

Abstract
Building 3-dimensional (3D) molecules is the starting point in molecular modeling. Conformer search and identification of a global energy minimum structure are often performed computationally during spectral analysis of data from NMR, IR, and VCD or during rational drug design through ligand-based, structure-based, and QSAR approaches. I herein report a convenient script that allows for automated building of 3D structures and conformer searching from 2-dimensional (2D) drawing of chemical structures. With this Bash shell script, which runs on Mac OS X and the Linux platform, the tasks are consecutively and iteratively executed without a 3D molecule builder via the command line interface of the free (academic) software OpenBabel, Balloon, and MOPAC2012. A large number of 2D chemical drawing files can be processed simultaneously, and the script functions with stereoisomers. Semi-empirical quantum chemical calculation ensures reliable ranking of the generated conformers on the basis of energy. In addition to an energy-sorted list of file names of the conformers, their Gaussian input files are provided for ab initio and density functional theory calculations to predict rigorous electronic energies, structures, and properties. This script is freely available to all scientists.
Keywords: 3D structure generation, conformer search, shell script, free software
Background
Building 3-dimensional (3D) structures of molecules is a starting point for computational analysis because physical, chemical, and biological properties of a molecule are related to its 3D structure. Observed physicochemical properties reflect the statistical average of all possible conformations, and are predominantly associated with the global energy minimum structure and its periphery. Thus, conformational space for flexible molecules is often explored after 3D structure generation to find minimum energy structures, i.e., conformers. Searching for conformers, finding the global energy minimum conformer and its periphery, and taking into account the Boltzmann distribution can help with the spectral assignment during spectroscopic analysis such as NMR, IR, and VCD. For rational drug design, searching for conformers is a crucial step because most drugs have an optimal bioactive conformation in the active site of their target biomolecule, and this conformation is likely to be close to that of low-energy conformers.
I herein report a free script for automated building of 3D structures and conformer searching based on 2-dimensional (2D) drawing of chemical structures. This is a Bash shell script, and runs on Mac OS X and the Linux platform. Without a 3D molecule builder, the tasks are consecutively and iteratively executed via the command line interface and free (academic) programs OpenBabel [1, 2], Balloon [3, 4], and MOPAC2012 [5, 6]. There are many commercial applications for conformer generation using a 3D molecule builder [7]. On the other hand, scientists are familiar with 2D skeletal formulas, and bench scientists routinely draw 2D chemical structures using molecule editor such as ChemDraw [8] or MarvinSketch [9]. The proposed script is convenient in this regard, and processes a large number of 2D chemical drawing files simultaneously. It also functions with stereoisomers. Energy-minimized conformers are generated using a force field, and are further processed using semi-empirical quantum chemical calculations to estimate their heats of formation. In addition, input files for ab initio and density functional theory (DFT) calculations are dumped to predict rigorous electronic energies, structures and properties. After completion of all energy calculations of the generated conformers, an energy-sorted list of file names of the conformers is provided.
Settings
OpenBabel is a freely available chemistry toolbox designed to accept inputs in all languages that are used for processing chemical data, and was installed in /usr/local/bin. The executable babel (version 2.3.1 for Mac and 2.3.2 for Linux) is used to convert file formats and to generate input files for MOAPC2012 and Gaussian09 [10] in the script.
Balloon is a freely available tool that generates 3D atomic coordinates from molecular connectivity via distance geometry, and conformer ensembles by using a multi-objective genetic algorithm (GA). This tool also considers stereochemistry of double bonds and tetrahedral chiral atoms. The executable balloon (version 1.4.1.1068 for Mac and Linux) was installed in the home directory, and is used for addition of hydrogen atoms to a skeletal formula, generation of a 3D model and its conformers, and energy minimization using a MMFF94-like force field.
MOPAC2012, a free academic program, is a general-purpose semi-empirical molecular orbital package for studying solid state and molecular structures and reactions. The executable MOPAC2012.exe (version 13.306 for Mac and Linux) was installed in /opt/mopac and is used for single point energy calculation by the PM7 method.
Input & Output
When the shell script confs.sh (see supplementary material) is run in a working directory, it reads the file name of a 2D chemical structure saved in MDL SDfile (.sdf) format (i.e., the atoms are recorded as coordinates), and creates a new directory with the file name. In the new directory, hydrogen atoms are added to the skeletal formula, and 100 3D conformers, minimized with the MMFF94-like force field, are generated as a single output molecule entry in the .mol2 file format. After each conformer put into consecutively numbered files, its single point energy calculation is performed using the PM7 method in MOPAC2012. After completion of the calculation, the heat of formation is printed in kcal/mol in a text (.txt) file. The script also dumps a short summary (.arc) file of computational results including the geometry, and its converted (_1scf.sdf) file. These (.mol2, .arc, and _1scf.sdf) files are accessible for visual verification of the geometry using a molecular viewer such as Jmol [11, 12], ViewDock in Chimera [13, 14], and DS Visualizer [15]. The Gaussian input (.com) file is also generated for geometry optimization and vibrational analysis through highlevel quantum chemical calculation. After completion of all single point energy calculations, an energy-sorted list of file names of the conformers is dumped with the extension .xls in the same directory. The lowest-energy conformer can be considered as the global energy minimum structure. As shown in (Figure 1 & Figure 2), the lowest-energy conformers being close to the X-ray crystal structures of the small molecules [16, 17] were predicted as a result of selecting adequate options and keywords for Balloon and MOPAC2012. These data would be confirmed through high-level ab initio and DFT calculation for the several low-energy conformers. The Boltzmann distribution of optimized conformers at certain temperature can be estimated from their energies [18].
Figure 1.
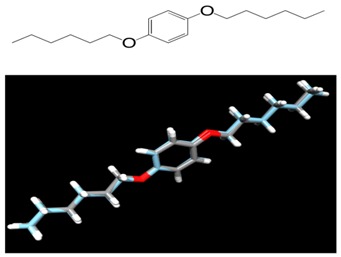
(Top) skeletal formula of 1,4-bis(hexyloxy)benzene; (bottom) superposition of the X-ray crystal structure (gray carbon) [16] with the lowest-energy conformer (cyan carbon). The superposition was done using DS Visualizer.
Figure 2.
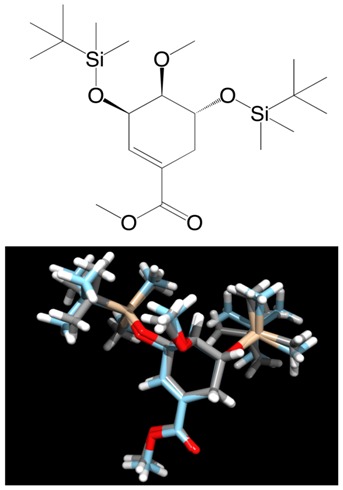
(Top) skeletal formula of (3R,4S,5R)-methyl 3,5- bis[(tert-butyldimethylsilyl)oxy]-4-methoxycyclohex-1 enecarboxylate; (bottom) superposition of the X-ray crystal structure (gray carbon) [17] with the lowest-energy conformer (cyan carbon). The superposition was done using DS Visualizer.
As mentioned above, this script runs on Mac OS X and the Linux platform, and can process a large number of chemical drawing files simultaneously.
Caveat & Conclusion
When working with any stereoisomer, a user must check the spatial arrangement of its atoms visually with one initial 3D model (_3d.sdf) generated by an auxiliary shell script check3d.sh (see supplementary material). Suitable arrangements of atoms and choices of bonds in a 2D chemical structure will result in a desired 3D model. The user should always check the initial 3D model using check3d.sh before running confs.sh. Open source MOPAC7 [19, 20] can be used in place of MOPAC2012, but the PM7 method cannot be processed in MOPAC7. The installation path names and Gaussian keywords in the script should be changed accordingly.
In conclusion, this script integrates excellent free scientific applications and has the following advantages: (i) 2D chemical drawing files are available as input; (ii) 3D molecules are built and conformers are sampled without a 3D molecule builder; (iii) time is saved because of batch processing via the command line, and (iv) accurate energies are provided along with input files for high-level quantum chemical calculations. I hope that this free script will be useful for scientists who study molecules using experimental and computational methods.
Supplementary material
Footnotes
Citation:Ishikawa, Bioinformation 9(19): 988-992 (2013)
References
- 1. http://openbabel.org/wiki/Main_Page.
- 2.O'Boyle NM, et al. J Cheminform. 2011;3:33. doi: 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. http://users.abo.fi/mivainio/balloon/index.php.
- 4.Vainio MJ, et al. J Chem Inf Model. 2009;49:492. doi: 10.1021/ci800315d. [DOI] [PubMed] [Google Scholar]
- 5. http://openmopac.net/MOPAC2012.html.
- 6.Stewart JJ, et al. J Mol Model. 2013;19:1. doi: 10.1007/s00894-012-1667-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen IJ, et al. J Chem Inf Model. 2008;48:1773. doi: 10.1021/ci800130k. [DOI] [PubMed] [Google Scholar]
- 8. http://www.cambridgesoft.com/Ensemble_for_Chemistry /ChemDraw/Default.aspx.
- 9. http://www.chemaxon.com/products/marvin/marvinske tch/
- 10. http://www.gaussian.com/index.htm.
- 11. http://jmol.sourceforge.net/
- 12.Herráez A, et al. Biochem Mol Biol Educ. 2006;34:255. doi: 10.1002/bmb.2006.494034042644. [DOI] [PubMed] [Google Scholar]
- 13. http://www.cgl.ucsf.edu/chimera/
- 14.Pettersen EF, et al. J Comput Chem. 2004;25:1605. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 15. http://accelrys.com/products/discoverystudio/ visualization.html.
- 16.Cheng H, et al. Acta Cryst. 2013;69:O1721. doi: 10.1107/S1600536813029024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu R, et al. Acta Cryst. 2013;69:O632. doi: 10.1107/S1600536813007551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mazzeo G, et al. J Nat Prod. 2013;76:588. doi: 10.1021/np300770s. [DOI] [PubMed] [Google Scholar]
- 19. http://www.webmo.net/support/binaries_osx.html.
- 20. http://www.webmo.net/support/mopac_linux.html.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.