

Abstract
BACKGROUND
Targeting tumor metabolism by energy restriction-mimetic agents (ERMAs) has emerged as a strategy for cancer therapy/prevention. Evidence suggests a mechanistic link between ERMA-mediated antitumor effects and epigenetic gene regulation.
METHODS
Microarray analysis showed that a novel thiazolidinedione-derived ERMA, CG-12, and glucose deprivation could suppress DNA methyltransferase (DNMT)1 expression and reactivate DNA methylation-silenced tumor suppressor genes in LNCaP prostate cancer cells. Thus, we investigated the effects of a potent CG-12 derivative, CG-5, vis-à-vis 2-deoxyglucose, glucose deprivation and/or 5-aza-deoxycytidine, on DNMT isoform expression (Western blotting, RT-PCR), DNMT1 transcriptional activation (luciferase reporter assay), and expression of genes frequently hypermethylated in prostate cancer (quantitative real-time PCR). Promoter methylation was assessed by pyrosequencing analysis. SiRNA-mediated knockdown and ectopic expression of DNMT1 were used to validate DNMT1 as a target of CG-5.
RESULTS
CG-5 and glucose deprivation upregulated the expression of DNA methylation-silenced tumor suppressor genes, including GADD45a, GADD45b, IGFBP3, LAMB3, BASP1, GPX3, and GSTP1, but also downregulated methylated tumor/invasion-promoting genes, including CD44, S100A4, and TACSTD2. In contrast, 5-aza-deoxycytidine induced global reactivation of these genes. CG-5 mediated these epigenetic effects by transcriptional repression of DNMT1, which was associated with reduced expression of Sp1 and E2F1. SiRNA-mediated knockdown and ectopic expression of DNMT1 corroborated DNMT1's role in the modulation of gene expression by CG-5. Pyrosequencing revealed differential effects of CG-5 versus 5-aza-deoxycytidine on promoter methylation in these genes.
CONCLUSIONS
These findings reveal a previously uncharacterized epigenetic effect of ERMAs on DNA methylation-silenced tumor suppressor genes, which may foster novel strategies for prostate cancer therapy.
Keywords: energy restriction-mimetic agent, prostate cancer, energy restriction, DNA methyltransferases, epigenetics
INTRODUCTION
Cells undergoing malignant transformation often exhibit a shift in cellular metabolism from oxidative phosphorylation to glycolysis, known as the Warburg effect, to gain growth advantage in the microenvironment (1,2)]. This enhanced glycolysis appears to be attributable to the dysregulation of multiple oncogenic signaling pathways (1), including those mediated by hypoxia-inducible factor 1 (3), Akt (4), c-Myc (5), and p53 (6), and enables cancer cells to adapt to low-oxygen environments, to produce biosynthetic building blocks needed for cell proliferation, to acidify the local environment to facilitate tumor invasion, and to generate NADPH and glutathione through the pentose phosphate shunt to increase resistance to oxidative stress (1,2). The Warburg effect is considered to be a fundamental property of neoplasia, and constitutes the basis for tumor imaging by [18F]2-fluoro-2-deoxyglucose positron emission tomography (7). From a therapeutic perspective, targeting glycolysis represents a relevant strategy for cancer prevention and treatment (2), of which the proof-of-concept is provided by the effective suppression of carcinogenesis in various animal models by dietary caloric restriction and natural product-based energy restriction-mimetic agents (ERMAs), such as 2-deoxyglucose (2-DG) and resveratrol.
Previously, based on the scaffold of thiazolidinediones, we developed a novel class of ERMAs, as represented by CG-12, that mimic the ability of 2-DG and glucose deprivation to elicit starvation-like cellular responses with high potency in cancer cells through the inhibition of glucose uptake (8). The suppression of energy metabolism by CG-12 leads to an intricate signaling network mediated by silent information regulator 1, AMP-activated protein kinase, and oxidative stress, the interplay among which culminates in autophagy and apoptosis in cancer cells. More recently, we demonstrated an epigenetic effect of CG-12 in cancer cells involving histone acetylation and H3 lysine 4 methylation, leading to the transcriptional activation of Kruppel-like factor 6 (KLF6) and a series of proapoptotic genes (9). In this study, we report the unique ability of CG-5, a structurally optimized CG-12 derivative (Fig. 1A) to suppress the expression of DNA methyltransferase (DNMT)1 and DNMT3A in prostate cancer cells, resulting in the reactivation of a series of DNA methylation-silenced tumor suppressor genes. Pyrosequencing analysis indicates that this effect was attributable to hypomethylation in the promoter regions of these tumor suppressor genes. In light of the important role of aberrant DNA methylation in carcinogenesis (10), our findings underscore the translational potential of this novel class of glucose uptake inhibitors in prostate cancer prevention and therapy.
Fig. 1.

CG-5, 2-DG, glucose starvation and 5-aza-dC differentially affect the expression levels of DNMT isoforms. (A) Left, structures of CG-12 and CG-5. Center, dose-dependent inhibitory effects of CG-5 versus CG-12 on the uptake of [3H]-2DG into LNCaP cells after 30 min of treatment. Point, mean; bars, SD (n = 3). Right, dose-dependent inhibitory effects of CG-5 versus CG-12 on the viability of LNCaP cells by MTT assays after 72 h of treatment. Point, mean; bars, SD (n = 6). (B) LNCaP cells were treated with CG-5, 2-DG and 5-aza-dC at the indicated concentrations in 10% FBS-supplemented medium for 48 h or glucose-free medium for various time intervals. The expression levels of DNMT1, DNMT3A and DNMT3B were determined by Western blotting (upper) and RT-PCR (lower). The percentages denote the relative intensities of mRNA and protein bands of treated samples to those of the respective DMSO vehicle-treated controls after normalization to the respective internal reference β-actin. Each value represents the average of three independent experiments.
MATERIALS AND METHODS
Detailed information on materials, reagents, their commercial sources, and experimental procedures are available in Supplementary Information.
Microarray analysis
Total RNA isolated from LNCaP cells exposed to 10 μM CG-12 or glucose-depleted medium for 48 h was submitted to the Microarray Shared Resource at The Ohio State University Comprehensive Cancer Center for microarray analysis of gene expression.
Glucose uptake assay
This assay was performed as we described previously (8) with modifications. Specifically, LNCaP cells were treated with test agents for 1.5 h, followed by exposure to [3H]2-DG in the presence of excess non-radioactive 2-DG for 30 min.
Cell viability assay
Cell viability was determined using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay as we described previously (8).
RNA interference and luciferase reporter assay
For siRNA experiments, cells were transfected with scrambled or DNMT1-specific siRNA. Knockdown of DNMT1 was confirmed by immunoblotting. For the DNMT1 promoter-luciferase reporter assay, luciferase activities were determined with the dual-luciferase system, which uses co-transfected herpes simplex virus thymidine kinase promoter-driven Renilla reniformis luciferase as an internal control.
Western blotting
Western blotting was performed as described previously (8). Relative differences in protein levels among experimental groups were determined by densitometry.
DNA methylation analysis by pyrosequencing
To determine methylation levels of candidate genes in response to drug treatment or glucose deprivation, the Pyrosequencing System (Qiagen) was used to detect methylated CpG sites in sequencing reactions (13).
Statistical analysis
Data from quantitative real time (qRT)-PCR, luciferase reporter assays and pyrosequencing were analyzed using Student's t test. Differences between group means were considered significant at P < 0.05.
RESULTS
Microarray analysis reveals the suppression of DNMT1 and DNMT3A expression and the upregulation of methylation-silenced genes by energy restriction in prostate cancer cells
Pursuant to our hypothesis that energy restriction mediates antitumor effects, in part, through epigenetic gene regulation, we examined the effect of 10 μM CG-12 versus glucose depletion on global gene expression in LNCaP cells via cDNA microarray analysis after 48 h of treatment. This microarray analysis showed that both treatments significantly reduced the gene expression of DNMT1, accompanied by a modest, but statistically significant, decrease in DNMT3B expression and no change in DNMT3A levels (Table 1).
Table 1.
Microarray analyses of the effects of 10 μM CG-12 versus glucose depletion on the expression of DNMTs in LNCaP cells after 48 h of treatment
| Gene Name | 10 μM CG-12 | Glucose Depletion | ||
|---|---|---|---|---|
| Fold Change | P value | Fold Change | P value | |
| DNMT1 | −2.208398 | 5.32E-06 | −2.1143294 | 6.45E-07 |
| DNMT3A | −1.023485 | 0.690570233 | −1.0391039 | 0.404186671 |
| DNMT3B | −1.468048 | 0.001498209 | −1.3947437 | 2.82E-04 |
The ability of CG-12 and glucose starvation to downregulate DNMT expression suggests a mechanistic link between energy restriction and epigenetic regulation of gene expression through changes in DNA methylation. Previously, a global survey of DNA methylation patterns in prostate cancer cell lines identified a number of cancer-related genes that were transcriptionally silenced due to aberrant promoter hypermethylation (14). Based on this report, we examined the microarray data for the effect of CG-12 versus glucose depletion on the expression of 13 genes reported to be silenced by DNA methylation (Table 2). Among these, BASP1, GADD45a, GADD45b, GPX3, GSTP1, IGFBP3, KRT7, LAMB3, PDLIM4, and THBS1 are tumor-suppressive genes, whereas CD44, S100A4, and TACSTD2 have been associated with tumorigenesis or aggressive phenotype of prostate cancer (15-17). It is noteworthy that CG-12 mimicked the ability of glucose starvation to activate the expression of GADD45a, GADD45b, and IGFBP3, while many other genes examined were not affected by either treatment, with the exception of THBS1, which was upregulated by CG-12.
Table 2.
Microarray analyses of the effects of 10 μM CG-12 versus glucose depletion on the expression of thirteen DNA methylation-silenced genes in LNCaP cells after 48 h of treatment
| Gene Name | Gene Description | Molecular Function | CG-12 | Glucose Depletion |
|---|---|---|---|---|
|
Fold change (P value) | ||||
| BASP1 (NM_006317_1) | Brain acid soluble protein 1 | Inhibition of Myc-induced cell transformation; a potential tumor suppressor (18) | 1.182303151 (0.027893026) | 0.920825697 (0.307789115) |
| CD44 (NM_000610_1) | Receptor for hyaluronic acid | Cell adhesion; markers for breast and prostate cancer stem cells (17) | n/a | n/a |
| GADD45a (NM_001924-1) | Growth arrest and DNA-damage-inducible protein 45a | Apoptosis, cell cycle arrest, and DNA repair; tumor suppressive (19) | 5.838104359 (3.73E-10) | 3.11320375 (1.21E-08) |
| GADD45b (AL050044_1) | Growth arrest and DNA-damage-inducible protein 45b | Apoptosis, cell cycle arrest, and DNA repair; reported tumor suppressor in hepatocellular carcinoma (20) | 5.333934097 (2.01E-08) | 4.483731849 (5.37E-09) |
| GPX3 (NM_002084) | Glutathione peroxidase 3 | Maintaining genomic integrity via the detoxification of reactive oxygen species; tumor suppressive (21) | 1.089147993 (0.370942845) | 0.946090435 (0.352881698) |
| GSTP1 (NM_000852_1) | Glutathione S-transferase pi | Conjunction and detoxification of carcinogens; tumor suppressive (22) | 1.192508872 (0.028969162) | 0.952208888 (0.362806007) |
| IGFBP3 (NM_000598_1) | Insulin-like growth factor binding protein 3 | Inhibiting cancer cell proliferation, adhesion, motility, and metastasis; tumor suppressive (23,24) | 5.195274579 (4.89E-08) | 2.137167525 (4.50E-07) |
| KRT7 (NM_005556_1) | Keratin 7 | Cytoskeletal organization and biogenesis (14) | 1.241685666 (0.018831416) | 1.04298615 (0.622715314) |
| LAMB3 (NM_000228_1) | Laminin, b3 | A component of the extracellular matrix involved in cell adhesion, growth, migration, proliferation, and differentiation; tumor suppressive (25) | n/a | 1.22357285 (0.031432552) |
| PDLIM4 (NM_003687_1) | PDZ and LIM domain 3 | An actin-binding protein; tumor suppressive (26) | 0.993684661 (0.94564817) | 0.981663174 (0.801008691) |
| S100A4 (NM_019554_1) | S100 calcium-binding protein A4 | A Ca2+-binding protein; promoting metastasis (27) | 0.900563555 (0.217905821) | 0.933789972 (0.206088795) |
| TACSTD2 (NM_002353_1) | Tumor-associated calcium signal transducer 2 | A marker of human prostate basal cells with stem cell characteristics; promoting tumorigenesis and invasion (15) | n/a | 0.907644973 (0.10922003) |
| THBS1 (NM_003246_1) | Thrombospondin-1 | Cell adhesion and motility; a p53 and Rb regulated angiogenesis inhibitor; tumor suppressive (28) | 2.609367236 (8.89E-08) | 0.986198525 (0.755628795) |
n/a: Gene was not listed in the array results.
Energy restriction suppresses the expression of DNMT1 and DNMT3A through transcriptional repression and proteasomal degradation, respectively
Our efforts to structurally optimize CG-12 led to the identification of CG-5, an active derivative in which the terminal methylcyclohexyl ring was replaced by a 3-pentyl moiety. This simple modification improved the potency of CG-5 relative to CG-12 in suppressing [3H]-2DG uptake (IC50, 6 μM versus 9 μM) and cell viability (IC50, 4.5 μM versus 6 μM) in LNCaP cells (Fig. 1A). Consequently, we used CG-5 to validate our microarray data by examining the dose-dependent suppressive effects of CG-5 and 2-DG vis-à-vis glucose starvation on the expression of DNMT1, DNMT3A, and DNMT3B, at both protein and mRNA levels, in LNCaP cells. In addition, the DNMT inhibitor 5-aza-dC was used as a control in light of its reported activity in suppressing DNMT1 expression via proteasomal degradation (29,30), and, to a lesser extent, DNMT3A through a yet unidentified mechanism (31).
Western blot analysis indicated that CG-5, 2-DG, and glucose depletion shared with 5-aza-dC the ability to decrease the expression levels of DNMT1 and, to a lesser extent, DNMT3A in a dose- or time-dependent manner (Fig. 1B, upper panel). Nevertheless, the mechanism underlying energy restriction-facilitated downregulation of DNMT1 was different from that of 5-aza-dC as CG-5, 2-DG, and glucose starvation decreased DNMT1 mRNA levels, while no significant changes were noted in response to 5-aza-dC (Fig. 1B, lower panel). In addition, consistent with the microarray findings, no changes in the mRNA level of DNMT3A were noted in response to any of these treatments, suggestive of a posttranslational effect on protein levels. As for DNMT3B, neither the protein nor mRNA expression level was affected by any of the treatments, which contrasted with the microarray data that showed a modest decrease in DNMT3B gene expression in response to energy restriction (Table 1). This discrepancy might have arisen from inherent systematic errors associated with microarrays (32).
To examine the mechanism by which CG-5 suppressed the mRNA expression of DNMT1, we assessed its effect on the promoter activity of DNMT1 by using a DNMT1 promoter-luciferase reporter construct. As shown, CG-5 diminished the luciferase activity in a dose-dependent manner (Fig. 2A), suggesting that CG-5 suppressed DNMT1 expression through transcriptional repression. As the core promoter region of DNMT1 contains three Sp1 (33) and four E2F (34) binding sites, we examined the effect of CG-5 on the expression of these transcription factors and their target genes, androgen receptor (AR) for Sp1 (35) and cyclins E and D3 for E2F1 (36,37).
Fig. 2.

CG-5 suppresses the expression of DNMT1 and DNMT3A through transcriptional repression and proteasomal degradation, respectively. (A) Dose-dependent, suppressive effect of CG-5 on DNMT1 promoter activity. LNCaP cells were transiently transfected with the DNMT1 promoter-luciferase reporter plasmid pGL3-DNMT1-Luc, and exposed to CG-5 at the indicated concentrations or DMSO vehicle control in 10% FBS-containing medium for 48 h. Column, mean (n = 3); error bars, SD. (B) Parallel Western blot and RT-PCR analyses of the dose-dependent suppressive effect of CG-5 on the expression levels of Sp1, E2F1, AR, and cyclins D3 and E. (C) The proteasome inhibitor MG132 protected cells from CG-5-induced ablation of DNMT3A, but not DNMT1. LNCaP cells were treated with CG-5 at the indicated concentrations for 36 h, followed by co-treatment with 10 μM MG132 for an additional 12 h. Cell lysates were analyzed by immunoblotting for DNMT1 and -3A.
In line with our previous findings with CG-12 (8,9), CG-5 facilitated a dose-dependent decrease in Sp1 protein level without affecting mRNA expression, suggestive of proteasomal degradation (Fig. 2B). It is noteworthy that the expression of E2F1, at both protein and mRNA levels, was also reduced suggesting a different mode of regulation from that of Sp1. Moreover, decreases in the expression of Sp1 and E2F1 were accompanied by parallel decreases in the expression of their respective targets, namely AR and cyclins D3 and E (Fig. 2B).
As for the CG-5-mediated inhibition of DNMT3A protein expression, a role for proteasomal degradation was supported by the ability of the proteasome inhibitor MG-132 to rescue DNMT3A protein expression from drug-induced suppression in LNCaP cells (Fig. 2C).
Similar findings regarding the ability of CG-5 to suppress the expression of DNMT1 and DNMT3A without disturbing that of DNMT3B was also noted in PC-3 and DU-145 cells (Fig. 3), indicating that this was not a cell line-specific effect. Moreover, the drug's effect on DNMT1 expression correlated with that on E2F1 and Sp1 expression in a dose-dependent manner, suggestive of a causal relationship.
Fig. 3.
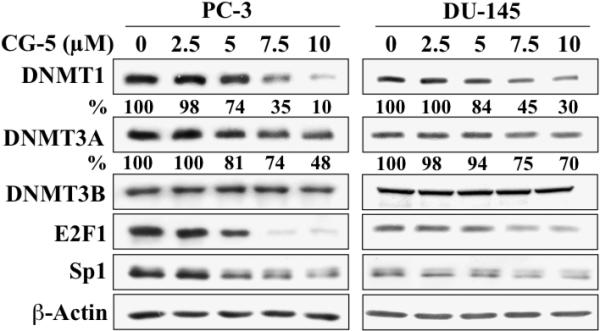
Western blot analysis of the dose-dependent effects of CG-5 on the expression of DNMT1, DNMT3A, DNMT3B, E2F1, and Sp1 in PC-3 and DU-145 cells. Cells were treated with CG-5 at the indicated concentrations for 48 h. The percentages denote the relative intensities of protein bands of treated samples to those of the respective DMSO vehicle-treated controls after normalization to the respective internal reference β-actin. Each value represents the average of three independent experiments.
Differential effects of energy restriction on the activation of DNA methylation-silenced genes
The 13 DNA methylation-silenced genes previously evaluated by microarray analysis (Table 2) were assessed by qRT-PCR for changes in expression in response to energy restriction. LNCaP cells were exposed to 5 μM CG-5 or 5 μM 5-aza-dC in 10% FBS-supplemented RPMI 1640 medium for 48 or 72 h, or to 10% FBS-supplemented glucose-free medium for 72 h. qRT-PCR analysis indicates that these treatments led to distinct patterns of activation of these epigenetically silenced genes (Fig. 4). 5-Aza-dC mediated varying degrees of activation of 12 of the 13 genes examined relative to the DMSO control (at 72 h: GSTP1, 510-fold; KRT7, 295-fold; CD44, 26-fold; TACSTD2, 23-fold; BASP1, 12-fold; LAMB3, 10-fold; IGFBP3, 9-fold; GPX3, S100A4, and THBS1, 8-fold; PDLIM4 and GADD45a, 3-fold), while no significant change in GADD45b mRNA expression was noted (Fig. 4A). In contrast, CG-5 activated 7 of the 13 genes with a distinct preference for the two DNA damage response genes GADD45a and GADD45b (81- and 31-fold, respectively), followed by IGFBP3 (12-fold), LAMB3 (11-fold), BASP1 (9-fold), GPX3 (5-fold), and GSTP1 (2-fold) at 72 h, while only modest increases (<2-fold) in the expression of KRT7 and THBS1 were noted (Fig. 4B). Moreover, CG-5 downregulated the mRNA levels of PDLIM4, S100A4, and TACSTD2 by 97%, 56%, and 95%, respectively. Although CG-5 caused a modest, but statistically insignificant, increase in CD44 mRNA expression at 48 h (1.16-fold), the treatment led to a 54% decrease (P < 0.05) at 72 h. It is noteworthy that two of these downregulated genes, S100A4 and TACSTD2, are associated with the promotion of tumorigenesis, tumor invasion, and metastasis (15,27), and that CD44 represents a putative marker for prostate cancer stem cells (38).
Fig. 4.
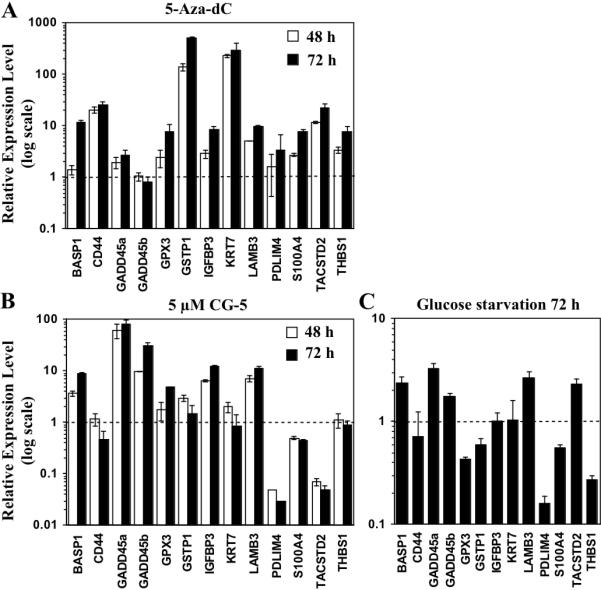
Effects of 5-aza-dC, CG-5, and glucose deprivation on the expression levels of methylation-silenced cancer-related genes. LNCaP cells were treated with (A) 5 μM CG-5 or (B) 5 μM 5-aza-dC in 10% FBS-supplemented medium for 48 or 72 h, or with (C) glucose-depleted medium for 72 h. The expression levels of thirteen target genes reported to be silenced by DNA hypermethylation in prostate cancer cells were quantitated by qRT-PCR. Column, mean (n = 3); error bars, SD.
As compared to CG-5-induced energy restriction, glucose deprivation showed a qualitatively similar, but muted effect on gene activation, which, in part, may be reflective of smaller decreases in the expression levels of DNMT1 and DNMT3A. Glucose-depleted medium shared the ability of CG-5 to activate GADD45a (3.3-fold), LAMB3 (2.7-fold), BASP1 (2.4-fold), and GADD45b (1.8-fold), as well as to downregulate the expression of PDLIM4 and S100A4, while having no significant impact on the mRNA expression of CD44 and KRT7 (Fig. 4C). However, in contrast to CG-5, glucose deprivation diminished the mRNA expression of GPX3, GSTP1, and THBS1, and increased that of TACSTD2.
Role of DNMT1 downregulation in CG-5-facilitated activation of epigenetically silenced genes
Given the greater suppressive effect of CG-5 on DNMT1 expression than on that of DNMT3A (81% and 26%, respectively; Fig. 1B, upper panel), we rationalized that DNMT1 downregulation played a major role in the CG-5-mediated activation of these methylation-silenced genes. This premise was corroborated by two lines of evidence. First, qRT-PCR analysis indicated that siRNA-mediated knockdown of DNMT1 in LNCaP cells mimicked the effects of CG-5 by activating, by at least 2-fold, many of the same genes, including IGFBP3 (7.5-fold), BASP1 (3.8-fold), LAMB3 (2.9-fold), and GSTP1 (2.3-fold), as well as sharply reducing the expression of PDLIM4 (Fig. 5A). Second, ectopic DNMT1 expression attenuated the effect of CG-5 on the expression of many of the 13 genes examined (Fig. 5B). With the exception of IGFBP3, DNMT1 overexpression diminished the extent of CG-5-mediated gene activation, returning the expression levels of many of these genes, such as BASP1, GADD45b, GPX3, and GSTP1, to the basal level or lower. DNMT1 overexpression also abrogated the suppressive effect of CG-5 on the expression of PDLIM4 and TACSTD2. However, ectopic DNMT1 expression had no significant effect on S100A4 expression.
Fig. 5.

Evidence that DNMT1 plays a pivotal role in CG-5-mediated regulation of methylation-silenced cancer-related gene expression. (A) siRNA-mediated knockdown of DNMT1 mimics the effect of CG-5 on the expression of the selected thirteen genes reported to be silenced by DNA hypermethylation in prostate cancer cells, as determined by qRT-PCR (right). LNCaP cells were transiently transfected with DNMT1 or control (Ctl) siRNA, and then treated with 5 μM CG-5 for 72 h. The expression levels of DNMT isoforms were analyzed by Western blotting to confirm the specificity of the knockdown (left). Column, mean (n = 3); error bars, SD. (B) Ectopic expression of DNMT1 protects cells from CG-5-mediated effects on the expression of the selected thirteen methylation-silenced genes, as determined by qRT-PCR (right). LNCaP cells were transiently transfected with the Flag-tagged DNMT1 or control (Ctl) vector, and then treated with 5 μM CG-5 for 48 h. The expression levels of DNMT isoforms following treatment with CG-5 or DMSO control were analyzed by Western blotting (left). Column, mean (n = 3); error bars, SD.
CG-5 alters CpG methylation in the promoter region of the 13 hypermethylated genes in LNCaP cells
To correlate the aforementioned changes in gene expression with the effects of 5-aza-dC, CG-5, and glucose deprivation on DNA methylation, we used pyrosequencing to analyze DNA methylation at CpG islands in the promoter regions of the aforementioned 13 genes in response to individual treatments (Fig. 6A - M). Pyrosequencing is the leading method for quantitative DNA methylation analysis, in part, due to its ability to identify differentially methylated positions in close proximity, thereby allowing concurrent quantification of multiple CpG sites in the promoter region (39). As neighboring CpG sites within a single promoter showed different degrees of methylation (Fig. 6A – M, right panels; each color-coded circle represents a single CpG site and each designated row represents a treatment condition), the average of all sites was used to represent the level of methylation for each gene (left panels).
Fig. 6.

Effects of CG-5, glucose deprivation, and 5-aza-dC on CpG island methylation in methylation-silenced cancer-related genes. LNCaP cells were treated with DMSO control (Ctl), 5 μM 5-aza-dC (dC), 5 μM CG-5 (CG) or glucose-depleted medium [G(-)] for 48 and 72 h. Pyrosequencing analysis of CpG island methylation in the promoter/first exon regions of the selected thirteen methylation-silenced genes was performed as described in Materials and Methods. A, BASP1; B, CD44; C, GADD45a; D, GADD45b; E, GPX3; F, GSTP1; G, IGFBP3; H, KRT7; I, LAMB3; J, PDLIM4; K, S100A4; L, TACSTD2; M, THBS1. Average methylation levels of individual CpG sites in the promoter/first exon of each of the genes examined under each condition are represented by the color-coded circles (right panels; dark blue, 100%; white, 0%; scale at top of each column). The average methylation level of all CpG sites within each of the promoters/first exons was used to represent the level of methylation for each of the thirteen genes under each treatment condition (left panel; column, mean (n = 3 - 19); error bars, SD). a and b: DMSO control for 48 and 72 h, respectively; c and d: 5 μM 5-aza-dC for 48 and 72 h, respectively; e and f: 5 μM CG-5 for 48 and 72 h, respectively; g and h: glucose-depleted medium for 48 and 72 h, respectively.
LNCaP cells were treated with DMSO (Fig. 6, control; a and b for 48 and 72 h, respectively), 5 μM 5-aza-dC (c and d), 5 μM CG-5 (e and f), or glucose-depleted medium (g and h), after which genomic DNA was collected for pyrosequencing analysis. As noted, the promoter and/or the first exon of each of these 13 genes contain multiple CpG sites, ranging from 3 to 19 sites. Not only did the methylation level among these sites vary within a single promoter/exon region (Fig. 6 A-M, right panels, a and b), but also the total methylation levels of the promoters/exons varied greatly among these genes (left panels, a and b). For example, while many of these genes were highly methylated, BASP1 and THBS1 showed only 10% and 20% CpG methylation, respectively, in control cells (Fig. 6A and M, respectively). Consistent with the qRT-PCR findings, 5-aza-dC, CG-5, and glucose-depleted medium exhibited differential effects on the DNA methylation patterns of these genes. 5-Aza-dC facilitated decreases in DNA methylation in all of the genes examined (all panels, c and d). These epigenetic changes correlated with activation of these genes in 5-aza-dC-treated LNCaP cells (Fig. 4A) with the exception of GADD45b (panel D), of which the mRNA levels remained unaltered after drug treatment. It is noteworthy that, while CG-5 mediated the hypomethylation and resulting activation of many tumor suppressor genes, it enhanced the DNA methylation of PDLIM4 (panel J) and the tumor-promoting genes S100A4 and TACSTD2 (panels K and L, respectively), resulting in the downregulated expression of these genes (Fig. 4B). The effects of glucose-depleted medium on DNA methylation of many of these genes paralleled those of CG-5, however, to a lesser extent. Nevertheless, glucose starvation contrasted with CG-5-induced energy restriction in its opposite effects on the DNA methylation pattern of GPX3, GSTP1, TACSTD2, and THBS1 (panels E, F, L, and M, respectively), which underlies the observed differences in the effects of these two treatments on the activation of these DNA methylation-silenced genes (Fig. 4C).
DISCUSSION
Aberrant promoter hypermethylation of critical pathway genes plays an important role in prostate carcinogenesis and tumor progression (40,41), thereby representing a therapeutically relevant target for cancer treatment (42). In this study, we demonstrated the high potency of the novel ERMA CG-5 relative to 2-DG in suppressing the expression of DNMT1 and, to a lesser extent, DNMT3A, which led to the reactivation of a series of DNA methylation-silenced tumor suppressor genes, including GADD45a, GADD45b, IGFBP3, LAMB3, BASP1, GPX3, and GSTP1, in prostate cancer cells through promoter hypomethylation.
The effect of CG-5 on DNA methylation profiles is largely associated with the reduction in the expression of DNMT1 as siRNA-mediated knockdown and ectopic expression of DNMT1 mimicked and diminished, respectively, the ability of CG-5 to modulate the expression of these silenced genes. Although CG-5 and 5-aza-dC share the ability to downregulate DNMT1 expression, the underlying mechanisms are distinctly different. Evidence suggests that CG-5 facilitated the downregulation of DNMT1 expression through transcriptional repression, which our data suggest is be associated with the reduced expression of Sp1 and E2F1. Our previous study demonstrated that β-transducin repeat-containing protein (β-TrCP)-dependent proteasomal degradation of Sp1 represents one of the energy restriction-associated cellular responses elicited by CG-12 (8), and is likely the mechanism by which Sp1 is suppressed in CG-5-treated cells. In contrast, CG-5-mediated suppression of E2F1 expression occurred at the transcriptional level.
The specificity with which CG-5 activates DNA methylation-silenced genes is noteworthy, and contrasts with the nonspecific reactivation of nearly of all the silenced genes examined by 5-aza-dC. For example, our data indicate that CG-5 reduced the basal expression levels of PDLIM4 and the three tumor/invasion-promoting genes, namely CD44, S100A4, and TACSTD2, while 5-aza-dC increased the expression of these genes by 8- to 26-fold. This target specificity was further confirmed by pyrosequencing analysis, which showed the differential effect of CG-5 versus 5-aza-dC on DNA methylation in the promoter regions of the 13 genes examined. While 5-aza-dC caused universal hypomethylation of all of these 13 genes, CG-5 enhanced the DNA methylation of PDLIM4 and the tumor-promoting genes S100A4 and TACSTD2. However, the suppressive effect of CG-5 on CD44 promoter methylation relative to the control (65.9% versus 67.42%; P = 0.033) represents an anomaly since CG-5 reduced CD44 expression.
Mechanistically, this differential regulation of DNA methylation-silenced genes is attributed not just to CG-5's effect on DNA hypomethylation alone, but also to its ability to affect the expression of transcription factors, such as Sp1 and E2F1, and histone-modifying enzymes (9). Together, these concerted actions underline a more complicated mode of epigenetic gene regulation than that of 5-aza-dC's inhibitory effect on DNMT activity alone.
CONCLUSIONS
It is well recognized that cancer cells undergo a metabolic shift to anaerobic glycolysis that provides growth advantages within the tumor microenvironment. Consequently, there is intense interest in targeting tumor metabolism as a therapeutic strategy, including small-molecule approaches. In this study, we demonstrate that epigenetic activation of DNA methylation-silenced tumor suppressor genes represents an important antitumor response to energy restriction. Moreover, our novel small-molecule ERMA, GC5, regulates the expression of these genes through modulation of DNA methylation and, perhaps by virtue of its concomitant effects on histone modifications, exhibits target gene specificity and a broader spectrum of antitumor gene activation that might offer therapeutic advantages over DNMT inhibitors.
Supplementary Material
Acknowledgments
Grant Sponsors: National Institutes of Health grant CA112250 and Department of Defense Prostate Cancer Research Program grant W81XWH-09-0198 to CSC; predoctoral fellowship to HYL from the Graduate Student Study Abroad Program of the National Science Council, Taiwan.
Footnotes
Disclosure Statement: The authors declare no conflicts of interest.
REFERENCES
- 1.Kroemer G, Pouyssegur J. Tumor cell metabolism: cancer's Achilles’ heel. Cancer Cell. 2008;13(6):472–482. doi: 10.1016/j.ccr.2008.05.005. [DOI] [PubMed] [Google Scholar]
- 2.Vander Heiden MG. Targeting cancer metabolism: a therapeutic window opens. Nat Rev Drug Discov. 2011;10(9):671–684. doi: 10.1038/nrd3504. [DOI] [PubMed] [Google Scholar]
- 3.Denko NC. Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat Rev Cancer. 2008;8(9):705–713. doi: 10.1038/nrc2468. [DOI] [PubMed] [Google Scholar]
- 4.Elstrom RL, Bauer DE, Buzzai M, Karnauskas R, Harris MH, Plas DR, Zhuang H, Cinalli RM, Alavi A, Rudin CM, Thompson CB. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res. 2004;64(11):3892–3899. doi: 10.1158/0008-5472.CAN-03-2904. [DOI] [PubMed] [Google Scholar]
- 5.Osthus RC, Shim H, Kim S, Li Q, Reddy R, Mukherjee M, Xu Y, Wonsey D, Lee LA, Dang CV. Deregulation of glucose transporter 1 and glycolytic gene expression by c-Myc. J Biol Chem. 2000;275(29):21797–21800. doi: 10.1074/jbc.C000023200. [DOI] [PubMed] [Google Scholar]
- 6.Matoba S, Kang JG, Patino WD, Wragg A, Boehm M, Gavrilova O, Hurley PJ, Bunz F, Hwang PM. p53 regulates mitochondrial respiration. Science. 2006;312(5780):1650–1653. doi: 10.1126/science.1126863. [DOI] [PubMed] [Google Scholar]
- 7.Kelloff GJ, Hoffman JM, Johnson B, Scher HI, Siegel BA, Cheng EY, Cheson BD, O'Shaughnessy J, Guyton KZ, Mankoff DA, Shankar L, Larson SM, Sigman CC, Schilsky RL, Sullivan DC. Progress and promise of FDG-PET imaging for cancer patient management and oncologic drug development. Clin Cancer Res. 2005;11(8):2785–2808. doi: 10.1158/1078-0432.CCR-04-2626. [DOI] [PubMed] [Google Scholar]
- 8.Wei S, Kulp SK, Chen CS. Energy restriction as an antitumor target of thiazolidinediones. J Biol Chem. 2010;285(13):9780–9791. doi: 10.1074/jbc.M109.065466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen CH, Huang PH, Chu PC, Chen MC, Chou CC, Wang D, Kulp SK, Teng CM, Wang Q, Chen CS. Energy restriction-mimetic agents induce apoptosis in prostate cancer cells in part through epigenetic activation of KLF6 tumor suppressor gene expression. J Biol Chem. 2011;286(12):9968–9976. doi: 10.1074/jbc.M110.203240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sharma S, Kelly TK, Jones PA. Epigenetics in cancer. Carcinogenesis. 2010;31(1):27–36. doi: 10.1093/carcin/bgp220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang J, Wei S, Wang DS, Wang YC, Kulp SK, Chen CS. Pharmacological exploitation of the peroxisome proliferator-activated receptor gamma agonist ciglitazone to develop a novel class of androgen receptor-ablative agents. J Med Chem. 2008;51(7):2100–2107. doi: 10.1021/jm701212m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25(4):402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 13.Tost J, Gut IG. DNA methylation analysis by pyrosequencing. Nat Protoc. 2007;2(9):2265–2275. doi: 10.1038/nprot.2007.314. [DOI] [PubMed] [Google Scholar]
- 14.Ibragimova I, Ibanez de Caceres I, Hoffman AM, Potapova A, Dulaimi E, Al-Saleem T, Hudes GR, Ochs MF, Cairns P. Global reactivation of epigenetically silenced genes in prostate cancer. Cancer Prev Res (Phila) 2010;3(9):1084–1092. doi: 10.1158/1940-6207.CAPR-10-0039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goldstein AS, Lawson DA, Cheng D, Sun W, Garraway IP, Witte ON. Trop2 identifies a subpopulation of murine and human prostate basal cells with stem cell characteristics. Proc Natl Acad Sci U S A. 2008;105(52):20882–20887. doi: 10.1073/pnas.0811411106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Herrlich P, Morrison H, Sleeman J, Orian-Rousseau V, Konig H, Weg-Remers S, Ponta H. CD44 acts both as a growth- and invasiveness-promoting molecule and as a tumor-suppressing cofactor. Ann N Y Acad Sci. 2000;910:106–118. doi: 10.1111/j.1749-6632.2000.tb06704.x. discussion 118-120. [DOI] [PubMed] [Google Scholar]
- 17.Li F, Tiede B, Massague J, Kang Y. Beyond tumorigenesis: cancer stem cells in metastasis. Cell Res. 2007;17(1):3–14. doi: 10.1038/sj.cr.7310118. [DOI] [PubMed] [Google Scholar]
- 18.Hartl M, Nist A, Khan MI, Valovka T, Bister K. Inhibition of Myc-induced cell transformation by brain acid-soluble protein 1 (BASP1). Proc Natl Acad Sci U S A. 2009;106(14):5604–5609. doi: 10.1073/pnas.0812101106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zerbini LF, Libermann TA. GADD45 deregulation in cancer: frequently methylated tumor suppressors and potential therapeutic targets. Clin Cancer Res. 2005;11(18):6409–6413. doi: 10.1158/1078-0432.CCR-05-1475. [DOI] [PubMed] [Google Scholar]
- 20.Qiu W, David D, Zhou B, Chu PG, Zhang B, Wu M, Xiao J, Han T, Zhu Z, Wang T, Liu X, Lopez R, Frankel P, Jong A, Yen Y. Down-regulation of growth arrest DNA damage-inducible gene 45beta expression is associated with human hepatocellular carcinoma. Am J Pathol. 2003;162(6):1961–1974. doi: 10.1016/s0002-9440(10)64329-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yu YP, Yu G, Tseng G, Cieply K, Nelson J, Defrances M, Zarnegar R, Michalopoulos G, Luo JH. Glutathione peroxidase 3, deleted or methylated in prostate cancer, suppresses prostate cancer growth and metastasis. Cancer Res. 2007;67(17):8043–8050. doi: 10.1158/0008-5472.CAN-07-0648. [DOI] [PubMed] [Google Scholar]
- 22.Cairns P, Esteller M, Herman JG, Schoenberg M, Jeronimo C, Sanchez-Cespedes M, Chow NH, Grasso M, Wu L, Westra WB, Sidransky D. Molecular detection of prostate cancer in urine by GSTP1 hypermethylation. Clin Cancer Res. 2001;7(9):2727–2730. [PubMed] [Google Scholar]
- 23.Massoner P, Colleselli D, Matscheski A, Pircher H, Geley S, Jansen Durr P, Klocker H. Novel mechanism of IGF-binding protein-3 action on prostate cancer cells: inhibition of proliferation, adhesion, and motility. Endocr Relat Cancer. 2009;16(3):795–808. doi: 10.1677/ERC-08-0175. [DOI] [PubMed] [Google Scholar]
- 24.Mehta H, Gao Q, Galet C, Paharkova V, Wan J, Said JW, Sohn J, Lawson G, Cohen P, Cobb L, Lee KW. IGFBP-3 is a Metastasis Suppression Gene in Prostate Cancer. Cancer Res. 2011 doi: 10.1158/0008-5472.CAN-10-4513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sathyanarayana UG, Padar A, Suzuki M, Maruyama R, Shigematsu H, Hsieh JT, Frenkel EP, Gazdar AF. Aberrant promoter methylation of laminin-5-encoding genes in prostate cancers and its relationship to clinicopathological features. Clin Cancer Res. 2003;9(17):6395–6400. [PubMed] [Google Scholar]
- 26.Vanaja DK, Grossmann ME, Cheville JC, Gazi MH, Gong A, Zhang JS, Ajtai K, Burghardt TP, Young CY. PDLIM4, an actin binding protein, suppresses prostate cancer cell growth. Cancer Invest. 2009;27(3):264–272. doi: 10.1080/07357900802406319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boye K, Maelandsmo GM. S100A4 and metastasis: a small actor playing many roles. Am J Pathol. 2010;176(2):528–535. doi: 10.2353/ajpath.2010.090526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li Q, Ahuja N, Burger PC, Issa JP. Methylation and silencing of the Thrombospondin-1 promoter in human cancer. Oncogene. 1999;18(21):3284–3289. doi: 10.1038/sj.onc.1202663. [DOI] [PubMed] [Google Scholar]
- 29.Ghoshal K, Datta J, Majumder S, Bai S, Kutay H, Motiwala T, Jacob ST. 5-Aza-deoxycytidine induces selective degradation of DNA methyltransferase 1 by a proteasomal pathway that requires the KEN box, bromo-adjacent homology domain, and nuclear localization signal. Mol Cell Biol. 2005;25(11):4727–4741. doi: 10.1128/MCB.25.11.4727-4741.2005. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 30.Patel K, Dickson J, Din S, Macleod K, Jodrell D, Ramsahoye B. Targeting of 5-aza-2'-deoxycytidine residues by chromatin-associated DNMT1 induces proteasomal degradation of the free enzyme. Nucleic Acids Res. 2010;38(13):4313–4324. doi: 10.1093/nar/gkq187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Palii SS, Van Emburgh BO, Sankpal UT, Brown KD, Robertson KD. DNA methylation inhibitor 5-Aza-2'-deoxycytidine induces reversible genome-wide DNA damage that is distinctly influenced by DNA methyltransferases 1 and 3B. Mol Cell Biol. 2008;28(2):752–771. doi: 10.1128/MCB.01799-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fang Y, Brass A, Hoyle DC, Hayes A, Bashein A, Oliver SG, Waddington D, Rattray M. A model-based analysis of microarray experimental error and normalisation. Nucleic Acids Res. 2003;31(16):e96. doi: 10.1093/nar/gng097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu S, Liu Z, Xie Z, Pang J, Yu J, Lehmann E, Huynh L, Vukosavljevic T, Takeki M, Klisovic RB, Baiocchi RA, Blum W, Porcu P, Garzon R, Byrd JC, Perrotti D, Caligiuri MA, Chan KK, Wu LC, Marcucci G. Bortezomib induces DNA hypomethylation and silenced gene transcription by interfering with Sp1/NF-kappaB-dependent DNA methyltransferase activity in acute myeloid leukemia. Blood. 2008;111(4):2364–2373. doi: 10.1182/blood-2007-08-110171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kimura H, Nakamura T, Ogawa T, Tanaka S, Shiota K. Transcription of mouse DNA methyltransferase 1 (Dnmt1) is regulated by both E2F-Rb-HDAC-dependent and -independent pathways. Nucleic Acids Res. 2003;31(12):3101–3113. doi: 10.1093/nar/gkg406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang CC, Wang YC, Wei S, Lin LF, Chen CS, Lee CC, Lin CC. Peroxisome proliferator-activated receptor gamma-independent suppression of androgen receptor expression by troglitazone mechanism and pharmacologic exploitation. Cancer Res. 2007;67(7):3229–3238. doi: 10.1158/0008-5472.CAN-06-2759. [DOI] [PubMed] [Google Scholar]
- 36.Ohtani K, DeGregori J, Nevins JR. Regulation of the cyclin E gene by transcription factor E2F1. Proc Natl Acad Sci U S A. 1995;92(26):12146–12150. doi: 10.1073/pnas.92.26.12146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ma Y, Yuan J, Huang M, Jove R, Cress WD. Regulation of the cyclin D3 promoter by E2F1. J Biol Chem. 2003;278(19):16770–16776. doi: 10.1074/jbc.M212702200. [DOI] [PubMed] [Google Scholar]
- 38.Palapattu GS, Wu C, Silvers CR, Martin HB, Williams K, Salamone L, Bushnell T, Huang LS, Yang Q, Huang J. Selective expression of CD44, a putative prostate cancer stem cell marker, in neuroendocrine tumor cells of human prostate cancer. Prostate. 2009;69(7):787–798. doi: 10.1002/pros.20928. [DOI] [PubMed] [Google Scholar]
- 39.Dejeux E, El abdalaoui H, Gut IG, Tost J. Identification and quantification of differentially methylated loci by the pyrosequencing technology. Methods Mol Biol. 2009;507:189–205. doi: 10.1007/978-1-59745-522-0_15. [DOI] [PubMed] [Google Scholar]
- 40.Li LC, Carroll PR, Dahiya R. Epigenetic changes in prostate cancer: implication for diagnosis and treatment. J Natl Cancer Inst. 2005;97(2):103–115. doi: 10.1093/jnci/dji010. [DOI] [PubMed] [Google Scholar]
- 41.Park JY. Promoter hypermethylation in prostate cancer. Cancer Control. 2010;17(4):245–255. doi: 10.1177/107327481001700405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Issa JP, Kantarjian HM. Targeting DNA methylation. Clin Cancer Res. 2009;15(12):3938–3946. doi: 10.1158/1078-0432.CCR-08-2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.