

Abstract
The protein kinase C family (PKC) regulates a variety of neural functions including neurotransmitter release. The selective activation of a wide range of PKC isoforms in different cells and domains is likely to contribute to the functional diversity of PKC phosphorylating activity. In this review, we describe the isoform localization, phosphorylation function, regulation and signalling of the PKC family at the neuromuscular junction. Data show the involvement of the PKC family in several important functions at the neuromuscular junction and in particular in the maturation of the synapse and the modulation of neurotransmission in the adult.
Keywords: electrical stimulation, immunofluorescence, isoforms, neuromuscular junction, neurotransmission, protein kinase C, synapse elimination
Introduction
Protein kinase C (PKC) comprises a family of serine-threonine protein kinases that are widely distributed in all cells and at high concentrations in neural tissues and regulate a variety of neural functions including neurotransmitter release. The PKC family is classified into three groups [conventional PKCs (cPKCs), novel PKCs (nPKCs) and atypical PKCs (aPKCs) isoforms] on the basis of their sequence motifs and cofactor requirements (Fig. 1A). The cPKCs (α, βI, βII, γ) require phosphatidylserine (PS), diacylglycerol (DAG) and Ca2+ if they are to be activated, nPKCs (δ, ε, η and θ) require only PS and DAG, and aPKCs (ζ and ι/λ) require only PS. These different isoforms exhibit distinct tissue and cell distributions, suggesting specific roles in a variety of cellular functions (Tanaka & Nishizuka, 1994; Mochly-Rosen & Gordon, 1998). The differential colocalization of an activated PKC isoform with its endogenous protein substrates, contributes to the functional diversity of the PKC isoforms (Mochly-Rosen, 1995). Intracellular PKC-binding proteins known as receptors for activated C-kinase (RACKs) are essential to achieve the cellular specific patterns of distribution of an individual activated PKC isoform (Mochly-Rosen et al. 1991a,b). Therefore, it is fundamental to identify the mechanisms that activate and compartmentalize PKC isoforms in order to understand the physiological functions of PKC. Many studies have been carried out on the mammalian neuromuscular junction (NMJ), which is a useful paradigm of synaptic structure and function (Fig. 1B). In this review, we describe the localization and relevant roles of the PKC family at the NMJ, both during development and in neurotransmission in the adult.
Fig. 1.

Protein kinase C and NMJ. (A) Serine/threonine kinase C family. Schematic diagram of primary structures of PKC family members showing domain composition and activators. The PKC family of isozymes consists of three classes: the classical (α, βI, βII, γ), novel (δ, ε, η and θ) and atypical (ζ and ι/λ). (B) Adult NMJ image. Double immunofluorescence NMJs labelled with syntaxin/neurofilament-200 nerve terminal, NT, in green) and α-BTX (AChR in red). (C1–3) Semithin (0.5 μm) cross-sections of the NMJ stained with toluidine blue (C1) and with a triple immunofluorescence method (C3; syntaxin/neurofilament-200-NT, in green, S-100 Schwann cell, SC, in blue and α-BTX -AChR in red). In (C2), the cellular components in C1 are delineated. Reproduced with permission from Lanuza et al. (2007). (D) Immunohistochemical staining for cPKC isoform βI at the adult NMJ. cPKCβI are labelled in green, the AChRs in red and the Schwann cells (SC, S-100) or the nerve terminals (NT, neurofilament-200/syntaxin) in blue. (D1) NMJ from a whole muscle immunolabelled. (D2–D3) Semithin cross-sections from a whole-mount multiple-immunofluorescent stained muscle. cPKCβI was found to be localized to the presynaptic terminals. Reproduced with permission from Besalduch et al. (2010). (D4) The diagram summarizes the localization of the PKC isoforms in the three cellular components of the NMJ (nerve terminal, muscle cell and Schwann cell). Scale bars: 10 μm.
PKC isoforms in the NMJ
Several isoforms of PKC are expressed in adult and newborn skeletal muscle (Nakano et al. 1992; Arakawa et al. 1993; Hilgenberg & Miles, 1995; Lanuza et al. 2000, 2010; Kim et al. 2002; Moraczewski et al. 2002; Brandt et al. 2003; Canto et al. 2004; Perrini et al. 2004; Rose et al. 2004; Van Ginneken et al. 2004; Vary et al. 2005; Besalduch et al. 2010). These isoforms are from conventional, novel and atypical families and are expressed in significant quantities. Most of these studies have demonstrated that PKCs have a role in insulin and exercise-mediated glucose transport and that a substantial amount of PKC is detected in the T-tubule membrane (Salvatori et al. 1993), which suggests that these extrasynaptic PKCs have a role in the metabolism and contraction of muscle fibers. It has been demonstrated that synaptic and extrasynaptic regions of muscle have a similar proportion of cPKC isoforms (Besalduch et al. 2010), which suggests the involvement of the PKC family also in neural functions.
The state of activation of PKC isoforms in basal conditions appears to be high, as the greatest proportion of total enzymatic activity of PKC in skeletal muscle is associated with the particulate fraction (Richter et al. 1987; Cleland et al. 1989; Besalduch et al. 2010). PKCs undergo translocation during activation, involving kinases moving from cytoplasmic to membrane domains, which leads to conformational changes in the enzyme and activation (Kraft et al. 1982; Kraft & Anderson, 1983; Oancea & Meyer, 1998; Tsuruno & Hirano, 2007). For example, the location of the enzyme in the skeletal muscle membrane fraction is associated with the phosphorylation, and therefore activation, of PKCs (Besalduch et al. 2010).
Several PKC isoforms from the cPKC and nPKC families have been described at the NMJ (Nakano et al. 1992; Arakawa et al. 1993; Hilgenberg & Miles, 1995; Lanuza et al. 2000, 2010; Perkins et al. 2001; Kim et al. 2002; Besalduch et al. 2010).
cPKC (α, βI, βII, γ) expression and localization at the NMJ
Initially, cPKC β-subspecies were demonstrated in the presynaptic (Nakano et al. 1992; Arakawa et al. 1993; Perkins et al. 2001) and postsynaptic cell (Nakano et al. 1992; Perkins et al. 2001), whereas cPKCα appeared to be widely expressed (Nakano et al. 1992; Hilgenberg & Miles, 1995; Lanuza et al. 2000; Kim et al. 2002). More recently, using high resolution immunohistochemistry in semithin cross-sections of skeletal muscle (Lanuza et al. 2007) a selective concentration and distribution of βI and βII isoforms at the NMJ was demonstrated (Besalduch et al. 2010). Furthermore, we found that the cPKC isoforms (except PKCγ) are selectively distributed in specific cell types at the adult NMJ (Fig. 1D). cPKCα and βI were localized to the presynaptic terminals, whereas cPKCβII and also cPKCα were associated with the postsynaptic muscle fibre and Schwann cells (Besalduch et al. 2010). Therefore, at present, cPKCβI is the cPKC isoform that is almost exclusively detected in the nerve terminals of the NMJ, suggesting that it has a specific role in transmitter release. cPKCα may also be involved in this function, although it is ubiquitously expressed and may play more than one role. Furthermore, cPKCα and βII have been detected in both postsynaptic muscle fibres and Schwann cells, agreeing with other studies that have demonstrated immunoreactivity for cPKCα and βII in the Schwann cells of sciatic nerves (Ekström et al. 1992; Roberts & McLean, 1997). Conversely, cPKCγ is restricted mainly to the brain and spinal cord (Nishizuka, 1995) and is almost absent from the NMJ and extrasynaptic muscle fibre (Besalduch et al. 2010).
nPKC (ε, θ) expression and localization at the NMJ
nPKCε is widely distributed in the central nervous system (Shirai et al. 2008) and muscle (Moraczewski et al. 2002; Vary et al. 2005) but more recently this isoform has been demonstrated in substantial quantities nPKCε at the rodent NMJ and is roughly twice as abundant during development compared with the adult (Lanuza et al. 2012). This may indicate some specific involvement in motor axon withdrawal mechanism during developmental synapse elimination at the NMJ. In the NMJ adult, nPKCε has been located in the motor nerve terminals but not in the Schwann or postsynaptic muscle cells (Lanuza et al. 2012). Immunohistochemistry performed in the central nervous system revealed that the enzyme is most abundantly expressed in nerve fibers and, more specifically, using electron microscopy, presynaptically (Saito et al. 1993; Tanaka & Nishizuka, 1994).
The calcium-independent isoform nPKCθ has been found to be both postsynaptically and presynaptically distributed at the NMJ (Hilgenberg & Miles, 1995; Lanuza et al. 2000, 2010; Kim et al. 2002), supporting the notion that nPKCθ acts in both compartments. It had been reported that nPKCθ was a postsynaptic isoform because of its predominant localization in skeletal muscle, but neural expression of nPKCθ is not surprising because it was reported that cell co-culture preparations of normal muscle with nerve derived from kinase KO spinal cord failed to demonstrate axonal competition and synaptic down-regulation (Li et al. 2004). In fact, studies in rodent ex vivo preparations have found that blocking PKC can increase ACh release from the weakest axons in developing polyinnervated synapses (Santafe et al. 2009b), supporting the notion that there is a PKC-dependent release inhibition mechanism that, when fully active in certain weak motor axons, can depress ACh release and even disconnect synapses.
Figure 1-D4 summarizes findings concerning cPKC and nPKC isoform localization and shows that PKCα, βI, ε and θ are located in the nerve terminals at the NMJ; cPKCα, βII and θ are located in the postsynaptic component of the NMJ, and cPKCα and βII are also localized in the Schwann cells.
Synaptic activity, neurotransmission and PKC isoforms
Presynaptic protein phosphorylation by PKC regulates transmitter release
Presynaptic protein phosphorylation by PKC is an important mechanism that regulates transmitter release (West et al. 1991; Numann et al. 1994; Byrne & Kandel, 1996; Catterall, 1999; Santafe et al. 2005). Investigation into PKC coupling to acetylcholine (ACh) release at the NMJ indicated that presynaptic PKC is involved in the modulation of neurotransmission in the adult NMJ because ACh release increases when PKC is highly activated by a phorbol ester (phorbol 12-myristate 13-acetate, PMA, Santafe et al. 2006). Nevertheless, PKC is uncoupled from the ACh release mechanism at rest in the ex-vivo muscle preparation because calphostin C (CaC), a potent inhibitor of PKC, is not able to reduce release in these conditions (Santafe et al. 2006). Furthermore, the action of PKC is dependent on the Ca2+ inflow through the P/Q-type voltage-dependent calcium channels during evoked activity, because the blocking of this channel with ω-Agatoxin IVA inhibited the PMA-potentiation (Santafe et al. 2006).
It is known that modulation of the ACh release can be carried out by mAChR autoreceptors (see Caulfield, 1993 for a review; Ganguly & Das, 1979; Abbs & Joseph, 1981; Wessler et al. 1987; Arenson, 1989). Investigations at the NMJ showed that M1 and M2 subtypes of muscarinic receptors are involved in ACh release from adult NMJ motor nerve terminals (Santafe et al. 2003). There are two opposing, though finely balanced, M1-M2 mAChR-operated mechanisms that tonically modulate transmitter release. PKC appears to be involved in this modulation because when an imbalance of the M1-M2 mAChRs function was experimentally produced with selective blockers, PKC was then able to stimulate transmitter release tonically, as evidenced by its inhibitory effect on release of CaC in these conditions (Santafe et al. 2006).
In conclusion, it seems that at least one calcium-dependent PKC in the adult NMJ mediates presynaptic mAChR modulation of transmitter release (Santafe et al. 2006; see also the diagram corresponding to the adult NMJ in Fig. 4A). Given the size and diversity of the PKC family, it is important to determine which PKC family member(s) are involved in this mediation.
Fig. 4.
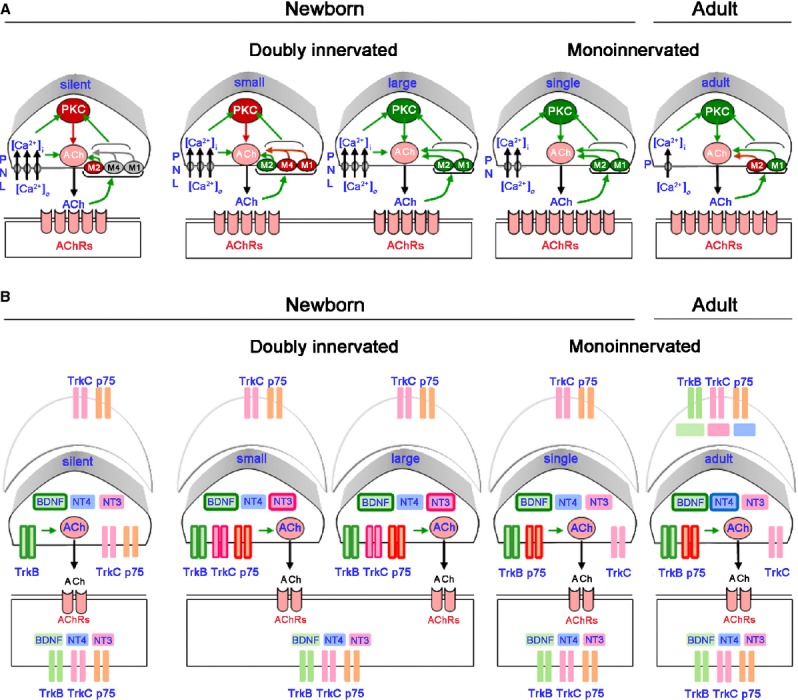
Presynaptic PKC signalling. (A) Functional relation between calcium inflows, voltage-dependent calcium channels (VDCC), presynaptic muscarinic acetylcholine receptors (mAChRs), and PKC activity in the modulation of the postnatal developmental activity dependent-synaptic elimination process. (B) Localization of neurotrophins (brain-derived neurotrophic factor (BDNF), neurotrophin (NT)-3 and NT-4 and its receptor proteins (p75NTR, trkB and trkC) in neonatal monoinnervated and multiple-innervated NMJ. Figure also shows that neurotrophins enhance neurotransmission and that this role is selective for the different types of nerve terminals depending on their developmental maturation. In the diagram the framed-neurotrophin name into a particular presynaptic component indicates that this neurotrophin is potentiating the ACh release in this specific nerve terminal type.
PKC isoforms involved in the activity-induced ACh release mechanism
There are several lines of suggesting that at least one calcium-dependent PKC isoform might be involved in modulating ACh release in conditions in which PKC is active. One important mechanism that activates PKC is synaptic activity (Besalduch et al. 2010). PKC couples to ACh release when continuous electrical stimulation imposes moderate activity on the NMJ (Besalduch et al. 2010). Figure 2A shows that when the nerve fibers innervating the NMJs were electrically stimulated at a low frequency (1 h of continuous electrical stimulation at 1 Hz), to introduce moderate, physiologically relevant activity, PKC was coupled to neurotransmitter release. The block of all PKC isoforms with CaC reduces approximately 40% of the evoked release in stimulated muscles, suggesting the involvement of PKC (see raw data in Fig. 2A). Further, electrical stimulation of peripheral nerve at a low frequency did not, by itself, change the end plate potential (EPP) amplitude (Fig. 2A). This result suggests to us that the PKC has a role in the maintenance of the release machinery in active synapses. Another well known mechanism that enhances neurotransmission is the increase of external calcium, suggesting that the process could be mediated by a calcium-dependent PKC. The increase in external calcium (from 2 to 5 mm) increased EPP size (100%); this effect may be due partly to the involvement of the coupling of PKC to neurotransmission, because in the presence of high calcium, the blocking of the kinase with CaC reduces evoked release (Fig. 2A). These results strongly suggest that at least one calcium-dependent PKC present at the NMJ might be involved in modulating ACh release in conditions in which PKC is active, and cPKC localized in the nerve terminal at the NMJ is a good candidate for this role. As previously stated, it has been demonstrated that both cPKCα and cPKCβI have a presynaptic location, with cPKCβI being exclusively located in the nerve terminal (Fig. 1D; Besalduch et al. 2010). It would be interesting to know whether cPKCα and -βI activity can be changed after electrical stimulation because, if it can, the isoform-specific activity of the cPKCs may affect the targeted modulation of neurotransmitter release and synaptic activity. Moreover, muscle contraction induced by synaptic activity could change the expression of these presynaptic cPKC isoforms in the membrane fraction of the muscle synaptic zone, as several studies have reported that electrical stimulation-induced contraction increases the translocation of total PKC activity to the particulate fraction (Richter et al. 1987; Cleland et al. 1989; Antipenko et al. 1999). Figure 2B shows that the stimulation of synaptic inputs significantly decreases the amount of the two presynaptic isoforms in the nerve terminal membrane. When there is muscle contraction, stimulation of synaptic inputs significantly increases the amount of the two isoforms in the nerve terminal membrane. Moreover, these changes in the synaptic membrane are accompanied by an increase in the phosphorylation of cPKC and of several proteins in the cPKC consensus sites, indicating that the increase in the PKC isoforms in the synaptic membranes in this experimental condition is in concordance with the phosphorylating action of the kinases. Therefore, in active synaptic zones, cPKCα and βI isoforms increase or decrease depending on whether the muscle cells contract (Besalduch et al. 2010). The level of the postsynaptic cPKCβII isoform in the synaptic membrane when the nerve fibres are stimulated (with and without contraction) increases or decreases in the membrane depending on whether the muscle cells contract or not, respectively (Fig. 2B). We suggest that a muscle cell contraction-dependent increase in the presynaptic isoforms (α and βI) in the synaptic zone, may require neurotrophic-positive feedback from the postsynaptic component as a result of postsynaptic contractile activity. The increase in α and βII isoforms in the postsynaptic site may be directly induced in the muscle cell when contracting.
Fig. 2.
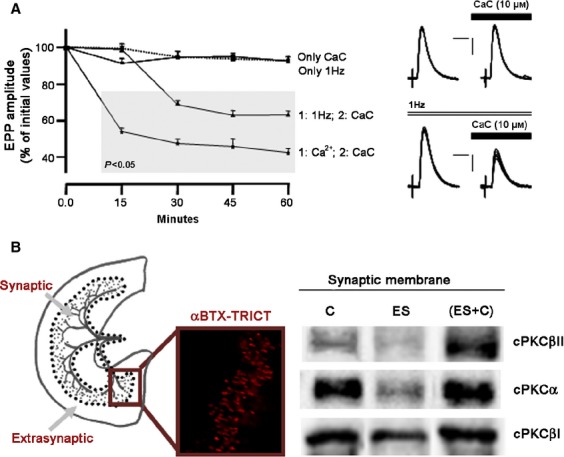
Effect of electrical stimulation and electrical stimulation -induced contraction on nPKCε, cPKCα and cPKC βI. (A) Time course of CaC effect on EPP amplitude when incubated solely (only CaC, 10 μm; dotted line) to the muscle, when a continuous stimulation at 1 Hz was applied previously (1 h) during the CaC incubation (1 : 1 Hz; 2 : CaC) and in the presence of high external calcium (1 : Ca2+; 2 : CaC). We can see also the EPP amplitude when continuous stimulation at 1 Hz was applied (only 1 Hz). Values are expressed as percentages (mean ± SE) with respect to initial amplitude. n = 10–15 single fibers by the kind of experiment. Points into the grey area: P < 0.05 with respect to initial values (0.0 min). On the right, raw data showing examples of the CaC effect on synaptic potentials with and without electrical stimulation. EPPs were recorded before and at 60 min of CaC application. Up: only CaC. Down: CaC with continuous stimulation at 1 Hz. Note that the following changes can be seen only in down: the EPP sizes were diminished and the EPP variances increased. Artefacts are modified for clarity. Scale bars: horizontal: 4 ms, vertical: 3 mV. (B) Drawing of the diaphragm muscle showing the innervated area of the muscle. Dotted lines indicate the place at which synaptic and extrasynaptic zones were separated. The fluorescent image shows the NMJs detected with TRITC-conjugated α-BTX. Note that NMJs are located at the synaptic area and no NMJs were detected in the extrasynaptic area. Western blotting analysis of cPKCα, βI, βII isoform immunoreactivity levels in diaphragm muscles from synaptic membranes under basal conditions (Control) and after electrical stimulation (1 Hz, 30 min) with (ES + C) and without (ES) muscle contraction. Experimental conditions resulted in a significant change (P < 0.05) from control values. Reproduced with permission from Besalduch et al. (2010).
In conclusion, increases in synaptic activity, by nerve stimulation, sufficient to trigger muscle contraction, couples PKC to transmitter release in the rat neuromuscular junction and increases the level of α, βI and βII isoforms in the membrane. The phosphorylation activity of these classical-PKCs also increases. It seems that the muscle needs to contract in order to maintain or increase cPKCs in the membrane. These results indicate that PKCα and/or βI isoforms potentially are directly involved in modulating calcium-dependent ACh release at the NMJ.
nPKCε, a novel type of PKC, is expressed at a high level in the brain and several neural functions of nPKCε, including neurotransmitter release, have been identified (see Shirai et al. 2008 for review). Presynaptic location of the nPKCε at the NMJ has been described (Lanuza et al. 2006b), which suggested that it may also have a role in neurotransmitter release. Functional data indicate that nPKCε is involved in the activity-induced ACh release mechanism at the NMJ and that the action of this isoform seems related to the cPKCβI isoform in an activity-dependent way that also needs muscle contraction (Lanuza et al. 2012).
PKC signallingin developmental activity-dependent synapse elimination
During the development of the nervous system there is an initial overproduction of synapses that is followed by an activity-dependent reduction in the number of connections, which refines the neuronal circuits. The elimination of synapses has been studied extensively at the NMJ and using this model it was demonstrated that both the stability of the postsynaptic receptors and presynaptic neuronal structures contribute to the process of elimination (Jansen et al. 1976; Sanes & Lichtman, 1999; Nelson et al. 2003). Figure 3 shows several examples of NMJs from a newborn animal that are polyinnervated (asterisk) and other NMJs that are monoinnervated. The synapse elimination process is accompanied by changes in the morphology of the AChR cluster in the postsynaptic component; Fig. 3 also shows a classification of the morphology of the AChR clusters into four maturity stages to visualize the maturation of the cluster. As normal maturation takes place, changes in the AChR distribution transform the uniform AChRs oval plaque at birth (M1) into an elongated oval plaque with hints of heterogeneities in receptor density (M2) that later change into clusters containing small areas of low AChR density appearing as holes (M3). This morphology leads to an increasingly structured pattern of fluorescently labelled independent primary gutters (M4) below the nerve terminals. A careful examination of the time course of loss of innervating axons and the disappearance of postsynaptic receptors shows that these mechanisms are gradual (finishing at the end of the 2nd to 3rd postnatal week at the rodent NMJ) and reveals that there is a degree of independence between these two processes. A considerable decrease in polyneuronal innervation occurs at a time when there is relatively little loss of the postsynaptic acetylcholine receptors (Lanuza et al. 2002). On the other hand, there can be a local receptor loss followed by a later loss of the corresponding nerve axon (Balice-Gordon & Lichtman, 1993). On the basis of some of these and other observations, and the competitive nature of nerve and receptor loss, several mechanisms have been postulated to understand the synapse elimination process (Liu et al. 1994; Nguyen & Lichtman, 1996; Chang & Balice-Gordon, 1997; Sanes & Lichtman, 1999; Herrera & Zeng, 2003; Nelson et al. 2003; Wyatt & Balice-Gordon, 2003; Buffelli et al. 2004). Data suggests that serine protein kinases activity could be one of the biological mechanisms that drive this synapse elimination process. Both serine protein kinases C and A play a role in activity-dependent synapse modification: PKC activation results in a decrease in synaptic strength, whereas the action of the PKA opposes or reverses the PKC effect (Jia et al. 1999; Lanuza et al. 2000, 2001, 2002, 2006a, 2010; Li et al. 2001, 2004; Santafe et al. 2001, 2002, 2003, 2007; Nelson et al. 2003).
Fig. 3.
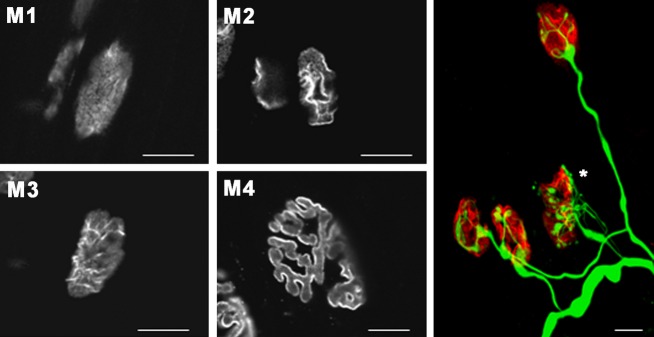
Pre- and postsynaptic changes during developmental activity-dependent synapse elimination. Synaptic AChR cluster morphologies in the neuromuscular junctions of the rat LAL muscle from birth to adulthood. AChRs were stained with rhodamine-conjugated α-bungarotoxin. Postsynaptic AChR clusters are classified from M1 to M4 types according their morphologic maturation. The colour image shows a confocal image showing several NMJs immunostained in green with neurofilament-200 and in red with α-bungarotoxin. Scale bars: 10 μm. Reproduced with permission from Lanuza et al. (2002).
PKC signalling– presynaptic
The functional relationships among PKC activity, calcium inflows, voltage-dependent calcium channels (VDCC) and presynaptic muscarinic acetylcholine receptors (mAChRs) in the modulation of the postnatal developmental activity-dependent synaptic elimination process has been extensively studied (Santafe et al. 2002, 2003, 2007, 2009b; Tomàs et al. 2011). The results support a specific presynaptic model of PKC signalling (Fig. 4A). When PKC is coupled to the neurotransmission mechanism, it enhances ACh release in all synaptic contacts, regardless of their state of developmental maturation (in green in Fig. 4A), excepting those that are ‘weaker’ competitor during synapse elimination (in red in Fig. 4A). In dually innervated endplates, the weak nerve terminal is potentiated by partially reducing calcium entry (P/Q-, N-, or L-type VDCC-specific block or 500 μm magnesium ions), M1- or M4-type selective mAChR block, or PKC block. Moreover, reducing calcium entry or blocking PKC or mAChRs results in the unmasking of functionally silent nerve endings that now recover neurotransmitter release (Santafe et al. 2009b). All these results show interactions between these molecules and indicate that there is a release inhibition mechanism based on an mAChR-PKC-VDCC intracellular cascade. When it is fully active in certain weak motor axons, it can depress ACh release and even disconnect synapses (Tomàs et al. 2011). We suggest that this mechanism plays a central role in the elimination of redundant neonatal synapses, because functional axonal withdrawal can indeed be reversed by mAChRs, VDCCs or PKC block.
Neurotrophins and their receptors – p75NTR and the tyrosine kinase receptor family (trks) – that modulate the synaptic activity of NMJ (Wang & Poo, 1997) have a differential spatial and temporal expression during neuromuscular synapse development (Garcia et al. 2010b,c,d, 2011). This particular configuration of neurotrophin signalling specifically contributes to the control of ACh release during synapse elimination at the NMJ, and could be related to the mechanism based on the previously described mAChR-PKC-VDCC intracellular cascade. Figure 4B represents the localization of neurotrophins [brain-derived neurotrophic factor (BDNF), neurotrophin-3 and 4 (NT-3 and NT-4] and its receptor proteins (p75NTR, trkB and trkC) in neonatal monoinnervated and multiple-innervated NMJ. Figure 4B also shows that neurotrophins enhance neurotransmission and that this role is selective for different nerve terminals depending on their developmental maturation. Therefore, BDNF potentiates ACh release at all synaptic contacts, regardless of their developmental maturation, NT-3 only potentiates neurotransmission in synaptic contacts that are competing and NT-4 only in the mature nerve terminals and not during synapse elimination (Garcia et al. 2010b,c,d, 2011).
Brain-derived neurotrophic factor is a member of the neurotrophin family that plays a role in neuronal proliferation, differentiation and survival during development (Barde et al. 1982; Wang et al. 1995) and also, rapidly potentiates both the spontaneous and evoked synaptic activity of the developing NMJs of Xenopus laevis studied in culture (Poo, 2001) and of adult rat (Garcia et al. 2010a). Therefore, it can be hypothesized that functional differences exist in BDNF signalling between axons that are competing during synapse elimination. There is evidence that exogenous BDNF transiently recruits functionally depressed silent terminals, and this effect seems to be mediated by trkB which may oppose the previously mentioned PKC-mediated ACh release depression (Garcia et al. 2010b). Thus, a balance between trkB, PKC and muscarinic pathways may contribute to the final functional suppression of the weaker and more inactive neuromuscular synapses during development, contributing to developmental synapse disconnection (Tomàs et al. 2011).
PKC signalling – postsynaptic
The maturation of the NMJ involves the transformation of postsynaptic AChR clusters. Although little is known about the regulation of the dynamics of stabilization– destabilization during the development of the postsynaptic apparatus, it has been described that the axons that will be eliminated become less efficient due to the fact that a progressive loss of quantal content is associated with a decreased density of postsynaptic AChRs (Balice-Gordon & Lichtman, 1993). It is known that PKC activity regulates the distribution of AChRs in the NMJ of chicken and mammals by dispersing them (Bursztajn et al. 1988; Ross et al. 1988; Wallace, 1988; Nimnual et al. 1998; Lanuza et al. 2000). Phorbol ester treatment of innervated mouse myotubes causes a reduction in the synaptic potentials, accompanied by a loss of AChR molecules from the functional aggregates (Lanuza et al. 2000), suggesting that phosphorylation by the PKC may drive dispersion. On the other hand, the inhibition of PKC activity in vivo significantly prevents the normal dispersion of AChRs associated with synapse elimination (Lanuza et al. 2002), suggesting that PKC activity regulates the postnatal disappearance of synaptic AChRs in neonatal muscles. Conversely, PMA accelerates the maturation of AChR clusters, suggesting that PKC activity regulates the postnatal redistribution of synaptic AChRs in neonatal muscle (Lanuza et al. 2002). Moreover, the delay in the maturity of the AChRs in postsynaptic clusters after exposure to CaC is accompanied by total prevention of synapse loss during the first postnatal week, indicating a pre- and postsynaptic component to the process (Lanuza et al. 2002).
In vitro experiments have shown that the activation of PKA and PKC has opposite effects on AChR stability in the myotube membrane (Li et al. 2001). Activation of PKA by cAMP totally blocks the dispersion stimulated by PMA, indicating that the action of PKC decreases the stability of the AChR in the membrane and that PKA activity stabilizes the AChR in the aggregates (Li et al. 2001). We also know that these pharmacological manipulations that affect receptor stability also produce changes in the phosphorylation state of the AChR. Thus, PKC activity increases the phosphorylation of the AChR delta subunit (which is the target of PKC), whereas PKA activity increases the phosphorylation of the AChR epsilon subunit (which is the target of PKA) (Lanuza et al. 2006a). We suggest that selective AChR-phosphorylation by PKC and PKA could be one of the causes of dispersion and stability, respectively. Perhaps a balance between the phosphorylating actions of the kinases PKC and PKA could determine the final stabilization of AChRs within clusters. In conclusion, a spatially specific and opposing action of PKC and PKA may result in activity-dependent alterations to synaptic connectivity at both presynaptic nerve terminals and postsynaptic AChR clusters during the initial process of synapse elimination. This balance between PKC and PKA actions could mediate the retention of active neural inputs and the loss of the inactive (or less active) inputs located over the common postsynaptic cell (Fig. 5). In a CNS context, PKC phosphorylation reactions involving central neurotransmitter receptors such as the various glutamate receptors are involved in a number of examples of plasticity (Roche et al. 1994; MacDonald et al. 2001; Sanderson & Dell'Acqua, 2011; Ren et al. 2013). Differential cellular localization of specific kinases and phosphatases that act to regulate receptor membrane insertion and stability may be a key determinant of effective synaptic circuitry in plasticity phenomena such as LTP (Kotecha & MacDonald, 2003; Sacktor, 2008; Anwyl, 2009).
Fig. 5.
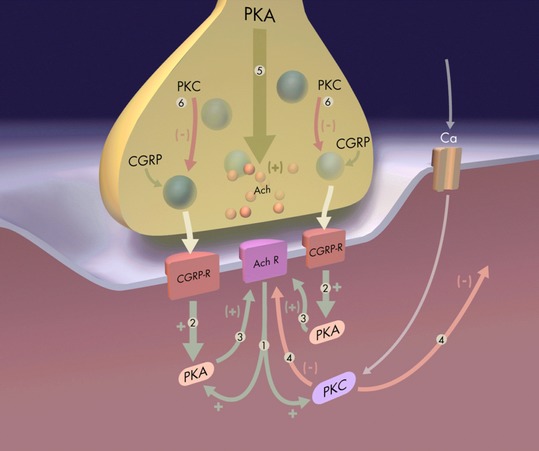
Postsynaptic PKC signalling. A spatially specific and opposing action of PKC and PKA may result in activity-dependent alterations to synaptic connectivity at both the nerve inputs and the postsynaptic AChR clusters in the initial process of synapse elimination. This balance between PKC and PKA actions could mediate the retention of active neural inputs and the loss of the inactive (or less active) inputs located over the common postsynaptic cell. The diagram represents these mechanisms: (1) Cholinergic activation of muscle produces an increase in PKC and PKA activity. (2) Muscle activation, due to peptidergic mechanisms (CGRP), produces an increase in PKA activity, which has a positive effect, increasing efficacy and producing a local synapse stabilization effect (3). (4) PKC activity has a general negative effect; it is widely distributed within the postsynaptic cell and acts on all synapses to reduce postsynaptic responsiveness. The positive, synapse-stabilizing effect, mediated by PKA (2–3), is more localized, counteracts the PKC effect and tends to preserve only stimulated inputs because of this local action. These effects of both PKA and PKC may be due, at least in part, to direct action of the kinases in phosphorylating the AChR at the posynaptic site. In addition, neural activity produces both positive (5) and negative (6) effects on transmitter and neuropeptide output on presynaptic site (Li et al., 2001).
nPKCθ is required for postnatal activity-dependent synapse elimination and maturation of the nAChR cluster
There are several indirect pieces of evidence showing that nPKCθ is an isoform of PKC that can play a specific role in NMJ postnatal development. First, the nPKCθ isoform has been shown to be strongly expressed in skeletal muscle and is under neural control (Hilgenberg & Miles, 1995; Hilgenberg et al. 1996). Secondly, overexpression of nPKCθ in myotubes inhibited agrin-induced AChR cluster formation and disrupted pre-existing AChR clusters (Miles & Wagner, 2003), suggesting that this isoform inhibits the function of agrin (clustering of the AChRs in the postsynaptic component (Sanes & Lichtman, 1999). Thirdly, the nPKCθ isoform is translocated to the membrane by PMA (Lanuza et al. 2000). Finally, nPKCθ is more highly expressed in postnatal development than in the adult (Lanuza et al. 2010). Therefore, it has been important to examine whether nPKCθ is related to the process of postnatal activity-dependent synapse elimination and maturation of AChR clusters using mice lacking this particular isoform.
When synapse elimination is analyzed in KO mice, a significant delay from P4 to P12 is found, indicates that nPKCθ could act in the initial process of synapse elimination (Lanuza et al. 2010). It has also been demonstrated that the elimination of synapses remains altered in co-cultures in which muscle cells do not express the isoform theta of the PKC and, surprisingly, synapse elimination also remained suppressed in co-cultures in which only the neurons were KO for nPKCθ, suggesting a coordinated pre- and postsynaptic role of this isoform (Li et al. 2004). In vivo results also show that the process of synaptic disconnection that takes place during the postnatal period in the NMJ depends at least, at the postsynaptic level, on the activation of the isoform theta of the PKC (Lanuza et al. 2010). Figure 6 shows several examples of NMJs from developing WT and KO muscles indicating that KO mice show a delay in presynaptic and postsynaptic maturation. In the KO polyinnervated P4 synapse shown in Fig. 6 (KO, P4), the cluster morphology corresponds to stage M1 and the cluster morphology of the WT (P4) corresponds to M3. Also, in the monoinnervated synapses (P8) shown in Fig. 6, the cluster morphology of the WT corresponds to stage M3, whereas the KO corresponds to M2, indicating that even after eliminating multiple innervation, the developmental delay present in the nAChR cluster morphology persists. Moreover, differences in the maturity of the presynaptic component have been also seen in both WT and KO mice, with a higher number of axonal inputs in the P4 KO synapse than in the P4 WT synapse (Fig. 6) and nerve terminal growth (axonal sprouts not apposed to nAChRs) observed in KO synapses (arrow in Fig. 6, P4). These examples and the quantitative analysis performed help to conclude that KO NMJs from different developing ages showed signs of being less mature than the WT NMJs with changes at nerve terminals and in postsynaptic receptors (Lanuza et al. 2010). Although the synapses are less mature there are no morphological abnormalities present even at the ultra-structural level. Moreover, NMJs in the adult KO mice have a normal appearance, indicating that PKCθ is critical in initial development of the NMJ but not for its final maturation (Fig. 6; Besalduch et al. 2011).
Fig. 6.

Confocal and electron microscopy images of NMJs from newborn and adult. Confocal microphotographs from wild type (WT) and knockout in nPKCθ (KO) NMJs at neonatal postnatal 4-day (P4, polyinnervated NMJs), postnatal 8-day (P8, monoinnervated NMJs) and adult were stained for AChRs (red) and axons and nerve terminals (neurofilament-200 protein in green). The selected examples of polyinnervated and monoinnervated NMJs from developing WT and KO muscles (P4, P8) show differences in the pre- and postsynaptic components that are in accordance with the quantitative measurements of the cluster morphology. Scale bars: 10 μm. The ultrastructure images of the KO NMJs during development and in the adult have the same structure than NMJs with comparable levels of maturation in WT muscles at P3, P6, P13 and adult. The NMJ from a P3 KO muscle shows the coexistence of several nerve terminal boutons (marked by asterisks in the drawing at the right of the picture) on a poorly defined, low-density, postsynaptic membrane without gutters. The NMJ from a P6 KO muscle shows an intermediary stage of axon separation. The NMJ from a P13 KO muscle shows advanced gutter formation and nerve terminal segregation and elimination. Nerve terminals (marked with # in the accompanying drawing) are engulfed by Schwann cell processes that also contain membrane debris. The right column shows drawings in which the three cellular components of the NMJ (axon terminal, Schwann cells and postsynaptic membrane density) have been delineated. Scale bar: 200 nm. Reproduced with permission from Lanuza et al. (2010) and Besalduch et al. (2011).
The target of PKCθ phosphorylation at the postsynaptic site
As mentioned above, pharmacological PKC manipulations that alter receptor stability also produce changes in the phosphorylation of the delta subunit of nAChRs (Lanuza et al. 2006a). It has been demonstrated that nPKCθ-dependent phosphorylation affects the serine phosphorylation sites of the nAChR delta subunit because there is a decrease of the phosphorylation of this subunit in the KO mice associated with the delay in maturity of nAChR clusters (Lanuza et al. 2010). Moreover, this variation in phosphorylation level comes about with no apparent change in the expression of the delta subunit of the AChR cluster in KO mice (Lanuza et al. 2010), indicating that the nPKCθ isoform of PKC is required to phosphorylate the delta-AChR subunit. So, AChR phosphorylation depends on the nPKCθ. These results and others suggest that the increased phosphorylation of AChR on serine residues in the major cytoplasmic domain of the delta subunit could promote the detachment of nAChRs from their anchorage to structural proteins which constrain the lateral mobility of clustered nAChR and favour the emergence of areas free of AChRs (holes: stages M3 and M4, Fig. 3).
Furthermore, KO muscles also show deficient phosphorylation of the epsilon AChR subunit (which is the target of PKA) during postnatal development, suggesting that nPKCθ may not only contribute to the phosphorylation of the delta subunit but also influence PKA phosphorylation on the epsilon subunit (Lanuza et al. 2010). It is known that PKA-specific phosphorylation of the epsilon subunit affects the physiology of the AChR (Nishizaki & Sumikawa, 1994). It is also known that the PKC-dependent AChR dispersion can be blocked or reversed by changes in PKA activity and that the activation of PKA results in greater stability of AChR clusters (Li et al. 2001) and increases the phosphorylation of the epsilon subunit of the AChR on the membrane of +cultured myotubes (Lanuza et al. 2006a). Thus, the correct phosphorylation of AChR delta and epsilon subunits would aid the AChR cluster to mature appropriately and this would also affect synapse loss during postnatal development. PKC would produce AChR instability and loss by its phosphorylation of the delta subunit, whereas PKA would reverse this effect and increase receptor stability by its phosphorylation of the epsilon subunit. PKC action would also, however, be required for PKA to produce that phosphorylation and receptor stabilization. This interaction, we propose, could induce a sharpening of the boundaries between areas of highly concentrated, stabilized AChR, (dominated by PKA phosphorylated receptors at pre/post synaptic appositions) and receptor-poor areas in which PKC (in the absence of PKA action) has destabilized the receptor. On the basis of this and previous studies, we hypothesize that the spatially specific and opposing action of nPKCθ and PKA may result in activity-dependent alterations to synaptic connectivity at both the nerve terminals and the AChR clusters.
Conclusion
In this review, we summarize the PKC signalling that plays critical roles in specific functional aspects of the NMJ: neurotransmission in the adult and maturation of the synapse in the neonate. PKC is a ubiquitous signalling molecule with effects that are dependent upon localization. Several isoforms of PKC are present within a single cell in the NMJ, each mediating unique intracellular functions. Studies using Western blotting or confocal microscopy reveal complex and specific localization of PKC isoforms in their inactive and their active state. This review focuses on cPKC (α, βI, βII, γ) and nPKC (ε, θ) subfamilies and provides evidence that there is a specific cellular distribution of these PKC isoforms in the NMJ that favours the functional diversity of these isoforms. Specifically, we demonstrate that the PKC family couples to transmitter release at the neuromuscular junction when synaptic activity increases. PKCα and/or βI isoforms could be directly involved in modulating calcium-depending ACh release at the NMJ. nPKCε is also involved in the activity-induced ACh release mechanism at the NMJ and the action of this isoform seems related to the cPKCβI isoform in an activity-dependent way that also requires muscle contraction. nPKCθ might play a role in developing NMJs, by affecting the phosphorylation and stability of the receptor clusters during the initial postnatal development.
Acknowledgments
This work was supported by a grant from MEC (SAF2011-23711) and a grant from the Catalan Government (Generalitat) (2009SGR0 1248).
Authors' contributions
M.A.L., M.M.S., N.G., P.G.N., J.T.: Concept, literature search, data interpretation. M.A.L., J.T.: Manuscript preparation. M.A.L., N.G.: Confocal microscopy. M.A.L., M.M.S., N.G., N.B., M.T., T.O.: Quantitative analysis. M.A.L., M.M.S.: Statistics. M.M.S., M.T., T.O.: Electrophysiological techniques. N.B., M.P.: Western blotting techniques. N.B., M.T., M.P.: Imunohistochemical techniques. N.B.: Electron microscopy. N.B., M.T., T.O., M.P.: Data collection.
References
- Abbs ET, Joseph DN. The effects of atropine and oxotremorine on acetylcholine release in rat phrenic nerve diaphragm preparations. Br J Pharmacol. 1981;73:481–483. doi: 10.1111/j.1476-5381.1981.tb10446.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antipenko A, Frías JA, Parra J, et al. Effect of chronic electrostimulation of rabbit skeletal muscle on calmodulin level and protein kinase activity. Int J Biochem Cell Biol. 1999;31:303–310. doi: 10.1016/s1357-2725(98)00112-5. [DOI] [PubMed] [Google Scholar]
- Anwyl R. Metabotropic glutamate receptor-dependent long-term potentiation. Neuropharmacology. 2009;56:735–740. doi: 10.1016/j.neuropharm.2009.01.002. [DOI] [PubMed] [Google Scholar]
- Arakawa M, Mizoguchi A, Masutani M, et al. Ultrastructural localization of protein kinase C beta-subspecies in the axon terminal of rat neuromuscular junction. Neurosci Res. 1993;16:125–130. doi: 10.1016/0168-0102(93)90079-6. [DOI] [PubMed] [Google Scholar]
- Arenson MS. Muscarinic inhibition of quantal transmitter release from the magnesium-paralysed frog sartorius muscle. Neuroscience. 1989;30:827–836. doi: 10.1016/0306-4522(89)90174-7. [DOI] [PubMed] [Google Scholar]
- Balice-Gordon RJ, Lichtman JW. In vivo observations of pre- and postsynaptic changes during the transition from multiple to single innervation at developing neuromuscular junctions. J Neurosci. 1993;13:834–855. doi: 10.1523/JNEUROSCI.13-02-00834.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barde YA, Edgar D, Thoenen H. Purification of a new neurotrophic factor from mammalian brain. EMBO J. 1982;1:549–553. doi: 10.1002/j.1460-2075.1982.tb01207.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besalduch N, Tomàs M, Santafe MM, et al. Synaptic activity-related classical protein kinase C isoform localization in the adult rat neuromuscular synapse. J Comp Neurol. 2010;518:211–228. doi: 10.1002/cne.22220. [DOI] [PubMed] [Google Scholar]
- Besalduch N, Santafe MM, Garcia N, et al. Transmitter release in the neuromuscular synapse of the protein kinase C theta-deficient adult mouse. J Comp Neurol. 2011;519:849–855. doi: 10.1002/cne.22551. [DOI] [PubMed] [Google Scholar]
- Brandt DT, Goerke A, Heuer M, et al. Protein kinase C delta induces Src kinase activity via activation of the protein tyrosine phosphatase PTP alpha. J Biol Chem. 2003;278:34073–34078. doi: 10.1074/jbc.M211650200. [DOI] [PubMed] [Google Scholar]
- Buffelli M, Busetto G, Bidoia C, et al. Activity-dependent synaptic competition at mammalian neuromuscular junctions. News Physiol Sci. 2004;19:85–91. doi: 10.1152/nips.01464.2003. [DOI] [PubMed] [Google Scholar]
- Bursztajn S, Schneider LW, Jong YJ, et al. Phorbol esters inhibit the synthesis of acetylcholine receptors in cultured muscle cells. Biol Cell. 1988;63:57–65. [PubMed] [Google Scholar]
- Byrne JH, Kandel ER. Presynaptic facilitation revisited: state and time dependence. J Neurosci. 1996;16:425–435. doi: 10.1523/JNEUROSCI.16-02-00425.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canto C, Suarez E, Lizcano JM, et al. Neuregulin signaling on glucose transport in muscle cells. J Biol Chem. 2004;279:12260–12268. doi: 10.1074/jbc.M308554200. [DOI] [PubMed] [Google Scholar]
- Catterall WA. Interactions of presynaptic Ca2+ channels and snare proteins in neurotransmitter release. Ann NY Acad Sci. 1999;868:144–159. doi: 10.1111/j.1749-6632.1999.tb11284.x. [DOI] [PubMed] [Google Scholar]
- Caulfield MP. Muscarinic receptors – characterization, coupling and function. Pharmacol Ther. 1993;58:319–379. doi: 10.1016/0163-7258(93)90027-b. [DOI] [PubMed] [Google Scholar]
- Chang Q, Balice-Gordon RJ. Nip and tuck at the neuromuscular junction: a role for proteases in developmental synapse elimination. BioEssays. 1997;19:271–275. doi: 10.1002/bies.950190402. [DOI] [PubMed] [Google Scholar]
- Cleland PJ, Appleby GJ, Rattigan S, et al. Exercise-induced translocation of protein kinase C and production of diacylglycerol and phosphatidic acid in rat skeletal muscle in vivo. Relationship to changes in glucose transport. J Biol Chem. 1989;264:17704–17711. [PubMed] [Google Scholar]
- Ekström PA, Bergstrand H, Edström A. Effects of protein kinase inhibitors on regeneration in vitro of adult frog sciatic sensory axons. J Neurosci Res. 1992;31:462–469. doi: 10.1002/jnr.490310308. [DOI] [PubMed] [Google Scholar]
- Ganguly DK, Das M. Effects of oxotremorine demonstrate presynaptic muscarinic and dopaminergic receptors on motor nerve terminals. Nature. 1979;278:645–646. doi: 10.1038/278645a0. [DOI] [PubMed] [Google Scholar]
- Garcia N, Santafe MM, Tomas M, et al. The interaction between tropomyosin-related kinase B receptors and presynaptic muscarinic receptors modulates transmitter release in adult rodent motor nerve terminals. J Neurosci. 2010a;30:16514–16522. doi: 10.1523/JNEUROSCI.2676-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia N, Santafe MM, Tomas M, et al. Involvement of Brain-derived neurotrophic factor (BDNF) in the functional elimination of synaptic contacts at polyinnervated neuromuscular synapses during development. J Neurosci Res. 2010b;88:1406–1419. doi: 10.1002/jnr.22320. [DOI] [PubMed] [Google Scholar]
- Garcia N, Santafe MM, Tomas M, et al. Neurotrophin-4 couples to locally modulated ACh release at the end of neuromuscular synapse maturation. Neurosci Lett. 2010c;468:72–74. doi: 10.1016/j.neulet.2009.10.066. [DOI] [PubMed] [Google Scholar]
- Garcia N, Santafe MM, Tomàs M, et al. Involvement of neurotrophin-3 (NT-3) in the functional elimination of synaptic contacts during neuromuscular development. Neurosci Lett. 2010d;473:141–145. doi: 10.1016/j.neulet.2010.02.040. [DOI] [PubMed] [Google Scholar]
- Garcia N, Tomàs M, Santafe MM, et al. Blocking p75 (NTR) receptors alters polyinnervation of neuromuscular synapses during development. J Neurosci Res. 2011;89:1331–1341. doi: 10.1002/jnr.22620. [DOI] [PubMed] [Google Scholar]
- Herrera AA, Zeng Y. Activity-dependent switch from synapse formation to synapse elimination during development of neuromuscular junctions. J Neurocytol. 2003;32:817–833. doi: 10.1023/B:NEUR.0000020626.29900.fb. [DOI] [PubMed] [Google Scholar]
- Hilgenberg L, Miles K. Developmental regulation of a protein kinase C isoform localized in the neuromuscular junction. J Cell Sci. 1995;108:51–61. doi: 10.1242/jcs.108.1.51. [DOI] [PubMed] [Google Scholar]
- Hilgenberg L, Yearwood S, Milstein S, et al. Neural influence on protein kinase C expression in skeletal muscle. J Neurosci. 1996;16:4994–5003. doi: 10.1523/JNEUROSCI.16-16-04994.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen JK, Van Essen DC, Brown MC. Formation and elimination of synapses in skeletal muscles of rat. Cold Spring Harb Symp Quant Biol. 1976;40:425–434. doi: 10.1101/sqb.1976.040.01.040. [DOI] [PubMed] [Google Scholar]
- Jia M, Li M, Dunlap V, et al. The thrombin receptor mediates functional activity-dependent neuromuscular synapse reduction via protein kinase C activation in vitro. J Neurobiol. 1999;38:369–381. [PubMed] [Google Scholar]
- Kim S, Bondeva T, Nelson PG. Activation of protein kinase C isozymes in primary mouse myotubes by carbachol. Brain Res Dev Brain Res. 2002;137:13–21. doi: 10.1016/s0165-3806(02)00362-0. [DOI] [PubMed] [Google Scholar]
- Kotecha SA, MacDonald JF. Signaling molecules and receptor transduction cascades that regulate NMDA receptor-mediated synaptic transmission. Int Rev Neurobiol. 2003;54:51–106. doi: 10.1016/s0074-7742(03)54003-x. [DOI] [PubMed] [Google Scholar]
- Kraft AS, Anderson WB. Phorbol esters increase the amount of Ca2+, phospholipid-dependent protein kinase associated with plasma membrane. Nature. 1983;301:621–623. doi: 10.1038/301621a0. [DOI] [PubMed] [Google Scholar]
- Kraft AS, Anderson WB, Cooper HL, et al. Decrease in cytosolic calcium/phospholipid-dependent protein kinase activity following phorbol ester treatment of EL4 thymoma cells. J Biol Chem. 1982;257:13193–13196. [PubMed] [Google Scholar]
- Lanuza MA, Li MX, Jia M, et al. Protein kinase C-mediated changes in synaptic efficacy at the neuromuscular junction in vitro: the role of postsynaptic acetylcholine receptors. J Neurosci Res. 2000;61:616–625. doi: 10.1002/1097-4547(20000915)61:6<616::AID-JNR5>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Lanuza MA, Garcia N, Santafe M, et al. Pertussis toxin-sensitive G-protein and protein kinase C activity are involved in normal synapse elimination in the neonatal rat muscle. J Neurosci Res. 2001;63:330–340. doi: 10.1002/1097-4547(20010215)63:4<330::AID-JNR1027>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Lanuza MA, Garcia N, Santafe M, et al. Pre- and postsynaptic maturation of the neuromuscular junction during neonatal synapse elimination depends on protein kinase C. J Neurosci Res. 2002;67:607–617. doi: 10.1002/jnr.10122. [DOI] [PubMed] [Google Scholar]
- Lanuza MA, Gizaw R, Viloria A, et al. Phosphorylation of the nicotinic acetylcholine receptor in myotube-cholinergic neuron cocultures. J Neurosci Res. 2006a;83:1407–1414. doi: 10.1002/jnr.20848. [DOI] [PubMed] [Google Scholar]
- Lanuza MA, Besalduch N, Garcia N, et al. PKA and PKC localization at the adult rat neuromuscular junction. 2006b. FENS Forum Abstracts. Vol. 3, A048.2.240. The Federation of European Neuroscience Societies. http://www.fns.org.
- Lanuza MA, Besalduch N, Garcia N, et al. Plastic embedded semithin cross-sections as a tool for high-resolution immunofluorescence analysis of the neuromuscular junction molecules: specific cellular location of protease-activated receptor-1. J Neurosci Res. 2007;85:748–756. doi: 10.1002/jnr.21192. [DOI] [PubMed] [Google Scholar]
- Lanuza MA, Besalduch N, González C, et al. Decreased phosphorylation of δ and ε subunits of the acetylcholine receptor coincides with delayed postsynaptic maturation in PKC θ deficient mouse. Exp Neurol. 2010;225:183–195. doi: 10.1016/j.expneurol.2010.06.014. [DOI] [PubMed] [Google Scholar]
- Lanuza MA, Besalduch N, Obis T, et al. New Orleans, LA: Society for Neuroscience; 2012. Exclusive presynaptic location and coupling to ACh release of the novel protein kinase C epsilon isoform in the adult rat neuromuscular synapse. Program No. 137.11. 2012 Neuroscience Meeting Planner. Online. [Google Scholar]
- Li MX, Jia M, Jiang H, et al. Opposing actions of protein kinase A and C mediate Hebbian synaptic plasticity. Nat Neurosci. 2001;4:871–872. doi: 10.1038/nn0901-871. [DOI] [PubMed] [Google Scholar]
- Li MX, Jia M, Yang LX, et al. The role of the theta isoform of protein kinase C (PKC) in activity-dependent synapse elimination: evidence from the PKC theta knock-out mouse in vivo and in vitro. J Neurosci. 2004;24:3762–3769. doi: 10.1523/JNEUROSCI.3930-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Fields RD, Fitzgerald S, et al. Proteolytic activity, synapse elimination, and the Hebb synapse. J Neurobiol. 1994;25:325–335. doi: 10.1002/neu.480250312. [DOI] [PubMed] [Google Scholar]
- MacDonald JF, Kotecha SA, Lu WY, et al. Convergence of PKC-dependent kinase signal cascades on NMDA receptors. Curr Drug Targets. 2001;2:299–312. doi: 10.2174/1389450013348452. [DOI] [PubMed] [Google Scholar]
- Miles K, Wagner M. Overexpression of nPKC theta is inhibitory for agrin-induced nicotinic acetylcholine receptor clustering in C2C12 myotubes. J Neurosci Res. 2003;71:188–195. doi: 10.1002/jnr.10467. [DOI] [PubMed] [Google Scholar]
- Mochly-Rosen D. Localization of protein kinases by anchoring proteins: a theme in signal transduction. Science. 1995;268:247–251. doi: 10.1126/science.7716516. [DOI] [PubMed] [Google Scholar]
- Mochly-Rosen D, Gordon AS. Anchoring proteins for protein kinase C: a means for isozyme selectivity. FASEB J. 1998;12:35–42. [PubMed] [Google Scholar]
- Mochly-Rosen D, Khaner H, Lopez J, et al. Intracellular receptors for activated protein kinase C. Identification of a binding site for the enzyme. J Biol Chem. 1991a;266:14866–14868. [PubMed] [Google Scholar]
- Mochly-Rosen D, Khaner H, Lopez J. Identification of intracellular receptor proteins for activated protein kinase C. Proc Natl Acad Sci USA. 1991b;88:3997–4000. doi: 10.1073/pnas.88.9.3997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moraczewski J, Nowotniak A, Wrobel E, et al. Differential changes in protein kinase C associated with regeneration of rat extensor digitorum longus and soleus muscles. Int J Biochem Cell Biol. 2002;34:938–949. doi: 10.1016/s1357-2725(02)00014-6. [DOI] [PubMed] [Google Scholar]
- Nakano S, Shimohama S, Saitoh T, et al. Localization of protein kinase C in human skeletal muscle. Muscle Nerve. 1992;15:496–499. doi: 10.1002/mus.880150414. [DOI] [PubMed] [Google Scholar]
- Nelson PG, Lanuza MA, Jia M, et al. Phosphorylation reactions in activity-dependent synapse modification at the neuromuscular junction during development. J Neurocytol. 2003;32:803–816. doi: 10.1023/B:NEUR.0000020625.70284.a6. [DOI] [PubMed] [Google Scholar]
- Nguyen QT, Lichtman JW. Mechanism of synapse disassembly at the developing neuromuscular junction. Curr Opin Neurobiol. 1996;6:104–112. doi: 10.1016/s0959-4388(96)80015-8. [DOI] [PubMed] [Google Scholar]
- Nimnual AS, Chang W, Chang NS, et al. Identification of phosphorylation sites on AChR-subunit associated with dispersal of AChR clusters on the surface of muscle cells. Biochemistry. 1998;37:14823–14832. doi: 10.1021/bi9802824. [DOI] [PubMed] [Google Scholar]
- Nishizaki T, Sumikawa K. A cAMP-dependent Ca2+ signalling pathway at the endplate provided by the gamma to epsilon subunit switch in ACh receptors. Brain Res Mol Brain Res. 1994;24:341–345. doi: 10.1016/0169-328x(94)90148-1. [DOI] [PubMed] [Google Scholar]
- Nishizuka Y. Protein kinase C and lipid signaling for sustained cellular responses. FASEB J. 1995;9:484–496. [PubMed] [Google Scholar]
- Numann R, Hauschka SD, Catterall WA, et al. Modulation of skeletal muscle sodium channels in a satellite cell line by protein kinase C. J Neurosci. 1994;14:4226–4236. doi: 10.1523/JNEUROSCI.14-07-04226.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oancea E, Meyer T. Protein kinase C as a molecular machine for decoding calcium and diacylglycerol signals. Cell. 1998;95:307–318. doi: 10.1016/s0092-8674(00)81763-8. [DOI] [PubMed] [Google Scholar]
- Perkins GA, Wang L, Huang LJ, et al. PKA, PKC, and AKAP localization in and around the neuromuscular junction. BMC Neurosci. 2001;2:17. doi: 10.1186/1471-2202-2-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrini S, Henriksson J, Zierath JR, et al. Exercise-induced protein kinase C isoform-specific activation in human skeletal muscle. Diabetes. 2004;53:21–24. doi: 10.2337/diabetes.53.1.21. [DOI] [PubMed] [Google Scholar]
- Poo MM. Neurotrophins as synaptic modulators. Nat Rev Neurosci. 2001;2:24–32. doi: 10.1038/35049004. [DOI] [PubMed] [Google Scholar]
- Ren SQ, Yan JZ, Zhang XY, et al. PKCλ is critical in AMPA receptor phosphorylation and synaptic incorporation during LTP. EMBO J. 2013;32:1365–1380. doi: 10.1038/emboj.2013.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter EA, Cleland PJ, Rattigan S, et al. Contraction-associated translocation of protein kinase C in rat skeletal muscle. FEBS Lett. 1987;217:232–236. doi: 10.1016/0014-5793(87)80669-5. [DOI] [PubMed] [Google Scholar]
- Roberts RE, McLean WG. Protein kinase C isozyme expression in sciatic nerves and spinal cords of experimentally diabetic rats. Brain Res. 1997;754:147–156. doi: 10.1016/s0006-8993(97)00062-0. [DOI] [PubMed] [Google Scholar]
- Roche KW, Tingley WG, Huganir RL. Glutamate receptor phosphorylation and synaptic plasticity. Curr Opin Neurobiol. 1994;4:383–388. doi: 10.1016/0959-4388(94)90100-7. [DOI] [PubMed] [Google Scholar]
- Rose AJ, Michell BJ, Kemp BE, et al. Effect of exercise on protein kinase C activity and localization in human skeletal muscle. J Physiol. 2004;561:861–870. doi: 10.1113/jphysiol.2004.075549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross A, Rapuano M, Prives J. Induction of phosphorylation and cell surface redistribution of acetylcholine receptors by phorbol ester and carbamylcholine in cultured chick muscle cells. J Cell Biol. 1988;107:1139–1145. doi: 10.1083/jcb.107.3.1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacktor TC. PKMzeta, LTP maintenance, and the dynamic molecular biology of memory storage. Prog Brain Res. 2008;169:27–40. doi: 10.1016/S0079-6123(07)00002-7. [DOI] [PubMed] [Google Scholar]
- Saito N, Itouji A, Totani Y, et al. Cellular and intracellular localization of epsilon-subspecies of protein kinase C in the rat brain; presynaptic localization of the epsilon-subspecies. Brain Res. 1993;607:241–248. doi: 10.1016/0006-8993(93)91512-q. [DOI] [PubMed] [Google Scholar]
- Salvatori S, Furlan S, Millikin B, et al. Localization of protein kinase C in skeletal muscle T-tubule membranes. Biochem Biophys Res Commun. 1993;196:1073–1080. doi: 10.1006/bbrc.1993.2360. [DOI] [PubMed] [Google Scholar]
- Sanderson JL, Dell'Acqua ML. AKAP signaling complexes in regulation of excitatory synaptic plasticity. Neuroscientist. 2011;17:321–336. doi: 10.1177/1073858410384740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanes JR, Lichtman JW. Development of the vertebrate neuromuscular junction. Annu Rev Neurosci. 1999;22:389–442. doi: 10.1146/annurev.neuro.22.1.389. [DOI] [PubMed] [Google Scholar]
- Santafe MM, Garcia N, Lanuza MA, et al. Calcium channels coupled to neurotransmitter release at dually innervated neuromuscular junctions in the newborn rat. Neuroscience. 2001;102:697–708. doi: 10.1016/s0306-4522(00)00507-8. [DOI] [PubMed] [Google Scholar]
- Santafe MM, Garcia N, Lanuza MA, et al. Decreased calcium influx into the neonatal rat motor nerve terminals can recruit additional neuromuscular junctions during the synapse elimination period. Neuroscience. 2002;110:147–154. doi: 10.1016/s0306-4522(01)00543-7. [DOI] [PubMed] [Google Scholar]
- Santafe MM, Salon I, Garcia N, et al. Modulation of ACh release by presynaptic muscarinic autoreceptors in the neuromuscular junction of the newborn and adult rat. Eur J Neurosci. 2003;17:119–127. doi: 10.1046/j.1460-9568.2003.02428.x. [DOI] [PubMed] [Google Scholar]
- Santafe MM, Lanuza MA, Garcia N, et al. Calcium inflow-dependent protein kinase C activity is involved in the modulation of transmitter release in the neuromuscular junction of the adult rat. Synapse. 2005;57:76–84. doi: 10.1002/syn.20159. [DOI] [PubMed] [Google Scholar]
- Santafe MM, Lanuza MA, Garcia N, et al. Muscarinic autoreceptors modulate transmitter release through protein kinase C and protein kinase A in the rat motor nerve terminal. Eur J Neurosci. 2006;23:2048–2056. doi: 10.1111/j.1460-9568.2006.04753.x. [DOI] [PubMed] [Google Scholar]
- Santafe MM, Garcia N, Lanuza MA, et al. Protein kinase C activity affects neurotransmitter release at polyinnervated neuromuscular synapses. J Neurosci Res. 2007;85:1449–1457. doi: 10.1002/jnr.21280. [DOI] [PubMed] [Google Scholar]
- Santafe MM, Garcia N, Lanuza MA, et al. Interaction between protein kinase C and protein kinase A can modulate transmitter release at the rat neuromuscular synapse. J Neurosci Res. 2009a;87:683–690. doi: 10.1002/jnr.21885. [DOI] [PubMed] [Google Scholar]
- Santafe MM, Garcia N, Lanuza MA, et al. Presynaptic muscarinic receptors, calcium channels, and protein kinase C modulate the functional disconnection of weak inputs at polyinnervated neonatal neuromuscular synapses. J Neurosci Res. 2009b;87:1195–1206. doi: 10.1002/jnr.21934. [DOI] [PubMed] [Google Scholar]
- Shirai Y, Adachi N, Saito N. Protein kinase Cepsilon: function in neurons. FEBS J. 2008;275:3988–3994. doi: 10.1111/j.1742-4658.2008.06556.x. [DOI] [PubMed] [Google Scholar]
- Tanaka C, Nishizuka Y. The protein kinase C family for neuronal signaling. Annu Rev Neurosci. 1994;17:551–567. doi: 10.1146/annurev.ne.17.030194.003003. [DOI] [PubMed] [Google Scholar]
- Tomàs J, Santafe MM, Lanuza MA, et al. Silent synapses in neuromuscular junction development. J Neurosci Res. 2011;89:3–12. doi: 10.1002/jnr.22494. [DOI] [PubMed] [Google Scholar]
- Tsuruno S, Hirano T. Persistent activation of protein kinase Calpha is not necessary for expression of cerebellar long-term depression. Mol Cell Neurosci. 2007;35:38–48. doi: 10.1016/j.mcn.2007.01.016. [DOI] [PubMed] [Google Scholar]
- Van Ginneken MM, Keizer HA, Wijnberg ID, et al. Immunohistochemical identification and fiber type specific localization of protein kinase C isoforms in equine skeletal muscle. Am J Vet Res. 2004;65:69–73. doi: 10.2460/ajvr.2004.65.69. [DOI] [PubMed] [Google Scholar]
- Vary TC, Goodman S, Kilpatrick LE, et al. Nutrient regulation of PKCepsilon is mediated by leucine, not insulin, in skeletal muscle. Am J Physiol Endocrinol Metab. 2005;289:684–694. doi: 10.1152/ajpendo.00613.2004. [DOI] [PubMed] [Google Scholar]
- Wallace B. Regulation of agrin-induced acetylcholine receptor aggregation by Ca2+ and phorbol ester. J Cell Biol. 1988;107:267–268. doi: 10.1083/jcb.107.1.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XH, Poo MM. Potentiation of developing synapses by postsynaptic release of neurotrophin-4. Neuron. 1997;19:825–835. doi: 10.1016/s0896-6273(00)80964-2. [DOI] [PubMed] [Google Scholar]
- Wang T, Xie K, Lu B. Neurotrophins promote maturation of developing neuromuscular synapses. J Neurosci. 1995;15:4796–4805. doi: 10.1523/JNEUROSCI.15-07-04796.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessler I, Karl M, Mai M, et al. Muscarine receptors on the rat phrenic nerve, evidence for positive and negative muscarinic feedback mechanisms. Naunyn-Schmiedeberg's Arch Pharmacol. 1987;335:605–612. doi: 10.1007/BF00166975. [DOI] [PubMed] [Google Scholar]
- West JW, Numann R, Murphy BJ, et al. A phosphorylation site in the Na+ channel required for modulation by protein kinase C. Science. 1991;254:866–868. doi: 10.1126/science.1658937. [DOI] [PubMed] [Google Scholar]
- Wyatt RM, Balice-Gordon RJ. Activity-dependent elimination of neuromuscular synapses. J Neurocytol. 2003;32:777–794. doi: 10.1023/B:NEUR.0000020623.62043.33. [DOI] [PubMed] [Google Scholar]