

Abstract
Chitinases are known to hydrolyze chitin polymers into smaller chitooligosaccharides. Chitinase from bacterium Serratia proteamaculans (SpChiD) is found to exhibit both hydrolysis and transglycosylation activities. SpChiD belongs to family 18 of glycosyl hydrolases (GH-18). The recombinant SpChiD was crystallized and its three-dimensional structure was determined at 1.49 Å resolution. The structure was refined to an R-factor of 16.2%. SpChiD consists of 406 amino acid residues. The polypeptide chain of SpChiD adopts a (β/α)8 triosephosphate isomerase (TIM) barrel structure. SpChiD contains three acidic residues, Asp149, Asp151 and Glu153 as part of its catalytic scheme. While both Asp149 and Glu153 adopt single conformations, Asp151 is observed in two conformations. The substrate binding cleft is partially obstructed by a protruding loop, Asn30 - Asp42 causing a considerable reduction in the number of available subsites in the substrate binding site. The positioning of loop, Asn30 - Asp42 appears to be responsible for the transglycosylation activity. The structure determination indicated the presence of sulfone Met89 (SMet89). The sulfone methionine residue is located on the surface of the protein at a site where extra domain is attached in other chitinases. This is the first structure of a single domain chitinase with hydrolytic and transglycosylation activities.
Keywords: Chitinase D, chitooligosaccharides, Serratia proteamaculans, hydrolysis, transglycosylation, constrained substrate binding cleft
Introduction
Chitinases (EC 3.2.1.14) hydrolyze chitin polymers and long chain chitooligomers into shorter oligomers and N-acetyl glucosamine monomers. Chitinases are found in microorganisms such as bacteria and fungi, plants and animals [1,2]. Depending upon the source, chitinases have been shown to play various important functional roles in the respective organisms. In the case of bacteria, chitinases degrade chitin for utilizing it as a source of carbon and nitrogen [3]. Fungal chitinases play a key role in hyphal growth and morphogenesis apart from serving nutritional requirement of fungi [4]. Plants produce chitinases to defend themselves from phytopathogenic fungi [5,6]. In addition to other unknown functions, the mammalian chitinases have been shown to be important biomarkers in asthma [7].
Chitinases belong to either family 18 or 19 of glycosyl hydrolases (GH-18 or GH-19) [2]. The families GH-18 and GH-19 differ from each other in structures and modes of action [8-10]. A majority of chitinases from bacterial origin and also a few from plants are grouped under family GH-18. The catalytic action of GH-18 chitinases involves the distortion of the stereochemistry of substrates leading to glycosidic bond cleavage [11-13]. The structures of chitinases of family GH-18, analyzed so far [14,15] as well as other biochemical studies of chitin hydrolysis [1,16] have shown that they form an oxazolinium ion intermediate. The catalytic domain in GH-18 chitinases consists of a common (β/α)8 triosephosphate isomerase (TIM)-barrel fold [8,9,17] as well as a highly conserved signature sequence of Asp - Xaa - Asp - Xaa - Glu motif. In this motif Glu acts as a catalytic proton donor while the second Asp is supposed to contribute to the stabilization of the essential distortion of the substrate [18].
A few chitinases have transglycosylation (TG) property along with hydrolytic activity [19-23]. TG is a reaction in which new glycosidic bonds are introduced between donor and acceptor sugar molecules [24,25]. However, the precise mechanism of this reaction has not yet been clearly understood. Nevertheless, the TG activity of GH-18 chitinases, as an enzymatic synthesis of oligosaccharides, is an important property to exploit for useful applications. Some efforts are being made to improve the TG activity of GH-18 family bacterial chitinases by introducing empirical mutations [21,24-26]. The structural details available for bacterial chitinase of family GH-18 are limited to chitinase having only hydrolytic activity [10,27,28]. For example chitinase from Bacillus cereus NCTU2 is also a single domain chitinase similar to SpChiD but it lacks TG activity [29]. The crystal structure of chitinase B from Serratia marcescens (SmChiB) [17] was the first structure of bacterial exo-chitinase in which the binding of chitin oligomers was shown to be regulated as it could not bind beyond the -3 subsite. As a result, the enzyme displayed chitotriosidase activity by degrading chitin chain from the non-reducing end [17]. Studies on chitinase A from Serratia marcescens (SmChiA) revealed a standard mode of catalytic action as observed generally in chitinases [13]. The amino acid sequence of chitinase D from Serratia proteamaculans (SpChiD) shows a high sequence identity of 79% with chitinase II from Klebsiella pneumoniae (KpChiII) whose crystal structure has been deposited recently (PDB code: 3QOK) but no report has been published on this structure so far.
In the present study, we report the crystal structure determination of chitinase SpChiD at 1.49 Å resolution. The structure determination showed that SpChiD folds into a single catalytic domain containing a (β/α)8 TIM barrel conformation. The substrate binding cleft in SpChiD has been found to be partially blocked by a 13-residue loop, Asn30 - Asp42. The presence of this loop in the cleft reduced the size of substrate binding cleft to less than half as compared to other chitinases [8,17]. The presence of loop, Asn30 - Asp42 in the cleft appears to be responsible for the TG activity of SpChiD [23]. The structure also revealed the modification of Met89 to sulfone methionine (SMet89).
Materials and methods
Bacterial strains, plasmids, culture conditions, and biochemicals
The plasmid pET-22b (+) and E. coli BL21 (DE3) (Novagen, Madison, USA) were used for heterologous expression. E. coli was grown in LB broth (1% peptone, 0.5% yeast extract, 1% NaCl) at 37°C. Ampicillin, at a working concentration of 100 μg/ml, was added to the LB broth as required. Isopropyl-β-D-thiogalactopyranoside (IPTG), ampicillin and all other chemicals were purchased from Calbiochem or Merck (Darmstadt, Germany). Ni-NTA His bind resin was procured from Novagen (Madison, USA) for protein purification.
Protein expression, isolation and purification
Expression of SpChiD protein was carried out as described previously [25]. The harvested culture pellet was processed for isolation of periplasmic fraction (PF) as described in pET manual (Novagen, Darmstadt, Germany). The PFs with desired protein were used for purification as described in Qiagen (Duesseldorf, Germany) manual using Ni-NTA affinity chromatography under native conditions. The recombinant protein bound to Ni-NTA matrix was eluted with different concentrations of imidazole containing buffers [25]. The purified fractions were electrophoresed using 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and visualized with the help of Coomassie brilliant blue. Fractions with high purity were concentrated using Amicon filters of 10 kDa cut-off (Millipore, Billerica, MA).
N-terminal sequence determination
The N-terminal sequence reported in the data base and the one obtained from the preliminary structure determination were found to be different. The information about signal peptide was not clearly mentioned and X-ray structure determination can sometimes miss the N-terminal residues due to flexible nature of N-terminal segments in many proteins. In order to remove this ambiguity in the N-terminal sequence of SpChiD, the samples of purified matured protein were blotted on polyvinylidene fluoride (PVDF) membrane. The blotted protein sample was subjected to Edman degradation and the sequence of the first 14 amino acid residues was determined with Protein Sequencer PPSQ-21A (Shimadzu, Kyoto, Japan).
Crystallization of SpChiD
SpChiD was crystallized using hanging drop vapor diffusion method in a 24-well plate. Each hanging drop was prepared by mixing 2.5 μl of SpChiD enzyme solution (10 mg/ml in 20 mM sodium phosphate buffer, pH 8.0) with equal volume of reservoir solution containing 2 M sodium formate and 0.1 M sodium acetate, pH 8.0 and equilibrated against 2 ml of the same reservoir solution. Crystals measuring 0.3 × 0.2 × 0.2 mm3, which were suitable for X-ray diffraction measurements, were obtained after two weeks.
X-ray intensity data collection
X-ray intensities were collected using a beamline, BM-14 at the European Synchrotron Radiation Facility (ESRF) in Grenoble (France). The diffraction intensities were recorded on MAR 300 CCD detector (Rayonix, Evanston, USA). Before mounting in the loop, the crystals were stabilized in a cryoprotecting solution consisting of 25% glycerol and 75% of well solution. The crystals were flash-frozen in a stream of nitrogen at 100K. The data were integrated and scaled with the help of HKL-2000 software package [30]. The details of data processing statistics are given in Table 1.
Table 1.
Summary of X-ray crystallographic data collection and refinement
| PDB entry | 4LGX |
|---|---|
| Data collection | |
| Space group | P21212 |
| Unit cell parameters (Å) | a = 75.3, b = 87.2, c = 59.5 |
| Resolution (Å) | 35.19 - 1.49 (1.52 - 1.49) |
| Rsym (%) | 5.0 (25.0) |
| I/σ (I) | 44.5 (6.1) |
| Completeness (%) | 100 (100) |
| Multiplicity | 8.1 (6.8) |
| Number of reflections measured | 524810 |
| Number of unique reflections | 64748 |
| Refinement | |
| Rcryst/Rfree | 0.162/0.176 |
| Number of protein atoms | 3164 |
| Number of water oxygen atoms | 593 |
| Number of acetate ion | 1 |
| Number of glycerol molecules | 2 |
| R.M.S. deviations in | |
| Bond length (Å) | 0.016 |
| Bond angles (°) | 1.6 |
| Dihedral angles (°) | 12.7 |
| Mean B factor (Å)2 for | |
| Main chain atoms | 11.3 |
| Side chain and water oxygen atoms | 16.7 |
| Overall | 14.4 |
| Ramachandran plot statistics | |
| Residues in the most favoured regions (%) | 91.0 |
| Residues in additionally allowed regions (%) | 9.0 |
Values in the parentheses are for the highest resolution shell. Data were collected from one crystal.
Structure determination and refinement
The structure was determined using molecular replacement method with program MOLREP [31] from the CCP4 suite [32]. The co-ordinates of chitinase KpChiII (PDB code: 3QOK) with which it has a high sequence identity (UniProt - A6T7R3) were used as a search model. The structure was refined with REFMAC 5.5 [33] from the CCP4 suite [32]. The protein chain tracing was carried out by building the model manually with the help of (2Fo - Fc) and (Fo - Fc) electron density maps. The model buildings were performed using programs O [34] and Coot [35]. While examining the electron densities in the Fourier (2Fo - Fc) and difference Fourier (Fo - Fc) maps, the conformations of several segments in the protein were found to be significantly different from that of the model protein (PDB: 3QOK). These were corrected manually and refined. After completing the fitting of the full protein chain into the electron density manually, further cycles of refinement were carried out using the manually improved coordinates. When the refinement reached the stage of value 0.251 for the Rcryst factor, the electron density maps (2Fo - Fc) and (Fo - Fc) were calculated again and reexamined critically. It revealed two conformations for the side chains of Ser110, Asp151 and Met220 (Figure 1A). An extra electron density with features of an acetate molecule was also observed in the proximity of active site (Figure 1B). However, the most striking observation pertained to detecting the extra electron density at the sulfur atom of residue, Met89. It clearly suggested that there could be two extra oxygen atoms which might be covalently attached to Sulfur atom of Met89 and the residue could be a modified methionine as Sulfone Met89 (SMet89) (Figure 1C). All the atoms of the side chain of SMet89 refined well with full occupancies. The presence of two extra oxygen atoms at Met89 was also confirmed by making mass spectrometry measurements which produced a 12 - residue segment Val88 - Met89 - Ala90 - Asp91 - Leu92 - Gln93 - Leu94 - Leu95 - Pro96 - Val97 - Leu98 - Arg99. The mass spectrometric data indicated its molecular weight to be nearly 1399.70 Da whereas the molecular weight of the same segment with Met89 instead of SMet89 should be 1367.71 Da.
Figure 1.
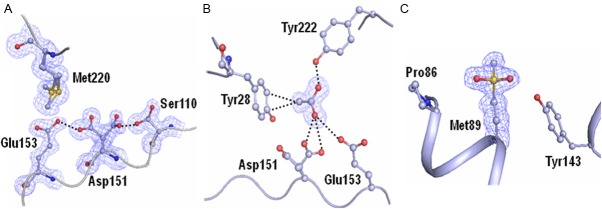
The initial electron densities from (2Fo - Fc) map at 1.3 σ cutoffs for (A) residues Ser110, Asp151, Glu153 and Met220. The side chains of residues Ser110, Asp151 and Met220 adopt two distinct conformations (B) acetate ion and (C) sulfone Met89 (SMet89).
Therefore, it was assumed that Met89 is modified to SMet89. The positions of 593 water oxygen atoms were determined. These were also included in the subsequent cycles of refinement. Finally, the refinement converged with values of 0.162 and 0.176 for Rcryst and Rfree factors, respectively. The stereochemical quality of the refined model was examined using PROCHECK [36] and the details of the refinement statistics are included in Table 1.
Results
N-terminal amino acid sequence
The sequence of the first 14 N-terminal amino acid residues of SpChiD was determined using Edman degradation [37,38]. The peaks for individual amino acids were clearly observed with reliabilities of 100% indicating the correctness of the determination. The sequence starting from the first residue at N-terminus SpChiD was found to be Ala1 - Gly2 - Met3 - Ala4 - His5 - Ala6 - Ala7 - Ser8 - Tyr9 - Leu10 - Ser11 - Val12 - Gly13 - Tyr14. It matched with the original sequence of SpChiD as reported in the data base UniProt - A8GFD6 [23] from residue number Ala21 to Tyr34. Hence, the sequence of the matured protein in the database begins from residue number 21 as Ala21 and ends at residue number 426 as Gln426. Thus, the protein SpChiD contains a total of 406 amino acid residues corresponding to the molecular weight of 44.69 kDa.
Sequence comparison
The amino acid sequence of SpChiD was compared with three other chitinases of family GH-18 whose structures have been reported [8,17] (PDB code: 3QOK) and which are representatives of different types of chitinases belonging to the same family (Figure 2). As reported in the database UniProt - A8GFD6 [23], the amino acid sequence of SpChiD consisted of 426 amino acid residues. However, as indicated by N-terminal sequence determination, the first amino acid is Ala1 which corresponds to Ala21 in the data base. In order to compare the sequence of SpChiD with the sequences of other structures, the sequence numbering in SpChiD was changed to Ala15 instead of Ala1 and thus the last residue was Gln420 instead of Gln426 (Figure 2). The BLAST algorithm has been used for the alignment of sequences and the relevant aspects were compared. It showed the highest sequence identity of 79% with chitinase II from Klebsiella pneumoniae (KpChiII, whose structure is not reported so far) while with other chitinases belonging to classes A (SmChiA) and B (SmChiB), it showed relatively low sequence identities of 23% and 25% respectively. It may be noted here that the residue methionine at position 89 which was found to be modified to sulfone methionine in SpChiD is not methionine at the corresponding positions in KpChiII, SmChiA and SmChiB. Instead, these were Gln, Lys and Arg respectively. The residues that form the substrate binding cleft in SpChiD are Thr69, Trp114, Trp160, Leu162, Ala169, Ala247, Arg278, Asp291, Lys322 and Asp323 which are highlighted in cyan. The active site residues are highlighted in green. The sequence of the segment, Asn30 - Asp42 (highlighted in grey) which is identical in SpChiD and KpChiII but it is completely different in SmChiA and SmChiB indicating differences in the structural and functional aspects involving this segment.
Figure 2.
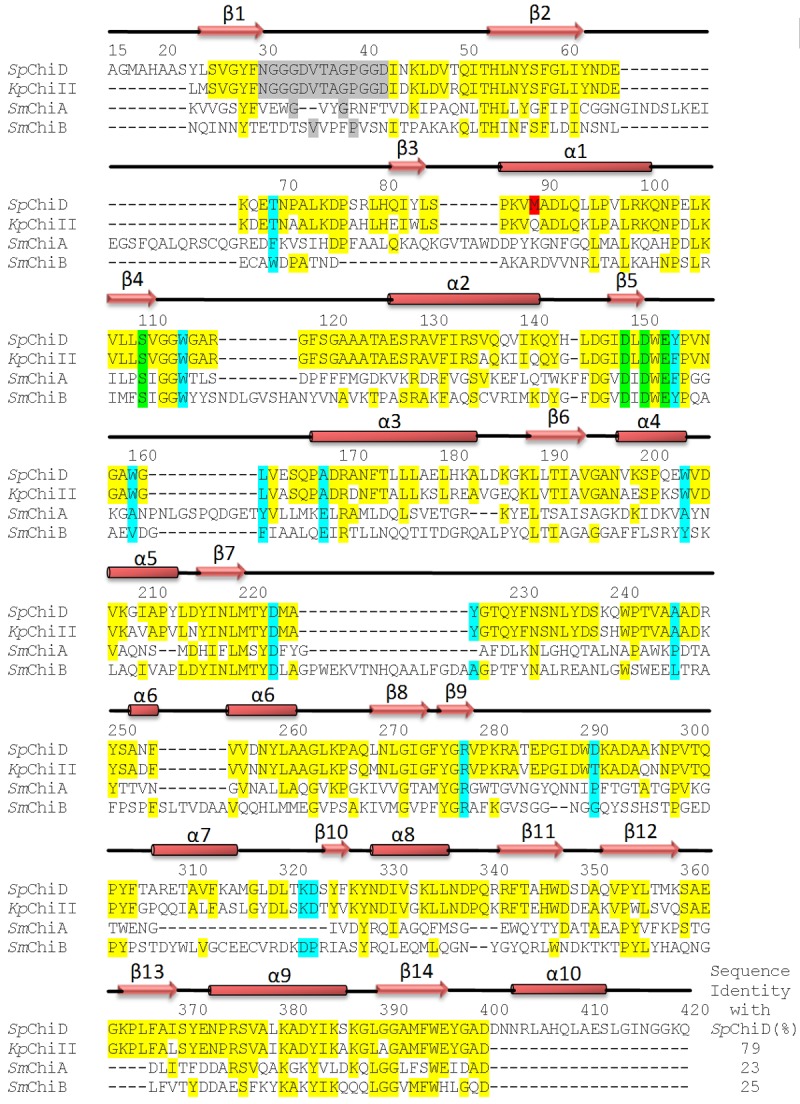
Multiple sequence alignment of chitinases, SpChiD (PDB: 4LGX), KpChiII (PDB: 3QOK), SmChiA (PDB: 1CTN) and SmChiB (PDB: 1E15). Since SpChiD is single domain protein consisting of only main catalytic TIM barrel domain so the comparison is restricted only to the main catalytic TIM barrel domain. The mature protein of SpChiD consists of residues from Ala15 to Gln420. The identical residues are highlighted in yellow. The residues of loop Asn30 - Asp42 are highlighted in grey. This is a unique loop as it protrudes into the substrate binding cleft in SpChiD. The residue sulfone Met89 (SMet89) shown in red is present only in SpChiD. The catalytic residues are shown green. The residues of substrate binding cleft are highlighted in cyan. The secondary structure elements such as α-helices and β-strands are also indicated.
Overall structure
The final model of SpChiD consisted of 3164 protein atoms from 398 out of a total of 406 amino acid residues in the protein for which the electron density was observed. The electron density was not observed for three residues Ala15 - Gly16 - Met17 at the N-terminus and five residues, Asn416 - Gly417 - Gly418 - Lys419 - Glu420 at the C-terminus. The positions of 593 water oxygen atoms, one acetate molecule and two glycerol molecules were also determined. SpChiD is a single domain protein with 406 amino acid residues (Figure 3). The structure is comprised of 14 β-strands and 10 α-helices out of which eight β-strands, β1 (residues Leu24 - Asn30), β2 (residues His53 - Tyr62), β4 (residues Lys106 - Val111), β5 (residues Ile148 - Asp151), β6 (residues Leu189 - Val194), β7 (residues Tyr216 - Met220), β8 (residues Leu269 - Gly274), and β14 (residues Gly390 - Glu396) and eight α-helices α1 (residues Pro86 - Gln101), α2 (residues Ala126 - Gln142), α3 (residues Pro167 - Ala183), α4 (residues Val198 - Trp204), α5 (residues Val207 - Tyr213), α6 (residues Ala252 - Ala261), α9 (residues Pro374 - Ser386) and α10 (residues Arg404 - Ser412) were part of the TIM barrel (Figure 3) while β-strands β3 (residues Gln81 - Leu84), β9 (residues Tyr276 - Val279), β10 (residues Tyr325 - Lys327), β11 (residues Phe343 - Asp348), β12 (residues Val353 - Lys359) and β13 (residues Lys364 - Ile369) are outside the TIM barrel. Similarly, α-helices α7 (residues Ala307 - Ala315) and α8 (residues Tyr328 - Leu336) were also deviated from TIM barrel. The structure also consisted of a 9 short 310-helices which are indicated as SH1, SH2, SH3, SH4, SH5, SH6, SH7, SH8 and SH9 (Figure 3). The side chains of three residues Ser110, Asp151 and Met220 showed two conformations with equal occupancies (Figure 1) indicating the unconstrained environment around them. As observed in other family GH-18 proteins [2,8,38], the structure of SpChiD adopts a perfect (β/α)8 - TIM barrel fold. However, the most striking feature of this structure pertains to the position of loop, Asn30 - Asp42 which intrudes into the substrate binding cleft and occupies nearly half of the space of substrate binding site (Figure 4). The residues at the front positions in the loop are Val35 and Thr36 which may provide a favourable chemical nature for interaction with the front residue of chitin oligomers in the cleft.
Figure 3.
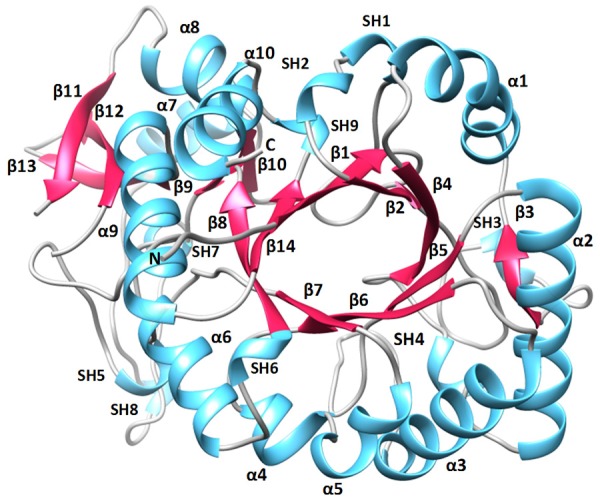
A view of the structure of SpChiD showing TIM barrel. The secondary structure elements α-helices, β-strands and short 310-helices have been labeled.
Figure 4.
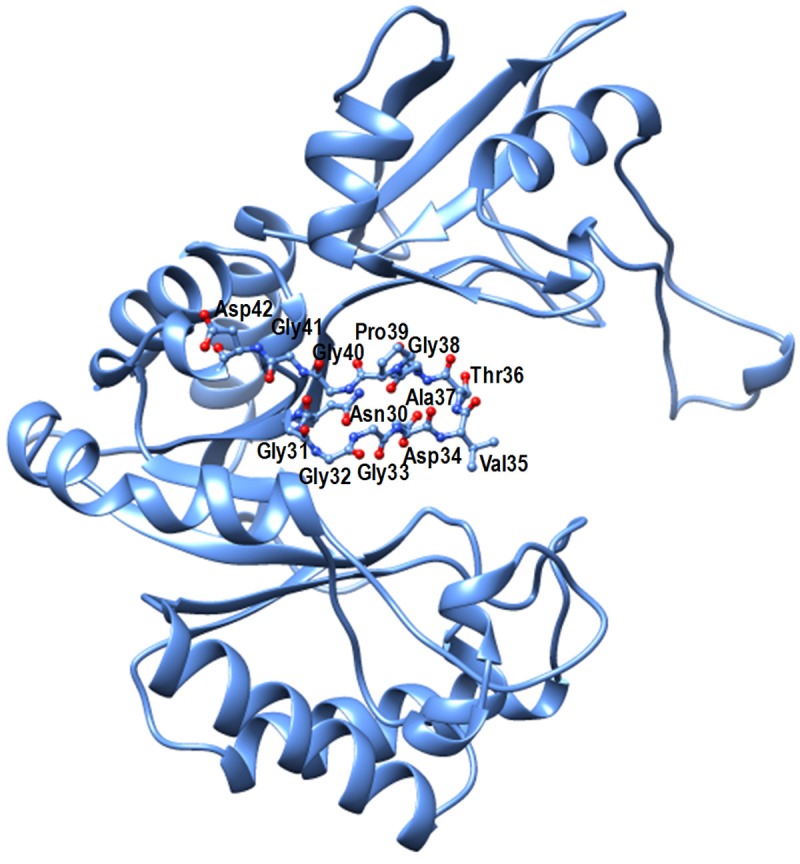
Structure of SpChiD showing the protruding loop Asn30 - Asp42 into the substrate binding cleft.
It is noteworthy that the superimposition of Cα traces of SpChiD on those of KpChiII (PDB code: 3QOK) with which it has a 79% amino acid sequence identity shows an r.m.s. shift of 0.7 Å for the Cα atoms indicating a high similarity between the two structures. Furthermore, the conformation of the novel loop, Asn30 - Asp42 is also identical in the structures of SpChiD and KpChiII as the r.m.s. shift for the Cα atoms of this segment is only 0.4 Å. Since the overall structures of SpChiD and KPChiII are very similar, the following studies will be performed in comparison with the coordinates of SpChiD.
Structure of the substrate binding cleft
The polypeptide chain of SpChiD folds part-wise into two distinct parts between which the substrate binding cleft is formed (Figure 5). The lower N-terminal part consists of residues from Ala15 to Glu220 while the upper part is made up of residues Met221 to Glu420. A loop Asn30 - Asp42 emerges from the back wall of the cleft and intrudes into the substrate binding site (Figure 6) thus reducing its length to less than half of those observed in SmChiA [8] and SmChiB [17]. The front face of the loop is equipped with residues, Val35 and Thr36. The lower side of the wall consists of residues, Thr69, Trp114, Trp160, Leu162 and Ala169, while the upper wall has residues, Trp226, Ala247, Arg278, Trp290, Asp291, Lys322 and Asp323 (Figure 7). The lower side also contains three active site acidic residues, Asp149, Asp151 and Glu153 (Figure 8). The side chain of Glu153 protrudes into the cleft. Its position with respect to the location of substrate is aligned in such a way that it could hydrolyze chitins to produce chitin monomers.
Figure 5.
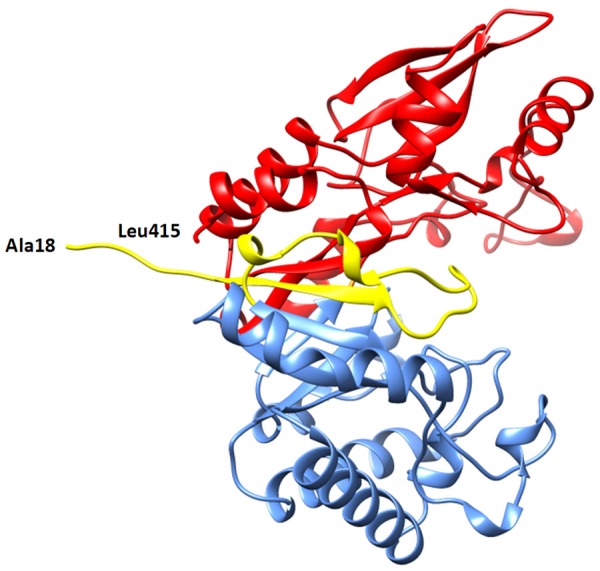
Showing independent folding of N- and C-regions. The substrate binding cleft is hindered partially by the loop Asn30 - Asp42.
Figure 6.
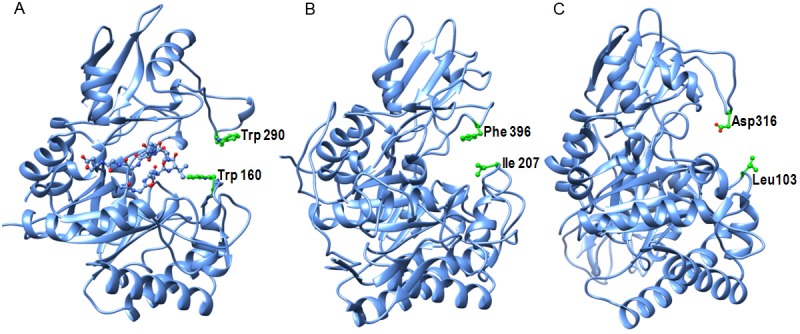
Showing views of the substrate binding clefts in (A) SpChiD, (B) SmChiA and (C) SmChiB. A loop Asn30 - Asp42 in SpChiD located in the substrate binding site is indicated in yellow. The residues on either side of the entrance to the substrate binding clefts are also indicated.
Figure 7.

View of the molecule showing the residues on either side of the cleft.
Figure 8.
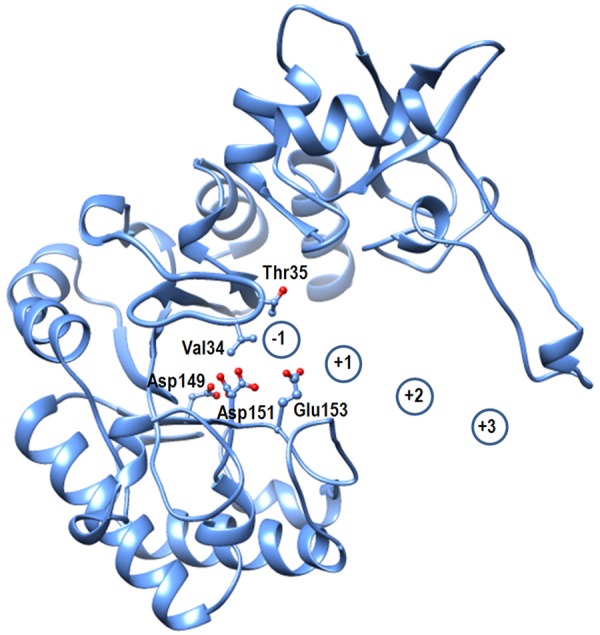
Showing substrate binding cleft with catalytic residues Ser110, Asp149, Asp151 and Glu153.
Catalytic site
The size of the substrate binding cleft in SpChiD has decreased to less than half as compared to those in SmChiA and SmChiB due to the insertion of a loop, Asn30 - Asp42 in the middle of the cleft (Figure 4). As a result of it, the number of vacant subsites in the cleft for substrate binding have reduced. The positions of catalytic residues Asp149, Asp151 and Glu153 are arranged in such a way that Glu153 is oriented appropriately to interact with the glycosidic linkage between residues at -1 and +1 subsites (Figure 8). In the present structure, Asp151 exist in two conformations. In one state Asp151 is oriented towards Asp149 and forms hydrogen bonds with it. In this arrangement Ser110 also forms a hydrogen bond with one of the carboxyl oxygen atoms. However, in the other state, Asp151 turns towards Glu153. In this orientation, Asp151 forms a hydrogen bond with Glu153. This orientation of Glu153 is appropriate to interact with disaccharide at the glycosidic linkage leading to hydrolysis of chitin by generating monomers.
Structure of novel loop and transglycosylation activity
SpChiD showed hyper TG activity [23], which was improved by mutagenesis [25]. The substrate binding cleft in SpChiD has an additional feature where the groove is blocked partially by a loop Asn30 - Asp42 which is part of the N-terminal segment, Ser25 - Ile51 (Figure 9). This segment consists of a β-strand, β1 (residues, 24 - 30) and two short 310-helices (SH1, residues 43 - 45 and SH2, residues 48 - 50). A part of this segment, Asn30 - Asp42 has been pushed into the substrate binding cleft. This is a unique feature of the structure of SpChiD and occurs due to specific amino acid sequence that induces a novel type1 β-turn that changes the path of the protein chain which is generally a β-strand in the structures of other chitinases [8,17]. The observed type1 β-turn at Asn30 - Gly31 - Gly32 - Gly33 with torsion angles of Φ1 = -92.3°, ψ1 = -36.7° and Φ2 = -90°, ψ2 = -10.3° is stabilized by a hydrogen bond Gly33NH - - - Oδ1 Asn30 = 2.80 Å. The sequences of the corresponding segments in SmChiA and SmChiB (Figure 2) seem to be unfavourable for forming such a tight type 1 β-turn and hence these segments remain parts of extended β-strands in other chitinases and hence do not move towards the cleft. The upper part of the N-terminal segment, Ser25 - Ile51 is sandwiched between α-helix, α1 (residues, 86 - 101) on one side and a β-strand, β14 (residues, 390 - 396), 310-helix SH9 (residues, 397 - 399) and α - helix, α10 (residues, 404 - 412) on the other side. This is firmly held at this position by several hydrogen bonds Lys87Nz - - - OGly31 = 2.78 Å, Asp91Oδ1 - - - NzLys87 = 3.00 Å, Lys87Nz - - - OGly41 = 2.72 Å, Asn44Nδ2 - - - Oδ1Asp42 = 3.12 Å, Asn44Nδ2 - - - Asp42Oδ1 = 2.90 Å. As seen from Figure 9, the loop Asn30 - Asp42 is internally well stabilized with the help of a number of intra - loop hydrogen bonds. This unique structure of SpChiD appears to be well designed to alter the nature of hydrolytic action of chitinase molecule and for introducing new function of TG. In this case, the substrate binding cleft is shortened and the last residue of the chitin oligomers is positioned in the cleft at -1 subsite. It is clear from the structure of the loop that it may contribute to hold the chitin residue at that position through a number of interactions from residues, Asp34, Val35 and Thr36. In addition to these, other proximal residues of the cleft such as Trp114, Arg278 and Asp323 also stabilize the chitin residue at this position. It appears that even after the hydrolysis of the chitin oligomer, the hydrolyzed shorter oligomers may remain bound to the protein because of additional interactions from the residues of the loop Asn30 - Asp42. Therefore, whenever the next oligomer slides into the cleft, the attachment of this residue to the oligomer might occur leading to the transglycosylated product. This is the first structural evidence of the TG activity in chitinases. However, there were a few earlier reports on this novel property in SpChiD and other similar chitinases which were based on biochemical and mutational studies [19-23].
Figure 9.
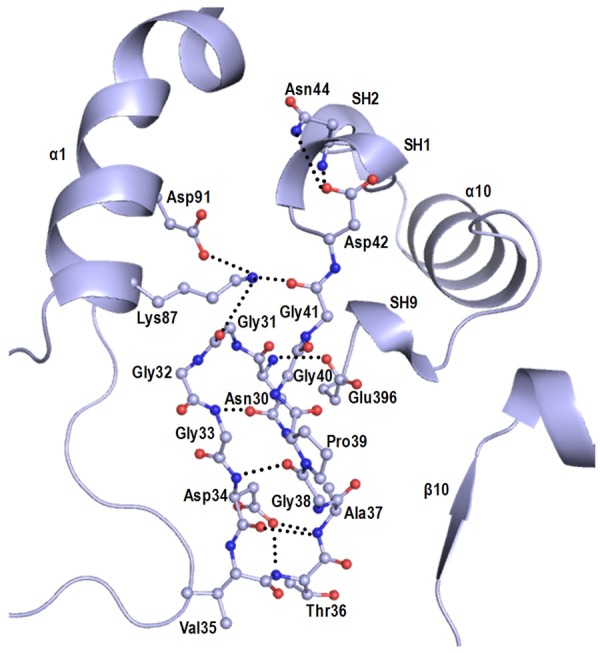
The loop Asn30 - Asp42 is internally stabilized but poorly connected to rest of the protein.
Discussion
SpChiD is a TIM barrel containing single domain protein which folds region-wise into two molecular halves where the lower molecular half (residues, Ala15 - Met200) is designated as N-region and the upper half (residues, Tyr221 - Glu420) is termed as C-region. In comparison, SmChiA contains two domains, N-terminal domain (residues, Ala24 - Asp132) and the main (β/α)8 TIM barrel catalytic domain (residues, Thr133 - Gly561) whereas SmChiB also contains two domains having main catalytic TIM barrel domain (residues, Thr3 - Gly442) and a C-terminal domain (residues, Val443 - Val498). The extra domains in both SmChiA and SmChiB with several aromatic residues provide additional binding support to polymeric substrates. In contrast, in the absence of extra domain SpChiD can only bind to oligomeric substrates and may not bind to polymeric substrates with efficiency. In the structure of SpChiD, sulfone methionine, SMet89 was observed at the site on the surface of protein which corresponds to the site in SmChiB structure where an extra domain was attached [17].
The deep substrate binding cleft in SpChiD is essentially formed at the interface of two molecular halves (Figure 5). However, it is partially filled by its own loop, Asn30 - Asp42 allowing a relatively smaller space for the binding of chitin oligomers unlike well formed grooves in other chitinases. At the entrance of the cleft in SpChiD, the residues Trp160 and Trp290 are situated on the opposite sides. In the cases of SmChiA and SmChiB, the corresponding residues are Leu103 and Asp316, and Ile207 and Phe396 respectively. The approximate nearest distance between the pairs of above residues at the entrance of the clefts were found to be 9.4 Å in SpChiD, 17.0 Å in SmChiA and 10.1 Å in SmChiB. The estimated length of the cleft in SpChiD was 11.0 Å while the corresponding distances in SmChiA and SmChiB were 21.0 Å and 23.1 Å respectively. The differences in the lengths and widths of the cleft due to the presence or absence of extra domains could be responsible for the preferences in sizes of substrates as well as in the nature of products.
The active site in SpChiD contains three acidic residues, Asp149, Asp151 and Glu153 as part of the Asp - X - Asp - X - Glu signature of glycosyl hydrolases. In this structure, an acetate molecule is also observed at the active site in the proximity of Asp151. It is noteworthy that the side chains of Ser110 and the middle acidic residue, Asp151 adopt two conformations in the present structure (Figure 10). The critical factor that seems to induce double conformation in Asp151 is the environment of Ser110 as the side chain of Ser110 is flanked by two aromatic residues Tyr28 and Phe58 on its two sides forming similar van der Waals contacts (Figure 10). The conformation of Ser110 with its side chain towards Asp149 corresponds to resting state while the other conformation when its side chain is turned towards Asp151 is considered as the intermediate state. The conformation the side chain of Ser110 in the resting state corresponds to the side chain torsion angle, x1 = -84°, when the side chain of Ser110 forms van der Waals contacts with the side chain of Tyr28. The conformation in the intermediate state corresponds to side chain torsion angle, x1 = 180° where the side chain of Ser110 forms van der Waals contacts with Phe58 (Figure 10). This indicates that the side chain of Ser110 can flip flop easily into opposite positions. It may also be noted that Ser110 Oγ forms H-bonds with Asp149 at both positions, resting state, (Ser110 OγH - - - Oδ2 Asp149 = 2.76 Å) and intermediate state, (Ser110 OγH - - -Oδ2 Asp149 = 2.69 Å). However, when Ser110 is in the intermediate state, its Oγ atom causes in severe steric constraints with the side chain of Asp151 in the resting state (resting state, x1 = -65°) at a distance of Ser110 Oγ - - -Oδ2 Asp151 = 2.23 Å. As a result, the side chain of Asp151 flips to the position corresponding to the intermediate state (x1 = -180°) where the distance between Ser110 Oγ and Oδ2 of Asp151 is 4.11 Å. In the new position, the side chain of Asp151 forms two bifurcated hydrogen bonds with the acetate OH group at distances 2.80 Å and 3.31 Å (Figure 10). The Oδ1 atom of the side chain of Asp151 forms a strong hydrogen bond (2.40 Å) with Glu153 O€2. The Glu153 O€2 is also hydrogen bonded to acetate OH at a distance of 3.00 Å. As a result of these interactions, the O€1 atom of Glu153 is appropriately placed to interact with the glycosidic bond between chitin residues at subsites +1 and -1. In this scheme, Ser110 protonates Asp151, which in turn protonates Glu153. The acetate molecule helps Glu153 to attain the correct orientation.
Figure 10.
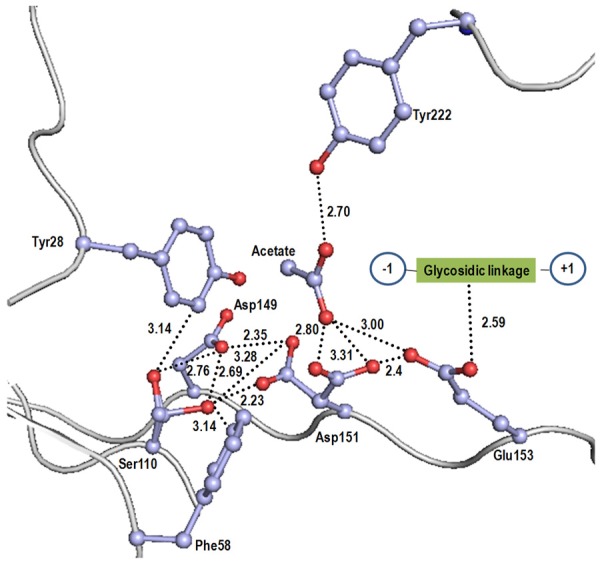
The residues at the active sites and interactions involving these residues. Acetate molecule helps in orienting Glu153 to the correct orientation. Acetate occupies the position of chitin residue at (i+2) position.
The presence of the loop, Asn30 - Asp42, in the substrate binding cleft and Trp160 and Trp290 at +2 position makes SpChiD as a favourable molecule for the TG activity. The mutant of SmChiA replacing Phe396 by Trp396, increased the TG activity many fold [24] with increased binding effects. Another important factor concerns with positions of Glu153 to avoid hydrolytic effect. It has been shown that mutation of Ser110 which is responsible for the cascade effect on attaining the correct orientation of Glu153 reduced hydrolytic activity and increased the TG activity of SpChiD [25]. A similar effect can also be achieved by mutating Asp151. However, the presence of loop Asn30 - Asp42 has natural effects in improving the TG activity of SpChiD. The structure determination has provided a deeper insight into the hydrolytic and transglycosylating activities of SpChiD. The novel structural features like the presence of a well stabilized Asn30 - Asp42 loop, sulfonation of Met89 and interactions of other residues with the loop region, provide an ideal platform for the activities of SpChiD.
Acknowledgements
The Synchrotron beamline BM-14 at the European Synchrotron Radiation Facility (ESRF) in Grenoble (France) is sponsored by the Department of Biotechnology, Ministry of Science and Technology, Government of India, New Delhi. This work was supported by the grant under Funds for Infrastructure in Science and Technology, Level II support to both departments from Department of Science and Technology, Government of India. TPS thanks Department of Biotechnology, Government of India for the award of Distinguished Biotechnology Research Professorship. AS and JM thank DBT and CSIR for the award of senior research fellowships respectively. We thank Dr. M. Lakshmi Kantam, Director, Indian Institute of Chemical Technology, Hyderabad, India for the mass spectrometric analysis.
Disclosure of conflict of interest
None.
References
- 1.Armand S, Tomita H, Heyraud A, Gey C, Watanabe T, Henrissat B. Stereochemical course of the hydrolysis reaction catalyzed by chitinases A1 and D from Bacillus circulans WL-12. FEBS Lett. 1994;343:177–80. doi: 10.1016/0014-5793(94)80314-5. [DOI] [PubMed] [Google Scholar]
- 2.Henrissat B, Davies G. Structural and sequence-based classification of glycoside hydrolases. Curr Opin Struct Biol. 1997;7:637–44. doi: 10.1016/s0959-440x(97)80072-3. [DOI] [PubMed] [Google Scholar]
- 3.Yu C, Lee AM, Bassler BL, Roseman S. Chitin utilization by marine bacteria. A physiological function for bacterial adhesion to immobilized carbohydrates. J Biol Chem. 1991;266:24260–7. [PubMed] [Google Scholar]
- 4.Takaya N, Yamazaki D, Horiuchi H, Ohta A, Takagi M. Intracellular chitinase gene from Rhizopus oligosporus: molecular cloning and characterization. Microbiology. 1998;144:2647–54. doi: 10.1099/00221287-144-9-2647. [DOI] [PubMed] [Google Scholar]
- 5.Felse PA, Panda T. Regulation and cloning of microbial chitinase genes. Appl Microbiol Biotechnol. 1999;51:141–51. doi: 10.1007/s002530051374. [DOI] [PubMed] [Google Scholar]
- 6.Xu JG, Zhao XM, Han XW, Du YG. Antifungal activity of oligochitosan against Phytophthora capsici and other plant pathogenic fungi in vitro. Pestic Biochem Physiol. 2007;87:220–8. [Google Scholar]
- 7.Shuhui L, Mok YK, Wong WS. Role of mammalian chitinases in asthma. Int Arch Allergy Immunol. 2009;149:369–77. doi: 10.1159/000205583. [DOI] [PubMed] [Google Scholar]
- 8.Perrakis A, Tews I, Dauter Z, Oppenheim AB, Chet I, Wilson KS, Vorgias CE. Crystal structure of a bacterial chitinase at 2.3 A resolution. Structure. 1994;2:1169–80. doi: 10.1016/s0969-2126(94)00119-7. [DOI] [PubMed] [Google Scholar]
- 9.Terwisscha van Scheltinga AC, Hennig M, Dijkstra BW. The 1.8 Å resolution structure of hevamine, a plant chitinase/lysozyme, and analysis of the conserved sequence and structure motifs of glycosyl hydrolase family 18. J Mol Biol. 1996;262:243–57. doi: 10.1006/jmbi.1996.0510. [DOI] [PubMed] [Google Scholar]
- 10.Malecki PH, Raczynska JE, Vorgias CE, Rypniewski W. Structure of a complete four-domain chitinase from Moritella marina, a marine psychrophilic bacterium. Acta Crystallogr D Biol Crystallogr. 2013;69:821–9. doi: 10.1107/S0907444913002011. [DOI] [PubMed] [Google Scholar]
- 11.Brameld KA, Goddard WA 3rd. The role of enzyme distortion in the single displacement mechanism of family 19 chitinases. J Amer Chem Soc. 1998;120:3571–80. doi: 10.1073/pnas.95.8.4276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Terwisscha van Scheltinga AC, Armand S, Kalk KH, Isogai A, Henrissat B, Dijkstra BW. Stereochemistry of chitin hydrolysis by a plant chitinase/lysozyme and X-ray structure of a complex with allosamidin: evidence for substrate assisted catalysis. Biochemistry. 1995;34:15619–23. doi: 10.1021/bi00048a003. [DOI] [PubMed] [Google Scholar]
- 13.Tews I, van Scheltinga Terwisscha AC, Perrakis A, Wilson KS, Dijkstra BW. Substrate-assisted catalysis unifies two families of chitinolytic enzymes. J Am Chem Soc. 1997;119:7954–9. [Google Scholar]
- 14.Papanikolau Y, Prag G, Tavlas G, Vorgias CE, Oppenheim AB, Petratos K. High resolution structural analyses of mutant chitinase A complexes with substrates provide new insight into the mechanism of catalysis. Biochemistry. 2001;40:11338–43. doi: 10.1021/bi010505h. [DOI] [PubMed] [Google Scholar]
- 15.Bortone K, Monzingo AF, Ernst S, Robertus JD. The structure of an allosamidin complex with the Coccidioides immitis chitinase defines a role for a second acid residue in substrate-assisted mechanism. J Mol Biol. 2002;320:293–302. doi: 10.1016/S0022-2836(02)00444-8. [DOI] [PubMed] [Google Scholar]
- 16.Sasaki C, Yokoyama A, Itoh Y, Hashimoto M, Watanabe T, Fukamizo T. Comparative study of the reaction mechanism of family 18 chitinases from plants and microbes. J Biochem. 2002;131:557–64. doi: 10.1093/oxfordjournals.jbchem.a003134. [DOI] [PubMed] [Google Scholar]
- 17.van Aalten DMF, Synstad B, Brurberg MB, Hough E, Riise BW, Eijsink VG, Wierenga RK. Structure of a two-domain chitotriosidase from Serratia marcescens at 1.9-A resolution. Proc Natl Acad Sci U S A. 2000;97:5842–7. doi: 10.1073/pnas.97.11.5842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van Aalten DMF, Komander D, Synstad B, Gaseidnes S, Peter MG, Eijsink VG. Structural insights into the catalytic mechanism of a family 18 exo-chitinase. Proc Natl Acad Sci U S A. 2001;98:8979–84. doi: 10.1073/pnas.151103798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xia G, Jin C, Zhou J, Yang S, Zhang S, Jin C. A novel chitinase having a unique mode of action from Aspergillus fumigatus YJ-407. Eur J Biochem. 2001;268:4079–85. doi: 10.1046/j.1432-1327.2001.02323.x. [DOI] [PubMed] [Google Scholar]
- 20.Aguilera B, Ghauharali-van der Vlugt K, Helmond MT, Out JM, Donker-Koopman WE, Groener JE, Boot RG, Renkema GH, van der Marel GA, van Boom JH, Overkleeft HS, Aerts JM. Transglycosidase activity of chitotriosidase: improved enzymatic assay for the human macrophage chitinase. J Biol Chem. 2003;278:40911–16. doi: 10.1074/jbc.M301804200. [DOI] [PubMed] [Google Scholar]
- 21.Aronson NN Jr, Halloran BA, Alexeyev MF, Zhou XE, Wang Y, Meehan EJ, Chen L. Mutation of a conserved tryptophan in the chitin-binding cleft of Serratia marcescens chitinase A enhances transglycosylation. Biosci Biotechnol Biochem. 2006;70:243–51. doi: 10.1271/bbb.70.243. [DOI] [PubMed] [Google Scholar]
- 22.Taira T, Fujiwara M, Dennhart N, Hayashi H, Onaga S, Ohnuma T, Letzel T, Sakuda S, Fukamizo T. Transglycosylation reaction catalyzed by a class V chitinase from cycad, Cycas revoluta: a study involving site-directed mutagenesis, HPLC, and real-time ESI-MS. Biochim Biophys Acta. 2009;1804:668–75. doi: 10.1016/j.bbapap.2009.10.015. [DOI] [PubMed] [Google Scholar]
- 23.Purushotham P, Podile AR. Synthesis of long-chain chitooligosaccharides by a hypertransglycosylating processive endochitinase of Serratia proteamaculans 568. J Bacteriol. 2012;194:4260–71. doi: 10.1128/JB.06473-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zakariassen H, Hansen MC, Jøranli M, Eijsink VGH, Sørlie M. Mutational effects on transglycosylating activity of family 18 chitinases and construction of a hypertransglycosylating mutant. Biochemistry. 2011;50:5693–703. doi: 10.1021/bi2002532. [DOI] [PubMed] [Google Scholar]
- 25.Madhuprakash J, Tanneeru K, Purushotham P, Guruprasad L, Podile AR. Transglycosylation by chitinase D from Serratia proteamaculans improved through altered substrate interactions. J Biol Chem. 2012;287:44619–27. doi: 10.1074/jbc.M112.400879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Suginta W, Vongsuwan A, Songsiriritthigul C, Svasti J, Prinz H. Enzymatic properties of wild-type and active site mutants of chitinase A from Vibrio carchariae, as revealed by HPLC-MS. FEBS J. 2005;272:3376–86. doi: 10.1111/j.1742-4658.2005.04753.x. [DOI] [PubMed] [Google Scholar]
- 27.Songsiriritthigul C, Pantoom S, Aguda AH, Robinson RC, Suginta W. Crystal structures of Vibrio harveyi chitinase A complexed with chitooligosaccharides: implications for the catalytic mechanism. J Struct Biol. 2008;162:491–9. doi: 10.1016/j.jsb.2008.03.008. [DOI] [PubMed] [Google Scholar]
- 28.Tsuji H, Nishimura S, Inui T, Kado Y, Ishikawa K, Nakamura T, Uegaki K. Kinetic and crystallographic analyses of the catalytic domain of chitinase from Pyrococcus furiosus- the role of conserved residues in the active site. FEBS J. 2010;277:2683–95. doi: 10.1111/j.1742-464X.2010.07685.x. [DOI] [PubMed] [Google Scholar]
- 29.Hsieh YC, Wu YJ, Chiang TY, Kuo CY, Shrestha KL, Chao CF, Huang YC, Chuankhayan P, Wu WG, Li YK, Chen CJ. Crystal structures of Bacillus cereus NCTU2 chitinase complexes with chitooligomers reveal novel substrate binding for catalysis: a chitinase without chitin binding and insertion domains. J Biol Chem. 2010;285:31603–15. doi: 10.1074/jbc.M110.149310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–26. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 31.Vagin A, Teplyakov A. Molecular replacement with MOLREP. Acta Crystallogr D Biol Crystallogr. 2010;66:22–5. doi: 10.1107/S0907444909042589. [DOI] [PubMed] [Google Scholar]
- 32.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr. 2011;67:235–42. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Murshudov GN, Skubak P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F, Vagin AA. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr D Biol Crystallogr. 2011;67:355–67. doi: 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A. 1991;47:110–9. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 35.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–32. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 36.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Cryst. 1993;26:283–91. [Google Scholar]
- 37.Edman P, Begg G. A protein sequenator. Eur J Biochem. 1967;1:80–91. doi: 10.1007/978-3-662-25813-2_14. [DOI] [PubMed] [Google Scholar]
- 38.Niall HD. Automated Edman degradation: the protein sequenator. Methods Enzymol. 1973;27:942–1010. doi: 10.1016/s0076-6879(73)27039-8. [DOI] [PubMed] [Google Scholar]