

Abstract
Medullary thyroid cancer (MTC) is a neuroendocrine tumor (NET) that is often resistant to standard therapies. Resveratrol suppresses MTC growth in vitro, but it has low bioavailability in vivo due to its poor water solubility and rapid metabolic breakdown, as well as lack of tumor-targeting ability. A novel unimolecular micelle based on a hyperbranched amphiphilic block copolymer was designed, synthesized, and characterized for NET-targeted delivery. The hyperbranched amphiphilic block copolymer consisted of a dendritic Boltorn® H40 core, a hydrophobic poly(L-lactide) (PLA) inner shell, and a hydrophilic poly(ethylene glycol) (PEG) outer shell. Octreotide (OCT), a peptide that shows strong binding affinity to somatostatin receptors, which are overexpressed on NET cells, was used as the targeting ligand. Resveratrol was physically encapsulated by the micelle with a drug loading content of 12.1%. The unimolecular micelles exhibited a uniform size distribution and spherical morphology, which were determined by both transmission electron microscopy (TEM) and dynamic light scattering (DLS). Cellular uptake, cellular proliferation, and Western blot analyses demonstrated that the resveratrol-loaded OCT-targeted micelles suppressed growth more effectively than non-targeted micelles. Moreover, resveratrol-loaded NET-targeted micelles affected MTC cells similarly to free resveratrol in vitro, with equal growth suppression and reduction in NET marker production. These results suggest that the H40-based unimolecular micelle may offer a promising approach for targeted NET therapy.
Introduction
Cancer is one of the leading causes of mortality in the world, and the worldwide incidence of cancer continues to increase.1-3 Medullary thyroid cancer (MTC) is a neuroendocrine tumor (NET) that arises from the calcitonin-secreting parafollicular cells of the thyroid gland. While MTC accounts for only 5% of all thyroid cancer diagnoses, it causes a disproportionate 13% of all deaths from thyroid cancer.4 Surgery is the only curative treatment; however, disseminated disease is common at presentation, preventing curative surgical resection and causing significant morbidity.4, 5 Thus, there is an urgent demand for new treatment modalities for metastatic disease. MTC does not respond to standard therapeutic modalities used for other well-differentiated thyroid cancers, such as radioactive iodine, due to its different parent cell of origin. Standard cytotoxic chemotherapy is the most frequently used systemic therapy. Unfortunately, although more and more cell- and tissue-specific therapies are being developed, most of the clinically used chemotherapeutics require high tissue concentrations that cause systemic toxicity.6
Nanomedicine offers extraordinary opportunities to address the challenges of conventional cancer therapy, such as improving the solubility and stability of anticancer drugs and providing a stimuli-responsive drug release profile at the pathological site, thereby optimizing the pharmacokinetics of the drug and enhancing the therapeutic efficacy.7-13 In addition, drug nanocarriers can specifically target cancerous tissues/cells due to their passive (i.e., the enhanced permeability and retention (EPR) effect10) and active (i.e., via specific tumor-targeting ligands) targeting abilities, thereby greatly enhancing the therapeutic outcomes while reducing any non-specific systemic toxicity.7, 10, 14-18 Encapsulation of anticancer drugs can also protect the drugs from premature degradation and non-specific interaction, which can improve the bioavailability of the compound.
Active tumor targeting in nanomedicine can be achieved by conjugating certain tumor-targeting ligands such as peptides, antibodies, or aptamers that recognize and bind specifically to receptors over-expressed by cancer cells.14-17 The majority of NETs overexpress somatostatin receptors (SSTRs types 1-5, a family of guanosine triphosphate-binding protein-coupled receptors). Treatment with somatostatin analogs such as octreotide (OCT, a cyclic octapeptide alcohol (D30 Phe-c(Cys-Phe-D-Trp-Lys-Thr-Cys)-Thr-ol)), pasireotide, and lanreotide have been shown to effectively suppress NET hormone secretion through binding of SSTRs.19-23
Polymer micelles, formed by self-assembly of amphiphilic block copolymers, have gained significant attention in drug and gene delivery due to the versatility of polymer chemistry and ease of fabrication.24 These micelles possess a core–shell structure. The hydrophobic core serves as a reservoir for pharmaceutical compounds with poor solubility and/or low stability in physiological environments, while the hydrophilic shell provides the nanocarriers with desirable solubility in aqueous solutions.24-30
Although polymer micelles offer many benefits in drug delivery, conventional multimolecular micelles composed of linear amphiphilic block copolymers suffer from limited in vivo stability due to the dynamic nature of self-assembly. A promising approach to overcome this instability lies in the development of unimolecular micelles formed by individual dendritic or hyperbranched amphiphilic copolymers.24-29 In contrast to conventional multimolecular polymer micelles, unimolecular micelles exhibit excellent in vivo stability and are relatively insensitive to polymer concentration or environmental fluctuations such as temperature, pH, etc. This improved stability is attributed to the covalent nature of the individual dendritic/hyperbranched amphiphilic block copolymer molecules that form the unimolecular micelles. Our recent studies have demonstrated that Boltorn® H40 (H40), a hyperbranched aliphatic polyester, serves as an effective inner core/macroinitiator for unimolecular micelles because of its biocompatibility, biodegradability, globular architecture, and high number of terminal functional groups.24, 31-35
Herein, an OCT-conjugated unimolecular micelle nanoplatform composed of a dendritic H40 core, a hydrophobic poly(L-lactide) (PLA) inner shell, and a hydrophilic poly(ethylene glycol) (PEG) outer shell has been designed, characterized, and evaluated for targeted neuroendocrine cancer therapy (Fig. 1). Resveratrol, a model anticancer drug, was physically encapsulated into the hydrophobic core of a unimolecular micelle consisting of H40 and PLA. Resveratrol is a dietary polyphenol found in grape skins and peanuts that has been previously shown to suppress cell growth and NET marker production in carcinoid and MTC cells.36, 37 However, due to its low water solubility, poor in vivo chemical stability (with a half-life of approximately 8 to 14 min), and extremely low bioavailability, resveratrol administration still remains a big challenge.38-41 Thus, resveratrol was selected as the model drug in this study in order to improve its solubility in aqueous solution, its in vivo chemical stability and bioavailability, and its tumor-targeting ability. Extensive in vitro studies including cellular uptake, cellular proliferation, and Western blot analysis were conducted to evaluate the potential of the H40-based unimolecular micelles as a promising drug nanocarrier for targeted NE cancer therapy.
Fig. 1.
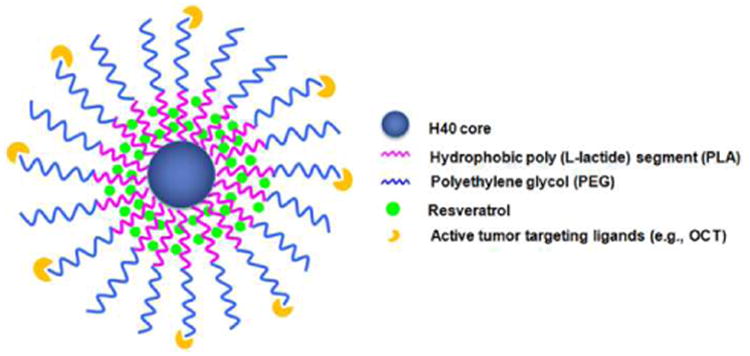
A schematic illustration of the H40-PLA-PEG-OCT nanocarriers for NET-targeted drug delivery.
Results and discussion
Synthesis and characterization of H40-PLA-PEG-OCT
H40-PLA-PEG-OCT was synthesized following the procedures shown in Scheme 1. First, H40-PLA was prepared by the ring-opening polymerization of L-lactide using H40 as a macro-initiator and Sn(Oct)2 as a catalyst under inert atmosphere at 120 °C for 24 h. The product was first purified by a neutral alumina column to remove the catalyst. Then the low molecular weight fraction was removed through precipitation in cold diethyl ether. The chemical structure of H40-PLA was confirmed by 1H NMR (Fig. 2A). The peaks located at (a) 1.54 to 1.56 ppm and (b) 5.10 to 5.16 ppm were assigned to the protons of methyl and methine groups in the PLA main chains, respectively. The signal at (c) 1.45 ppm and (d) 4.32 to 4.35 ppm corresponded to the terminal methyl and methine protons of PLA (HOCHCH3) in H40-PLA. The peaks at 1.18 to 1.22 ppm and around 4.20 ppm were assigned to the protons of the methyl groups and methylene groups of H40, respectively, confirming the dendritic structure of the H40-PLA polymer. By calculating the relative intensity of the peak at 1.45 ppm, which originated from the methyl group adjacent to the hydroxyl end group, and the peak at 1.56 ppm, which originated from the methyl groups present in the polymer chain, the molecular weight (Mn) and degree of polymerization (DP) of the PLA arms were found to be about 936 Da and 13, respectively. The average number of arms per H40-PLA molecule was estimated via comparing the molecular weights of H40 and H40-PLA as determined by a GPC equipped with triple detectors, which is shown in Table 1. The average number of arms per H40-PLA molecule was estimated to be 25 based on the molecular weight measurements shown in Table 1.42 This result agrees well with those reported by Kreutzer et al. and our previous studies.31, 43-45
Scheme 1.

The synthesis scheme of the hyperbranched H40-PLA-PEG-OCT amphiphilic block copolymers.
Fig. 2.
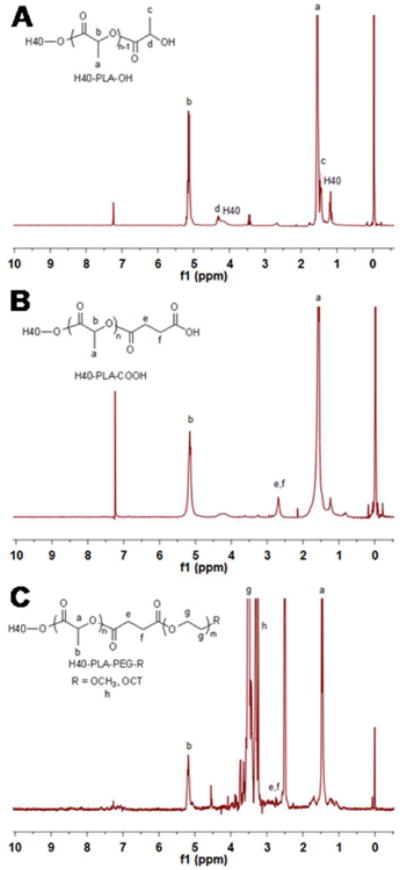
1H NMR spectrum of (A) H40-PLA, (B) H40-PLA-COOH, and (C) H40-PLA-PEG-OCH3/OCT.
Table 1. Gel permeation chromatography of H40, H40-PLA and H40-PLA-MPEG.
| Sample | Mn | Mw | Mw/Mn |
|---|---|---|---|
| H40a | 2833 | 5100 | 1.80 |
| H40-PLA | 26208 | 36456 | 1.39 |
| H40-PLA-MPEG | 150310 | 248949 | 1.66 |
Perstorp data sheet.
The hydroxyl terminal groups of H40-PLA were subsequently converted into carboxyl terminal groups by reacting H40-PLA with succinic anhydride in the presence of DMAP as the catalyst. The product was precipitated with diethyl ether and dialyzed against deionized water for purification. The 1H NMR spectrum of H40-PLA-COOH (Fig. 2B) showed a new peak at 2.65 ppm (methylene groups of succinic anhydride, e, f), which confirmed the formation of H40-PLA-COOH. Lastly, in order to conjugate the OCT for NET-specific targeting, OCT was first conjugated onto a heterobifunctional PEG derivative, i.e., OH-PEG-NHS (Mw: 5000 Da), to form OH-PEG-OCT. Then, OH-PEG-OCT was conjugated onto H40-PLA-COOH via an esterification reaction between the hydroxyl group and the carboxyl group in the presence of DCC and DMAP. The feed molar ratio of MPEG, HO-PEG-OCT, and H40-PLA-COOH was set at 7:3:1. In the 1H NMR spectrum of H40-PLA-PEG-OCH3/OCT (Fig. 2C), in addition to the peaks from PLA, peaks at (g) 3.65 ppm and (h) 3.38 ppm were observed due to the methylene protons of oxyethylene units and methyl protons of MPEG, respectively. The appearance of a group of NMR peaks ranging from 6.8 to 8.0 ppm can be attributed to the protons of octreotide.
Fourier transform infrared spectroscopy (FTIR) analyses provided additional information about the hyperbranched amphiphilic block copolymer H40-PLA-PEG-OCH3/OCT. As shown in Fig. 3, the peak at 2891 cm−1 was assigned to the antisymmetric C–H stretching of CH2. The strong peak at 1760 cm−1 was attributed to the characteristic absorption of C=O stretching due to the presence of a hyperbranched polyester H40 core and PLA blocks. The absorption peak located at 1342 cm−1 could be attributed to CH2 wagging and C–C stretching in the PEG blocks. The strongest peak at 1105 cm−1 was assigned to C–O–C stretching. The C–O–C stretching was also verified at 962 cm−1. The FTIR results further testified to the successful formation of H40-PLA-PEG-OCH3/OCT.46
Fig. 3.

FTIR spectra ofH40-PLA-PEG-OCH3/OCT.
Micellar properties of hyperbranched H40-PLA-PEG-OCT copolymers
The resveratrol-loaded H40-PLA-PEG-OCT copolymers formed stable unimolecular micelles in an aqueous solution due to their large number of amphiphilic arms (∼25) and their globular architecture. An inner hydrophobic core was formed by hydrophobic H40-PLA and an outer hydrophilic shell was formed by the hydrophilic PEG layer. Previous studies indicate that the cellular internalization process and in vivo performance of drug nanocarriers depend in large part on the stability and size of the nanoparticles.10 The size distribution histogram of the targeted H40-PLA-PEG-OCT micelles measured by DLS is shown in Fig. 4A. The average hydrodynamic diameter of the targeted micelles was about 68 nm. Fig. 4B shows the TEM images of the unimolecular micelles stained by phosphotungstic acid. The diameter of the dried spherical unimolecular micelles ranged from 18 to 30 nm.
Fig. 4.
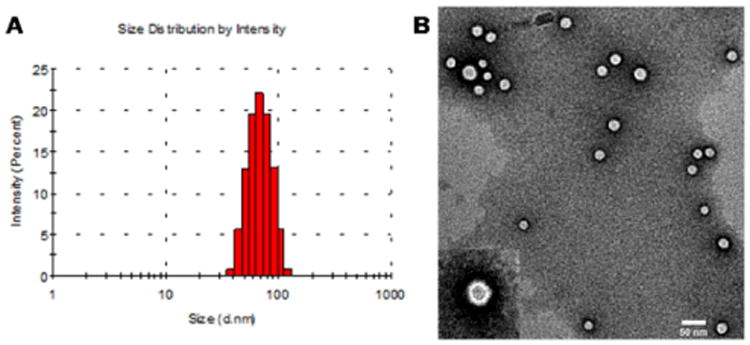
The (A) size distribution and (B) morphology of unimolecular micelles as measured by DLS and TEM, respectively.
Drug loading level and in vitro drug release
The amount of resveratrol incorporated into the unimolecular micelles was 12.1 wt.% as measured by HPLC analysis using the absorption peak of resveratrol at 306 nm. Resveratrol release behaviors from the H40-PLA-PEG-OCT unimolecular micelles were studied in either an acetate buffer (pH 5.3) or a phosphate buffer (pH 7.4) solution at 37 °C. pH 7.4 mimics the physiological conditions in the bloodstream during circulation; while pH 5.3 mimics the acidic endosome/lysosome compartments after the unimolecular micelles are taken up by the cells via endocytosis. As shown in Fig. 5, the release profiles at both pH 7.4 and pH 5.3 exhibited an initial rapid release followed by a sustained slow release over a prolonged time (nearly one week). During the first 8 h, 35.8 and 30.1% of resveratrol was released from the micelles at a pH 7.4 and 5.3, respectively. Thereafter, resveratrol was released in a more sustained manner from the micelles. After four days, 81.1 and 72.5% of resveratrol was released at pH 7.4 and pH 5.3, respectively. It has been reported that resveratrol in its original free form is metabolized quickly in vivo.39 Encapsulating resveratrol inside of the hydrophobic cores of the unimolecular micelles can enhance the in vivo stability of resveratrol while providing controlled and sustained drug release.
Fig. 5.

In vitro resveratrol release profiles of resveratrol-loaded unimolecular micelles at a pH of 5.3 and 7.4.
Micelle cellular uptake
The proficiency of micelle intracellular uptake was studied using flow cytometry. Based on DOX's fluorescent properties, these experiments were conducted to monitor micelle–cellular interactions and were not related to the anticancer properties of the micelles. Flow cytometry results were analyzed to select the live, single-cell population, and the DOX-positive population was determined based on unlabeled media controls. These controls showed minimal fluorescence levels at both time points, consistent with background fluorescence. After 4 h of treatment, targeted micelles showed a nearly 3-fold greater uptake compared to non-targeted micelles (Fig. 6). Similarly, cells treated for 6 h showed a nearly 3-fold greater uptake of targeted vs. non-targeted micelles (Fig. 6). Furthermore, longer treatment resulted in a higher uptake of targeted micelles compared to the control, with a 12-fold higher uptake at 6 h compared to only a 4-fold higher uptake at 4 h (Fig. 6). These findings indicate that octreotide conjugation on micelles improved the cellular uptake in TT cells considerably, with this difference being more dramatic after a longer period of treatment. Previous studies have shown that non-targeted micelles were taken up by the cells through non-specific endocytosis, while targeted micelles were taken up by the cells via receptor-mediated endocytosis.24, 45, 47 OCT-targeted micelles executed their cellular uptake by binding to the over expressed somatostatin receptors on the membranes of TT cells.
Fig. 6.

Flow cytometry analysis of TT medullary thyroid cancer cells treated with control media and DOX-labeled targeted and non-targeted micelles, both with an equal dose of DOX (2 ug/mL). Cells were treated for (A) 4 h or (B) 6 h, and then analyzed on an LSRII flow cytometer. The control media results were used to set the gate for DOX+ cells, and this gate was used to assess DOX positive cells, indicating micelle uptake. Percentages of single, live cells included within the DOX-positive gate are reported.
Cellular growth suppression with resveratrol-loaded micelle treatment
The ability of resveratrol-loaded micelles to limit TT cell proliferation was assessed using MTT assays. Resveratrol-loaded micelles (targeted and non-targeted) were compared to three controls: empty targeted micelles, free resveratrol, and culture media, at two concentrations (50 μM and 100 μM). Resveratrol-loaded targeted micelles suppressed cell growth significantly more than non-targeted resveratrol-loaded micelles with 6 days of treatment (50 μM, p=0.02; 100 μM, p=0.04) (Fig. 7). At days 4 and 6, targeted resveratrol-loaded micelles suppressed cell growth to equivalent levels as free resveratrol (p=1.00), while non-targeted resveratrol-loaded micelles at 50 μM had significantly weaker growth suppression than free resveratrol at 6 days (p=0.003; 100 μM, p=0.13) (Fig. 7). These compiled results indicate that resveratrol-loaded targeted micelles have anti-tumor growth properties equivalent to free resveratrol in vitro, which are significantly greater than all other micelle formulations by day 6 of treatment.
Fig. 7.

MTT analysis of TT cells treated with control media, DMSO, targeted empty micelles (M-T Empty), resveratrol-loaded non-targeted micelles (M-NT Resv), resveratrol-loaded targeted micelles (M-T Resv), and free resveratrol (Resv) at a concentration of (A) 50 μM or (B) 100 uM in all treatments containing resveratrol. The concentration of targeted empty micelles was calculated to be the same in micelles/mL as the treatments containing resveratrol. M-T Resv treatments suppressed growth significantly more than M-NT Resv at day 6 at both concentrations (50 μM, * p = 0.02; 100 μM, # p = 0.04).
Reduction of neuroendocrine tumor marker production
After demonstrating cell growth suppression, the ability of resveratrol-loaded micelle treatments to limit production of NET markers was evaluated using standard Western blot analysis. Previous studies have shown that achaete-scute complex-like1 (ASCL1) is a highly expressed NET marker that is important for determining cell fate and hormone production in TT cells.48-50 The NET marker chromogranin A (CgA) is a secretory glycoprotein, and changes in the expression levels of this protein correspond closely with changes in the expression levels of other NET markers.51, 52 Therefore, these markers can be used as an indication of the broader hormone-producing activity of TT cells. As expected, free resveratrol treatment in TT cells after four days resulted in decreased expression of both ASCL1 and CgA (Fig. 8). Both targeted and non-targeted resveratrol-loaded micelles also decreased ASCL1 and CgA expression on par with free resveratrol (Fig. 8). Targeted empty micelle treatments did not show any reduction of NET marker expression compared to control cells, indicating that somatostatin receptor activation via OCT was not sufficient to suppress NET marker production alone. Combined with demonstrated tumor cell growth suppression, this reduction in NET marker production in vitro suggests that resveratrol-loaded micelles could reduce both tumor growth and symptom-inducing endocrine hormone production in MTC effectively.
Fig. 8.

Western blot analysis of TT cells treated for 4 days with control media, targeted empty micelles (M-T Empty), resveratrol-loaded non-targeted micelles (M-NT Resv), resveratrol-loaded targeted micelles (M-T Resv), and free resveratrol (Resv) at a concentration of 50 μM in all treatments containing resveratrol and at equivalent micelles/mL concentration in the targeted empty micelles.
Experimental
Materials
Boltorn H40 (a hyperbranched polyester with 64 hydroxyl terminal groups per molecule; Mn: 2833 Da) was provided by PerstorpPolyols Inc., USA, and purified with acetone and tetrahydrofuran (THF). L-lactide was purchased from Sigma-Aldrich and recrystallized from ethyl acetate before use. Succinic anhydride, 4-dimethylamino pyridine (DMAP), N-hydroxysuccinimide (NHS), 1,3-dicyclohexylcarbodiimide (DCC), and dichloromethane (DCM) were purchased from Sigma-Aldrich (Milwaukee, WI, USA) and used without further purification. THF, triethylamine (TEA), dimethyl sulfoxide (DMSO), and dimethyl formamide (DMF) were purchased from Sigma-Aldrich and were distilled before use. The heterobifunctional PEG derivatives, succinimidyl (NHS)-PEG114-OH or methoxy (-OCH3))-PEG114-OH, both of which have a Mw of 5000, were acquired from JenKem Technology (Allen, TX, USA). Doxorubicin hydrochloride (DOX·HCl) (Tecoland Corporation, Irvine, CA, USA), resveratrol (Enzo Life Sciences International, Inc.) and octreotide acetate (Aroz Technologies, USA) are commercially available. Human TT MTC cells were obtained from the Biochemistry Department of the Medical College of Wisconsin. The cell culture media included RPMI 1640 (Invitrogen Life Technologies, Carlsbad, CA), Fetal Bovine Serum (Sigma, St. Louis, MO), Penicillin (10,000 IU/mL), and Streptomycin (10,000 μg/mL; Invitrogen). Trypsin (Invitrogen) in phosphate buffered saline (Invitrogen) or Cellstripper™ (Mediatech, Inc, Manassas, VA) were used as cell dissociation buffers. Cell proliferation assays were performed using 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) (Sigma) and DMSO (Fischer Scientific, Pittsburg, PA), and measured with a μQuant spectrophotometer (Bio-Tek Instruments, Winooski, VT). Cellular uptake of micelles was measured using flow cytometry on an LSR II flow cytometer (BD Biosciences, San Jose, CA) with FACSDiva (version 6.2, BD Biosciences) and FlowJo (v. 9.6.2 for Mac (2013), Treestar, Inc., Ashland, OR). Immunoblot assays were performed using the following mouse and rabbit antibodies: MASH for ASCL1 (BD Pharmingen, San Diego, CA), CgA (Zymed Laboratories, San Francisco, CA), and G3PDH (Trevigen, Gaithersburg, MD). The secondary horseradish peroxidase-linked goat anti-rabbit and anti-mouse antibodies were purchased from Cell Signaling Technology, Beverly, MA. Proteins were detected with the Immun-star (Bio-Rad Laboratories, Hercules, CA), Pico, or Femto (Pierce Protein Research Products, Rockford, IL) chemiluminescence kits.
Synthesis of H40-PLA
H40-PLA was prepared by the ring-opening polymerization of L-lactide using H40 as a macroinitiator and Sn(Oct)2 as a catalyst. A 50 mL Schlenk flask was charged with H40 (400 mg, 9.04 mmol of hydroxyl groups) under an inert atmosphere and placed in an oil bath at 120 °C in order to melt it and facilitate its mixing with L-lactide. L-lactide (4.00 g, 27.8 mmol) was slowly introduced into the flask and a catalytic amount ([catalyst]/[monomer] of 1:1000) of Sn(Oct)2 (9 μL, 27.8 μmol) was added afterwards. The polymerization reaction mixture was stirred for 24 h. The resulting mixture was dissolved in THF and passed through a neutral alumina column. Next, the mixture was concentrated and precipitated into cold diethyl ether to yield a white H40-PLA powder. The final product was dried under vacuum for 24 h.
Synthesis of carboxyl-functionalized H40-PLA (H40-PLA-COOH)
H40-PLA-COOH was prepared by reacting H40-PLA (1.00g, 38.1 μmol) with succinic anhydride (0.38 g, 3.8 mmol) in the presence of DMAP (0.70 g, 5.7 mmol) as a catalyst. The reaction was carried out in anhydrous DCM (10 mL) for 48 h at room temperature with constant stirring. Thereafter, the formed product was precipitated with cold diethyl ether and dried under a vacuum. The impurities and unreacted materials of the product were removed by dialysis against deionized water using cellulose tubing (molecular weight cut-off of 3500 Da). After 48 h of dialysis, the product was separated out using the freeze-drying method.
Synthesis of HO-PEG-OCT
The reaction was conducted by reacting HO-PEG-NHS (111.5 mg, 0.022 mmol) with OCT (25 mg, 0.025 mmol) in the presence of TEA (200 μL) in freshly distilled anhydrous DMF for 24 h. Afterwards, the products were dialyzed against DI water and then freeze-dried.
Synthesis of H40-PLA-PEG-OCH3 (Non-targeted micelles)
H40-PLA-COOH (30 mg, 1.04 μmol) and MPEG-OH (methoxy (-OCH3) PEG, 157 mg, 31.3 μmol) were dissolved in 10 mL anhydrous DCM, which was treated with DCC (6.5 mg, 31.3 μmol) and DMAP (0.4 mg, 3.1 μmol) at about 0°C. The reaction was carried out at room temperature for 48 h under stirring. After the by-product dicyclohexylcarbodiurea was removed by filtration, the product was precipitated with cold diethyl ether. The precipitate was then dialyzed against deionized water for 48 h using cellulose tubing (molecular weight cut-off, 12 kDa) and freeze-dried.
Synthesis of H40-PLA-PEG-OCH3/Octreotide (Targeted micelles)
H40-PLA-COOH (30 mg, 1.04 μmol), MPEG-OH (109.2 mg, 21.84 μmol), and OCT-PEG-OH (55.3 mg, 9.4 μmol) were dissolved in 10 mL anhydrous DCM, which was treated with DCC (19.2 mg, 31.3 μmol) and DMAP (0.4 mg, 3.1 μmol) at about 0° C. The reaction was carried out at room temperature for 24 h under stirring. After the by-product dicyclohexylcarbodiurea was removed by filtration, the product was precipitated with cold diethyl ether. The products were dialyzed against DI water, followed by freeze drying.
Preparation of resveratrol-loaded targeted micelles
The polymer (H40-PLA-MPEG/OCT) (50 mg) and resveratrol (20 mg) were dissolved in 5 mL of CH3CN under stirring. With this mixture, 15 mL of deionized water was added dropwise and stirred for 24 h in darkness. Thereafter, the mixture was dialyzed against deionized water using dialysis tubing with a molecular weight cut-off of 2 kDa for 24 h followed by freeze drying. Resveratrol-loaded non-targeted micelles were prepared following a similar procedure using H40-PLA-MPEG polymers.
Preparation of DOX-loaded targeted micelles
DOX-loaded micelles were created to take advantage of DOX's fluorescent properties in experiments measuring cellular uptake. These micelles were used only in experiments that did not assess the anti-tumor properties of the micelles or resveratrol. Doxorubicin∙HCl (15 mg) was first treated with 2 moles excess of triethylamine. Afterwards, these were mixed with the hyperbranched amphiphilic block copolymer (H40-PLA-PEG-OCT, 50 mg) and dissolved in 5 mL of DMF while stirring. With this mixture, 15 mL of deionized water was added dropwise. Thereafter, the mixture was dialyzed against deionized water using a dialysis tubing (molecular weight cut-off of 2 kDa) for 24 h and then freeze-dried. DOX-loaded non-targeted micelles were prepared following a similar procedure using H40-PLA-MPEG polymers.
Characterization
The hyperbranched amphiphilic block copolymers and the resulting unimolecular micelles were characterized using various techniques.24 The structures of the intermediate and final polymer products were confirmed by nuclear magnetic resonance (NMR) spectroscopy. 1H NMR spectra were recorded at 25 °C on a Bruker DPX 300 spectrometer using D2O, CDCl3, or DMSO as the solvent. The molecular weight of the polymers was determined at 35 °C using gel permeation chromatography (GPC) equipped with a refractive index detector, a viscometer detector, and a light scattering detector (Viscotek, USA). DMF (with 10 mmol/L LiBr) was used as a mobile phase with a flow rate of 1 mL/min. The hydrodynamic size and size distribution of the unimolecular micelles were determined by dynamic light scattering (DLS) (ZetaSizer Nano ZS90, Malvern Instrument, USA), at a polymer concentration of 0.05 mg/mL. The morphology and size of the dried unimolecular micelles were measured using a transmission electron microscope (TEM) at 75 kV (Hitachi H-600, Japan). The TEM samples were prepared by depositing a drop of micelle solution (0.05 mg/mL) containing 0.8 wt.% of phosphotungstic acid onto a 200 mesh copper grid coated with carbon and dried at room temperature. The drug loading content of resveratrol, defined as the weight percentage of resveratrol in the resveratrol-loaded unimolecular micelles, was measured by high performance liquid chromatography (HPLC, Hitachi) in triplicate. A standard curve for quantification of the amount of resveratrol was determined by HPLC. The Hitachi HPLC was equipped with a UV detector and a reverse-phase C18 column (150×4.6 mm) with a non-gradient mobile phase of water (0.1% H3PO4) and acetonitrile (v/v 60/40) at a constant flow rate of 0.6 mL/min. The resveratrol peak was measured at a wavelength of 306 nm. The DOX loading content in the DOX-loaded unimolecular micelles was quantified by a Varian Cary 300 Bio UV-visible spectrophotometer in triplicate. The calibration curve of absorbance for different concentrations of DOX was determined at 485 nm. To determine the DOX loading content, a weighed quantity (25 mg) of DOX-loaded micelles was extracted with ethanol at room temperature for 48 h under uniform stirring. After centrifugation, the supernatant containing DOX was assayed by a UV–visible spectrophotometer at a wavelength of 485 nm.
In vitro drug release study
Drug release studies were performed in either an acetate buffer (pH 5.3) or a phosphate buffer (pH 7.4) solution by a dialysis method. Briefly, 5 mg of freeze-dried resveratrol-loaded unimolecular micelles were suspended in 10 mL of donor medium (an acetate buffer (pH 5.3) or a phosphate buffer (pH 7.4)) and placed in a dialysis bag with a molecular weight cut-off of 100-500 Da. The dialysis bag was immersed in 100 mL of the receptor medium (an acetate buffer (pH 5.3) or a phosphate buffer (pH 7.4) containing 0.2% Tween 80) and kept in a horizontal laboratory shaker with continuous stirring (100 rpm) at 37 °C. At selected time points, samples of 10 mL of release medium were withdrawn and used to quantify the amount of resveratrol released using HPLC. The same amount of fresh medium was added back to the release medium. The drug release studies were performed in triplicate for each sample. The cumulative amount of drug released was calculated and plotted against time.
Cell culture
TT cells were maintained in 1640 RPMI media with 16% fetal bovine serum, 100 IU/mL penicillin, and 100 μg/mL streptomycin at 37 °C with 5% carbon dioxide in a humidified environment. Cells were passaged using 0.05% Trypsin in phosphate buffered saline.
Cellular uptake of micelles
The cellular uptake of control cell media only and DOX-loaded targeted and non-targeted micelles was measured using flow cytometry. TT cells were plated into 100 mm culture dishes in standard media and incubated overnight. They were then treated in serum-free RPMI 1640 media with DOX-loaded targeted and non-targeted micelles (2 μg/mL DOX). Prior to application to cells, the micelles in media, along with the micelle-free media control, were sonicated for 30 minutes at 40 kHz with the temperature maintained between 23 and 27 °C (Bransonic 3510R-DTH, Branson Ultrasonics Corporation, Danbury, CT). Cells were then treated for 4 or 6 h. After treatment, cells were washed with phosphate buffered saline and then removed from culture plates with Cellstripper™ and concentrated by centrifugation to 5-10 × 105 cells/mL. Using the LSRII Flow Cytometer, a minimum of 5 × 104 cells were analyzed for each treatment, and DOX-positive cells were detected using 530LP and 585/42 filters on a log scale compared to media controls.
Cell proliferation assay
The ability of resveratrol-loaded unimolecular micelles to suppress cell proliferation was measured using 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) assays. TT cells were seeded into 96-well culture plates with 1.3 × 104 cells per well and incubated overnight to allow cell adhesion. Cells were then treated with resveratrol-loaded targeted and non-targeted micelles, and three controls:empty targeted micelles, free resveratrol, and media. In free resveratrol and resveratrol-loaded micelle aliquots, the concentration of resveratrol was either 50 or 100 μM. These doses were chosen based on the half-maximal inhibition concentration (IC50) in TT cell proliferation assays performed with resveratrol alone after 4 or 6 days of treatment (data not shown). In the resveratrol-free aliquot, the micelle concentration was set to be equivalent to the resveratrol-loaded samples. Treatment media was changed every 2 days, and MTT assays were performed on day 0, 2, 4, and 6. At each time point, treatment media was removed from the plates, and 50 μL of serum-free RPMI 1640 with 0.25 mg/mL MTT was applied for 3.5 h. Following incubation, 150 μL of DMSO was added to each well and mixed for 5 minutes. Absorbance at 540 nm was read for each well with a μQuant spectrophotometer, and the average absorbance and percent growth compared to controls were calculated.
Immunoblot analysis
TT cells were seeded into 100 mm culture plates and incubated overnight. Culture media was removed and replaced with aliquots of resveratrol-loaded non-targeted and targeted micelles, empty targeted micelles, free resveratrol, and media controls, at a concentration of 50 μM of resveratrol or the equivalent, as described above. Plates were incubated for 4 days, with treatment media changed after 2 days. Total cellular protein lysates were isolated and concentrations were quantified using a bicinchoninic acid assay (Thermo Scientific, Waltham, MA) per the manufacturer's instructions. Protein lysates (30 μg) were denatured and resolved on 10% SDS-PAGE, and Western blot analysis was carried out as previously described.53 Briefly, separated proteins were transferred to nitrocellulose membranes (Bio-Rad Laboratories, Hercules, CA), blocked in milk (5% nonfat dry milk, 0.05% Tween 20 in phosphate buffered saline), and incubated with primary antibodies. The primary antibody dilutions were as follows: MASH for ASCL1 (1:2000), CgA (1:3000), and G3PDH (1:3000). Blot membranes were then incubated in the appropriate amounts of secondary antibodies— horseradish peroxidase goat anti-rabbit or anti-mouse—that were then detected using Immun-star, Pico, or Femto kits per the manufacturer's instructions.
Statistical analysis
Continuous variables in the MTT analysis were compared using one-way analysis of variance (ANOVA) with Tukey HSD correction for between-group comparisons (IBM SPSS, version 20.0; SPSS Inc., Chicago, IL). P-values less than or equal to 0.05 were considered significant.
Conclusions
OCT-conjugated unimolecular micelles formed by amphiphilic hyperbranched block copolymers have been developed as nanocarriers for targeted neuroendocrine tumor therapy. Resveratrol, a hydrophobic anticancer drug, has been physically loaded into the hydrophobic segments of micelles in order to improve its solubility and bioavailability. Cellular uptake, cellular proliferation, and NET marker expression studies revealed that the resveratrol-loaded targeted micelles showed higher cellular uptake and suppressed growth significantly more than resveratrol-loaded non-targeted micelles. We envision that this OCT-conjugated unimolecular micelle would serve as a new therapeutic nanoplatform for effective NET-targeted delivery of a variety of hydrophobic therapeutic agents.
Acknowledgments
This work was supported in part by the National Science Foundation (DMR 1032187), the NIH 1K25CA166178, the IEDR award from the Wisconsin Alumni Research Foundation (WARF), the NIH RO1 CA121115, the American Cancer Society Research Scholars Grant, the American Cancer Society MEN2 Thyroid Cancer professorship, the University of Wisconsin Carbone Cancer Center and Wisconsin Institutes for Discovery pilot project grant, the American College of Surgeons Resident Research Fellowship, and the NIH T32 Research Grant in Surgical Oncology CA090217.
Contributor Information
Renata Jaskula-Sztul, Email: sztul@surgery.wisc.edu.
Shaoqin Gong, Email: sgong@engr.wisc.edu.
References
- 1.Chabner BA, Roberts TG., Jr Nat Rev Cancer. 2005;5:65–72. doi: 10.1038/nrc1529. [DOI] [PubMed] [Google Scholar]
- 2.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 3.Siegel R, Naishadham D, Jemal A. CA Cancer J Clin. 2012;62:10–29. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- 4.Pitt SC, Moley JF. Semin Oncol. 2010;37:567–579. doi: 10.1053/j.seminoncol.2010.10.010. [DOI] [PubMed] [Google Scholar]
- 5.Sippel RS, Kunnimalaiyaan M, Chen H. Oncologist. 2008;13:539–547. doi: 10.1634/theoncologist.2007-0239. [DOI] [PubMed] [Google Scholar]
- 6.Gu FX, Karnik R, Wang AZ, Alexis F, Levy-Nissenbaum E, Hong S, Langer RS, Farokhzad OC. Nano Today. 2007;2:14–21. [Google Scholar]
- 7.Ferrari M. Nat Rev Cancer. 2005;5:161–171. doi: 10.1038/nrc1566. [DOI] [PubMed] [Google Scholar]
- 8.Williams DF. Biomaterials. 2009;30:5897–5909. doi: 10.1016/j.biomaterials.2009.07.027. [DOI] [PubMed] [Google Scholar]
- 9.Jain RK, Stylianopoulos T. Nat Rev Clin Oncol. 2010;7:653–664. doi: 10.1038/nrclinonc.2010.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nat Nanotechnol. 2007;2:751–760. doi: 10.1038/nnano.2007.387. [DOI] [PubMed] [Google Scholar]
- 11.Krol S, Macrez R, Docagne F, Defer G, Laurent S, Rahman M, Hajipour MJ, Kehoe PG, Mahmoudi M. Chem Rev. 2013;113:1877–1903. doi: 10.1021/cr200472g. [DOI] [PubMed] [Google Scholar]
- 12.Guo R, Li LL, Zhao WH, Chen YX, Wang XZ, Fang CJ, Feng W, Zhang TL, Ma X, Lu M, Peng SQ, Yan CH. Nanoscale. 2012;4:3577–3583. doi: 10.1039/c2nr30425b. [DOI] [PubMed] [Google Scholar]
- 13.Fan X, Jiao G, Zhao W, Jin P, Li X. Nanoscale. 2013;5:1143–1152. doi: 10.1039/c2nr33158f. [DOI] [PubMed] [Google Scholar]
- 14.Bae YH, Park K. J Controlled Release. 2011;153:198–205. doi: 10.1016/j.jconrel.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang X, Peng X, Wang Y, Wang Y, Shin DM, El-Sayed MA, Nie S. ACS nano. 2010;4:5887–5896. doi: 10.1021/nn102055s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu MK, Park J, Jon S. Theranostics. 2012;2:3–44. doi: 10.7150/thno.3463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zetter BR. Nat Biotechnol. 1997;15:1243–1244. doi: 10.1038/nbt1197-1243. [DOI] [PubMed] [Google Scholar]
- 18.Teow Y, Valiyaveettil S. Nanoscale. 2010;2:2607–2613. doi: 10.1039/c0nr00204f. [DOI] [PubMed] [Google Scholar]
- 19.Bauer W, Briner U, Doepfner W, Haller R, Huguenin R, Marbach P, Petcher TJ, Pless J. Life Sci. 1982;31:1133–1140. doi: 10.1016/0024-3205(82)90087-x. [DOI] [PubMed] [Google Scholar]
- 20.Chang CC, Liu DZ, Lin SY, Liang HJ, Hou WC, Huang WJ, Chang CH, Ho FM, Liang YC. Food Chem Toxicol. 2008;46:3116–3121. doi: 10.1016/j.fct.2008.06.084. [DOI] [PubMed] [Google Scholar]
- 21.Chen CH, Liu DZ, Fang HW, Liang HJ, Yang TS, Lin SY. Biosci Biotechnol Biochem. 2008;72:1586–1594. doi: 10.1271/bbb.80096. [DOI] [PubMed] [Google Scholar]
- 22.Mezo G, Manea M. Expert Opin Drug Deliv. 2010;7:79–96. doi: 10.1517/17425240903418410. [DOI] [PubMed] [Google Scholar]
- 23.Xiao Y, Jaskula-Sztul R, Javadi A, Xu W, Eide J, Dammalapati A, Kunnimalaiyaan M, Chen H, Gong S. Nanoscale. 2012;4:7185–7193. doi: 10.1039/c2nr31853a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xiao Y, Hong H, Javadi A, Engle JW, Xu W, Yang Y, Zhang Y, Barnhart TE, Cai W, Gong S. Biomaterials. 2012;33:3071–3082. doi: 10.1016/j.biomaterials.2011.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gong C, Deng S, Wu Q, Xiang M, Wei X, Li L, Gao X, Wang B, Sun L, Chen Y, Li Y, Liu L, Qian Z, Wei Y. Biomaterials. 2013;34:1413–1432. doi: 10.1016/j.biomaterials.2012.10.068. [DOI] [PubMed] [Google Scholar]
- 26.Nishiyama N, Kataoka K. Pharmacol Ther. 2006;112:630–648. doi: 10.1016/j.pharmthera.2006.05.006. [DOI] [PubMed] [Google Scholar]
- 27.Torchilin VP. Pharm Res. 2007;24:1–16. doi: 10.1007/s11095-006-9132-0. [DOI] [PubMed] [Google Scholar]
- 28.Xiao L, Xiong X, Sun X, Zhu Y, Yang H, Chen H, Gan L, Xu H, Yang X. Biomaterials. 2011;32:5148–5157. doi: 10.1016/j.biomaterials.2011.03.071. [DOI] [PubMed] [Google Scholar]
- 29.Zhao B, Zhao Y, Huang Y, Luo L, Song P, Wang X, Chen S, Yu K, Zhang X, Zhang Q. Biomaterials. 2012;33:2508–2520. doi: 10.1016/j.biomaterials.2011.11.078. [DOI] [PubMed] [Google Scholar]
- 30.Gao X, Wang B, Wei X, Men K, Zheng F, Zhou Y, Zheng Y, Gou M, Huang M, Guo G, Huang N, Qian Z, Wei Y. Nanoscale. 2012;4:7021–7030. doi: 10.1039/c2nr32181e. [DOI] [PubMed] [Google Scholar]
- 31.Prabaharan M, Grailer JJ, Pilla S, Steeber DA, Gong SQ. Biomaterials. 2009;30:5757–5766. doi: 10.1016/j.biomaterials.2009.07.020. [DOI] [PubMed] [Google Scholar]
- 32.Prabaharan M, Grailer JJ, Pilla S, Steeber DA, Gong SQ. Biomaterials. 2009;30:3009–3019. doi: 10.1016/j.biomaterials.2009.02.011. [DOI] [PubMed] [Google Scholar]
- 33.Li XJ, Qian YF, Liu T, Hu XL, Zhang GY, You YZ, Liu SY. Biomaterials. 2011;32:6595–6605. doi: 10.1016/j.biomaterials.2011.05.049. [DOI] [PubMed] [Google Scholar]
- 34.Pang Y, Liu JY, Su Y, Zhu BS, Huang W, Zhou YF, Zhu XY, Yan DY. Science China-Chemistry. 2010;53:2497–2508. [Google Scholar]
- 35.Xu W, Siddiqui IA, Nihal M, Pilla S, Rosenthal K, Mukhtar H, Gong S. Biomaterials. 2013;34:5244–5253. doi: 10.1016/j.biomaterials.2013.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Truong M, Cook MR, Pinchot SN, Kunnimalaiyaan M, Chen H. Ann Surg Oncol. 2011;18:1506–1511. doi: 10.1245/s10434-010-1488-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pinchot SN, Jaskula-Sztul R, Ning L, Peters NR, Cook MR, Kunnimalaiyaan M, Chen H. Cancer. 2011;117:1386–1398. doi: 10.1002/cncr.25652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Santos AC, Veiga F, Ribeiro AJ. Expert Opin Drug Deliv. 2011;8:973–990. doi: 10.1517/17425247.2011.581655. [DOI] [PubMed] [Google Scholar]
- 39.Walle T, Hsieh F, DeLegge MH, Oatis JE, Walle UK. Drug Metab Dispos. 2004;32:1377–1382. doi: 10.1124/dmd.104.000885. [DOI] [PubMed] [Google Scholar]
- 40.Walle T. Ann N Y Acad Sci. 2011;1215:9–15. doi: 10.1111/j.1749-6632.2010.05842.x. [DOI] [PubMed] [Google Scholar]
- 41.Delmas D, Aires V, Limagne E, Dutartre P, Mazué F, Ghiringhelli F, Latruffe N. Ann N Y Acad Sci. 2011;1215:48–59. doi: 10.1111/j.1749-6632.2010.05871.x. [DOI] [PubMed] [Google Scholar]
- 42.Xu W, Siddiqui IA, Nihal M, Pilla S, Rosenthal K, Mukhtar H, Gong S. Biomaterials. 2013;34:5244–5253. doi: 10.1016/j.biomaterials.2013.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kreutzer G, Ternat C, Nguyen TQ, Plummer CJG, Månson JAE, Castelletto V, Hamley IW, Sun F, Sheiko SS, Herrmann A, Ouali L, Sommer H, Fieber W, Velazco MI, Klok HA. Macromolecules. 2006;39:4507–4516. [Google Scholar]
- 44.Xiao Y, Hong H, Javadi A, Engle JW, Xu W, Yang Y, Zhang Y, Barnhart TE, Cai W, Gong S. Biomaterials. 2012;33:3071–3082. doi: 10.1016/j.biomaterials.2011.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang X, Grailer JJ, Pilla S, Steeber DA, Gong S. Bioconjugate Chem. 2010;21:496–504. doi: 10.1021/bc900422j. [DOI] [PubMed] [Google Scholar]
- 46.Su Yl, Wang J, Liu Hz. Langmuir. 2002;18:5370–5374. [Google Scholar]
- 47.Prabaharan M, Grailer JJ, Pilla S, Steeber DA, Gong S. Biomaterials. 2009;30:3009–3019. doi: 10.1016/j.biomaterials.2009.02.011. [DOI] [PubMed] [Google Scholar]
- 48.Sippel RS, Carpenter JE, Kunnimalaiyaan M, Chen H. Surgery. 2003;134:866–871. doi: 10.1016/s0039-6060(03)00418-5. discussion 871-863. [DOI] [PubMed] [Google Scholar]
- 49.Chen H, Udelsman R, Zeiger M, Ball D. Oncol Rep. 1997;4:775–778. doi: 10.3892/or.4.4.775. [DOI] [PubMed] [Google Scholar]
- 50.Chen H, Kunnimalaiyaan M, Van Gompel JJ. Thyroid. 2005;15:511–521. doi: 10.1089/thy.2005.15.511. [DOI] [PubMed] [Google Scholar]
- 51.Sippel RS, Carpenter JE, Kunnimalaiyaan M, Lagerholm S, Chen H. Am J Physiol Gastrointest Liver Physiol. 2003;285:G245–254. doi: 10.1152/ajpgi.00420.2002. [DOI] [PubMed] [Google Scholar]
- 52.Zarebczan B, Pinchot SN, Kunnimalaiyaan M, Chen H. Am J Surg. 2011;201:329–332. doi: 10.1016/j.amjsurg.2010.08.018. discussion 333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kunnimalaiyaan M, Traeger K, Chen H. Am J Physiol Gastrointest Liver Physiol. 2005;289:G636–642. doi: 10.1152/ajpgi.00146.2005. [DOI] [PubMed] [Google Scholar]