

Abstract
In Dahl salt-sensitive rats (Dahl SS), glomerular capillary pressure (PGC) increases in response to high salt intake and this is accompanied by significant glomerular injury compared to spontaneously hypertensive rats (SHR) with similar blood pressure. PGC is controlled mainly by afferent arteriolar (Af-Art) resistance, which is regulated by the vasoconstrictor tubuloglomerular feedback (TGF) and the vasodilator connecting tubule glomerular feedback (CTGF). We hypothesized that Dahl SS have a decreased TGF response and enhanced TGF resetting compared to SHR, and that these differences are due in part to an increase in CTGF. In vivo, using micropuncture we measured stop-flow pressure (PSF, a surrogate of PGC). TGF was calculated as the maximal decrease in PSF caused by increasing nephron perfusion, TGF resetting as the attenuation in TGF induced by high salt diet, and CTGF as the difference in TGF response before and during CTGF inhibition with benzamil. Compared to SHR, Dahl SS had 1) lower TGF responses in normal (6.6±0.1 vs. 11.0±0.2 mm Hg; P<0.001) and high-salt diets (3.3±0.1 vs. 10.1±0.3 mmHg; P<0.001), 2) greater TGF resetting (3.3±0.1 vs. 1.0±0.3 mmHg; P<0.001), and 3) greater CTGF (3.4±0.4 vs. 1.2±0.1 mmHg; P<0.001). We conclude that Dahl SS have lower TGF and greater CTGF than SHR, and that CTGF antagonizes TGF. Furthermore, CTGF is enhanced by a high-salt diet and contributes significantly to TGF resetting. Our findings may explain in part the increase in vasodilatation, PGC, and glomerular damage in salt-sensitive hypertension during high salt intake.
Keywords: Dahl salt-sensitive, Spontaneously Hypertensive Rats, CTGF, TGF, stop-flow pressure, benzamil, salt-resistant
Introduction
There is evidence that in hypertension, glomerular capillary pressure (PGC) greatly influences the progression of renal nephrosclerosis 1,2. In African-Americans with salt-sensitive hypertension, high salt intake causes an abnormal renal hemodynamic response and an increase in estimated PGC3. In Dahl salt-sensitive rats (Dahl SS), PGC increases in response to high salt intake and this is accompanied by significantly greater glomerular injury compared to spontaneously hypertensive rats (SHR) with similar blood pressure 4-6. PGC is controlled by both afferent (Af-Art) and efferent arteriolar resistance. Af-Art resistance is regulated by mechanisms similar to other arterioles, including sympathetic nerve activity, angiotensin II, nitric oxide, eicosanoids, and myogenic response. In addition, Af-Art resistance is also regulated by two intrinsic renal autoregulatory mechanisms, namely tubuloglomerular feedback (TGF) and connecting tubule glomerular feedback (CTGF). TGF is initiated by increases in NaCl in the macula densa and causes Af-Art constriction, while CTGF is initiated by increases in NaCl in the connecting tubule and causes Af-Art dilatation 7 (see Figure 1). CTGF is initiated by Na entry via the epithelial Na channel (ENaC) in the connecting tubule and is blocked by the ENaC inhibitor benzamil. CTGF is mediated by prostaglandin E2 and epoxyeicosatrienoic acids 8-11.
Figure 1. Schematic representation of TGF and CTGF.
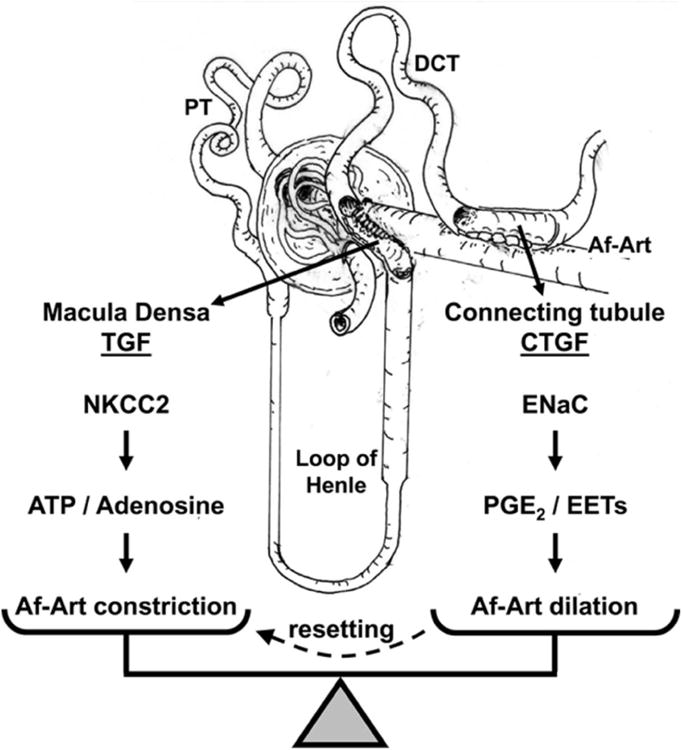
The macula densa triggers TGF when Na is reabsorbed via the Na/K/2Cl cotransporter type 2 (NKCC2), by releasing ATP which is broken down to adenosine, which in turn causes constriction of the Af-Art. The connecting tubule triggers CTGF when Na is reabsorbed via the epithelial sodium channel (ENaC), by releasing epoxyeicosatrienoic acids (EETs) and prostaglandin E2 (PGE2), which cause dilation of the Af-Art. PT indicates proximal tubule, and DCT distal convoluted tubule.
During high salt intake, if TGF were to remain unchanged, it would cause a decrease in PGC and glomerular filtration due to enhanced distal delivery of NaCl, and thus decrease the renal natriuretic response to high salt intake. However, this does not occur because TGF resets, so that a greater amount of NaCl is required to elicit the same vasoconstriction 12. In addition to high salt, TGF resetting occurs in response to physiological and pathophysiological conditions such as volume expansion, diabetes, and unilateral nephrectomy 13-16. The mechanisms that mediate TGF resetting are not completely understood.
In vivo NaCl in the lumen of the distal nephron regulates Af-Art resistance via the combined effect of TGF and CTGF 10. Thus, the observation that TGF is attenuated or reset in certain conditions could reflect an increase in CTGF that counteracts TGF. In fact, we have recently reported that CTGF partly mediates acute TGF resetting induced by sustained perfusion of single nephrons at the high end of the physiological tubular flow range 17.
The roles of TGF resetting and CTGF in hypertensive rats have not been well characterized. Here we studied for the first time the role of CTGF in the regulation of PGC and salt-induced TGF resetting in Dahl SS and SHR. We hypothesized that Dahl SS have a decreased TGF response and enhanced TGF resetting compared to SHR, and that these differences are due in part to an increase in CTGF. To test this hypothesis, we used Dahl SS, Dahl salt resistant rats (Dahl SR), SHR and Wistar Kyoto rats (WKY) fed a normal or high-salt diet and performed micropuncture of individual nephrons to measure stop-flow pressure (PSF, a surrogate of PGC). TGF was calculated as the decrease in PSF caused by increasing nephron perfusion.
Methods
Male Dahl SS, Dahl SR, SHR, and WKY weighing 307.9 ± 2.6 g were fed either a normal (0.23% NaCl) or high salt diet (4% NaCl) for two weeks. In micropuncture experiments in vivo, two consecutive stop-flow pressure (PSF) curves were performed. In half of the experiments, the ENaC blocker benzamil was added during the second PSF curve to inhibit CTGF. TGF was calculated as the maximal decrease in PSF caused by increasing nephron perfusion, TGF resetting as the attenuation in TGF induced by high salt diet, and CTGF as the difference in TGF response before and during CTGF inhibition with benzamil. An expanded Methods section is available online at http://hyper.ahajournals.org.
Results
1) Dahl SS and Dahl SR fed normal salt diet: TGF response and role of CTGF
Dahl SS (black circles) had an attenuated TGF response compared to Dahl SR (white circles). These differences reached statistical significance when the tubules were perfused at a rate of 20 nL/min or greater (Figure 2). In rats fed a normal salt diet, blocking CTGF potentiated the TGF response in both Dahl SR and Dahl SS (Figures 3A and 3B). Although this potentiation was somewhat greater in Dahl SS, it was not of statistical significance (Figure 3C). In time control experiments we confirmed that two consecutive TGF responses were reproducible in Dahl SR and Dahl SS with no time effect (see supplemental figures S1A and S1B).
Figure 2. TGF response in Dahl SR and Dahl SS fed normal salt diet.
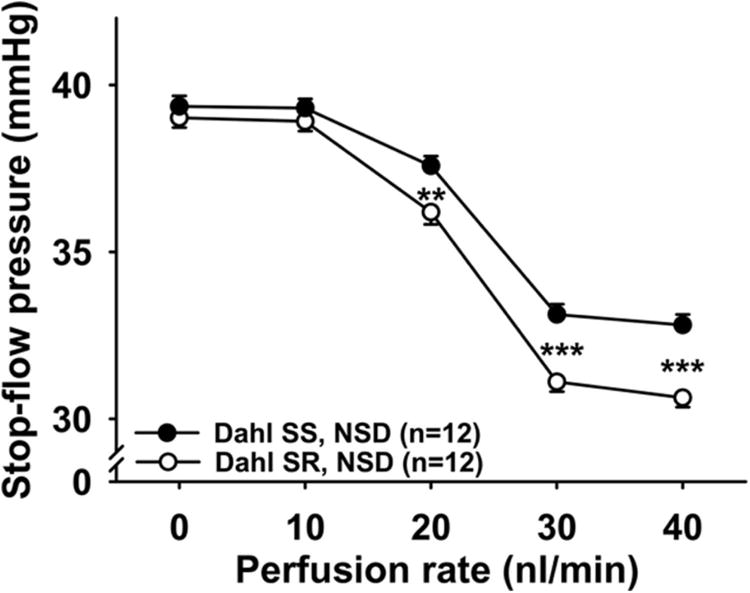
TGF induced by increased perfusion rates in the late proximal tubule in Dahl SR (○) and Dahl SS (●) on a normal salt diet (NSD). When the tubules were perfused at 20, 30, and 40 nL/min, TGF was significantly attenuated in Dahl SS. ** P < 0.01, *** P < 0.001, Dahl SR vs. Dahl SS.
Figure 3. Role of CTGF in TGF response in Dahl SR and Dahl SS fed normal salt diet.
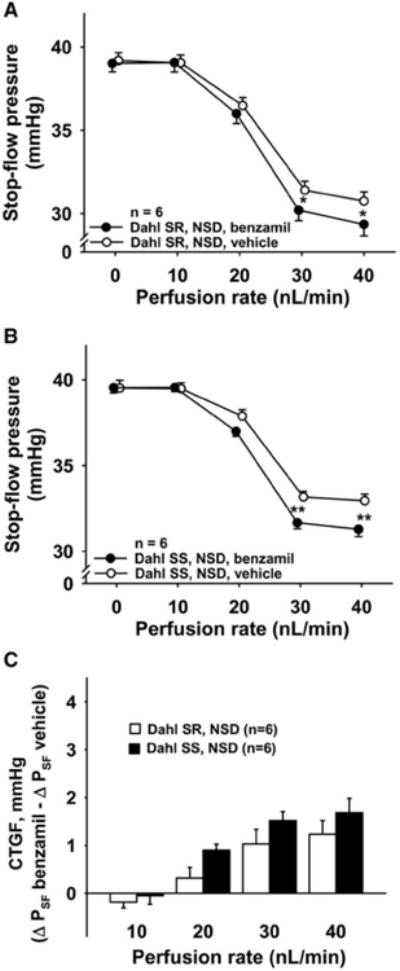
In rats fed a normal salt diet (NSD), two TGF responses were elicited, first in the presence of CTGF (○, vehicle), then during inhibition of CTGF (●, benzamil 1μM). Inhibition of CTGF potentiated the TGF response in both Dahl SR (Panel A) and Dahl SS (Panel B). Panel C: Both Dahl SR (open bars) and Dahl SS (closed bars) showed CTGF, but there were no differences between strains. * P < 0.05, ** P < 0.01, vehicle vs. benzamil.
SHR and WKY fed normal salt diet: TGF response and role of CTGF
SHR (black circles) had a greater TGF response compared to WKY (white circles). These differences reached statistical significance when the tubules were perfused at a rate of 20 nL/min or greater (Figure 4A). These data were normalized to baseline PSF since basal pressure was significantly higher in the SHR (see absolute numbers in Figure 4B). Inhibition of CTGF with benzamil potentiated TGF response in WKY when the tubules were perfused at a rate of 30 nL/min or greater (Figure 5A). However, in SHR inhibition of CTGF did not potentiate the TGF response (Figure 5B), suggesting that SHR fed a normal salt diet have little or no CTGF. WKY tended to have greater CTGF than SHR (P < 0.01 for the overall ANOVA group comparison). When CTGF in SHR was compared to that of WKY at each individual flow rate, P values were < 0.05 at 30 and 40 nL/min, however, these differences did not reach statistical significance after adjustment for multiple comparisons (Figure 5C). In time control experiments we confirmed that two consecutive TGF responses were reproducible in WKY and SHR with no time effect (see supplemental figures S2A and S2B).
Figure 4. TGF response in WKY and SHR fed normal salt diet.
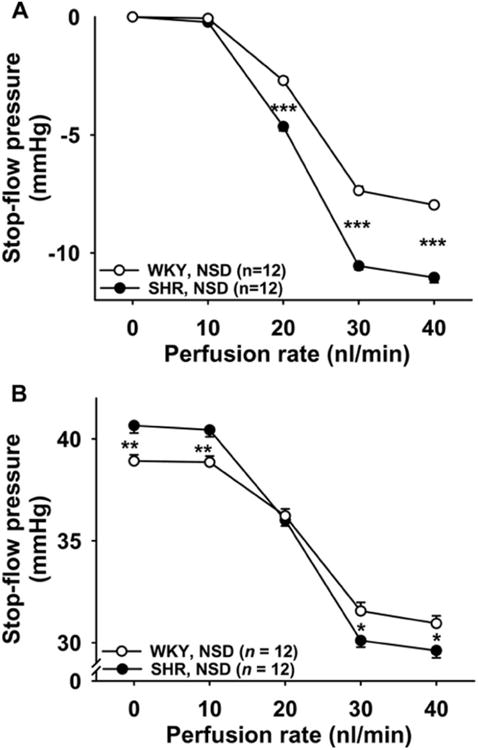
TGF induced by increased perfusion rates in the late proximal tubule in WKY (○) and SHR (●) on a normal salt diet (NSD). When the tubules were perfused at 20, 30, and 40 nL/min, TGF was significantly enhanced in SHR. * P < 0.05, ** P < 0.01, *** P < 0.001, WKY vs. SHR Data are presented as delta from baseline (Panel A) and absolute numbers (Panel B).
Figure 5. Role of CTGF in TGF response in WKY and SHR fed normal salt diet.
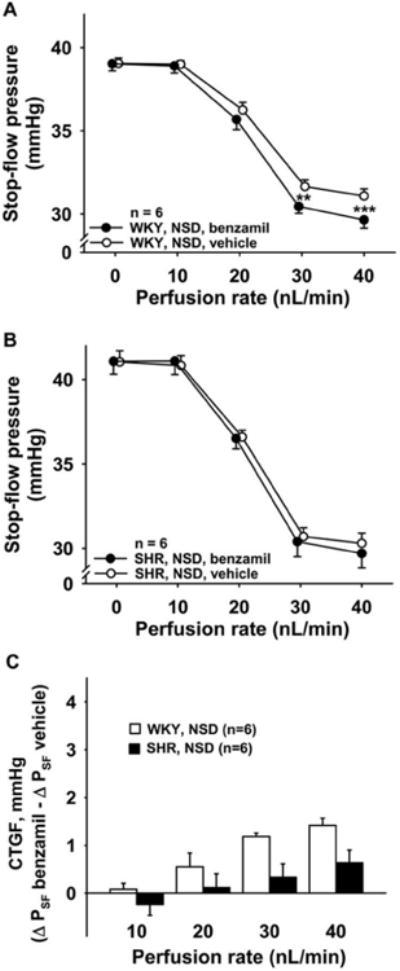
In rats fed a normal salt diet (NSD), two TGF responses were elicited, first in the presence of CTGF (○, vehicle), then during inhibition of CTGF (●, benzamil 1μM). Inhibition of CTGF potentiated the TGF response in WKY (Panel A) but not in SHR (Panel B). Panel C: SHR (closed bars) tended to have a smaller CTGF response compared to WKY (open bars). P < 0.01 for the overall comparison between strains (ANOVA) and P < 0.05 at the 30 and 40 nL/min perfusion rates, but not statistically significant after adjustment for multiple comparisons.
2) Dahl SS and Dahl SR: TGF resetting induced by high-salt diet (two weeks), role of CTGF in TGF resetting
When the rats were fed a high-salt diet (4% NaCl), TGF responses were attenuated in both Dahl SR and Dahl SS. These differences reached statistical significance when the tubules were perfused at 30 and 40 nL/min (Figure 6, panels A and B). However, the resetting was greater in Dahl SS than in Dahl SR (Figure 6C). Inhibition of CTGF with benzamil in Dahl SR and Dahl SS fed a high-salt diet led to a potentiation of TGF responses when the tubules were perfused at 30 and 40 nL/min (Figure 7, panels A and B). An inter-strain comparison showed that when fed a high-salt diet, Dahl SS had a greater CTGF response than Dahl SR (Figure 7C). In Dahl SS, the percentage of resetting due to CTGF was 53%, while in Dahl SR it was only 21% (supplemental Figure S3).
Figure 6. TGF resetting induced by high-salt diet (two weeks) in Dahl SR and Dahl SS.
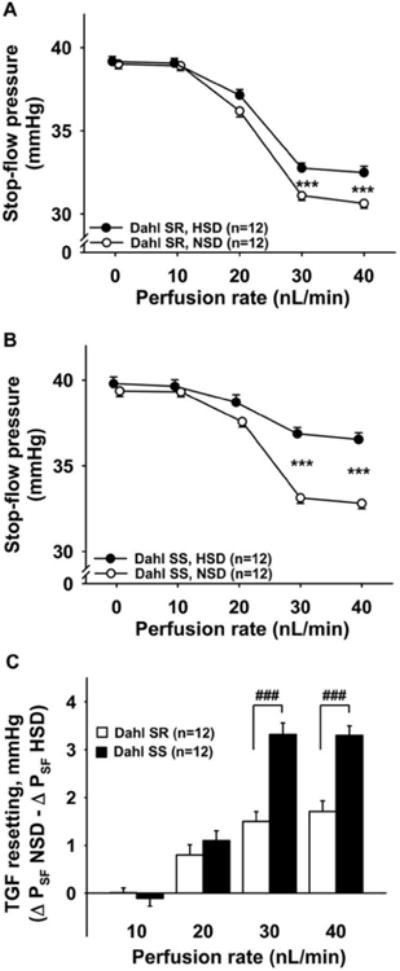
TGF resetting was calculated as the difference in ΔPSF on a normal salt diet (NSD) minus ΔPSF on a high-salt diet (HSD). Panels A and B show that a high-salt diet causes TGF resetting in Dahl SR (Panel A) and Dahl SS (Panel B). Panel C shows the inter-strain comparison of TGF resetting. Dahl SS (closed bars) have significantly higher TGF resetting than Dahl SR (open bars). *** P < 0.001, NSD vs. HSD; ###P < 0.001, Dahl SR vs. Dahl SS.
Figure 7. Role of CTGF in TGF response in Dahl SR and Dahl SS fed high-salt diet.

In rats fed a high-salt diet (HSD), two TGF responses were elicited, first in the presence of CTGF (○, vehicle), then during inhibition of CTGF (●, benzamil 1μM). Inhibition of CTGF potentiated the TGF response in both Dahl SR (Panel A) and Dahl SS (Panel B). Panel C: CTGF response in Dahl SR (open bars) and SS (closed bars). Dahl SS had greater CTGF responses than Dahl SR when fed a high-salt diet. **P < 0.01 ***P < 0.001, vehicle vs. benzamil. ##P < 0.01, Dahl SR vs. Dahl SS.
3) SHR and WKY: TGF resetting induced by high-salt diet (two weeks), role of CTGF in TGF resetting
When WKY rats were fed a high-salt diet, TGF responses were attenuated, these differences reached statistical significance when the tubules were perfused at 30 and 40 nL/min (Figure 8A). When SHR were fed a high-salt diet, there was a small decrease in TGF response but it did not reach statistical significance (Figure 8B). Resetting was significantly greater in WKY than in SHR (Figure 8C). Inhibition of CTGF with benzamil in WKY and SHR fed a high-salt diet led to potentiation of TGF responses when the tubules were perfused at 30 and 40 nL/min (Figure 9, panels A and B). An inter-strain comparison showed that when fed a high-salt diet, the CTGF response was greater in WKY than SHR (Figure 9C).
Figure 8. TGF resetting induced by high-salt diet (two weeks) in WKY and SHR.
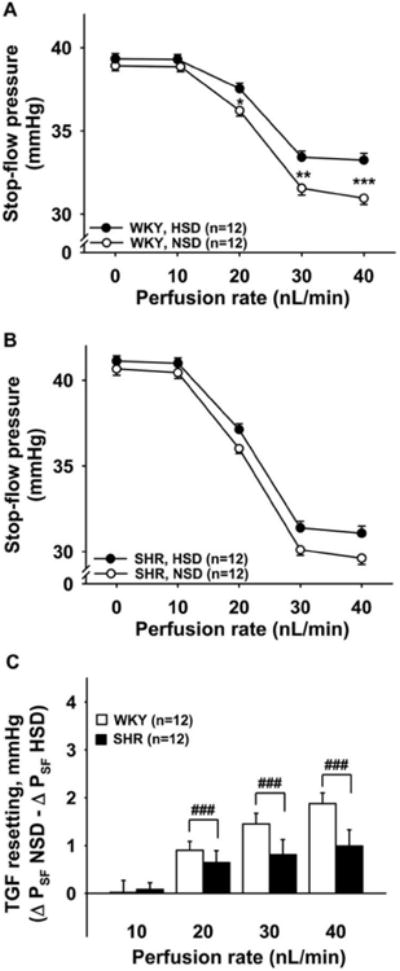
TGF resetting was calculated as the difference in ΔPSF on a normal salt diet (NSD) minus ΔPSF on a high-salt diet (HSD). Panels A and B show that a high-salt diet causes TGF resetting in WKY (Panel A) but no significant TGF resetting in SHR (Panel B). Panel C shows the inter-strain comparison of TGF resetting. SHR (closed bars) have significantly lower TGF resetting than WKY (open bars). *P < 0.05, **P < 0.01, ***P < 0.001, NSD vs. HSD; ###P < 0.001, WKY vs. SHR.
Figure 9. Role of CTGF in TGF response in WKY and SHR fed high-salt diet.
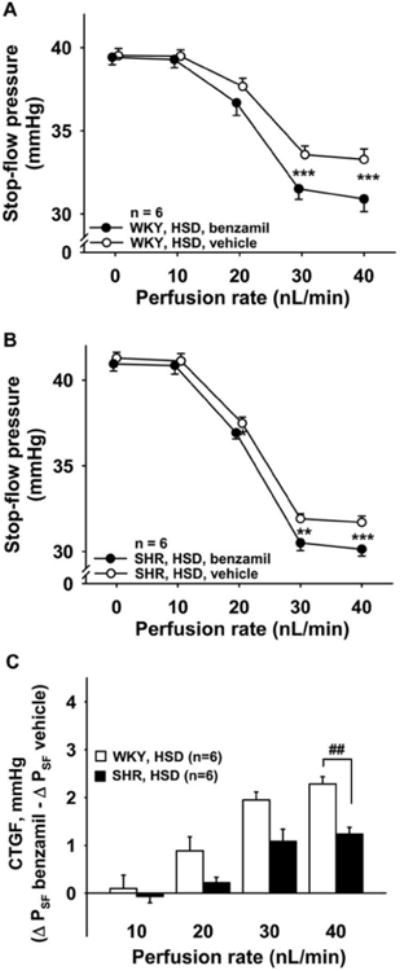
In rats fed a high-salt diet (HSD), two TGF responses were elicited, first in the presence of CTGF (○, vehicle), then during inhibition of CTGF (●, benzamil 1μM). Inhibition of CTGF potentiated the TGF response in both WKY (Panel A) and SHR (Panel B). Panel C: CTGF response in WKY (open bars) and SHR (closed bars). SHR had lower CTGF responses than WKY when fed a high-salt diet. P < 0.05, ** P < 0.01, *** P < 0.001, vehicle vs. benzamil. ###P < 0.001, WKY vs. SHR.
4) Dahl SS and SHR on normal and high-salt diet: Comparison of TGF response, TGF resetting and role of CTGF
Dahl SS rats on a normal salt diet had a significantly lower TGF response than SHR on normal salt diet (Figure 10, white circles vs. white triangles). TGF resetting induced by high salt intake was significantly greater in Dahl SS than in SHR (3.3 ± 0.1 vs. 1.0 ± 0.3 mm Hg; P < 0.001, Figure 10, delta between white and black circles vs. delta between white and black triangles). Inhibition of CTGF with benzamil potentiated TGF in Dahl SS to a greater extent than in SHR (3.4 ± 0.4 vs. 1.2 ± 0.1 mm Hg; P < 0.001, Figure 11, delta between black and white circles vs. delta between black and white triangles). These data suggest that TGF responses are much lower in Dahl SS than in SHR and that TGF resetting and the role of CTGF are more pronounced in Dahl SS than in SHR.
Figure 10. TGF resetting induced by high-salt diet (two weeks) in Dahl SS and SHR.
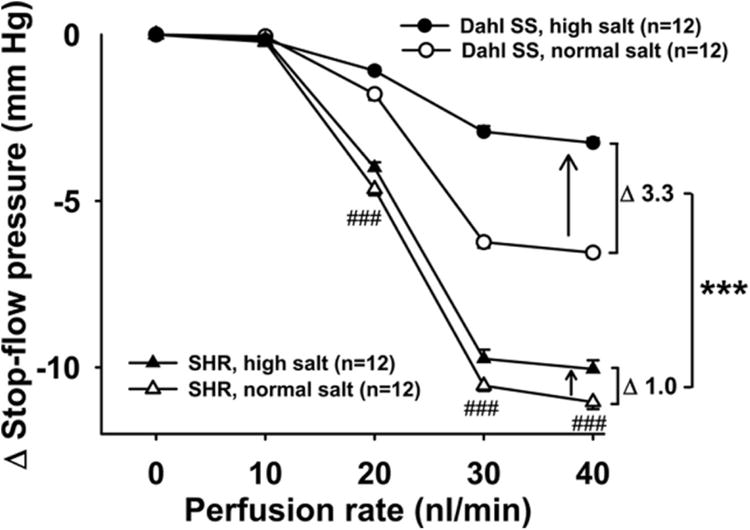
TGF resetting was calculated as the difference in ΔPSF on a normal salt diet (NSD) minus ΔPSF on a high-salt diet (HSD). Dahl SS have an attenuated TGF response compared to SHR, even when fed a normal salt diet. ###P < 0.001, Dahl SS normal salt vs. SHR normal salt. Furthermore, when fed a high-salt diet for two weeks, the difference between strains becomes even greater. This is because high salt attenuates the response in both strains, but to a greater extent in Dahl SS. ***P < 0.001, TGF resetting in Dahl SS vs. TGF resetting in SHR.
Figure 11. Role of CTGF in TGF response in Dahl SS and SHR fed high-salt diet.
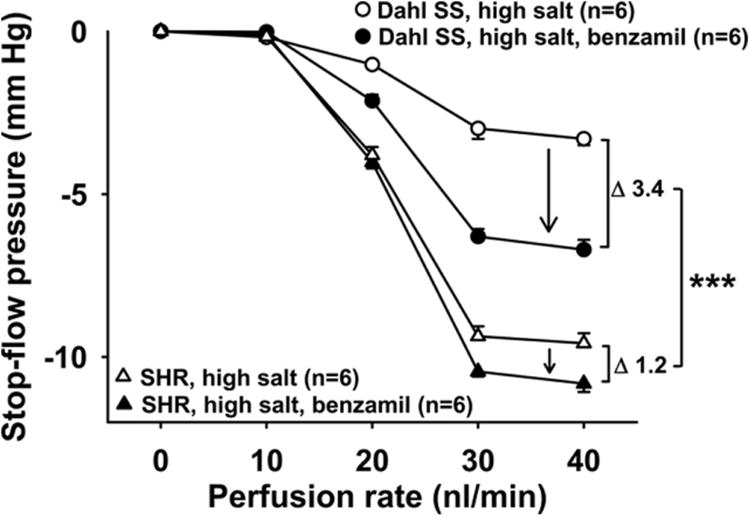
In rats fed a high-salt diet (HSD), two TGF responses were elicited, first in the presence of CTGF (○, vehicle), then during inhibition of CTGF (●, benzamil 1μM). Inhibition of CTGF potentiated the TGF response by 13 ± 2% in SHR (1.2 ± 0.1 mm Hg in absolute numbers) and by 83 ± 22 % in Dahl SS (3.4 ± 0.4 mm Hg in absolute numbers). Dahl SS had significantly greater CTGF than SHR when fed high salt. *** P < 0.001, CTGF in Dahl SS vs. CTGF in SHR.
Discussion
In humans, susceptibility to hypertension-induced renal damage varies, with African-Americans at high risk 18. African-Americans often have salt-sensitive hypertension 19, and high salt intake causes an abnormal renal hemodynamic response and an increase in estimated PGC3. Thus their enhanced susceptibility to renal damage may be related to the increased PGC associated with salt sensitivity, as salt sensitivity in humans predicts higher microalbuminuria in the short term 20 and higher mortality on long-term follow-up studies 21.
In animal models, there is substantial evidence that in hypertension PGC greatly influences the progression of renal nephrosclerosis 1,2,4,22. It is well known that at similar levels of systemic hypertension, Dahl SS but not SHR develop glomerular injury. Furthermore, these differences in glomerular pathology occur because Dahl SS but not SHR develop glomerular hypertension 4. In SHR, glomerular capillary pressure in cortical nephrons is normal in spite of severe systemic hypertension because of a marked increase in preglomerular arteriolar resistance 5. Thus glomeruli of SHR are protected from systemic hypertension. On the other hand, hypertensive Dahl SS display increased glomerular blood flow and glomerular capillary pressure due to decreased preglomerular arteriolar resistance 6. The present study is the first to explore whether differences in glomerular hemodynamics between these strains may be due to differences in CTGF.
TGF responses in Dahl rats have not been well characterized. Wilcox and Welch reported that Dahl SS fed a low-salt diet had an attenuated TGF response compared to Sprague-Dawley rats 23. On the other hand, Karlsen et al. found no differences in TGF between Dahl SS and Sprague-Dawley rats 24. We have previously shown that Sprague-Dawley rats have CTGF, that CTGF antagonizes TGF, and that CTGF at least partially mediates TGF resetting induced acutely by volume expansion 10,17. In the current work, we have not included Sprague-Dawley rats; rather, we have compared the Dahl SS to its genetic control and to SHR (to contrast these two models of hypertension). We believe our current work will help clarify the TGF response in Dahl rats, as well as provide the first studies of CTGF in hypertension. In our study, Dahl SS had an attenuated TGF response compared to Dahl SR on both normal and high-salt diets. Dahl SS fed a high-salt diet had greater TGF resetting and greater CTGF than Dahl SR. In Dahl SS inhibition of CTGF decreased TGF resetting by 53 % while in Dahl SR it did so by only 21%. Collectively, these results suggest that in Dahl SS fed a high-salt diet, CTGF is increased, leading to higher PSF and PGC. In this way, higher CTGF may participate in the development of nephrosclerosis. In this study, we used PSF as a surrogate for PGC since Dahl SS do not have superficial glomeruli that can be directly punctured to measure PGC 25.
In contrast to Dahl SS, we found that SHR fed a normal salt diet have a much greater TGF response than WKY or Dahl SS and that TGF resetting induced by high salt intake was minimal or nonexistent, consistent with previous reports 26,27. Furthermore, in SHR antagonism of CTGF to the TGF response and its contribution to TGF resetting were also minimal, indicating that a diminished CTGF may at least partially explain the enhancement in TGF and reduced TGF resetting. These data suggest that a decrease in CTGF in SHR increases preglomerular vascular resistance and helps maintain a normal PGC, thus preventing renal damage, in stark contrast to Dahl SS.
Our findings in WKY also expand our understanding of the role of CTGF in TGF resetting in normotensive rats. We previously reported that CTGF partly mediates TGF resetting in normotensive Sprague-Dawley rats induced acutely by sustained perfusion of the nephron at a high flow rate for 30 minutes 17. Here we found for the first time that CTGF also partly mediates TGF resetting that was induced chronically by high salt intake over two weeks. However, CTGF does not completely explain TGF resetting, as some TGF resetting still occurred even when CTGF was blocked. Likely, this represents intrinsic mechanisms of the macula densa that reset TGF, as previously described 12.
The mechanism by which CTGF is enhanced in Dahl SS remains unknown, but may relate to the fact that CTGF is initiated by Na transport in the CNT via ENaC 8. In spite of their high blood pressure and low serum aldosterone levels, Dahl SS fed high salt have increased ENaC mRNA, protein, and sodium transport compared to Dahl SR 28-33. Furthermore, Pavlov et al. recently measured ENaC single-channel activity by patch clamp studies in split-open cortical collecting ducts and found higher ENaC activity in Dahl SS on a high-salt diet compared to either a normal diet or to Dahl SR 34. In addition, CTGF is mediated by prostaglandin E2 (PGE2) and epoxyeicosatrienoic acids (EETs) 9,11. In Dahl SS, high salt intake increases renal cortical COX-2 expression 35 and urinary PGE2 excretion 36. Thus, it is possible that in Dahl SS, increased ENaC and PGE2 may cause enhanced CTGF, enhanced PGC, and enhanced glomerular damage. Conversely, attenuation of CTGF in SHR may be due to decreased EETs, since soluble epoxide hydrolase, the enzyme that metabolizes EETs, is increased in the kidney in this strain 37. Therefore, decreased levels of EETs, which partly mediate CTGF, could explain the decrease in CTGF in SHR.
In summary, our studies provide direct evidence that a high-salt diet causes TGF resetting, and that CTGF mediates TGF resetting induced by a high-salt diet, at least in part. Hypertensive Dahl SS have lower TGF compared to either SHR or Dahl SR, and because of an increased TGF resetting in Dahl SS these differences become exaggerated on a high-salt diet. These differences are due in part to a greater CTGF in Dahl SS fed a normal salt diet, as well as greater enhancement of CTGF by a high-salt diet. Our findings may help explain the excessive increase in PGC and glomerular damage observed in salt-sensitive hypertension.
Perspectives
An increase in CTGF may explain the higher glomerular pressure and renal damage in salt-sensitive hypertensive individuals, such as African-Americans, the elderly, and the diabetic. ENaC-blocking drugs (potassium-sparing diuretics), by blocking CTGF and decrease glomerular perfusion pressure, could be useful in preventing hypertensive nephrosclerosis. Our studies may also help explain the beneficial effects seen with mineralocorticoid receptor blockers and suggest new targets for prevention of renal damage.
Supplementary Material
Novelty and Significance.
What is New?
CTGF is a novel mechanism of regulation of afferent arteriole resistance.
We show that CTGF is responsible for part of the TGF resetting induced by chronic high salt intake.
In salt-sensitive hypertension, CTGF is augmented and explains more than 50% of TGF resetting.
What is Relevant?
In the United States, over one fourth of adults diagnosed with hypertension have moderate to severe chronic kidney disease, and hypertension is the second leading cause of end-stage renal disease (ESRD). In the last two decades, the incidence of hypertension-induced ESRD has nearly doubled, even as control of hypertension increased from 24% to 50%. Despite adequate blood pressure control, renal function declines more often in individuals with salt-sensitive hypertension, including African-Americans, the elderly, and people with diabetes mellitus, all of whom are more susceptible to developing hypertensive renal damage. The cause of this enhanced susceptibility is related to the increased PGC associated with salt-sensitivity, as salt-sensitivity in humans predicts higher microalbuminuria in the short term and higher mortality on long-term follow-up studies.
Summary
Our study shows that CTGF, which is a novel mechanism of regulation of afferent arteriole resistance, is responsible for more than 50% of the increase in PGC in salt-sensitive hypertension. Understanding the mechanism that increases PGC in salt-sensitive hypertension may lead to both prevention and better treatment of renal disease in salt-sensitive hypertension.
Acknowledgments
None.
Source(s) of Funding: HL028982-31
Footnotes
Conflict(s) of Interest/Disclosure(s): None.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Raij L. End-organ susceptibility as a determinant of renal disease in hypertension. Kidney Int. 2003;64:1923–1932. doi: 10.1046/j.1523-1755.2003.00291.x. [DOI] [PubMed] [Google Scholar]
- 2.Tolins JP, Shultz P, Raij L. Mechanisms of hypertensive glomerular injury. Am J Cardiol. 1988;62:54G–58G. doi: 10.1016/0002-9149(88)90033-1. [DOI] [PubMed] [Google Scholar]
- 3.Campese VM, Parise M, Karubian F, Bigazzi R. Abnormal renal hemodynamics in black salt-sensitive patients with hypertension. Hypertension. 1991;18:805–812. doi: 10.1161/01.hyp.18.6.805. [DOI] [PubMed] [Google Scholar]
- 4.Raij L, Azar S, Keane WF. Role of hypertension in progressive glomerular immune injury. Hypertension. 1985;7:398–404. [PubMed] [Google Scholar]
- 5.Azar S, Johnson MA, Scheinman J, Bruno L, Tobian L. Regulation of glomerular capillary pressure and filtration rate in young Kyoto hypertensive rats. Clin Sci. 1979;56:203–209. doi: 10.1042/cs0560203. [DOI] [PubMed] [Google Scholar]
- 6.Azar S, Limas C, Iwai J, Weller D. Single nephron dynamics during high sodium intake and early hypertension in Dahl rats. Jpn Heart J. 1979;20:138–140. [Google Scholar]
- 7.Ren Y, Garvin JL, Liu R, Carretero OA. Cross-talk between arterioles and tubules in the kidney. Pediatr Nephrol. 2009;24:31–35. doi: 10.1007/s00467-008-0852-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ren Y, Garvin JL, Liu R, Carretero OA. Crosstalk between the connecting tubule and the afferent arteriole regulates renal microcirculation. Kidney Int. 2007;71:1116–1121. doi: 10.1038/sj.ki.5002190. [DOI] [PubMed] [Google Scholar]
- 9.Ren Y, D'Ambrosio MA, Garvin JL, Wang H, Carretero OA. Possible mediators of connecting tubule glomerular feedback. Hypertension. 2009;53:319–323. doi: 10.1161/HYPERTENSIONAHA.108.124545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang H, Garvin JL, D'Ambrosio MA, Ren Y, Carretero OA. Connecting tubule glomerular feedback antagonizes tubuloglomerular feedback in vivo. Am J Physiol Renal Physiol. 2010;299:F1374–F1378. doi: 10.1152/ajprenal.00403.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ren Y, D'Ambrosio MA, Wang H, Garvin JL, Carretero OA. Participation of Prostaglandin E2 and EP4 receptors in Connecting Tubule Glomerular Feedback (CTGF) [abstract] AHA Council for High Blood Pressure Research. 2012;60:A33. [Google Scholar]
- 12.Schnermann J, Briggs JP. The macula densa is worth its salt. J Clin Invest. 1999;104:1007–1009. doi: 10.1172/JCI8539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kaufman JS, Hamburger RJ, Flamenbaum W. Tubuloglomerular feedback: effect of dietary NaCl intake. Am J Physiol. 1976;231:1744–1749. doi: 10.1152/ajplegacy.1976.231.6.1744. [DOI] [PubMed] [Google Scholar]
- 14.Fu Y, Roman RJ, Lu D, Zhu X, Liu R. High salt intake-induced TGF suppression is partially mediated by primary cilia on the macula densa [abstract] Hypertension. 2010;56:e159. [Google Scholar]
- 15.Schnermann J. Juxtaglomerular cell complex in the regulation of renal salt excretion. Am J Physiol. 1998;274:R263–R279. doi: 10.1152/ajpregu.1998.274.2.R263. [DOI] [PubMed] [Google Scholar]
- 16.Blantz RC, Thomson SC, Peterson OW, Gabbai FB. Physiologic adaptations of the tubuloglomerular feedback system. Kidney Int. 1990;38:577–583. doi: 10.1038/ki.1990.245. [DOI] [PubMed] [Google Scholar]
- 17.Wang H, D'Ambrosio MA, Garvin JL, Ren Y, Carretero OA. Connecting tubule glomerular feedback mediates acute tubuloglomerular feedback resetting. Am J Physiol Renal Physiol. 2012;302:F1300–F1304. doi: 10.1152/ajprenal.00673.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.United States Renal Data System. USRDS 2012 Annual Data Report. Bethesda, MD: National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), National Institutes of Health (NIH), U.S. Department of Health and Human Services (DHHS); 2012. [Google Scholar]
- 19.Weinberger MH, Miller JZ, Luft FC, Grim CE, Fineberg NS. Definitions and characteristics of sodium sensitivity and blood pressure resistance. Hypertension. 1986(8):II127–II134. doi: 10.1161/01.hyp.8.6_pt_2.ii127. [DOI] [PubMed] [Google Scholar]
- 20.Bigazzi R, Bianchi S, Baldari D, Sgherri G, Baldari G, Campese VM. Microalbuminuria in salt-sensitive patients. A marker for renal and cardiovascular risk factors. Hypertension. 1994;23:195–199. doi: 10.1161/01.hyp.23.2.195. [DOI] [PubMed] [Google Scholar]
- 21.Weinberger MH, Fineberg NS, Fineberg SE, Weinberger M. Salt sensitivity, pulse pressure, and death in normal and hypertensive humans. Hypertension. 2001;37:429–432. doi: 10.1161/01.hyp.37.2.429. [DOI] [PubMed] [Google Scholar]
- 22.Hayakawa H, Raij L. Nitric oxide synthase activity and renal injury in genetic hypertension. Hypertension. 1998;31:266–270. doi: 10.1161/01.hyp.31.1.266. [DOI] [PubMed] [Google Scholar]
- 23.Wilcox CS, Welch WJ. TGF and nitric oxide: effects of salt intake and salt-sensitive hypertension. Kidney Int Suppl. 1996;55:S9–13. [PubMed] [Google Scholar]
- 24.Karlsen FM, Leyssac PP, Holstein-Rathlou NH. Tubuloglomerular feedback in Dahl rats. Am J Physiol. 1998;274:R1561–R1569. doi: 10.1152/ajpregu.1998.274.6.R1561. [DOI] [PubMed] [Google Scholar]
- 25.Vallon V. Micropuncturing the nephron. Pflugers Arch. 2009;458:189–201. doi: 10.1007/s00424-008-0581-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brannstrom K, Morsing P, Arendshorst WJ. Exaggerated tubuloglomerular feedback activity in genetic hypertension is mediated by ANG II and AT1 receptors. Am J Physiol. 1996;270:F749–F755. doi: 10.1152/ajprenal.1996.270.5.F749. [DOI] [PubMed] [Google Scholar]
- 27.Ushiogi Y, Takabatake T, Haberle DA. Blood pressure and tubuloglomerular feedback mechanism in chronically salt-loaded spontaneously hypertensive rats. Kidney Int. 1991;39:1184–1192. doi: 10.1038/ki.1991.150. [DOI] [PubMed] [Google Scholar]
- 28.Kakizoe Y, Kitamura K, Ko T, Wakida N, Maekawa A, Miyoshi T, Shiraishi N, Adachi M, Zhang Z, Masilamani S, Tomita K. Aberrant ENaC activation in Dahl salt-sensitive rats. J Hypertens. 2009;27:1679–1689. doi: 10.1097/HJH.0b013e32832c7d23. [DOI] [PubMed] [Google Scholar]
- 29.Shehata MF. Regulation of the epithelial sodium channel [ENaC] in kidneys of salt-sensitive Dahl rats: insights on alternative splicing. Int Arch Med. 2009;2:28. doi: 10.1186/1755-7682-2-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aoi W, Niisato N, Sawabe Y, Miyazaki H, Tokuda S, Nishio K, Yoshikawa T, Marunaka Y. Abnormal expression of ENaC and SGK1 mRNA induced by dietary sodium in Dahl salt-sensitively hypertensive rats. Cell Biol Int. 2007;31:1288–1291. doi: 10.1016/j.cellbi.2007.03.036. [DOI] [PubMed] [Google Scholar]
- 31.Shehata MF. The epithelial sodium channel alpha subunit (alpha ENaC) alternatively spliced form “b” in Dahl rats: what's next? Int Arch Med. 2010;3:14. doi: 10.1186/1755-7682-3-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Husted RF, Takahashi T, Stokes JB. IMCD cells cultured from Dahl S rats absorb more Na+ than Dahl R rats. Am J Physiol. 1996;271:F1029–F1036. doi: 10.1152/ajprenal.1996.271.5.F1029. [DOI] [PubMed] [Google Scholar]
- 33.Husted RF, Takahashi T, Stokes JB. The basis of higher Na+ transport by inner medullary collecting duct cells from Dahl salt-sensitive rats: implicating the apical membrane Na+ channel. J Membr Biol. 1997;156:9–18. doi: 10.1007/s002329900182. [DOI] [PubMed] [Google Scholar]
- 34.Pavlov TS, Levchenko V, O'Connor PM, Ilatovskaya DV, Palygin O, Mori T, Mattson DL, Sorokin A, Lombard JH, Cowley AW, Jr, Staruschenko A. Deficiency of Renal Cortical EGF Increases ENaC Activity and Contributes to Salt-Sensitive Hypertension. J Am Soc Nephrol. 2013;24:1053–1062. doi: 10.1681/ASN.2012080839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chandramohan G, Bai Y, Norris K, Rodriguez-Iturbe B, Vaziri ND. Effects of dietary salt on intrarenal angiotensin system, NAD(P)H oxidase, COX-2, MCP-1 and PAI-1 expressions and NF-kappaB activity in salt-sensitive and -resistant rat kidneys. Am J Nephrol. 2008;28:158–167. doi: 10.1159/000110021. [DOI] [PubMed] [Google Scholar]
- 36.Jaimes EA, Zhou MS, Pearse DD, Puzis L, Raij L. Upregulation of cortical COX-2 in salt-sensitive hypertension: role of angiotensin II and reactive oxygen species. Am J Physiol Renal Physiol. 2008;294:F385–F392. doi: 10.1152/ajprenal.00302.2007. [DOI] [PubMed] [Google Scholar]
- 37.Yu Z, Xu F, Huse LM, Morisseau C, Draper AJ, Newman JW, Parker C, Graham L, Engler MM, Hammock BD, Zeldin DC, Kroetz DL. Soluble epoxide hydrolase regulates hydrolysis of vasoactive epoxyeicosatrienoic acids. Circ Res. 2000;87:992–998. doi: 10.1161/01.res.87.11.992. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.