

Abstract
Mitochondrial alterations have been documented for many years in the brains of Parkinson’s disease (PD), a disorder that is characterized by the selective loss of dopamine neurons. Recent studies have demonstrated that Parkinson’s disease-associated proteins are either present in mitochondria or translocated into mitochondria in response to stress, further reinforcing the importance of the mitochondrial function in the pathogenesis of Parkinson’s disease. Exposure to environmental chemicals such as pesticides and heavy metals has been suggested as risk factors in the development of Parkinson’s disease. It has been reported that a number of environmental agents including tobacco smoke and perfluorinated compounds, pesticides, as well as metals (Mn2+ and Pb2+) modulate mitochondrial function. However the exact mechanism of mitochondrial alteration has not been defined in the context of the development and progression of Parkinson’s disease. The complexity of the mammalian system has made it difficult to dissect the molecular components involved in the pathogenesis of Parkinson’s disease. In the present study we used the nematode Caenorhabditis elegans (C. elegans) model of neuron degeneration and investigated the effect of environmental chemicals on mitochondrial biogenesis and mitochondrial gene regulation. Chronic exposure to low concentration (2 or 4 μM) of pesticide rotenone, resulted in significant loss of dopamine neuron in C. elegans, a classic feature of Parkinson’s disease. We then determined if the rotenone-induced neuron degeneration is accompanied by a change in mitochondria biogenesis. Analysis of mitochondrial genomic replication by quantitative PCR showed a dramatic decrease in mitochondrial DNA (mtDNA) copies of rotenone-treated C. elegans compared to control. This decreased mitochondrial biogenesis occurred prior to the development of loss of dopamine neurons, and was persistent. The inhibition of mtDNA replication was also found in C. elegans exposed to another neuron toxicant Mn2+ at the concentration 50 or 100 mM. We further examined the mitochondrial gene expression and found significant lower level of mitochondrial complex IV subunits COI and COII in C. elegans exposed to rotenone. These results demonstrate that environmental chemicals cause persistent suppression of mitochondrial biogenesis and mitochondrial gene expression, and suggest a critical role of modifying mitochondrial biogenesis in toxicants-induced neuron degeneration in C. elegans model.
Keywords: Caenorhabditis elegans, C. elegans, neuron degeneration, mitochondrial suppression, environmental chemicals
Introduction
Environmental exposure has been linked positively with neurodegenerative disorders such as Parkinson’s disease (PD). Parkinson’s disease is the second most prevalent neurodegenerative disorder, characterized by the selective loss of dopamine neurons in the substantia nigra [1]. Although the role of the dopamine neurons in idiopathic pathogenesis of Parkinson’s disease remains unclear, studies suggest both genetic and environmental factors interact and contribute to the development of the Parkinson’s disease [2]. Exposures to pesticides and heavy metals have been suggested to be associated with neuron toxicity and increased Parkinson’s disease in humans [3,4]. Chronic exposure to the heavy metals, Mn2+, Cu2+, and Pb2+, for instance, has been reported to increase the likelihood of Parkinson’s disease [5]. Similarly, epidemiological studies have suggested that exposure to pesticides including rotenone, maneb and paraquat has been associated with increased risk of development of Parkinson’s disease [3,4]. While the precise molecular mechanisms by which these environmental toxicants induce dopamine neuron degeneration are unknown, impact on mitochondrial function has been proposed to be a crucial event in the etiology and development of Parkinson’s disease [6]. Due, in part, to the complexity of the vertebrate brain, the exact molecular mechanism behind the mitochondrial alteration has not been fully described in vivo which makes it difficult to dissect pathologic pathways in the development of Parkinson’s disease and neurodegenerative disorders [7].
The role for the mitochondria as a target of environmental agents has been widely implicated into the mechanism of chronic exposure to environmental toxicants. Both mitochondrial bioenergetics and mitochondrial genetic alterations have been frequently observed in response to these environmental agents [8]. The molecular mechanisms of the effects of environmental chemicals on mitochondria include interfering with ATP production, inducing oxidative stress, and interrupting cellular homeostasis and cell death pathways [9,10]. Both animal studies and epidemiological investigation implicate mitochondrial dysfunction into the etiology and pathologies of Parkinson’s disease [6]. Characterization of Parkinson’s disease-associated genes Parkin, Pink1, LRRK2, DJ-1, and a-Synuclein reveals all these major Parkinson’s disease proteins are either expressed in mitochondria, or translocated into mitochondria under stress [11-13], suggesting the role of interaction between PD genetic alteration with mitochondria in the mechanism of the Parkinson’s disease development. A recent study in a genome-wide analysis revealed that the genes controlling bioenergetics were underexpressed in Parkinson’s disease patients, while activation of nuclear-encoded subunits of the mitochondrial respiratory chain blocked the dopaminergic neuron loss induced by genetic mutation [14], further supporting the importance of mitochondrial function in the mechanism of Parkinson’s disease and neurodegenerative disorders. Moreover, the mode of action or targeting sites on mitochondria varies dramatically among the different chemicals studied. Because of its short life cycle and ease to culture in the laboratory, C elegans has been utilized in the study of Parkinson’s disease and mechanisms of toxicity. C elegans has well conserved mitochondrial functions, and share a lot of similarities in mitochondrial genome and gene regulation as in human. A number of human mitochondrial genes have homologs in the C. elegans [15]. The structure and function of the mitochondrial electron transport chain as well as many pathways of intermediate metabolism such as the Krebs cycle and many other signal transduction pathways are very well conserved between higher mammals and C. elegans [16]. The availability of the complete mtDNA and nuclear DNA sequences and well developed forward and reverse genetic approaches facilitate the rapid investigation of the gene functions in this nematode. These features have made C elegans an excellent model to study mitochondrial gene regulation and function and the contribution of mitochondrial alteration in the mechanisms of neurotoxicity induced by environmental chemicals.
In the present study we investigated the effect of environmental chemicals on the mitochondrial genomic regulation using the C. elegans. We selected rotenone, a classic inhibitor of mitochondrial complex I as a model compound to examine [17]. The experiments were designed to chronically exposeto the worms to low concentrations (2 and 4 μM), and developed loss of DA neuron, characterized of Parkinsonism. The effect of rotenone on mitochondrial DNA replication and gene expression was examined at different time points prior to development of neuron degeneration. MtDNA replication was also examined in the C. elegans exposed to 50 or 100 mM of MnCl2, which has been demonstrated to induce neuron degeneration in previous studies [18]. We found both rotenone and Mn2+ induce an early and persistent decrease in mitochondrial DNA content and suppress mitochondrial gene expression in C. elegans. These studies show that the C. elegans provides opportunities for new insights into further investigating the mechanism of chemically induced neuron degeneration.
Materials and methods
C. elegans strain and maintenance
The C. elegans strain BY250 (Pdat-1:GFP) was kindly provided by Dr. Blakely of Vanderbilt University. The C. elegans worms were cultured on bacterial lawns containing either OP50 or NA22 bacteria on NGM or 8P plates, respectively, at 20°C according to standard methods [19,20].
Toxicant exposures
Synchronized L1 stage worms were obtained by hypochlorite treatment of gravid adults followed by incubation of the embryos in M9 buffer for 18 h, and then washed three times with dH2O as described in previous studies [21,22]. Chronic exposure protocol was utilized for rotenone treatment. NGM plates were pre-coated with rotenone in DMSO to achieve a concentration of 2 or 4 μM. Rotenone at these concentrations did not cause any mortality throughout the course of experiments. However, higher concentrations (>10 μM) delayed the development of worms into the adult stage, and thus affected the examination of dopamine neurons. Washed L1 worms were then loaded on the 2 or 4 μM rotenone-coated plates until the further experiments. An equal volume of DMSO was applied to NGM plates as a control. For MnCl2 exposure, L1 worms (at 10 worms/μl) were incubated at 50 or 100 mM concentration at room temperature for 30 min. Incubation tubes were gently mixed every 10 min. Following the incubation, the worms were washed three times with dH2O, and then transferred onto NGM plates to culture until further experiments. The concentration of MnCl2 was selected based on a previous report that MnCl2 at the 100 mM concentration caused dopamine neuron degeneration [18].
Examination of neuron degeneration
Neuron degeneration in rotenone-exposed worms was examined under a dissecting fluorescence microscope (Leica MZ 16FA, Switzerlad). Specifically, following toxicant exposure, worms were washed once with dH2O. About 50 worms were picked using a worm pick and immobilized with 2% sodium azide that was pre-dropped on 2% agarose pad on a microscope slide. A coverslip was placed on top of the worms and DA neurons were examined with the fluorescence microscope. The four cephalic dendrites (CEPs) were examined from nerve ring to the tip of the nose. Worms were scored as positive for neuron degeneration when GFP in any part of the four CEPs was missing (Nass et al., 2002). The experiments were performed in triplicates, and the results were expressed presented as mean ± standard deviation. Imaging was recorded using a Nikon camera that was connected to the fluorescence micro- scope.
Genomic DNA extraction
Genomic DNA was isolated by phenol/chloroform method. Worm pellets were resuspended in 2 volume of lysis buffer containing 0.1 M Tris-HCl pH 8.5, 0.1 M NaCl, 50 mM EDTA pH 8.0, and 1% SDS, and digested with proteinase K at 65°C for 60 minutes. DNA was separated from proteins and other impurities by TE saturated Phenol:Chloroform. Following spinning in the tube in a bench top centrifuge at 4,000 g for 2 minutes, aqueous layer was collected and transferred to a new 15 mL conical tube. DNA was precipitated with 95% EtOH and washed with ice cold 70% EtOH. After air-dry, DNA was resuspended in DNAase free water and stored at -80°C for further experiments.
RNA extraction and cDNA synthesis
Total RNA was extracted from synchronized C. elegans population using Trizol reagent as described previously with slight modifications [23]. Specifically, worms were collected from treatment or control plates by washing with water. The worm pellets were resuspended in Trizol (1 ml/100 μl compact worm pellet), and frozen and thawed at 37°C and -80°C twice. Chloroform was used to separate RNA from protein and other materials. And total RNA was precipitated with isopropyl alcohol. The RNA pellet was washed once with 70% ethanol and air-dried. The resultant RNA was dissolved in RNase-free water, and stored at -80°C for further analysis. The RNA concentrations were measured using an ND-1000 spectrophotometer (Nanodrop Technology, Wilmington, DE). cDNA was synthesized from RNA using Invitrogen cDNA synthesis kit per manufacturer’s protocol.
Quantitative polymerase chain reaction (qPCR)
The mitochondrial DNA content was determined by amplification of a short mitochondrial DNA target with 7900HT Fast Real-time PCR Systems (Applied Biosystems). Amplification of mitochondrial and nuclear targets was performed using primers described previously, which are specific for C. elegans DNA [24]. Primers for nuclear target (9.3 kb) are, forward primer, 5’-TGGCTGGAACGAACCGAACCAT-3’; and reverse primer, 5’-GGCGGTTGTGGAGTGTGGGAAG-3’; and for mitochondrial target, forward primer, 5’-CACACCGGTGAGGTCTTTGGTT-3’; and reverse primer, 5’-TGTCCTCAAGGCTACCACCTTCTTCA-3’. Two pairs of primers specific for C. elegans mitochondrial genes COI and COII were designed for mitochondrial gene expression using Primer 3 software. Mitochondrial gene COI primers are forward 5’-GGGATTTTCACGGGTGTTAC-3’, and reverse primer 5’-CCCGTGTAGTCCTGCAAAAT-3’. The primers for COII are forward 5’-TGTTATTCATGCTTGGGCATT-3’, and reverse primer 5’-TGCTCCACAAATCTCTGAACAT-3’. The real-time PCR reactions were performed in triplicate for each gene. DNA (5 ng) was used to amplify mitochondrial regions, whereas 5 ng of DNA was used to amplify nuclear gene GAPDH using primers described previously [18]. Mitochondrial DNA content was expressed as mtDNA/nDNA ratio. The mitochondrial gene expression level was presented as a fold change relative to the control.
Statistical analysis
Data were presented as mean ± SD of three individual experiments. Differences between treatment and control were evaluated using one-way ANOVA. A p value of < 0.05 was considered as significance.
Results
Chronic exposure to rotenone caused neuron degeneration in the C. elegans
Rotenone is a broad-spectrum insecticide. It is a mitochondrial respiratory complex I inhibitor, and has been demonstrated to induce the symptoms in mammalian models similar to human Parkinson’s disease [25]. In the present study, we continually exposed C. elegans to rotenone and examined its effect on dopamine neurons correlating with mitochondrial biogenesis. The transparent nature of C. elegans and the strain (BY250) expressing GFP dopamine neuron allowed us to examine dopamine neurons under a dissecting fluorescence microscope [22]. The synchronized L1 worms were plated on the NGM plates containing 2 or 4 μM of rotenone, and grew into adults when the examination of the dopamine neurons was performed under a dissecting fluorescence microscope. The concentration of 4 μM was optimized as the highest concentration in the present study, and higher concentrations induced significant inhibition of worm growth, and therefore were not suitable for chronic exposure. As shown in Figure 1, exposed adult worms displayed significant loss of dopamine neurons in a concentration-dependent manner. The rotenone-induced neuron degeneration was characterized by the missing of a part, or whole CEP of the DA neurons (Figure 1D and 1F), and about 35% of worms exposed to 4 μM of rotenone exhibited DA neuron lesions (Figure 1G). However, no abnormal DA neurons were found at the early stage (24 or 48 h rotenone exposure), indicating a late onset of neuron degeneration in response to the toxicant exposure.
Figure 1.
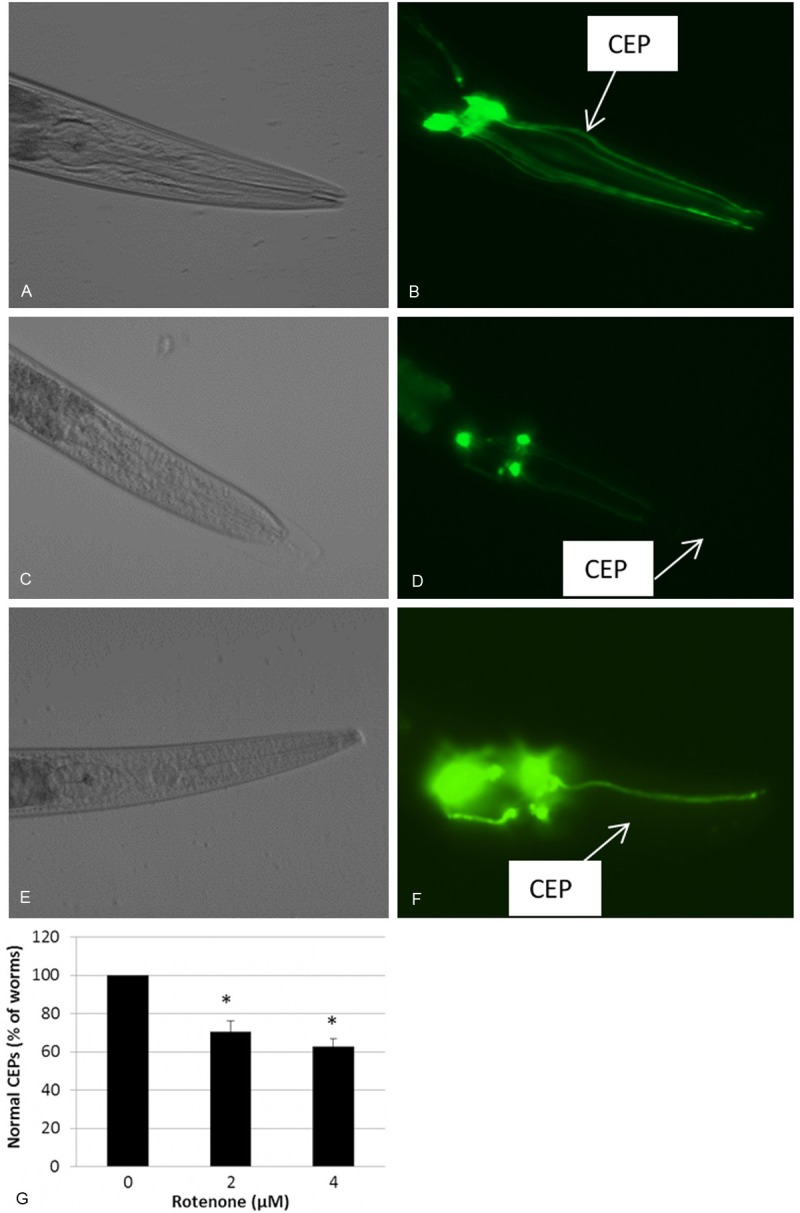
Chronic exposure to rotenone caused dopamine (DA) neuron degeneration. Synchronized L1 nematodes were grown on NGM plates containing 2 or 4 μM of rotenone, or a same volume of DMSO (control). Worms were examined for DA neuron degeneration as described in the Materials and Methods. (B, D and F) Representative images showing GFP-expressing DA neurons (CEPs) within the head of a control worm exposed to DMSO (control), 2, or 4 μM of rotenone. Arrows indicate the presence of CEPs in control (B), while a part missing or loss of the whole CEP in 2 (D) or 4 μM (F) rotenone exposed animals, respectively. (A, C and E) DIC image of the corresponding animals shown in B, D, and F. (G) Quantification of neuron degeneration. *, indicates significant difference compared to control (p < 0.05).
Mitochondrial DNA replication is suppressed by rotenone and Mn2+
The effect of rotenone exposure on mitochondrial DNA content was also assessed. The temporal pattern between mitochondrial biogenic change and neuron degeneration was examined using mitochondrial DNA copy numbers after 24 and 48 h exposure, respectively. These two exposure times did not produce DA neuron degeneration. The mtDNA copies were determined by qPCR. Since the number of mitochondria per cell and the total DNA content in each mitochondrion can be highly variable during the worm development, it is necessary to normalize qPCR results using mitochondrial copy number to nuclear DNA content. The primers employed here specifically for amplification of a short fragment of mitochondrial DNA and nuclear DNA were reported in a previous study [24]. As shown in the Figure 2A and 2B, dramatic decrease in relative mtDNA level was found in the worms exposed to rotenone for 24 and 48 h, respectively, indicating a persistent suppression of mtDNA replication. We also examined another toxicant metal Mn2+ that has been reported to be toxic to DA neurons in C. elegans [18]. As shown in Figure 3, a significant deccrease in mtDNA content was found in the worms treated with with 50 and 100 mM of Mn2+. These results suggest an important role of mtDNA biogenesis in the process of dopamine neuron damage and degeneration in C. elegans.
Figure 2.
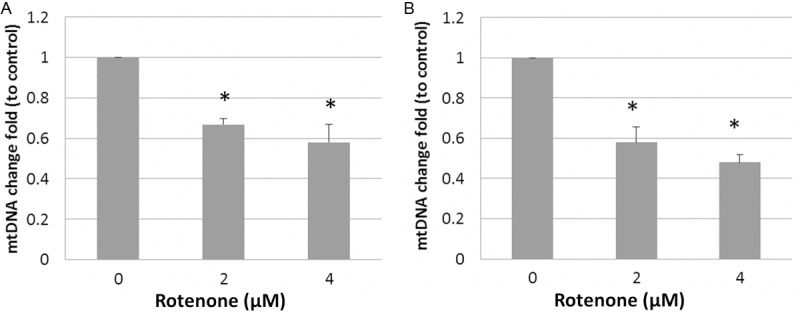
Rotenone-induced decrease in mtDNA content determined by qPCR. Synchronized L1 animals were exposed to 2 and 4 μM rotenone for 24 h or 48 h. Genomic DNA was extracted. mtDNA copy number was quantitated by real-time PCR. Relative mtDNA content was calculated as a fold change relative to nuclear DNA content. Data was expressed as the man ± SD of three individual replicates. *, indicates significant difference compared to control (p < 0.05).
Figure 3.
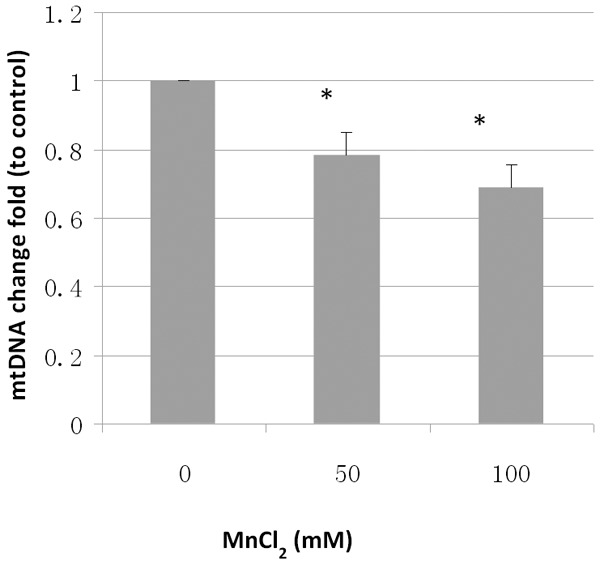
Mn2+-induced decrease in MtDNA content determined by qPCR. Synchronized L1 animals were exposed to 50 and 100 mM MnCl2 24 h. Genomic DNA was extracted. mtDNA copy number was quantitated by real-time PCR. Relative mtDNA content was calculated as a fold change relative to nuclear DNA content. Data was expressed as the man ± SD of three individual replicates. *, indicates significant difference compared to control (p < 0.05).
Decreased expression of mitochondrial gene expression induced by rotenone exposure
To determine the mitochondrial gene expression, we designed primers specific to mitochondrial complex IV subunits I and II (COI and COII), respectively. Consistent with the level of mtDNA content, the expression level of both COI and COII was significant lower in worms treated with 2 or 4 μM rotenone when compared to control (Figure 4).
Figure 4.
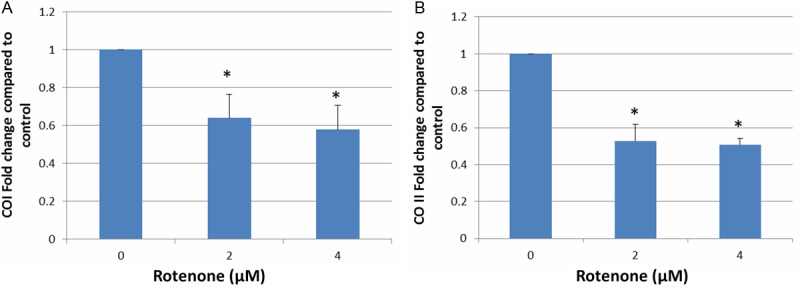
Suppression of mitochondrial gene expression. Synchronized L1 animals were grown on the NGM plates containing 2 and 4 μM rotenone for 48 h. Following the treatment, RNA was isolated using Trizol, and cDNA was synthesized using commercial kit per manufacturer’s instruction as described in the Materials and Methods. Two sets of primers designed specifically for COI and COII were employed for the qPCR. Mitochondrial gene expression level was calculated as a fold change relative to nuclear gene expression level (GAPDH). Data was expressed as the man ± SD of three individual replicates. *, indicates significant difference compared to control (p < 0.05).
Discussion
Mitochondrial dysfunction and oxidative stress have long been implicated as pathophysiologic mechanisms underlying Parkinson’s disease [3,26]. It has been suggested that the inhibition of mitochondrial complex I to be a leading mechanism although the exact molecular basis underlying mitochondrial dysfunction in the development of PD has not been fully elucidated.
Utilizing the C. elegans model, we report here that chronic exposure to low concentration of rotenone, a classic inhibitor of mitochondrial complex I, causes dopamine neuronal cell death, a major feature of human Parkinson’s disease and neurodegenerative disorder. Epidemiologic investigations have demonstrated that Parkinson’s disease is positively associated with use of pesticide rotenone [3,27], although a causative linkage between pesticide rotenone use and Parkinson’s disease has not been established. Rotenone has been widely tested in mammalian systems to induce symptoms similar to those observed in Parkinson’s disease [25], yet the mitochondrial mechanism of neuron toxicity has never been fully elucidated. Declined mitochondrial bioenergetics and increased oxidative stress are two major cellular signaling widely studied, which are closely associated with mitochondrial alterations to be implicated into the chemical toxicity mechanism. However, other studies have suggested that increased cellular oxidative stress rather than the compromised mitochondrial bioenergetics is responsible for rotenone toxicity [28]. Moreover, the hypothesis of mitochondrial complex I inhibition has been challenged by a recent study that demonstrated dopamine neuron death induced by a classic Parkinson’s disease drug 1-methyl-4-phenylpyridinium (MPP+) and pesticides (rotenone and paraquat) are independent on their effect on mitochondrial function [29]. Thus, a role for the mitochondria in Parkinson’s disease requires further investigation.
This study shows that for the first time rotenone exposure caused a persistent suppression of mitochondrial DNA replication in the C. elegans model. In addition, the decrease of mitochondrial DNA biogenesis was also found in C. elegans exposed to Mn2+, a metal that has been demonstrated to induce neuron degeneration in C. elegans [18]. This indicates the depression of mtDNA replication might be a common mechanism implicated in the process of toxicants-induced neuron damage. The fact that the suppression of this mitochondrial biogenesis occurred prior to the loss of dopamine neurons supports the critical role of these organelle in the development of neurodegenerative disease. Although the data presented here does not suggest rotenone exposure is causatively responsible for Parkinson’s disease in humans, it supports the concept that C. elegans is a good model for dissecting mitochondrial-mediated pathways implicated in the mechanism of environmentally induced neurodegenerative disorders.
The observed decrease in the mitochondrial content and mitochondrial gene expression may be the consequence of direct inhibition of mitochondrial replication regulated by nuclear encoded mitochondrial genomic machinery. Rotenone has been shown to induce increased oxidative stress in mammalian cells as well as in invertebrate models [30,31]. Studies have demonstrated the increased oxidative stress to be a mechanism to modulate mtDNA biogenesis [32]. In addition, rotenone may also induce DNA damage affecting PCR amplification of mtDNA. However, we were not able to dissect the precise mechanism of suppression of mitochondrial DNA biogenesis associated with rotenone exposure. Nevertheless, the depression of mtDNA biogenesis may result in a compromised mitochondrial function. It has been reported recently that a similar amount of damage to mtDNA caused higher extent in the inhibition of mitochondrial function in C. elegans than in cultured mammalian cells [33,34], indicating the important role of regulating and maintaining mtDNA integrity in C. elegans. Further identification of regulator(s) of mitochondrial biogenesis may offer new insights of the rotenone-induced neuron degeneration in the C. elegans.
It is worthy to note that rotenone exposure may directly inhibit mitochondrial respiratory activity resulting in the decrease in ATP production. However, in the present study we intended to investigate the effect of the environmental toxicants on mitochondrial biogenesis rather than the bioenergetics although in many causes these two events are integrated in mitochondrial function. Moreover, as mentioned above, studies in vertebrate system suggest that the compromised mitochondrial bioenergetics is not responsible for Parkinson’s disease symptom [28]. It has been reported that heavy metal Mn2+ causes inhibition of mitochondrial respiration in C. elegans [18]. In a recent study we reported that the impaired mitochondrial function is also associated with neuron degeneration in C. elegans induced by another metal Al3+ [35]. Further investigation of the effect of these metals on mitochondrial biogenesis and the regulation of mitochondrial gene expression may help dissecting molecular basis behind the compromised mitochondrial bioenergetics, thus further the understanding of the role of mitochondrial alteration in the development of neuron degeneration and pathologies of human Parkinson’s disease.
In summary, we demonstrated that chronic exposure to environmental toxicants rotenone and Mn2+ caused a persistent inhibition of mitochondrial biogenesis. Identification of molecular regulator(s) of toxicants-induced alteration in mitochondrial biogenesis is needed for furthering our understanding of the mitochondrial contribution in the process, etiology, and pathologies of Parkinson’s disease and neurodegenerative disorders.
Acknowledgements
This study was supported in part by a pilot grant from Indiana University School of Public Health.
Disclosure of conflict of interest
None.
References
- 1.Thomas B, Beal MF. Molecular insights into Parkinson’s disease. F1000 Med Rep. 2011;3:7. doi: 10.3410/M3-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dutheil F, Beaune P, Tzourio C, Loriot MA, Elbaz A. Interaction between ABCB1 and professional exposure to organochlorine insecticides in Parkinson disease. Arch Neurol. 2010;67:739–45. doi: 10.1001/archneurol.2010.101. [DOI] [PubMed] [Google Scholar]
- 3.Tanner CM, Kamel F, Ross GW, Hoppin JA, Goldman SM, Korell M, Marras C, Bhudhikanok GS, Kasten M, Chade AR, Comyns K, Richards MB, Meng C, Priestley B, Fernandez HH, Cambi F, Umbach DM, Blair A, Sandler DP, Langston JW. Rotenone, paraquat, and Parkinson’s disease. Environ Health Perspect. 2011;119:866–72. doi: 10.1289/ehp.1002839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang A, Costello S, Cockburn M, Zhang X, Bronstein J, Ritz B. Parkinson’s disease risk from ambient exposure to pesticides. Eur J Epidemiol. 2011;26:547–55. doi: 10.1007/s10654-011-9574-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weisskopf MG, Weuve J, Nie H, Saint-Hilaire MH, Sudarsky L, Simon DK, Hersh B, Schwartz J, Wright RO, Hu H. Association of cumulative lead exposure with Parkinson’s disease. Environ Health Perspect. 2010;118:1609–13. doi: 10.1289/ehp.1002339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Winklhofer KF, Haass C. Mitochondrial dysfunction in Parkinson’s disease. Biochim Biophys Acta. 2010;1802:29–44. doi: 10.1016/j.bbadis.2009.08.013. [DOI] [PubMed] [Google Scholar]
- 7.Dimitriadi M, Hart AC. Neurodegenerative disorders: insights from the nematode Caenorhabditis elegans. Neurobiol Dis. 2010;40:4–11. doi: 10.1016/j.nbd.2010.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meyer JN, Leung MC, Rooney JP, Sendoel A, Hengartner MO, Kisby GE, Bess AS. Mitochondria as a target of environmental toxicants. Toxicol Sci. 2013;134:1–17. doi: 10.1093/toxsci/kft102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wallace KB, Starkov AA. Mitochondrial targets of drug toxicity. Annu Rev Pharmacol Toxicol. 2000;40:353–88. doi: 10.1146/annurev.pharmtox.40.1.353. [DOI] [PubMed] [Google Scholar]
- 10.Wallace DC, Fan W, Procaccio V. Mitochondrial energetics and therapeutics. Annu Rev Pathol. 2010;5:297–348. doi: 10.1146/annurev.pathol.4.110807.092314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276:2045–7. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 12.Bonifati V, Rizzu P, van Baren MJ, Schaap O, Breedveld GJ, Krieger E, Dekker MC, Squitieri F, Ibanez P, Joosse M, van Dongen JW, Vanacore N, van Swieten JC, Brice A, Meco G, van Duijn CM, Oostra BA, Heutink P. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science. 2003;299:256–9. doi: 10.1126/science.1077209. [DOI] [PubMed] [Google Scholar]
- 13.Zimprich A, Muller-Myhsok B, Farrer M, Leitner P, Sharma M, Hulihan M, Lockhart P, Strongosky A, Kachergus J, Calne DB, Stoessl J, Uitti RJ, Pfeiffer RF, Trenkwalder C, Homann N, Ott E, Wenzel K, Asmus F, Hardy J, Wszolek Z, Gasser T. The PARK8 locus in autosomal dominant parkinsonism: confirmation of linkage and further delineation of the disease-containing interval. Am J Hum Genet. 2004;74:11–9. doi: 10.1086/380647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zheng B, Liao Z, Locascio JJ, Lesniak KA, Roderick SS, Watt ML, Eklund AC, Zhang-James Y, Kim PD, Hauser MA, Grünblatt E, Moran LB, Mandel SA, Riederer P, Miller RM, Federoff HJ, Wüllner U, Papapetropoulos S, Youdim MB, Cantuti-Castelvetri I, Young AB, Vance JM, Davis RL, Hedreen JC, Adler CH, Beach TG, Graeber MB, Middleton FA, Rochet JC, Scherzer CR Global PD Gene Expression (GPEX) Consortium. PGC-1alpha, a potential therapeutic target for early intervention in Parkinson’s disease. Sci Transl Med. 2010;2:52ra73. doi: 10.1126/scitranslmed.3001059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Okimoto R, Macfarlane JL, Clary DO, Wolstenholme DR. The mitochondrial genomes of two nematodes, Caenorhabditis elegans and Ascaris suum. Genetics. 1992;130:471–98. doi: 10.1093/genetics/130.3.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leung MC, Williams PL, Benedetto A, Au C, Helmcke KJ, Aschner M, Meyer JN. Caenorhabditis elegans: an emerging model in biomedical and environmental toxicology. Toxicol Sci. 2008;106:5–28. doi: 10.1093/toxsci/kfn121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sherer TB, Betarbet R, Testa CM, Seo BB, Richardson JR, Kim JH, Miller GW, Yagi T, Matsuno-Yagi A, Greenamyre JT. Mechanism of toxicity in rotenone models of Parkinson’s disease. J Neurosci. 2003;23:10756–64. doi: 10.1523/JNEUROSCI.23-34-10756.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Settivari R, Levora J, Nass R. The divalent metal transporter homologues SMF-1/2 mediate dopamine neuron sensitivity in caenorhabditis elegans models of manganism and parkinson disease. J Biol Chem. 2009;284:35758–68. doi: 10.1074/jbc.M109.051409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hope I. C. elegans: A Practical Approach. New York: Oxford University Press; 1999. [Google Scholar]
- 21.Jiang GC, Tidwell K, McLaughlin BA, Cai J, Gupta RC, Milatovic D, Nass R, Aschner M. Neurotoxic potential of depleted uranium effects in primary cortical neuron cultures and in Caenorhabditis elegans. Toxicol Sci. 2007;99:553–65. doi: 10.1093/toxsci/kfm171. [DOI] [PubMed] [Google Scholar]
- 22.Nass R, Hall DH, Miller DM 3rd, Blakely RD. Neurotoxin-induced degeneration of dopamine neurons in Caenorhabditis elegans. Proc Natl Acad Sci U S A. 2002;99:3264–9. doi: 10.1073/pnas.042497999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Novillo A, Won SJ, Li C, Callard IP. Changes in Nuclear Receptor and Vitellogenin Gene Expression in Response to Steroids and Heavy Metal in Caenorhabditis elegans. Integr Comp Biol. 2005;45:61–71. doi: 10.1093/icb/45.1.61. [DOI] [PubMed] [Google Scholar]
- 24.Hunter SE, Jung D, Di Giulio RT, Meyer JN. The QPCR assay for analysis of mitochondrial DNA damage, repair, and relative copy number. Methods. 2010;51:444–51. doi: 10.1016/j.ymeth.2010.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Caboni P, Sherer TB, Zhang N, Taylor G, Na HM, Greenamyre JT, Casida JE. Rotenone, deguelin, their metabolites, and the rat model of Parkinson’s disease. Chem Res Toxicol. 2004;17:1540–8. doi: 10.1021/tx049867r. [DOI] [PubMed] [Google Scholar]
- 26.Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat Neurosci. 2000;3:1301–6. doi: 10.1038/81834. [DOI] [PubMed] [Google Scholar]
- 27.Dhillon AS, Tarbutton GL, Levin JL, Plotkin GM, Lowry LK, Nalbone JT, Shepherd S. Pesticide/environmental exposures and Parkinson’s disease in East Texas. J Agromedicine. 2008;13:37–48. doi: 10.1080/10599240801986215. [DOI] [PubMed] [Google Scholar]
- 28.Betarbet R, Poisik O, Sherer TB, Greenamyre JT. Differential expression and ser897 phosphorylation of striatal N-methyl-d-aspartate receptor subunit NR1 in animal models of Parkinson’s disease. Exp Neurol. 2004;187:76–85. doi: 10.1016/j.expneurol.2003.12.012. [DOI] [PubMed] [Google Scholar]
- 29.Choi WS, Kruse SE, Palmiter RD, Xia Z. Mitochondrial complex I inhibition is not required for dopaminergic neuron death induced by rotenone, MPP+, or paraquat. Proc Natl Acad Sci U S A. 2008;105:15136–41. doi: 10.1073/pnas.0807581105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Girish C, Muralidhara Propensity of Selaginella delicatula aqueous extract to offset rotenone-induced oxidative dysfunctions and neurotoxicity in Drosophila melanogaster: Implications for Parkinson’s disease. Neurotoxicology. 2012;33:444–56. doi: 10.1016/j.neuro.2012.04.002. [DOI] [PubMed] [Google Scholar]
- 31.Parameshwaran K, Irwin MH, Steliou K, Pinkert CA. Protection by an antioxidant of rotenone-induced neuromotor decline, reactive oxygen species generation and cellular stress in mouse brain. Pharmacol Biochem Behav. 2012;101:487–92. doi: 10.1016/j.pbb.2012.02.011. [DOI] [PubMed] [Google Scholar]
- 32.Koopman WJ, Nijtmans LG, Dieteren CE, Roestenberg P, Valsecchi F, Smeitink JA, Willems PH. Mammalian mitochondrial complex I: biogenesis, regulation, and reactive oxygen species generation. Antioxid Redox Signal. 2010;12:1431–70. doi: 10.1089/ars.2009.2743. [DOI] [PubMed] [Google Scholar]
- 33.Bess AS, Crocker TL, Ryde IT, Meyer JN. Mitochondrial dynamics and autophagy aid in removal of persistent mitochondrial DNA damage in Caenorhabditis elegans. Nucleic Acids Res. 2012;40:7916–31. doi: 10.1093/nar/gks532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Leung MC, Rooney JP, Ryde IT, Bernal AJ, Bess AS, Crocker TL, Ji AQ, Meyer JN. Effects of early life exposure to ultraviolet C radiation on mitochondrial DNA content, transcription, ATP production, and oxygen consumption in developing Caenorhabditis elegans. BMC Pharmacol Toxicol. 2013;14:9. doi: 10.1186/2050-6511-14-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.VanDuyn N, Settivari R, LeVora J, Zhou S, Unrine J, Nass R. The metal transporter SMF-3/DMT-1 mediates aluminum-induced dopamine neuron degeneration. J Neurochem. 2013;124:147–57. doi: 10.1111/jnc.12072. [DOI] [PMC free article] [PubMed] [Google Scholar]