass) was utilized to exchange solutions from the well. The ceramic package was put into a PC board linked to a switch matrix unit, which was hooked to the Source Measuring Units (SMU’s) inputs of the Keithley SCS-4200 system. For a particular device, the source-drain current (IDS) was measured while the gate voltage (Vg) was swept, and the IDS-Vg curves recorded. Each device was swept 5 times, with the average curve and standard deviation of each measurement being extracted.
For electronic studies of primary antibody adsorption, the antibodies were flowed at a rate of 20uL/min for 5 min, then stopped and allowed to react with the given surface chemistries for 1 hour using the conjugation protocol above, and finally were measured in 0.01x PBS, pH 7.4. For target analyte detection using nanowires, the analytes were dissolved in 1x PBS/ 0.05% Tween-20 to their respective concentrations and flowed over the devices using a flow rate of 20uL/min for 5 minutes. The antibodies were allowed to bind for 30 minutes, then rinsed in 1x PBS/0.05% Tween-20 for 1min, 0.1x PBS for 1min, and finally measured in 0.01x PBS, pH 7.4.
Fluorescence and Bright Field Imaging
Bright field and fluorescent images for the binding specificity and primary antibody adsorption were taken with a Nikon Eclipse FN-1 microscope using a DS-R1 CCD camera (8-bit imaging) and Mercury lamp. Bright field images were taken with exposure times between 10–20ms using an automatic exposure mode. Fluorescent images were taken at an exposure time of 1s with a 1.2s automatic shutter. Texas Red labeled antibodies were conjugated to thermally oxidized silicon wafer pieces (1um thick via wet oxidation at 1000°C) using the bioconjugation and rinsing procedures highlighted above, and imaged using a Y2E/C filter cube at an exposure of 400ms and gain of 1.5x. Similarly, Texas Red modified target immunoglobulins (4 moles dye/mole antibody) were imaged on nanowires and thermal oxide substrates using the same exposure as above for direct comparison.
Results and Discussion
Each chip was fabricated with different device widths and structures for sensing and application of biases to the solution. Figure 1A and B show optical micrographs of the device and platinum (Pt) release window areas before etching, with photoresist patterned to expose the device areas for etching. The color of the device release window area is due to the constructive and destructive interference of incident wavelengths of light through the dielectric layers, giving us a pink color before starting the device release. A schematic of the etching process with the device release window is shown in Figure 1C. Briefly, a CF4 RIE etch is used to remove the passivation layer, and a 50:1 BOE wet etch applied to thin the gate oxide. Using a wet etch for the final few nanometers helps to remove any RIE damage the gate oxide may have been exposed to.
Figure 1.

Optical micrographs of the device release windows and surrounding titanium/platinum leads for silicon nanowires (A), nanoplates, and the Pt window (B). The light hue surrounding the release window areas is due to the thickness of the patterned photoresist. A schematic of the device and Pt window cross sections are shown in (C) illustrating the etching process to release the devices and Pt window. The legend for the different layers is below the cross sections.
In order to determine the time duration of the RIE etch for device release, the different etch rates of SiN, SiO2, and Pt were determined. Sacrificial chips were etched for various times in CF4, and the etch depth measured with a stylus profilometer. Moreover, optical images were taken of the release window to document the color at each etch depth. Figure 2A shows the etch depth vs. etch time for the device release window (blue circle) and the platinum release window (black square). On top of the graph are optical micrographs of the nanoplate devices to indicate the color observed at each etch step. It is well known that the etch rates of SiN, thermal SiO2, and Pt are quite different in CF4 plasmas, leading to three different etching regions, with their order of etching displayed in Figure 2A. For the device release window, PECVD nitride (region 1) and thermal SiO2 (region 2b) are encountered, while for the platinum release window only PECVD nitride and platinum (region 2a) are encountered. We linearly fit each of the etching regions in Figure 2A and extracted the etching rates (see Table S1 in supporting information). PECVD silicon nitride is known to etch quite fast in CF4, with a selectivity against thermal SiO2 between 3:1 and 4:1, and platinum is known to act as an etch stop. These etch rates agree well with our findings. Moreover, the different release window areas etch at similar rates for PECVD nitride, indicating minimal issues due to loading effect. By linearly extrapolating the fits of the different etching regions, we can estimate an RIE stop point time (dashed black line) before beginning the wet etch of the SiO2 gate oxide, which is close to 300s.
Figure 2.
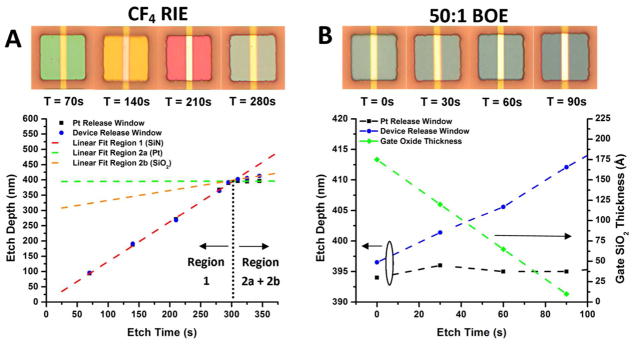
The etch depth of the device and platinum release windows vs. RIE etch time is plotted in (A) with linear fits (dashed lines) to each of the etching regions. The extrapolated area where the PECVD SiN has been entirely etched is shown by the black dashed line. The etch depth of the device and platinum release windows for the 50:1 BOE etch is plotted in (B) with the extracted gate oxide thickness. Optical images illustrating the color of the underlying nitride/oxide for each release time step is on top of the graphs for (A) and (B).
After a 300s CF4 RIE etch, 50:1 BOE was used to remove any local RIE damage and thin the gate oxide. The etch depth was monitored with a stylus profilometer as before, and the etching information displayed in Figure 2B. The color of the device release window is also monitored, and displayed on top of Figure 2B. Similar to the CF4 RIE etch, platinum (black square) does not etch in BOE and acts as a good reference etch stop, displayed in Figure 2B as no change in height above the platinum is observed. The approximate gate oxide thickness (green diamond) is extrapolated from the depth change from the profilometer and the grown oxide thickness of 175A˚́, determined by AFM. These protocols were used successfully to determine the etch depth and approximate gate oxide thickness for further experiments.
For immunological sensing of various protein analytes, the surface must be functionalized with a primary antibody and blocked effectively in order to maximize the signal to noise ratio. With regards to ISFET’s this point is particularly important, since maximizing the charge density to interface while minimizing steric hindrance is paramount to the efficiency of the device. In order to determine an optimum linking protocol, a monofunctional aminosilane was vapor deposited and various linker groups (for chemical structures and deposition method see supporting information) containing NHS esters or aldehydes were reacted to link amine or sulfhydryl containing groups on the primary antibody. The antibodies were then reacted and chips blocked according to the procedures outlined in the experimental section. In order to determine the best linker chemistries, fluorescence images were taken on functionalized blank SiO2 surfaces and corresponding electrical response on the silicon nanowires, displayed in Figure 3.
Figure 3.

Fluorescence intensity bar graph (part A) on thermally oxidized silicon surfaces showing the effect of conjugation linker on primary antibody fixation (Goat anti-mouse IgG) and subsequent specific/nonspecific target binding (Mouse IgG vs. Rabbit IgG). Each antibody and immunoglobulin was Texas Red labeled in order for direct comparison. The threshold voltage shift (Vt) of the silicon nanowires with respect to the primary antibody concentration and the linker chemistry is shown in (B). Fluorescence micrographs of silicon nanowires after Texas Red labeled antibodies were deposited at 100 ng/mL on the various linker chemistries are in (C).
Our model system was composed of Goat Anti-Mouse IgG with mouse IgG as the target and Rabbit IgG as the nonspecific binder. Each was either labeled with Texas Red (4 moles dye/ mole protein) or left unlabeled. The intensity of primary antibody attachment was first investigated using a concentration of 10 ug/mL, a common coating concentration for ELISA assays. A non-competitive binding assay was then done using an unlabeled primary antibody, where either a specific or nonspecific binder (Texas Red labeled) was added at a concentration of 1 ug/mL. The fluorescence intensities for each of the linker chemistries are in Figure 3A. From the intensities, we calculated the non-competitive binding affinity towards the specific and non-specific targets and the percent coverage of the target analyte (see Table S3 in the supporting information). Even though the fluorescent intensity for the DSC linker is the greatest, the specificity is the highest with the BS(PEG)5 linker. The glutaraldehyde linker displayed the worst specificity and antibody linkage, outside of the bare APDMS surface. We attribute the high specificity of the BS(PEG)5 to the inert nature of the poly(ethylene glycol) spacer units. We believe the poor linkage density of the glutaraldehyde layer is due to the labile nature of the Schiff base formation for primary antibody coverage. As a result more aldehyde groups are accessible, even after blocking, for reacting with nonspecific analytes. The maximum percent coverage of the specific analyte is within error for each of the reaction chemistries, indicating the primary antibodies attach in similar conformations on the surface with the same binding affinities.
Since the coating conditions for large scale areas (100μm-1mm) may vary at nanoscale dimensions, we characterized the response of the silicon nanowires to concentrations of reacted primary antibody with each linker. Each primary antibody concentration was reacted for 1 hour, which has been demonstrated to be enough time to reach saturation points for protein adsorption to monolayers. The devices were rinsed after incubation, and the threshold voltage was extracted. The change in threshold voltage was then plotted for each of the linker chemistries in Figure 3B. From the curves, we observe that saturation is nearly achieved at 100 ng/mL antibody concentration for all layers, justifying 10 ug/mL as being more than enough to reach a saturation linkage amount for the sensor in a 1 hour reaction time. When the threshold voltage shifts are normalized similar to the fluorescent intensities, we notice how important the distance to the surface becomes, as the normalized electrical response of the varying linker chemistries is greater than the fluorescence. Moreover, the effect doesn’t scale proportionally to the fluorescence. We attribute this to the surface charge density having a more pronounced effect, leading towards saturation faster, than does fluorescence. This effect has been observed with DNA FET’s, as probe adsorption densities can change an order of magnitude (1011 molecules/cm2 to 1012 molecules/cm2) with only 10–20 mV changes due to ion concentration redistribution around the layer. 46 As a control, we took fluorescence micrographs after 100ng/mL adsorption for 1 hour of each of the different linker chemistries near the release windows. The fluorescence micrographs are displayed in Figure 3C. The fluorescence intensity trend correlates with the electrical response, with the highest fluorescence indeed being observed with DSC and tailing off towards the APDMS. We have summarized the intensities of the primary antibody fluorescence and electrical response in Table S2 of the supporting information. The difference in the fluorescence intensity near the nanowires is not due to a change in the linkage density, but rather to fluorescence interference contrast by the underlying silicon layer.57
Silicon nanowires are known to display higher pH sensitivities than planar ISFET’s due to their increased surface area to volume ratio and are of high interest to enzyme modified reactions which generate hydrogen ions as the metabolic product. The sensing based upon pH changes on FET’s is currently being used in genomics and semiconductor sequencing by such companies as DNA Electronics58 and Ion Torrent59. For enzyme modified nanowires this requires access to pH sensitive groups on the surface, the linkage monolayer, or the protein, which will alter the surface potential. We have looked at pH sensitivity of the silicon nanowires before monolayer deposition, after monolayer deposition, and after protein deposition and blocking. The threshold voltage was monitored vs. the pH of a 1 mM Robinson buffer (See Figure 3 in supporting information). The range is quite linear for the bare SiO2 dielectric and the APDMS monolayer, but there is a distinct inflection point in the protein curve between pH 6 and 7. We attribute this to the protein being near the isolectric point, with different buffering capacities above and below the isoelectric point, leading to different pH sensitivities.60,61 In particular, the isoelectric point of goat IgG’s has been determined experimentally to be 6.3,62 which agrees well with our inflection point observation for the goat anti-mouse IgG antibody on the surface of the nanowire FET. We linearly fit the curves starting at pH 7 and constructed a table of the pH sensitivities. Overall, the highest pH sensitivity was conferred by the APDMS layer, as amines are known to have a higher buffering capacity than hydroxyl groups. However, the devices reacted with primary antibody had the lowest pH sensitivity, with only 32mV/pH unit. The advantage of this decrease in pH sensitivity for the devices is less drift due to slight pH changes in the buffer solution. However, the disadvantage would be decreased signal for proton generating enzymes. In our case, the change is beneficial since our detection is strictly due to antibody-antigen binding.
The high specificity of the silicon nanowires was demonstrated using competing antigens of similar structure to the analyte of interest, namely Mouse IgG. As negative controls, polyethylene oxide (PEO) MW 100,000 and the sensing buffer (0.01x PBS) were used. The competing antigens utilized in the experiments included Human and Rabbit IgG, as well as Mouse IgA. The Id-Vg curves for silicon nanowires were taken in 0.01x PBS after incubating the devices with 10 ng/mL of the antigens for 1 hour, and are in Figure 4A. The corresponding shift in the threshold voltages with respect to the curve for 0.01X PBS is in Figure 4B, with the calculated device specificity towards Mouse IgG in Figure 4C. The Id-Vg curves show very little shift due to the nonspecific binding entities, with a high uniformity as these curves are averaged over 5 nanowires (N=5). The error bars represent a pooled standard deviation for each of the 5 devices, comprised of their individual standard deviations. The affinity of the goat anti-mouse IgG towards the analyte of interest ranges between 15–26 times depending on the competing immunoglobulin. This result agrees well with the fluorescence specificity in Figure 2, verifying that nanowires can provide specificities as good if not better than standard ELISA or fluorescent assays.
Figure 4.
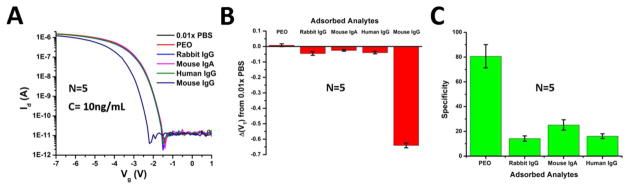
The Id-Vg response of nonspecific binding analytes vs. mouse IgG at a concentration of 10ng/mL is in (A). The change in the threshold voltage using 0.01x PBS as a reference for each of the biological analytes is plotted in (B) and the extracted specificity towards mouse IgG in (C). Device measurements were taken in 0.01x PBS, pH 7.4. The error bars represent a pooled standard deviation over the 5 measurements.
The aforementioned protocols for functionalizing nanowires have shown that they can lead to highly stable measurements and provide high binding specificity. However, the limit of detection for nanowires using this BS(PEG)5 protocol or similar linking chemistries has been seldom studied. Moreover, the dependence on the gate oxide thickness has been poorly characterized. The gate oxide thickness, and thus gate capacitance, directly affects the amount the surface potential changes the current through the underlying conducting silicon channel. In Figure 5A we plot the normalized Id-Vg curves of nanowire devices with different gate oxide thicknesses. The extracted subthreshold slope (S.S) for each gate oxide thickness is included as a table in Figure 5A. As the gate oxide thickness increases, the subthreshold slope becomes poorer and the devices threshold becomes more positive. This is in accordance with classical models for subthreshold swing in accumulation mode SOI MOSFET’s. 63,64 The shift to the right of the threshold voltage with thicker gate oxides is due to extra fixed negative charge and interface traps in the SiO2 dielectric. We set the threshold voltage at 1× 10−3 on the normalized curve, denoted by a dashed line.
Figure 5.
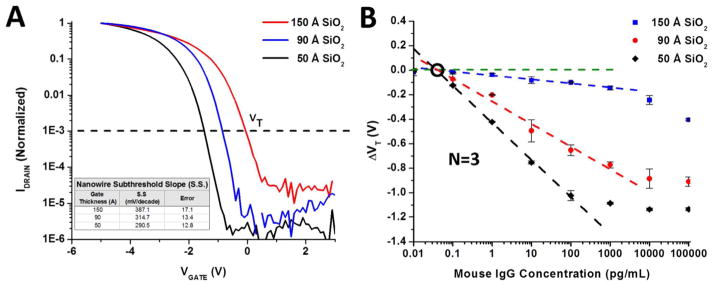
The representative Id-Vg curves of three different silicon oxide gate dielectric thicknesses (normalized) are shown in (A). The subthreshold slopes (S.S) of each of the devices (inset table) show better S.S. with decreasing oxide thickness, and where the VT for the device is taken at 1×10−3, indicated by the dashed line. The change in the threshold voltage versus Mouse IgG concentration is shown in (B) for the three different oxide thickness devices shown in A. Measurements were taken in 0.01x PBS, pH 7.4. Linear regressions were performed on the suitable dynamic range of each oxide thickness (dashed lines) and the limit of detection extracted for each of the thicknesses.
The sensitivity of the devices versus the thickness of the gate oxide is shown in Figure 5B. Concentrations of mouse IgG were titrated in for 30 min binding periods, then rinsed and measured in 0.01x PBS. The threshold voltage was extracted at the normalized value in Figure 5A for each gate oxide thickness, and plotted vs. the mouse IgG analyte concentration. The nanowires also appear to have a Vt response which scales linearly with respect to a logarithmic increase in mouse IgG concentration. This response appears to span at least 4 orders of magnitude from 0.1–100 pg/mL for the 50 Ǻ gate oxide, while the 90 Ǻ and 150 Ǻ oxide have a linear response from 0.1–1000 pg/mL A linear fit (dashed lines) to the semi-log plot was performed for each of the oxide thickness in its linear dynamic range, and the critical values extracted from this fit are shown in Table 1. The average of three devices was used for each gate oxide thickness. In agreement with theory, the largest gate oxide thickness showed the poorest sensitivity with a limit of detection (LOD) of 0.15pg/mL, while the 90 Ǻ and 50 Ǻ thicknesses had LOD’s of 0.125 and 0.0536 pg/mL, respectively. Similarly, the sensitivity to charge (portrayed in the slope) of the oxide thicknesses increase in magnitude with decreasing oxide thickness. The apparent dynamic range of the sensor also does appear to shift towards higher concentrations with increasing gate oxide thickness. We attribute this to the fact that trapped and mobile charges in the thicker oxides reduce the influence of the electric field generated by the proteins, thus reducing their influence on the conductance of the channel. Although analytes may very well be binding on the thinner oxide, the channel is effectively saturated.
Table 1.
Table of values for the linear regression of the nanowire response to Mouse IgG vs. the gate oxide thickness from Figure 5B. The limit of detection was taken at the 95% confidence level.
| TOX(A˚́) | Slope(mV/log (CMouse IgG )) | Mouse IgG at VT = 0(pg/mL) | LOD 95% C.I. (pg/mL) |
|---|---|---|---|
| 50 | −302.0 | 0.0383 | 0.0536 |
| 90 | −180.1 | 0.0477 | 0.125 |
| 150 | −32.5 | 0.0578 | 0.150 |
Conclusions
In this work we successfully demonstrated a functionalization and device release protocol for silicon nanowires which is capable of sensing large immunoglobulins to sub pg/mL levels. By using a combined dry and wet-etch technique, we were able to tailor the thickness of the gate oxide, thus changing the working dynamic range of the sensor. This method also yielded highly stable nanowires in solution capable of long-term measurements, with several wires yielding the same characteristics. We evaluated several different linking chemistries which may be utilized for protein attachment by amine and thiol groups, both by fluorescence and the electrical response of silicon nanowires. The maximum amount of primary antibody and target antigen binding occurs with disuccinimidyl carbonate (DSC) linker, yet the best ratio of specific: nonspecific binding in a non-competitive environment is conferred with the BS(PEG)5 linker. The differences between the normalized fluorescence response and electrical response were also characterized, demonstrating the electrical output is more sensitive than the fluorescent output using the same reaction conditions. In order to verify the results, fluorescence images were taken on the nanowires of the primary antibody reaction and showed a similarly scaling trend.
Using BS(PEG)5 as the model linking chemistry, the affinity of the nanowires for nonspecific binding was also explored. The silicon nanowires demonstrated excellent resistance to non-specific binders, yielding non-competitive binding ratios of Mouse IgG and Rabbit IgG (calculated from the threshold voltage shift) similar to those extracted from fluorescence. This result, given the output sensitivity being more on the nanowires, is quite encouraging and indicates the affinity of the antibody to the different protins being even better than that confirmed optically. Finally, the dose response of the nanowires to the target antigen (mouse IgG) versus the gate SiO2 thickness was explored. The subthreshold slope of the devices demonstrated the proper scaling trend for increasing gate oxide thicknesses, and was used as a control to verify the oxide thickness. We demonstrate the sensitivities of the different oxide thicknesses to the antigen, showing the device sensitivity changes with the oxide thickness, as does the dynamic range of the sensor. The thinnest gate oxide demonstrated limits of detection to 0.054 pg/mL using the BS(PEG)5 functionalization protocol, with a dynamic range spanning 4 orders of magnitude.
We have provided an in-depth analysis of linker chemistries on silicon nanowires. Utilizing the best protocol chosen from these findings, another study into different gate oxide thicknesses and their dose response to a model protein was undertaken, giving an even better in-depth perspective into the capabilities of nanowires as a point-of-care diagnostic platform. Gate dielectrics have now turned towards high-k platforms, which can offer even better sensitivities without sacrificing device characteristics, such as increasing gate leakage. We hope by using new high-k platforms we can achieve even better sensitivity and more stable devices in the future, in hopes of bringing CMOS compatible platforms to the point-of-care market.
Supplementary Material
Acknowledgments
We would like to acknowledge the use of the Micro and Nanotechnology Laboratory at UIUC for FET processing, and the Materials Research Laboratory at UIUC for materials characterization. B.R.D. was a trainee supported by the Midwestern Cancer Nanotechnology Training Center (NIH-NCI R25 CA154015) at UIUC. We would also like to acknowledge support from NIH R01-CA20003 and National Science Foundation (NSF) Grant EEC-0425626 (NSF Nanoscale Science and Engineering Center at Ohio State University).
Footnotes
Supporting Information. The process flow for fabrication of the silicon nanowires and the formation of the silane monolayers are described here. A picture of the device and packaging setup, the chemical structures of the silane and linkers, and the pH sensitivities of the monolayer, linkers, and proteins are also outlined. Tables of values for the fluorescence and electrical measurements of the functionalization process are also described here. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Moore GE. Proceedings of the IEEE. 1998;86:82–85. [Google Scholar]
- 2.Barrettino D. Kos. 2006. pp. 4359–4362. [Google Scholar]
- 3.Baccar ZM, Jaffrezic-Renault N, Martelet C, Jaffrezic H, Marest G, Plantier A. Sens Actuators, B. 1996;32:101–105. doi: 10.1016/0925-4005(96)80117-1. [DOI] [Google Scholar]
- 4.Bratov A, Abramova N, Domínguez C, Baldi A. Anal Chim Acta. 2000;408:57–64. doi: 10.1016/s0003-2670(99)00871-5. [DOI] [Google Scholar]
- 5.Cazalé A, Sant W, Launay J, Ginot F, Temple-Boyer P. Sens Actuators, B. 2013;177:515–521. doi: 10.1016/j.snb.2012.11.054. [DOI] [Google Scholar]
- 6.Hajji B, Temple-Boyer P, Launay J, Do Conto T, Martinez A. Microelectronics Reliability. 1999;40:783–786. [Google Scholar]
- 7.Ho KI, Lu TF, Chang CP, Lai CS, Yang CM. Christchurch. 2009. pp. 1128–1131. [Google Scholar]
- 8.Lu TF, Yang CM, Wang JC, Ho KI, Chin CH, Pijanowska DG, Jaroszewicz B, Lai CS. J Electrochem Soc. 2011;158:J91–J95. doi: 10.1149/1.3543922. [DOI] [Google Scholar]
- 9.Rochefeuille S, Jimenez C, Tingry S, Seta P, Desfours JP. Materials Science and Engineering C. 2002;21:43–46. doi: 10.1016/s0928-4931(02)00056-5. [DOI] [Google Scholar]
- 10.Senillou A, Jaffrezic-Renault N, Martelet C, Griffe F. Materials Science and Engineering C. 1998;6:59–63. doi: 10.1016/s0928-4931(98)00037-x. [DOI] [Google Scholar]
- 11.Janata J. Electroanalysis. 2004;16:1831–1835. doi: 10.1002/elan.200403070. [DOI] [Google Scholar]
- 12.Covington JA, Gardner JW, Briand D, De Rooij NF. Sens Actuators, B. 2001;77:155–162. doi: 10.1016/s0925-4005(01)00687-6. [DOI] [Google Scholar]
- 13.Abramova N, Bratov A. Sensors. 2009;9:7097–7110. doi: 10.3390/s90907097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sant W, Temple-Boyer P, Echanié;Launay J, Martinez A. Sens Actuators, B. 2011;160:59–64. doi: 10.1016/j.snb.2011.07.012. [DOI] [Google Scholar]
- 15.Antonisse MMG, Snellink-Ruël BHM, Engbersen JFJ, Reinhoudt DN. Sens Actuators, B. 1998;47:9–12. [Google Scholar]
- 16.Reinhoudt DN. Sensors and Actuators: B Chemical. 1995;24:197–200. [Google Scholar]
- 17.Diallo AK, Djeghlaf L, Mazenq L, Launay J, Sant W, Temple-Boyer P. Biosens Bioelectron. 2013;40:291–296. doi: 10.1016/j.bios.2012.07.063. [DOI] [PubMed] [Google Scholar]
- 18.Sant W, Pourciel ML, Launay J, Do Conto T, Martinez A, Temple-Boyer P. Sens Actuators, B. 2003;95:309–314. doi: 10.1016/s0925-4005(03)00430-1. [DOI] [Google Scholar]
- 19.Burgmair M, Eisele I. 1. Vol. 1. Orlando, FL: 2002. pp. 439–442. [Google Scholar]
- 20.Wilson DM, Hoyt S, Janata J, Booksh K, Obando L. IEEE Sensors Journal. 2001;1:256–274. doi: 10.1109/7361.983465. [DOI] [Google Scholar]
- 21.Koch S, Woias P, Meixner LK, Drost S, Wolf H. Biosens Bioelectron. 1999;14:413–421. doi: 10.1016/s0956-5663(99)00008-1. [DOI] [PubMed] [Google Scholar]
- 22.Besselink GAJ, Schasfoort RBM, Bergveld P. Biosens Bioelectron. 2003;18:1109–1114. doi: 10.1016/s0956-5663(02)00243-9. [DOI] [PubMed] [Google Scholar]
- 23.Eijkel JCT, Olthuis W, Bergveld P. Biosens Bioelectron. 1997;12:991–1001. doi: 10.1016/s0956-5663(97)00054-7. [DOI] [Google Scholar]
- 24.Estrela P, Stewart AG, Yan F, Migliorato P. Electrochim Acta. 2005;50:4995–5000. doi: 10.1016/j.electacta.2005.02.075. [DOI] [Google Scholar]
- 25.Marrakchi M, Dzyadevych SV, Biloivan OA, Martelet C, Temple P, Jaffrezic-Renault N. Materials Science and Engineering C. 2006;26:369–373. doi: 10.1016/j.msec.2005.10.052. [DOI] [Google Scholar]
- 26.Olthuis W, Luo J, Bergveld P. Biosens Bioelectron. 1994;9:743–751. doi: 10.1016/0956-5663(94)80073-1. [DOI] [Google Scholar]
- 27.Estrela P, Stewart AG, Keighley SD, Migliorato P. Journal of the Korean Physical Society. 2006;48:S22–S26. [Google Scholar]
- 28.Berney H, West J, Haefele E, Alderman J, Lane W, Collins JK. Sens Actuators, B. 2000;68:100–108. [Google Scholar]
- 29.Mascini M, Palchetti I, Marrazza G. Anal Bioanal Chem. 2001;369:15–22. doi: 10.1007/s002160000629. [DOI] [PubMed] [Google Scholar]
- 30.Mikkelsen SR. Electroanalysis. 1996;8:15–19. [Google Scholar]
- 31.Souteyrand E. Analusis. 1999;27:639–646. [Google Scholar]
- 32.Souteyrand E, Cloarec JP, Martin JR, Wilson C, Lawrence I, Mikkelsen S, Lawrence MF. J Phys Chem B. 1997;101:2980–2985. [Google Scholar]
- 33.Fang Y, Patolsky F, Lieber CM. Biophys J. 2007:551a. [Google Scholar]
- 34.Zheng GF, Lu W, Lieber CM. Abstr Pap Am Chem Soc. 2006:231. [Google Scholar]
- 35.Zheng GF, Patolsky F, Lieber CM. Abstr Pap Am Chem Soc. 2005;229:U782–U782. [Google Scholar]
- 36.Zheng GF, Patolsky F, Cui Y, Wang WU, Lieber CM. Nat Biotechnol. 2005;23:1294–1301. doi: 10.1038/Nbt1138. [DOI] [PubMed] [Google Scholar]
- 37.Wang WU, Chen C, Lin KH, Fang Y, Lieber CM. Proc Natl Acad Sci U S A. 2005;102:3208–3212. doi: 10.1073/pnas.0406368102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cui Y, Wei QQ, Park HK, Lieber CM. Science. 2001;293:1289–1292. doi: 10.1126/science.1062711. [DOI] [PubMed] [Google Scholar]
- 39.Fritz J, Cooper EB, Gaudet S, Sorger PK, Manalis SR. Proc Natl Acad Sci U S A. 2002;99:14142–14146. doi: 10.1073/pnas.232276699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lu W, Lieber CM. Nat Mater. 2007;6:841–850. doi: 10.1038/nmat2028. [DOI] [PubMed] [Google Scholar]
- 41.Stern E, Vacic A, Rajan NK, Criscione JM, Park J, Ilic BR, Mooney DJ, Reed MA, Fahmy TM. Nat Nanotechnol. 2010;5:138–142. doi: 10.1038/Nnano.2009.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stern E, Vacic A, Li C, Ishikawa FN, Zhou CW, Reed MA, Fahmy TM. Small. 2010;6:232–238. doi: 10.1002/smll.200901551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stern E, Klemic JF, Routenberg DA, Wyrembak PN, Turner-Evans DB, Hamilton AD, LaVan DA, Fahmy TM, Reed MA. Nature. 2007;445:519–522. doi: 10.1038/Nature05498. [DOI] [PubMed] [Google Scholar]
- 44.Nair PR, Alam MA. IEEE Trans Electron Devices. 2007;54:3400–3408. doi: 10.1109/Ted.2007.909059. [DOI] [Google Scholar]
- 45.Elfstrom N, Juhasz R, Sychugov I, Engfeldt T, Karlstrom AE, Linnros J. Nano Lett. 2007;7:2608–2612. doi: 10.1021/Nl0709017. [DOI] [PubMed] [Google Scholar]
- 46.Poghossian A, Cherstvy A, Ingebrandt S, Offenhäusser A, Schöning MJ. Sens Actuators, B. 2005;111–112:470–480. doi: 10.1016/j.snb.2005.03.083. [DOI] [Google Scholar]
- 47.Scarano S, Mascini M, Turner APF, Minunni M. Biosens Bioelectron. 2010;25:957–966. doi: 10.1016/j.bios.2009.08.039. [DOI] [PubMed] [Google Scholar]
- 48.Marquette CA, Corgier BP, Heyries KA, Blum LJ. Front Biosci. 2008;13:382–400. doi: 10.2741/2687. [DOI] [PubMed] [Google Scholar]
- 49.Jonkheijm P, Weinrich D, Schröder H, Niemeyer CM, Waldmann H. Angewandte Chemie - International Edition. 2008;47:9618–9647. doi: 10.1002/anie.200801711. [DOI] [PubMed] [Google Scholar]
- 50.Voue M, Goormaghtigh E, Homble F, Marchand-Brynaert J, Conti J, Devouge S, De Coninck J. Langmuir. 2007;23:949–955. doi: 10.1021/la061627j. [DOI] [PubMed] [Google Scholar]
- 51.Muck A, Svatoš A. Talanta. 2007;74:333–341. doi: 10.1016/j.talanta.2007.09.012. [DOI] [PubMed] [Google Scholar]
- 52.Brétangol F, Valsesia A, Ceccone G, Colpo P, Gilliland D, Ceriotti L, Hasiwa M, Rossi F. Plasma Processes Polym. 2006;3:443–455. doi: 10.1002/ppap.200600015. [DOI] [Google Scholar]
- 53.Korf U, Henjes F, Schmidt C, Tresch A, Mannsperger H, Löbke C, Beissbarth T, Poustka A. In: Werther M, Seitz H, editors. Vol. 110. 2008. pp. 153–175. [DOI] [PubMed] [Google Scholar]
- 54.Bally M, Halter M, Vörös J, Grandin HM. Surf Interface Anal. 2006;38:1442–1458. doi: 10.1002/sia.2375. [DOI] [Google Scholar]
- 55.Dorvel B, Reddy B, Block I, Mathias P, Clare SE, Cunningham B, Bergstrom DE, Bashir R. Adv Funct Mater. 2010;20:87–95. doi: 10.1002/adfm.200901688. [DOI] [Google Scholar]
- 56.Chen S, Nyholm L, Jokilaakso N, Karlstrm AE, Linnros J, Smith U, Zhang SL. Electrochem Solid-State Lett. 2011;14:J34–J37. doi: 10.1149/1.3584082. [DOI] [Google Scholar]
- 57.Lambacher A, Fromherz P. Applied Physics a-Materials Science & Processing. 1996;63:207–216. [Google Scholar]
- 58.Kalofonou M, Toumazou C. Sens Actuators, B. 2013;178:572–580. doi: 10.1016/j.snb.2012.12.054. [DOI] [Google Scholar]
- 59.Merriman B, Torrent I, Rothberg JM. Electrophoresis. 2012;33:3397–3417. doi: 10.1002/elps.201200424. [DOI] [PubMed] [Google Scholar]
- 60.Bergveld P, Vanhal REG, Eijkel JCT. Biosens Bioelectron. 1995;10:405–414. [Google Scholar]
- 61.Olthuis W, Luo J, Bergveld P. Biosens Bioelectron. 1994;9:743–751. [Google Scholar]
- 62.Gao HL, Li CY, Ma FX, Wang K, Xu JJ, Chen HY, Xia XH. Phys Chem Chem Phys. 2012;14:9460–9467. doi: 10.1039/c2cp40594f. [DOI] [PubMed] [Google Scholar]
- 63.Colinge JP, Flandre D, Van De Wiele F. Solid-State Electron. 1994;37:289–294. doi: 10.1016/0038-1101(94)90080-9. [DOI] [Google Scholar]
- 64.Colinge JP, Van de Wiele F, Flandre D. IEEE; Palm Springs, CA, USA: 1993. pp. 146–147. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.