

Abstract
Epigenetics refers to a heritable change in the pattern of gene expression that is mediated by a mechanism specifically not due to alterations in the primary nucleotide sequence. Well known epigenetic mechanisms encompass DNA methylation, chromatin remodeling (histone modifications) and RNA interference. Functionally, epigenetics provides an extra layer of transcriptional control and plays a crucial role in normal physiological development, as well as in pathological conditions. Aberrant DNA methylation is implicated in immune dysfunction, inflammation and insulin resistance. Epigenetic changes may be responsible for “metabolic memory” and development of micro- and macrovascular complications of diabetes. MicroRNAs are critical in the maintenance of glomerular homeostasis and hence RNA interference may be important in the progression of renal disease. Recent studies have shown that epigenetic modifications orchestrate the epithelial-mesenchymal transition and eventually fibrosis of the renal tissue. Oxidative stress, inflammation, hyperhomocysteinemia and uremic toxins could induce epimutations in chronic kidney disease. Epigenetic alterations are associated with inflammation and cardiovascular disease in patients with chronic kidney disease. Reversible nature of the epigenetic changes gives an unique opportunity to halt or even reverse the disease process through targeted therapeutic strategies.
The nucleus has to take care of the inheritance of the heritable characters, while the surrounding cytoplasm is concerned with accommodation or adaptation to the environment.
Ernst Haeckel
The Human Genome Project has revealed thousands of genes and millions of sequence variations that might influence human health 1. Completion of the International HapMap Project and the recent advances in genotyping technologies has led to a surge in genome wide association studies (GWAS) 2. These studies have certainly broadened our understanding of the genetic basis of many complex traits 2;3 often implicating previously unsuspected biological pathways. However, for most common traits studied, known gene polymorphisms explain only a fraction of associated risk, suggesting that sequence variations in the human genome are only part of the puzzle leading to the evolution of the nascent field of epigenetics. The term ‘epigenetics’ was first used by Conrad Waddington to explain the interactions of genes with their environment 4, and is increasingly being examined as one of the important determinants of complex human diseases 5;6. This review provides an overview of recent advances in the field of epigenetics with specific reference to chronic kidney disease (CKD).
The New Frontier of Genetics-Epigenetics
Epigenetics refers to a heritable change in the pattern of gene expression that is mediated by mechanisms other than alterations in the primary nucleotide sequence of genes 7. (Table 1) Epigenetic changes regulate gene expression, silence the activity of transposable elements and determine gene dosage in the case of chromosome X inactivation and genomic imprinting7. Epigenetic mechanisms such as DNA methylation, histone acetylation/deacetylation, histone methylation and RNA interference are dynamic processes and regulate gene expression patterns in normal and diseased state (Figure 1). However, the inclusion of the requirement for heritable transmission of patterns of gene expression through cell division, suggests that these changes are only partially dynamic. A brief overview of these important epigenetic modifications is given below.
Table 1.
Definitions and description of terms
| Terms | Definitions |
|---|---|
| CpG Islands | The genomic regions that contain a high frequency of CpG sites (300 to 3000 bp) mostly in promoters of mammalian genes. |
| DNA methylation | Addition of methyl groups to DNA, to the 5-carbon of cytosine with specific effect of reducing gene expression. |
| DNA Hypermethylation | An increased level of DNA methylation acts as an alternative to mutations to disrupt the gene function and can predispose to genetic alterations through inactivating genes. |
| DNA Hypomethylation | A decreased level of DNA methylation, which acts as an alternative to mutations to disrupt the gene function and can predispose to genetic alterations through inactivating genes involved in DNA repair. |
| Genomic Imprinting | Genomic imprinting is a distinct subset of epigenetic regulation in which the activity of a gene is reversibly modified depending on the sex of the parent that transmits it. |
| Histone | Histones are the primary protein components of chromatin. H2A, H2B, H3, and H4 form the core histones, which assemble to form the nucleosome. Each nucleosome winds around 146 base pairs of DNA. The linker histone H1 locks the DNA into place forming higher-order nucleosome structures. |
| Histone modification | Histones are subject to post-translational modifications at their N-terminal tails by enzymatic processes such as: methylation, citrullination, acetylation, phosphorylation, sumoylation, ubiquitination, and ADP-ribosylation. Modification of these proteins control chromatin states, which can be open (transcriptionally active) or closed (inactive). |
| microRNA | These may originate as single-stranded RNA molecules which form hairpin duplexes cleaved into small dimers of 21–23 nucleotides in length, which regulate gene expression by cleaving target transcripts, or arresting translation. |
| ncRNA | A non-coding RNA (ncRNA) is a functional RNA molecule that is not translated into a protein. Non-coding RNA genes include highly abundant and functionally important RNAs such as transfer RNA (tRNA) and ribosomal RNA (rRNA), as well as RNAs such as microRNAs. |
| Transposable elements | A small mobile DNA element, often of retroviral origin, that moves within the genome, usually by copying itself to a second site but sometimes by splicing itself out of its original site and inserting in a new location. |
Figure 1. Methylation of the Cysteine.
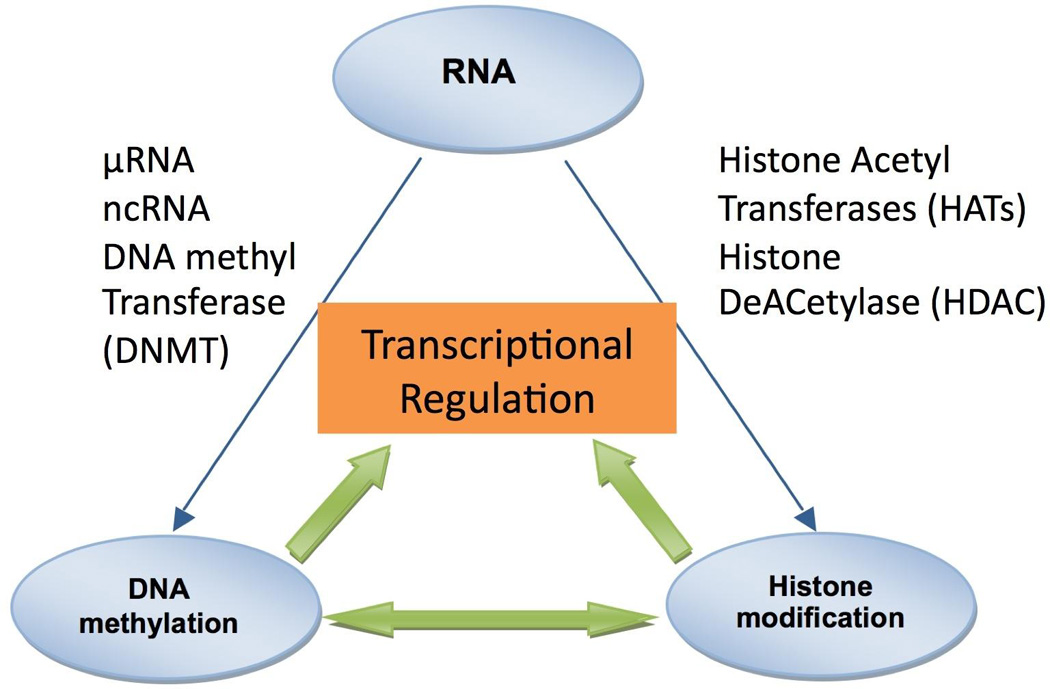
Epigenetic gene silencing refers to non-mutational gene inactivation that can be faithfully propagated from precursor cells to clones of daughter cells. Methylation of the C5 position of cytosine residues (CpG) in DNA has long been recognized as a fundamental epigenetic silencing mechanism. CpG methylation has recently been linked to an even more general mechanism of epigenetic silencing and histone modifications. Cells can inhibit the expression of individual genes by interfering with the mRNA being transcribed. Interaction between DNA methylation, histone modifications and RNA interference results in epigenetic silencing of gene. Micro RNA (µRNA) and non-coding RNA (ncRNA) play an important role in regulation of gene expression.
DNA Methylation
Many mammalian gene promoters are rich in CpG dinucleotude clusters known as CpG islands 8. Covalent addition of a methyl group to the 5 position of the nucleotide cytosine is common throughout the genome, but when added to CpG dinucleotides in promoter region CpG islands generally leads to loss of the associated gene expression. The mechanism of gene silencing by DNA methylation may be related to stearic hindrance of the transcriptional machinery, recruitment of repressors, or alteration in chromatin configuration 9 (Figure 2). Most recent studies have focused on this latter mechanism of gene repression. Several studies have shown that DNA hypermethylation plays an important role in inactivation of genes such as E-cadherin, p16, MDR1 and pi-class glutathione S-transferase10–13. On the other hand, DNA hypomethylation correlated with activation of several oncogenes, although direct mechanisms for altered expression have not been demonstrated. Rather, the association of hypomethylation leading to genomic instability due to generation of genomic mobile elements 14 or chromosomal break sites is supported by mechanistic evidence. Recent studies suggest that the induction and maintenance of DNA methylation are catalyzed by DNA methyltransferase-1 and DNA methyltransferase-3b and responsible for maintaining abnormal promoter methylation in diseased cells15. Furthermore, DNA methyltransferases interact directly with histone deacetylases to recruit them to gene promoters.
Figure 2. A: Epigenetic Modifications of Chromatin Structure.


Nucleosomes are formed and organized into chromatin as result of DNA strands wrapped around histone octamers. Modifications of histone occur at multiple sites due to methylation, acetylation and phosphorylation reactions. DNA methylation occurs at 5-position of cytosine residues (CpG sites) in a reaction catalyzed by DNA methyltransferases. Together, these modifications provide a unique epigenetic signature that regulates chromatin organization and gene expression.
B: Epigenetic Changes Associated with Disease States
In normal, healthy tissues, promoter regions of actively transcribed genes are without DNA methylation at CpG dinucleotides (open circles) within CpG islands, and histones are modified with predominantly active marks (Lysine 4 methylation, MeK4, and Acetylation of Lysine 9). The transcriptional start site is open and free of nucleosomes. This state is maintained by enzymes which modify the histone tails (histone acetyltransferases, HATs, and histone demethylases (HDemethylases). Intra and intergenic regions have predominately methylated CpGs and inactive histone modifications (Lysine 9 and Lysine 27 methylation). In diseased states, methylation of CpG sites within CpG islands is associated with repressive chromatin marks, with these changes resulting from the presence of histone deacetylaces (HDACs), histone methyltranserferases (H Mtases). Repressive chromatin marks lead to compaction of chromatin and nucleosome occupancy at the transcriptional start site, preventing gene transcription.
Chromatin Remodeling (Histone Modification)
Regulation of higher order chromatin structure is directly coupled with the expression of the genetic information and is recognized as an important factor in the functional integrity as well as origin and evolution of genes, genome, chromosomes and organism as a whole. Chromosomes are composed of euchromatin and heterochromatin domains. The latter generally is inaccessible to DNA binding factors and is transcriptionally silent. As depicted in Figure 2, gene expression is modulated by the accessibility of chromatin through the wrapping of the DNA around octamural globular histone proteins to form ‘nucleosomes’. Genes are activated if nucleosomes on chromatin are opened or otherwise inactivated if closed 16. The accessibility of chromatin is reversibly regulated by the epigenetic status of DNA methylation and histone modifications. Transcriptionally active regions of DNA, are highly unmethylated, rich in acetylated histones and accessible to transcription factors while the inactive regions are comprised of methylated DNA, deacetylated histones with compacted nucleosomes and unfavorable configuration to transcriptional machinery 17. This inactivation of DNA is linked to histone deacetylation through binding of methyl-CpG binding proteins to methylated promoter regions. Methylation of lysine 9 on the N-terminus of histone protein H-3 is also a characteristics of inactive DNA, while methylation of Lysine 4 on H-3 is a feature of activated DNA, and constitute some of the more well characterized components of the increasingly complicated “histone code” that regulates gene expression 18 . The heterochromatic state is epigenetically inherited and changes in heterochromatin state allow a transition from DNA sequence-specific genetic control to an adaptive sequence- independent dynamic epigenetic control 19.
RNA Interference
Fire and Mello, among others, discovered that double-stranded RNA could specifically silence the function of an endogenous gene 20. MicroRNAs are a class of endogenous, small, non-coding 21- or 22-nucleotide (nt) RNAs that have been implicated in the regulation of multiple biologic processes. The first evidence for gene disruption called RNA interference (RNAi) by double-stranded RNA (dsRNA) came from elegant experiments conducted on Caenorhabditis elegans 20. Thus, RNA interference and related RNA silencing pathways have emerged as new mechanisms for the regulation of the structure and activity of genes. Both RNA interference and microRNA decrease the level of the target protein within cells; RNA interference decreases the steady-state mRNA levels, whereas microRNA usually impairs the efficiency with which mRNA is translated into protein 21.
These short double-stranded RNAs can also cause a transcriptional silencing through methylation of homologous DNA promoter sequences 22 and/or formation of heterochromatin 23. Knockout of the microRNA-producing enzyme Dicer1 in mice is lethal, with Dicer1-null embryos depleted of stem cells 24. It appears that the patterns of microRNA expression are tightly regulated and play crucial roles in cell proliferation, apoptosis, and differentiation. Indeed, the links between RNA-silencing factors and inherited or acquired genetic disorders are increasingly being identified 25. For instance, miRNAs have been shown to regulate adipocyte differentiation 26, insulin secretion 27 and immune function28.
Emerging trends in analyzing epigenetic modifications
The capacity to analyze DNA methylation patterns of the entire genome will enable us to understand how DNA methylation influences chromatin function, and the role in normal development as well as disease state. The methods available to study the epigenetic changes have increased exponentially over the past decade. Detailed description of the methods is beyond the scope of this review and the reader is referred to these reviews29–31. An overview of the methodology is given below
The methods for detection of DNA methylation are based on one of the three primary ways to distinguish methylated cytosine from unmethylated cytosines: bisulfite conversion, digestion with methylation-sensitive restriction enzymes, and affinity purification of methylated DNA. Sodium bisulfite treatment selectively deaminates cytosine but not 5-methyl cytosine to uracil. The resulting sequence differences between a methylated and unmethylated cytosine can be determined by direct sequencing (specific restriction digestion32, nucleotide extension assays33, primer specific PCR34 or pyrosequencing35. Affinity purification takes advantage of the methyl-binding domain, which binds to methylated CpG sites36. This technique measures the density of methylation in a given region, and can be used for genome wide analyses when combined with genomic DNA arrays or with high-throughput sequencing. This analysis, however does not provide the base pair resolution of bisulfite based approaches.
Commercial oligonucleotide arrays are currently available for large-scale analysis of DNA methylation37. The most recent and exciting technique is the deep sequencing, which provides a quantitative measure of methylation abundance rather than relative measure of methylation abundance delivered by the array-based methods38;39.
Recruitment of methyl-CpG binding proteins (MeCP2) by DNA methylation silences gene expression partly by affecting the histone deacetylase (HDAC) activity and chromatin remodeling40 Immunoprecipitation (ChIP) using antibodies specific for a histone modifications and microarray analysis is performed to analyze the histone modifications such as H3 and H4 acetylation, H3 Lys 4 dimethylation (H3-di-meK4) and trimethylation (H3-tri-meK4) 41;42
Epigenetics, Developmental Biology and Stem cell Research
During development, cells undergo major epigenetic reprogramming 43. Because the DNA sequence of the human genome is the same in all cells in a given organism, the epigenome must vary from tissue to tissue, controlling the differential expression of genes and conferring specific identity to each cell type. De Bustos et al44 generated chromosome-wide methylation profiles of different tissues and noted very different methylation profiles from different organs, confirming the existence of tissue-specific epigenetic modification patterns across chromosome 1. Other investigators have shown that methylation profiles of the same tissue correlate better across individuals than do profiles of different tissues from the same individual 45;46.
Genomic parental imprinting is a process involving acquisition of DNA hypermethylation in one allele of a gene early on in the male and female germ line that leads to monoallelic expression47. Phenotypic plasticity determined by genetics is altered by epigenetic changes. Fraga et al48 demonstrated that although monozygotic twins were indistinguishable during the early years of life, approximately one-third of monozygotic twins display epigenetic differences in DNA methylation and patterns of histone modification with aging. This provides additional evidence that different phenotypes can originate from the same genotype through epigenetic drift which must result from external factors, including exposures.
The explosion of stem cell research has unveiled another area of importance for epigenetics. As for normal differentiation, the reprogramming of somatic cells cannot alter the primary DNA sequence, and thus must involve epigenetic preprogramming as cells progress through several stages before the full pluripotent state is attained. This involves distinct epigenetic changes including the reactivation of critical endogenous pluripotency-related genes, establishment of appropriate bivalent chromatin domains and DNA hypomethylation of genomic heterochromatic regions 49. The process of artificial somatic cell reprogramming by epigenetic approaches is highly relevant to research directed towards kidney regeneration50;51. Therefore, an understanding of epigenetics is vital to the advancement of stem cell therapies.
Genes, Environmental Interactions and Epigenetics
Environmental signals could modify the intracellular pathways that directly remodel the "epigenome" 52 (Figure 3). Imbalances in dietary nutrients could lead to hypomethylation and genetic instability 53. Methyl groups are acquired through the diet and donated to DNA through the folate and methionine pathways. Indeed, global 5-methyl cytosine content is influenced by nutritional availability of folate 54. Infections, especially viruses, are known to trigger DNA methylation 55. Loss of methylation and the resultant weakening of transcriptional repression can lead to re-expression of normally silenced genes, such as imprinted genes, and potentially harmful expression of inserted viral genes and repeat elements 56. Epidemiological studies have suggested a link between tobacco exposure and aberrant DNA methylation 57. Sensitivity to environmental exposure may vary depending on the underlying genetic variants that predispose to epigenetic changes 58, providing an explanation for the interactions between genotype and enviroment. For instance, polymorphism in the methylenetetrahydrofolate reductase gene is associated with altered DNA methylation in response to diet, alcohol consumption and disease manifestation59;60. Thus, DNA methylation patterns fluctuate in response to changes in diet, inherited genetic polymorphisms and exposure to environmental exposure.
Figure 3. Genetic-Epigenetic Phenotype.

Each cell has its own epigenetic signature which reflects genotype and environmental influence and is ultimately reflected in the phenotype of the cell and organism. Thus most genetic findings must be considered in an epigenetic and environmental context. The contribution from traditional genetics cannot be unequivocally realized until the complementary epigenetics and environmental changes are considered.
Epigenetic Basis for Human Diseases
Epigenetics plays a very important role in pathogenesis of a number of human diseases 5;61. Epigenetic-related diseases have the following characteristics: (a) a heritability that could not be fully explained by strict genetic inheritance patterns; (b) evidence of the influence of imprinting i.e. maternal diet or other in utero exposure influences the development of the disease in offspring well after adulthood and (c) an increase in prevalence with aging. An epigenetic model of complex disease has been proposed 19;61, which suggests that with progressive accumulation of epimutations over the life of the individual, a critical threshold is reached, beyond which the genome, cell, or tissue can no longer be able to function normally, leading to the disease phenotype. Recently, Feinberg colleagues proposed a proposed the common disease genetic and epigenetic model, which provides an epidemiologic framework that endeavors to integrate epigenetic changes with genetic variation in the context of age-related susceptibility to disease 62;63.
Is Epigenetics, the Epicenter of CKD phenotype?
The field of epigenetics of CKD is in its infancy. However, the plethora of metabolic alterations and co-existing inflammation CKD could incite diverse epigenetic changes. Emerging science indicates that epimutations may be involved in the initiation as well as progression of renal disease.
RNA interference and renal homeostasis
There is accumulating evidence that RNA interference is highly important for the development and progression of renal disease64. MicroRNAs are critical in the maintenance of glomerular homeostasis and RNA interference may be important in the progression of renal disease 65. When Dicer, an enzyme that generates microRNA was inactivated in mouse podocytes, the mice developed proteinuria and died subsequently from renal failure 66. The glomeruli demonstrated foot process effacement, podocyte apoptosis, mesangial expansion and glomerulosclerosis65. Similarly interruption of microRNA biogenesis in mouse podocytes resulted in proteinuria, podocyte dedifferentiation and crescent formation leading to end-stage kidney diseases 67;68.
Is renal fibrosis an epigenetic phenomenon?
Emerging data suggest that a strikingly large percentage of patients with acute kidney injury (AKI) do not completely recover renal function, and that this population contribute to the growing epidemiology of chronic kidney disease and end-stage renal disease (ESRD)69. Yang et al70 observed that in severe AKI there is a marked increase in the number of proximal tubule epithelial cells arresting in the G2/M phase of the cell cycle. These cells activate c-jun NH(2)-terminal kinase (JNK) signaling, which acts to up-regulate production of profibrotic cytokines such as transforming growth factor-β1 (TGF-β1) and connective tissue growth factor. Thus, cell cycle arrest converts normal epithelial cells to cells that promotes the activation of fibroblasts. Epithelial-mesenchymal transition is a process by which differentiated epithelial cells undergo phenotypic transition to the matrix producing fibroblasts and myofibroblasts71;72. Interestingly, Bechtel et al73 demonstrated that hypermethylation of RAS protein activator like-1 (RASAL1) is associated with the perpetuation of fibroblast activation and fibrogenesis in the kidney. RASALI encodes an inhibitor of the RAS oncoprotein, which is involved in cellular signal transduction regulating cell growth, differentiation and survival. RASAL1 hypermethylation is mediated by the methyltransferase Dnmt1 in renal fibrogenesis, and kidney fibrosis is ameliorated in Dnmt1(+/−) heterozygous mice. Thus, preliminary evidence suggests that epigenetic modifications orchestrates the cellular reprogramming leading to glomerular and interstitial fibrosis through transcriptional regulation74.
Homocysteinemia and epigenetics in CKD
Hyperhomocysteinemia with elevated S-adenosylhomocysteine levels has been reported in CKD and ESRD patients75. Analysis of a subpopulation of the Framingham Heart Study has identified the role of methenyltetrahydrofolate synthase gene in the progression of CKD76. The homocysteine precursor S-adenosylhomocysteine, a powerful competitive inhibitor of S-andenosyl methionine dependent methyltransferases is increased in various models of hyperhomocysteinemia including uremia 77, providing a mechanism for altered DNA methylation. Indeed, increased S-adenosylhomocysteine levels leading to DNA hypomethylation has also been reported in CKD patients with vascular disease 78. Thus, it is reasonable to suggest that epigenetic modifications may be an important risk factor for progression of renal disease as well as CVD in patients with CKD.
DNA methylation and CVD in ESRD
Progressive decline in renal function is associated with inflammation, augmented oxidative stress, accumulation of diverse toxins and deranged metabolism, all of which could result in altered epigenetic modifications 79 (Figure 4). While Nanayakkara et al 80 did not find any association between DNA methylation (as measured by 5-methylcytosine/total cytosine ratio in peripheral blood leukocyte) and renal function parameters or markers of atherosclerosis in early stages of CKD, Stenvinkel et al 78 reported global DNA hypermethylation (as defined by HpaII/Msp1 ratio using the Luminometric assay) in patients with and without CKD. The Cox regression model demonstrated that DNA hypermethylation associated with both all-cause and cardiovascular mortality. It is possible that the observed hypermethylation in CKD patients is due to interleukin (IL)-6 induced upregulation of the DNA methyltransferase gene expression81.
Figure 4. Chronic Kidney Disease Phenotype and Epigenetics.
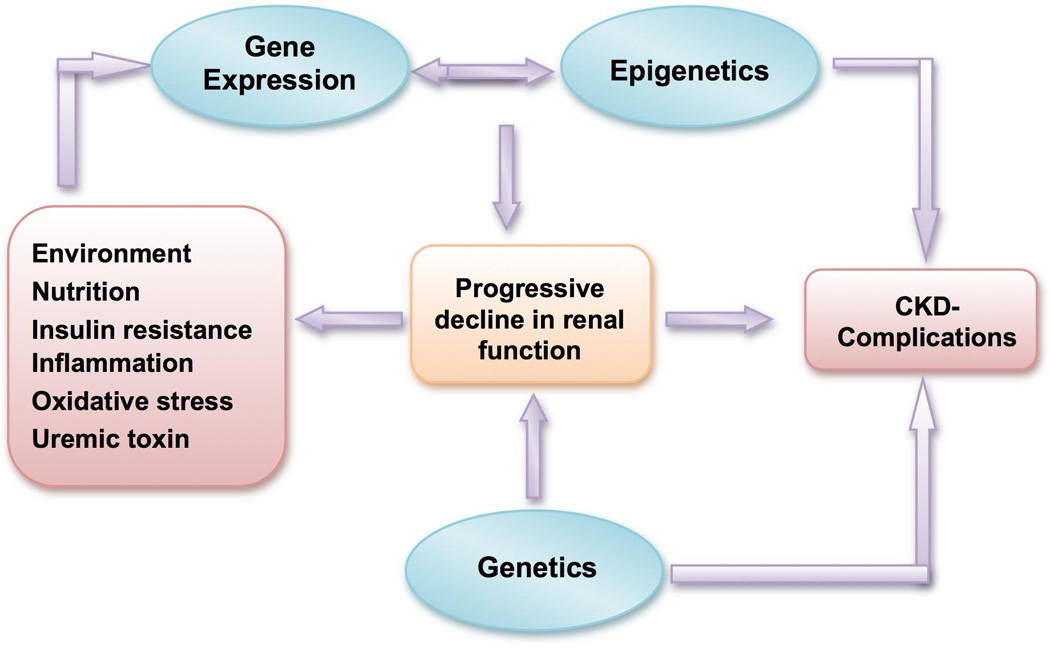
Progressive decline in renal function and its complications could be the result of genetic predisposition interacting with the epigenetic changes. Uremia induced adverse environmental factors may alter the epigenetic marks on the DNA or chromatin. A number of biochemical pathways and signaling cascades could be disturbed by aberrant methylation, which could contribute to the spectrum of abnormalities seen in patients with chronic kidney disease (CKD).
Epigenetic Regulation of Immune Response
Data from our laboratory has reported that patients with CKD have abnormal immune response and activation of pro-inflammatory cytokines 82. The activation of immune response is a highly coordinated multi-step process that involves epigenetic changes 83;84. Dynamic adaptations in both methylation and acetylation are essential to mount an immune response against a specific antigen 85. Lymphoid progenitor cells are committed to become either T or B cells and then to CD4 or CD8 and to T-helper (Th)1 or Th2 lineages through expression of CD4 and CD8 and the T cell receptor-CD3 complex. The orderly expression of transcription factors and other signaling molecules determines sequential gene expression that is regulated by epigenetic control factors 86;87. Impaired gene silencing may lead to a disorganized immune activation and excessive activation of cytokines 88. For instance, inactivation of suppressors of cytokine signaling (SOCS) gene by hypermethylation can induce aberrant inflammatory response through suppressors of cytokine signaling 89. It has also been suggested that IL-6 might modulate epigenetic changes in cells via regulation of DNA methyltransferase gene expression 81. Thus, chronic inflammation may be one of the mechanisms whereby reduced renal function affects DNA methylation in CKD.
Role of epigenetics in the metabolic memory in diabetes
Diabetic nephropathy is now the most common reported cause of ESRD in developed nations 90. Polymorphisms in genes involved in insulin secretion and response have been shown to be associated with diabetes. Interestingly, there is evidence that prenatal glucose and insulin levels influence the risk of developing type-2 diabetes mellitus later in life, independent of genetic predisposition 91. Several genes involved in glucose metabolism including facilitative glucose transporter-4 92 and uncoupling protein-2 93 exhibit differential DNA methylation in their promoters. Indeed, DNA methylation is likely to be involved in the propagation of insulin resistance in insulin target tissues94–97.
The Diabetes Control and Complications Trial and its follow-up study the Epidemiology of Diabetes Intervention reported a sustained effect of prior glucose control on subsequent clinical outcome, despite change in glycemic status98. This phenomenon of “metabolic memory” was first described in 1987 by Engerman99, who reported that the incidence of retinopathy in dogs that were switched to good glycemic control after 2.5 years of poor glucose control was similar to those animals that were exposed to poor glycemic control over the five years of the study. Although accumulation of advanced glycation end products has been touted as a potential mechanism for metabolic memory, epigenetic modifications is proposed as an alternative explanation100;101. El-Osta et al100 showed that transient exposure to hyperglycemia induced epigenetic changes in the promoter of the nuclear factor kappaB subunit p65 in aortic endothelial cells both in vitro and in nondiabetic mice leading to increased p65 gene expression. This was associated with mobilization of set7/9 protein, a histone methyltransferance that methylates the fourth lysine residue of the H3 histone tail.
Epigenetic Modifications and Atherosclerosis
CVD is the most important cause of death in patients with CKD 102. Newman proposed that aberrant DNA methylation may be involved in atherogenesis 103. Cells with abnormal DNA methylation assume a gene expression pattern that favors proliferation and dedifferentiation, leading to a transformed cellular phenotype. Interestingly, lipoproteins induce DNA hypermethylation in cultured endothelial cells 104. Early atherogenesis is associated with rearrangements of DNA methylation patterns involving both hypo- and hypermethylation. However, in advanced phases of the disease, cellular proliferation results in a predominant DNA hypomethylation 105. Hyperhomocysteinemia is associated with an increased risk of CVD 106. Indeed, circulating homocysteine levels were significantly correlated with the extent of DNA methyl-group loss in advanced atherosclerosis 107.
A marked reduction in the content of methylated CpG dinucleotides in extracellular superoxide dismutase gene is observed in atherosclerotic aortas 108. Estrogen receptor-α displays higher levels of DNA methylation in atheromas 109. Inhibition of monocarboxylate transport expression by DNA methylation has also been implicated in the development of atherosclerotic lesions 110. Epigenetic alterations in the chromatin structure have been shown to regulate myocardial gene expression. Genes known to be essential in maintaining homeostatic cardiac physiology are modulated by DNA methylation 111.
Mitochondrial transcription factor A (TFAM), which is essential for mtDNA transcription and replication. Studies in transgenic mice model show that overexpression of TFAM gene inhibit left ventricular remodeling after myocardial infarction112. The promoter of human TFAM contains CpG dinucleotides and is regulated by DNA methylation113. Investigators have shown that transgenic mice with deleted mt-TFAM allele developed dilated cardiomypathy 101,102. Thus, epigenetic alteration could be a key factor behind accelerated atherosclerosis and abnormal cardiac remodeling in patients with CKD.
The Promise of Epigenetics - Emerging Therapeutics Options
Because the gene itself is not mutated by methylation nor is the chromatin irreversibly changed epigenetic gene regulation is theoretically amenable to intervention. The epigenome normally displays more developmental and temporal variability, it is more susceptible to environmental influences. Thus, the importance of avoiding adverse environmental exposure and dietary modification in the management of epigenetic diseases could not be overemphasized. In an interventional study, Ingrosso et al 114 examined the effect of folate administration on DNA methylation in ESRD patients with hyperhomocysteinaemia. Preliminary findings from this study support that hyperhomocysteinaemia induced DNA hypomethylation could be reversed by administration of folate103.
A number of epigenetic drugs are in various stages of development, however, they lack target specificity 115. These nucleoside analogs and non-nucleoside analogues are known as DNA methylation inhibitors 115. Myelodysplastic syndrome and secondary acute myeloid leukemia that are resistant to standard chemotherapy have been treated with DNA methyltransferase inhibitors such as 5-azacytidine and decitabine with good prognosis 116. The histone deacytylase inhibitors comprise a large, diverse class of drugs that include short-chain fatty acids, hydroxamic acids, benzamides, and cyclic tetrapeptides.
All trans retinoic acid and 13-cis-retinoic acid induce hyperacetylation. Use of these medications is also being explored in the management of pauci-immune vasculitis. Theoretically, RNA interference (siRNA) might be used to treat any disease that is linked to over- expression of a specific gene. Thus, this technique might be relevant to genes involved in inflammation, proliferation and fibrosis. Indeed, nucleic acid based interventions are being explored for atherosclerosis and progressive renal diseases 117.
Perspective on Epigenetic Research in Nephrology
Despite the explosion in the science of epigenetics and emerging evidence that this phenomenon is important in the pathogenesis of diverse complex disease traits, it remains the domain of the oncologists. Although there is cautious foray in to the arena of epigenetics by other fields of medicine, such research is predominantly bench research with few pioneering publications exploring clinical significance of epigenetics in renal disease.
Efforts are underway to perform simultaneously gene expression studies performed together with epigenomics analysis in order to identify new diagnostic and prognostic markers for progressive renal disease. Lack of large scale epigenetic studies may be because of the uncertainty regarding the utility of traditional DNA source from peripheral blood mononuclear cells (PBMC) for the study of epigenetic epidemiology.
Researchers have detected the presence of methylated DNA in serum/plasma from patients with various types of malignancies118;119. It is presumed that circulating DNA is derived from DNA released from tumor cells from the site of disease activity120. It is reasonable to speculate that in systemic diseases such as diabetes, CKD and atherosclerosis DNA derived from PBMC could be used as a viable surrogate for localized organ specific diseases78;80;96;121. Study of longitudinal changes in epigenetic modifications could be highly relevant, especially when demonstrated to precede the change in cellular or clinical phenotype73;109. Demonstrating such change could indicate causality and potentially lead to targeted interventions.
Genetics of common diseases can be explained by interplay of genetic and epigenetic variation122. For instance, genetic variations could introduce or remove CpG sites, which are susceptible to methylation. Loss of imprinting of insulin-like growth factor −2 (IGF2) gene is noted in 10% of individuals with no clinical manifestation of Beckwith–Wiedeman syndrome, an imprinted growth disorder. Specific haplotypes within the IGF2 gene have been associated with loss of methylation at the locus in Beckwith–Wiedeman syndrome patients suggesting that epigenetic and genetic variation may act synergistically to influence a phenotype 123. Thus, future studies should consider the interaction between genotype and epigenotype in determining phenotype and therapeutic consequences.
Conclusion
Epigenetics has revolutionized the field of genetics leading to re-evaluation of the traditional concepts of heritability and genetics. Chromosomal instability as a result of DNA hypomethylation and silencing of genes due to hypermethylation has been reported in a variety of disorders beyond cancer. Inflammation and metabolic stress encountered in CKD could promote epigenetic changes leading to altered gene expression and abnormal cellular function. Advances in understanding of epigenetic modifications in the progression of chronic kidney diseases could be of great significance in predicting the pace of disease progression, developing targeted therapeutic strategies in preventing the progression of CKD, and providing an effective treatment of uremia-related complications.
Abbreviations
- 5-AC
5-azacytidine
- AKI
Acute kidney injury
- CKD
Chronic kidney disease
- CVD
Cardiovascular disease
- DNMT-1
DNA methyltransferase-1
- DNMT3b
DNA methyltransferase-3b
- ESRD
End stage renal disease
- GWAS
Genome wide association studies
- LV Disease
Left ventricular disease
- MeCP
Methyl-cytosine binding proteins
- MTHFR
Methylenetetrahydrofolate reductase
- MDR1
Multidrug resistance gene-1
- PBMC
Peripheral blood mononuclear cells
- RASAL1
RAS protein activator like-1
- SAH
S-adenosylhomocysteine
- SOCS
Suppressors of cytokine signaling
- TFAM
Mitochondrial transcription factor A
REFERENCES
- 1.Collins FS, Patrinos A, Jordan E, et al. New goals for the U.S. Human Genome Project 1998–2003. Science. 1998;282:682–689. doi: 10.1126/science.282.5389.682. [DOI] [PubMed] [Google Scholar]
- 2.Kruglyak L. The road to genome-wide association studies. Nat Rev Genet. 2008;9:314–318. doi: 10.1038/nrg2316. [DOI] [PubMed] [Google Scholar]
- 3.Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Waddington CH. The Strategy of the Genes. London: Geo Allen & Unwin; 1957. [Google Scholar]
- 5.Feinberg AP. Genome-scale approaches to the epigenetics of common human disease. Virchows Arch. 2010;456:13–21. doi: 10.1007/s00428-009-0847-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Egger G, Liang G, Aparicio A, Jones PA. Epigenetics in human disease and prospects for epigenetic therapy. Nature. 2004;429:457–463. doi: 10.1038/nature02625. [DOI] [PubMed] [Google Scholar]
- 7.Callinan PA, Feinberg AP. The emerging science of epigenomics. Hum Mol Genet. 2006;15(Spec No 1):R95–R101. doi: 10.1093/hmg/ddl095. [DOI] [PubMed] [Google Scholar]
- 8.Takai D, Jones PA. Comprehensive analysis of CpG islands in human chromosomes 21 and 22. Proc Natl Acad Sci U S A. 2002;99:3740–3745. doi: 10.1073/pnas.052410099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bird AP, Wolffe AP. Methylation-induced repression--belts, braces, and chromatin. Cell. 1999;99:451–454. doi: 10.1016/s0092-8674(00)81532-9. [DOI] [PubMed] [Google Scholar]
- 10.Graff JR, Herman JG, Lapidus RG, et al. E-cadherin expression is silenced by DNA hypermethylation in human breast and prostate carcinomas. Cancer Res. 1995;55:5195–5199. [PubMed] [Google Scholar]
- 11.Jarrard DF, Bova GS, Ewing CM, et al. Deletional, mutational, and methylation analyses of CDKN2 (p16/MTS1) in primary and metastatic prostate cancer. Genes Chromosomes Cancer. 1997;19:90–96. [PubMed] [Google Scholar]
- 12.Dwivedi RS, Wang LJ, Mirkin BL. S-adenosylmethionine synthetase is overexpressed in murine neuroblastoma cells resistant to nucleoside analogue inhibitors of S-adenosylhomocysteine hydrolase: a novel mechanism of drug resistance. Cancer Res. 1999;59:1852–1856. [PubMed] [Google Scholar]
- 13.Qiu YY, Mirkin BL, Dwivedi RS. MDR1 hypermethylation contributes to the progression of neuroblastoma. Mol Cell Biochem. 2007;301:131–135. doi: 10.1007/s11010-006-9404-3. [DOI] [PubMed] [Google Scholar]
- 14.Wolffe AP, Matzke MA. Epigenetics: regulation through repression. Science. 1999;286:481–486. doi: 10.1126/science.286.5439.481. [DOI] [PubMed] [Google Scholar]
- 15.Herman JG, Baylin SB. Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med. 2003;349:2042–2054. doi: 10.1056/NEJMra023075. [DOI] [PubMed] [Google Scholar]
- 16.Rountree MR, Bachman KE, Herman JG, Baylin SB. DNA methylation, chromatin inheritance, and cancer. Oncogene. 2001;20:3156–3165. doi: 10.1038/sj.onc.1204339. [DOI] [PubMed] [Google Scholar]
- 17.Peterson CL, Laniel MA. Histones and histone modifications. Curr Biol. 2004;14:R546–R551. doi: 10.1016/j.cub.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 18.Jenuwein T. Re-SET-ting heterochromatin by histone methyltransferases. Trends Cell Biol. 2001;11:266–273. doi: 10.1016/s0962-8924(01)02001-3. [DOI] [PubMed] [Google Scholar]
- 19.Jiang YH, Bressler J, Beaudet AL. Epigenetics and human disease. Annu Rev Genomics Hum Genet. 2004;5:479–510. doi: 10.1146/annurev.genom.5.061903.180014. [DOI] [PubMed] [Google Scholar]
- 20.Fire A, Xu S, Montgomery MK, et al. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–811. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- 21.Filipowicz W, Bhattacharyya SN, Sonenberg N. Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nat Rev Genet. 2008;9:102–114. doi: 10.1038/nrg2290. [DOI] [PubMed] [Google Scholar]
- 22.Suzuki M, Sunaga N, Shames DS, et al. RNA interference-mediated knockdown of DNA methyltransferase 1 leads to promoter demethylation and gene re-expression in human lung and breast cancer cells. Cancer Res. 2004;64:3137–3143. doi: 10.1158/0008-5472.can-03-3046. [DOI] [PubMed] [Google Scholar]
- 23.Lippman Z, Martienssen R. The role of RNA interference in heterochromatic silencing. Nature. 2004;431:364–370. doi: 10.1038/nature02875. [DOI] [PubMed] [Google Scholar]
- 24.Bernstein E, Kim SY, Carmell MA, et al. Dicer is essential for mouse development. Nat Genet. 2003;35:215–217. doi: 10.1038/ng1253. [DOI] [PubMed] [Google Scholar]
- 25.Meister G, Tuschl T. Mechanisms of gene silencing by double-stranded RNA. Nature. 2004;431:343–349. doi: 10.1038/nature02873. [DOI] [PubMed] [Google Scholar]
- 26.Esau C, Kang X, Peralta E, et al. MicroRNA-143 regulates adipocyte differentiation. J Biol Chem. 2004;279:52361–52365. doi: 10.1074/jbc.C400438200. [DOI] [PubMed] [Google Scholar]
- 27.Poy MN, Eliasson L, Krutzfeldt J, et al. A pancreatic islet-specific microRNA regulates insulin secretion. Nature. 2004;432:226–230. doi: 10.1038/nature03076. [DOI] [PubMed] [Google Scholar]
- 28.Chen CZ, Li L, Lodish HF, Bartel DP. MicroRNAs modulate hematopoietic lineage differentiation. Science. 2004;303:83–86. doi: 10.1126/science.1091903. [DOI] [PubMed] [Google Scholar]
- 29.Zilberman D, Henikoff S. Genome-wide analysis of DNA methylation patterns. Development. 2007;134:3959–3965. doi: 10.1242/dev.001131. [DOI] [PubMed] [Google Scholar]
- 30.Murrell A, Rakyan VK, Beck S. From genome to epigenome. Hum Mol Genet. 2005;14(Spec No 1):R3–R10. doi: 10.1093/hmg/ddi110. [DOI] [PubMed] [Google Scholar]
- 31.Laird PW. The power and the promise of DNA methylation markers. Nat Rev Cancer. 2003;3:253–266. doi: 10.1038/nrc1045. [DOI] [PubMed] [Google Scholar]
- 32.Xiong Z, Laird PW. COBRA: a sensitive and quantitative DNA methylation assay. Nucleic Acids Res. 1997;25:2532–2534. doi: 10.1093/nar/25.12.2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gonzalgo ML, Jones PA. Rapid quantitation of methylation differences at specific sites using methylation-sensitive single nucleotide primer extension (Ms-SNuPE) Nucleic Acids Res. 1997;25:2529–2531. doi: 10.1093/nar/25.12.2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Herman JG, Graff JR, Myohanen S, et al. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci U S A. 1996;93:9821–9826. doi: 10.1073/pnas.93.18.9821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Uhlmann K, Brinckmann A, Toliat MR, et al. Evaluation of a potential epigenetic biomarker by quantitative methyl-single nucleotide polymorphism analysis. Electrophoresis. 2002;23:4072–4079. doi: 10.1002/elps.200290023. [DOI] [PubMed] [Google Scholar]
- 36.Cross SH, Charlton JA, Nan X, Bird AP. Purification of CpG islands using a methylated DNA binding column. Nat Genet. 1994;6:236–244. doi: 10.1038/ng0394-236. [DOI] [PubMed] [Google Scholar]
- 37.Rauch T, Li H, Wu X, Pfeifer GP. MIRA-assisted microarray analysis, a new technology for the determination of DNA methylation patterns, identifies frequent methylation of homeodomain-containing genes in lung cancer cells. Cancer Res. 2006;66:7939–7947. doi: 10.1158/0008-5472.CAN-06-1888. [DOI] [PubMed] [Google Scholar]
- 38.Bentley DR. Whole-genome resequencing. Curr Opin Genet Dev. 2006;16:545–552. doi: 10.1016/j.gde.2006.10.009. [DOI] [PubMed] [Google Scholar]
- 39.Braslavsky I, Hebert B, Kartalov E, Quake SR. Sequence information can be obtained from single DNA molecules. Proc Natl Acad Sci U S A. 2003;100:3960–3964. doi: 10.1073/pnas.0230489100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fuks F, Hurd PJ, Wolf D, et al. The methyl-CpG-binding protein MeCP2 links DNA methylation to histone methylation. J Biol Chem. 2003;278:4035–4040. doi: 10.1074/jbc.M210256200. [DOI] [PubMed] [Google Scholar]
- 41.Ren B, Robert F, Wyrick JJ, et al. Genome-wide location and function of DNA binding proteins. Science. 2000;290:2306–2309. doi: 10.1126/science.290.5500.2306. [DOI] [PubMed] [Google Scholar]
- 42.Schubeler D, MacAlpine DM, Scalzo D, et al. The histone modification pattern of active genes revealed through genome-wide chromatin analysis of a higher eukaryote. Genes Dev. 2004;18:1263–1271. doi: 10.1101/gad.1198204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Morgan HD, Santos F, Green K, et al. Epigenetic reprogramming in mammals. Hum Mol Genet. 2005;14(Spec No 1):R47–R58. doi: 10.1093/hmg/ddi114. [DOI] [PubMed] [Google Scholar]
- 44.De BC, Ramos E, Young JM, et al. Tissue-specific variation in DNA methylation levels along human chromosome 1. Epigenetics Chromatin. 2009;2:7. doi: 10.1186/1756-8935-2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ball MP, Li JB, Gao Y, et al. Targeted and genome-scale strategies reveal gene-body methylation signatures in human cells. Nat Biotechnol. 2009;27:361–368. doi: 10.1038/nbt.1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Eckhardt F, Lewin J, Cortese R, et al. DNA methylation profiling of human chromosomes 6, 20 and 22. Nat Genet. 2006;38:1378–1385. doi: 10.1038/ng1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Esteller M. Aberrant DNA methylation as a cancer-inducing mechanism. Annu Rev Pharmacol Toxicol. 2005;45:629–656. doi: 10.1146/annurev.pharmtox.45.120403.095832. [DOI] [PubMed] [Google Scholar]
- 48.Fraga MF, Ballestar E, Paz MF, et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci U S A. 2005;102:10604–10609. doi: 10.1073/pnas.0500398102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mikkelsen TS, Hanna J, Zhang X, et al. Dissecting direct reprogramming through integrative genomic analysis. Nature. 2008;454:49–55. doi: 10.1038/nature07056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Romano G. Artificial reprogramming of human somatic cells to generate pluripotent stem cells: a possible alternative to the controversial use of human embryonic stem cells. Drug News Perspect. 2008;21:440–445. doi: 10.1358/dnp.2008.21.8.1272126. [DOI] [PubMed] [Google Scholar]
- 51.Brodie JC, Humes HD. Stem cell approaches for the treatment of renal failure. Pharmacol Rev. 2005;57:299–313. doi: 10.1124/pr.57.3.3. [DOI] [PubMed] [Google Scholar]
- 52.Zhang TY, Meaney MJ. Epigenetics and the environmental regulation of the genome and its function. Annu Rev Psychol. 2010;61:439–443. doi: 10.1146/annurev.psych.60.110707.163625. [DOI] [PubMed] [Google Scholar]
- 53.Friso S, Choi SW. Gene-nutrient interactions in one-carbon metabolism. Curr Drug Metab. 2005;6:37–46. doi: 10.2174/1389200052997339. [DOI] [PubMed] [Google Scholar]
- 54.Rampersaud GC, Kauwell GP, Hutson AD, et al. Genomic DNA methylation decreases in response to moderate folate depletion in elderly women. Am J Clin Nutr. 2000;72:998–1003. doi: 10.1093/ajcn/72.4.998. [DOI] [PubMed] [Google Scholar]
- 55.Heller H, Kammer C, Wilgenbus P, Doerfler W. Chromosomal insertion of foreign (adenovirus type 12, plasmid, or bacteriophage lambda) DNA is associated with enhanced methylation of cellular DNA segments. Proc Natl Acad Sci U S A. 1995;92:5515–5519. doi: 10.1073/pnas.92.12.5515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Holliday R. Genomic imprinting and allelic exclusion. Dev Suppl. 1990:125–129. [PubMed] [Google Scholar]
- 57.Kim DH, Nelson HH, Wiencke JK, et al. p16(INK4a) and histology-specific methylation of CpG islands by exposure to tobacco smoke in non-small cell lung cancer. Cancer Res. 2001;61:3419–3424. [PubMed] [Google Scholar]
- 58.Sutherland JE, Costa M. Epigenetics and the environment. Ann N Y Acad Sci. 2003;983:151–160. doi: 10.1111/j.1749-6632.2003.tb05970.x. [DOI] [PubMed] [Google Scholar]
- 59.Feil R. Environmental and nutritional effects on the epigenetic regulation of genes. Mutat Res. 2006;600:46–57. doi: 10.1016/j.mrfmmm.2006.05.029. [DOI] [PubMed] [Google Scholar]
- 60.Fenech M. The role of folic acid and Vitamin B12 in genomic stability of human cells. Mutat Res. 2001;475:57–67. doi: 10.1016/s0027-5107(01)00079-3. [DOI] [PubMed] [Google Scholar]
- 61.Bjornsson HT, Fallin MD, Feinberg AP. An integrated epigenetic and genetic approach to common human disease. Trends Genet. 2004;20:350–358. doi: 10.1016/j.tig.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 62.Feinberg AP. Epigenetics at the epicenter of modern medicine. JAMA. 2008;299:1345–1350. doi: 10.1001/jama.299.11.1345. [DOI] [PubMed] [Google Scholar]
- 63.Bjornsson HT, Sigurdsson MI, Fallin MD, et al. Intra-individual change over time in DNA methylation with familial clustering. JAMA. 2008;299:2877–2883. doi: 10.1001/jama.299.24.2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Harvey SJ, Jarad G, Cunningham J, et al. Podocyte-specific deletion of dicer alters cytoskeletal dynamics and causes glomerular disease. J Am Soc Nephrol. 2008;19:2150–2158. doi: 10.1681/ASN.2008020233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ho JJ, Marsden PA. Dicer cuts the kidney. J Am Soc Nephrol. 2008;19:2043–2046. doi: 10.1681/ASN.2008090986. [DOI] [PubMed] [Google Scholar]
- 66.Shi S, Yu L, Chiu C, et al. Podocyte-selective deletion of dicer induces proteinuria and glomerulosclerosis. J Am Soc Nephrol. 2008;19:2159–2169. doi: 10.1681/ASN.2008030312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Harvey SJ, Jarad G, Cunningham J, et al. Podocyte-specific deletion of dicer alters cytoskeletal dynamics and causes glomerular disease. J Am Soc Nephrol. 2008;19:2150–2158. doi: 10.1681/ASN.2008020233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ho J, Ng KH, Rosen S, et al. Podocyte-specific loss of functional microRNAs leads to rapid glomerular and tubular injury. J Am Soc Nephrol. 2008;19:2069–2075. doi: 10.1681/ASN.2008020162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cerda J, Lameire N, Eggers P, et al. Epidemiology of acute kidney injury. Clin J Am Soc Nephrol. 2008;3:881–886. doi: 10.2215/CJN.04961107. [DOI] [PubMed] [Google Scholar]
- 70.Yang L, Besschetnova TY, Brooks CR, et al. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat Med. 2010;16:535–543. 1p. doi: 10.1038/nm.2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kalluri R, Neilson EG. Epithelial-mesenchymal transition and its implications for fibrosis. J Clin Invest. 2003;112:1776–1784. doi: 10.1172/JCI20530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rashid A, Issa JP. CpG island methylation in gastroenterologic neoplasia: a maturing field. Gastroenterology. 2004;127:1578–1588. doi: 10.1053/j.gastro.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 73.Bechtel W, McGoohan S, Zeisberg EM, et al. Methylation determines fibroblast activation and fibrogenesis in the kidney. Nat Med. 2010;16:544–550. doi: 10.1038/nm.2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Liu Y. New insights into epithelial-mesenchymal transition in kidney fibrosis. J Am Soc Nephrol. 2010;21:212–222. doi: 10.1681/ASN.2008121226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Perna AF, Ingrosso D, Satta E, et al. Homocysteine metabolism in renal failure. Curr Opin Clin Nutr Metab Care. 2004;7:53–57. doi: 10.1097/00075197-200401000-00010. [DOI] [PubMed] [Google Scholar]
- 76.Kottgen A, Kao WH, Hwang SJ, et al. Genome-wide association study for renal traits in the Framingham Heart and Atherosclerosis Risk in Communities Studies. BMC Med Genet. 2008;9:49. doi: 10.1186/1471-2350-9-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Perna AF, Ingrosso D, Zappia V, et al. Enzymatic methyl esterification of erythrocyte membrane proteins is impaired in chronic renal failure. Evidence for high levels of the natural inhibitor S-adenosylhomocysteine. J Clin Invest. 1993;91:2497–2503. doi: 10.1172/JCI116485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Stenvinkel P, Karimi M, Johansson S, et al. Impact of inflammation on epigenetic DNA methylation - a novel risk factor for cardiovascular disease? J Intern Med. 2007;261:488–499. doi: 10.1111/j.1365-2796.2007.01777.x. [DOI] [PubMed] [Google Scholar]
- 79.Rao M, Wong C, Kanetsky P, et al. Cytokine gene polymorphism and progression of renal and cardiovascular diseases. Kidney Int. 2007 doi: 10.1038/sj.ki.5002391. [DOI] [PubMed] [Google Scholar]
- 80.Nanayakkara PW, Kiefte-de Jong JC, Stehouwer CD, et al. Association between global leukocyte DNA methylation, renal function, carotid intima-media thickness and plasma homocysteine in patients with stage 2–4 chronic kidney disease. Nephrol Dial Transplant. 2008;23:2586–2592. doi: 10.1093/ndt/gfn040. [DOI] [PubMed] [Google Scholar]
- 81.Hodge DR, Xiao W, Clausen PA, et al. Interleukin-6 regulation of the human DNA methyltransferase (HDNMT) gene in human erythroleukemia cells. J Biol Chem. 2001;276:39508–39511. doi: 10.1074/jbc.C100343200. [DOI] [PubMed] [Google Scholar]
- 82.Raj DSC, Dominic EA, Pai A, et al. Skeletal muscle, cytokines and oxidative stress in End-stage renal disease. Kidney Int. 2005;68:2338–2344. doi: 10.1111/j.1523-1755.2005.00695.x. [DOI] [PubMed] [Google Scholar]
- 83.Fitzpatrick DR, Wilson CB. Methylation and demethylation in the regulation of genes, cells, and responses in the immune system. Clin Immunol. 2003;109:37–45. doi: 10.1016/s1521-6616(03)00205-5. [DOI] [PubMed] [Google Scholar]
- 84.Bergman Y, Cedar H. A stepwise epigenetic process controls immunoglobulin allelic exclusion. Nat Rev Immunol. 2004;4:753–761. doi: 10.1038/nri1458. [DOI] [PubMed] [Google Scholar]
- 85.Wilson CB, Makar KW, Perez-Melgosa M. Epigenetic regulation of T cell fate and function. J Infect Dis. 2002;185(Suppl 1):S37–S45. doi: 10.1086/338001. [DOI] [PubMed] [Google Scholar]
- 86.Boyer LA, Lee TI, Cole MF, et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell. 2005;122:947–956. doi: 10.1016/j.cell.2005.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Avni O, Lee D, Macian F, et al. T(H) cell differentiation is accompanied by dynamic changes in histone acetylation of cytokine genes. Nat Immunol. 2002;3:643–651. doi: 10.1038/ni808. [DOI] [PubMed] [Google Scholar]
- 88.Armenante F, Merola M, Furia A, Palmieri M. Repression of the IL-6 gene is associated with hypermethylation. Biochem Biophys Res Commun. 1999;258:644–647. doi: 10.1006/bbrc.1999.0566. [DOI] [PubMed] [Google Scholar]
- 89.He B, You L, Uematsu K, et al. SOCS-3 is frequently silenced by hypermethylation and suppresses cell growth in human lung cancer. Proc Natl Acad Sci U S A. 2003;100:14133–14138. doi: 10.1073/pnas.2232790100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Liu Y, Freedman BI. Genetics of progressive renal failure in diabetic kidney disease. Kidney Int Suppl. 2005:S94–S97. doi: 10.1111/j.1523-1755.2005.09917.x. [DOI] [PubMed] [Google Scholar]
- 91.Dabelea D, Hanson RL, Lindsay RS, et al. Intrauterine exposure to diabetes conveys risks for type 2 diabetes and obesity: a study of discordant sibships. Diabetes. 2000;49:2208–2211. doi: 10.2337/diabetes.49.12.2208. [DOI] [PubMed] [Google Scholar]
- 92.Yokomori N, Tawata M, Onaya T. DNA demethylation during the differentiation of 3T3-L1 cells affects the expression of the mouse GLUT4 gene. Diabetes. 1999;48:685–690. doi: 10.2337/diabetes.48.4.685. [DOI] [PubMed] [Google Scholar]
- 93.Carretero MV, Torres L, Latasa U, et al. Transformed but not normal hepatocytes express UCP2. FEBS Lett. 1998;439:55–58. doi: 10.1016/s0014-5793(98)01335-0. [DOI] [PubMed] [Google Scholar]
- 94.Goh KP, Sum CF. Connecting the Dots: Molecular and Epigenetic Mechanisms in Type 2 Diabetes. Curr Diabetes Rev. 2010 doi: 10.2174/157339910791658899. [DOI] [PubMed] [Google Scholar]
- 95.Ling C, Poulsen P, Simonsson S, et al. Genetic and epigenetic factors are associated with expression of respiratory chain component NDUFB6 in human skeletal muscle. J Clin Invest. 2007;117:3427–3435. doi: 10.1172/JCI30938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wren JD, Garner HR. Data-mining analysis suggests an epigenetic pathogenesis for type 2 diabetes. J Biomed Biotechnol. 2005;2005:104–112. doi: 10.1155/JBB.2005.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Jiang MH, Fei J, Lan MS, et al. Hypermethylation of hepatic Gck promoter in ageing rats contributes to diabetogenic potential. Diabetologia. 2008;51:1525–1533. doi: 10.1007/s00125-008-1034-8. [DOI] [PubMed] [Google Scholar]
- 98.Nathan DM, Cleary PA, Backlund JY, et al. Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N Engl J Med. 2005;353:2643–2653. doi: 10.1056/NEJMoa052187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Engerman RL, Kern TS. Progression of incipient diabetic retinopathy during good glycemic control. Diabetes. 1987;36:808–812. doi: 10.2337/diab.36.7.808. [DOI] [PubMed] [Google Scholar]
- 100.El-Osta A, Brasacchio D, Yao D, et al. Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia. J Exp Med. 2008;205:2409–2417. doi: 10.1084/jem.20081188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Brasacchio D, Okabe J, Tikellis C, et al. Hyperglycemia induces a dynamic cooperativity of histone methylase and demethylase enzymes associated with gene-activating epigenetic marks that coexist on the lysine tail. Diabetes. 2009;58:1229–1236. doi: 10.2337/db08-1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Culleton BF, Larson MG, Evans JC, et al. Prevalence and correlates of elevated serum creatinine levels: the Framingham Heart Study. Arch Intern Med. 1999;159:1785–1790. doi: 10.1001/archinte.159.15.1785. [DOI] [PubMed] [Google Scholar]
- 103.Newman PE. Can reduced folic acid and vitamin B12 levels cause deficient DNA methylation producing mutations which initiate atherosclerosis? Med Hypotheses. 1999;53:421–424. doi: 10.1054/mehy.1998.0794. [DOI] [PubMed] [Google Scholar]
- 104.Lund G, Andersson L, Lauria M, et al. DNA methylation polymorphisms precede any histological sign of atherosclerosis in mice lacking apolipoprotein E. J Biol Chem. 2004;279:29147–29154. doi: 10.1074/jbc.M403618200. [DOI] [PubMed] [Google Scholar]
- 105.Zaina S, Lindholm MW, Lund G. Nutrition and aberrant DNA methylation patterns in atherosclerosis: more than just hyperhomocysteinemia? J Nutr. 2005;135:5–8. doi: 10.1093/jn/135.1.5. [DOI] [PubMed] [Google Scholar]
- 106.Brattstrom L, Wilcken DE. Homocysteine and cardiovascular disease: cause or effect? Am J Clin Nutr. 2000;72:315–323. doi: 10.1093/ajcn/72.2.315. [DOI] [PubMed] [Google Scholar]
- 107.Castro R, Rivera I, Struys EA, et al. Increased homocysteine and S-adenosylhomocysteine concentrations and DNA hypomethylation in vascular disease. Clin Chem. 2003;49:1292–1296. doi: 10.1373/49.8.1292. [DOI] [PubMed] [Google Scholar]
- 108.Laukkanen MO, Mannermaa S, Hiltunen MO, et al. Local hypomethylation in atherosclerosis found in rabbit ec-sod gene. Arterioscler Thromb Vasc Biol. 1999;19:2171–2178. doi: 10.1161/01.atv.19.9.2171. [DOI] [PubMed] [Google Scholar]
- 109.Ying AK, Hassanain HH, Roos CM, et al. Methylation of the estrogen receptor-alpha gene promoter is selectively increased in proliferating human aortic smooth muscle cells. Cardiovasc Res. 2000;46:172–179. doi: 10.1016/s0008-6363(00)00004-3. [DOI] [PubMed] [Google Scholar]
- 110.Zhu S, Goldschmidt-Clermont PJ, Dong C. Inactivation of monocarboxylate transporter MCT3 by DNA methylation in atherosclerosis. Circulation. 2005;112:1353–1361. doi: 10.1161/CIRCULATIONAHA.104.519025. [DOI] [PubMed] [Google Scholar]
- 111.D'Cruz LG, Baboonian C, Phillimore HE, et al. Cytosine methylation confers instability on the cardiac troponin T gene in hypertrophic cardiomyopathy. J Med Genet. 2000;37:E18. doi: 10.1136/jmg.37.9.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ikeuchi M, Matsusaka H, Kang D, et al. Overexpression of mitochondrial transcription factor a ameliorates mitochondrial deficiencies and cardiac failure after myocardial infarction. Circulation. 2005;112:683–690. doi: 10.1161/CIRCULATIONAHA.104.524835. [DOI] [PubMed] [Google Scholar]
- 113.Choi YS, Kim S, Pak YK. Mitochondrial transcription factor A (mtTFA) and diabetes. Diabetes Res Clin Pract. 2001;54(Suppl 2):S3–S9. doi: 10.1016/s0168-8227(01)00330-8. [DOI] [PubMed] [Google Scholar]
- 114.Ingrosso D, Cimmino A, Perna AF, et al. Folate treatment and unbalanced methylation and changes of allelic expression induced by hyperhomocysteinaemia in patients with uraemia. Lancet. 2003;361:1693–1699. doi: 10.1016/S0140-6736(03)13372-7. [DOI] [PubMed] [Google Scholar]
- 115.Ptak C, Petronis A. Epigenetics and complex disease: from etiology to new therapeutics. Annu Rev Pharmacol Toxicol. 2008;48:257–276. doi: 10.1146/annurev.pharmtox.48.113006.094731. [DOI] [PubMed] [Google Scholar]
- 116.Griffiths EA, Gore SD. DNA methyltransferase and histone deacetylase inhibitors in the treatment of myelodysplastic syndromes. Semin Hematol. 2008;45:23–30. doi: 10.1053/j.seminhematol.2007.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Fukuda N. Development of gene therapies for cardiovascular and renal diseases by nucleic acid medicines. Med Chem. 2006;2:13–19. doi: 10.2174/157340606775197723. [DOI] [PubMed] [Google Scholar]
- 118.Esteller M, Sanchez-Cespedes M, Rosell R, et al. Detection of aberrant promoter hypermethylation of tumor suppressor genes in serum DNA from non-small cell lung cancer patients. Cancer Res. 1999;59:67–70. [PubMed] [Google Scholar]
- 119.Muller HM, Widschwendter A, Fiegl H, et al. DNA methylation in serum of breast cancer patients: an independent prognostic marker. Cancer Res. 2003;63:7641–7645. [PubMed] [Google Scholar]
- 120.Garcia-Olmo D, Garcia-Olmo DC. Functionality of circulating DNA: the hypothesis of genometastasis. Ann N Y Acad Sci. 2001;945:265–275. doi: 10.1111/j.1749-6632.2001.tb03895.x. [DOI] [PubMed] [Google Scholar]
- 121.Sharma P, Senthilkumar RD, Brahmachari V, et al. Mining literature for a comprehensive pathway analysis: a case study for retrieval of homocysteine related genes for genetic and epigenetic studies. Lipids Health Dis. 2006;5:1. doi: 10.1186/1476-511X-5-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ulrich CM, Curtin K, Samowitz W, et al. MTHFR variants reduce the risk of G:C->A:T transition mutations within the p53 tumor suppressor gene in colon tumors. J Nutr. 2005;135:2462–2467. doi: 10.1093/jn/135.10.2462. [DOI] [PubMed] [Google Scholar]
- 123.Murrell A, Heeson S, Cooper WN, et al. An association between variants in the IGF2 gene and Beckwith-Wiedemann syndrome: interaction between genotype and epigenotype. Hum Mol Genet. 2004;13:247–255. doi: 10.1093/hmg/ddh013. [DOI] [PubMed] [Google Scholar]