

Abstract
The development of new synthetic fluorination reactions has important implications in medicinal, agricultural and materials chemistries. Given the prevalence and accessibility of alcohols, methods to convert alcohols to trifluoromethanes are desirable. However, this transformation typically requires four-step processes, specialty chemicals, and/or stoichiometric metals to access the trifluoromethyl-containing product. A two-step copper-catalyzed decarboxylative protocol for converting allylic alcohols to trifluoromethanes is reported. Preliminary mechanistic studies distinguish this reaction from previously reported Cu-mediated reactions.
New synthetic methods for the efficient incorporation of trifluoromethanes into organic molecules are important for the fields of agricultural chemistry,1 medicinal chemistry,2 chemical biology,2a–b and materials science.3 As a result, many elegant and important preparations of trifluoromethanes have emerged in recent years.4 However, many simple, useful, and important transformations have not been achieved.
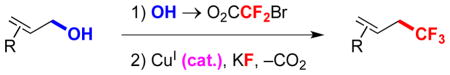
The ability to convert alcohols into trifluoromethanes using a simple, mild and robust catalytic system represents a desirable transformation. Alcohols are found in materials, bioactive molecules, and many chemical libraries, are readily accessed by many synthetic methods, and provide a wide variety of substrates for synthetic transformations. However, alcohols rarely serve as precursors to trifluoromethanes. Most commonly, the conversion of alcohols into trifluoromethanes requires four-step sequences that: 1) involve undesirable manipulation of oxidation states; 2) require excess time and labor to conduct; 3) generate excess waste; 4) lead to diminished overall yields (eq 1).5 Alternatively, alcohols can be converted to halides,6 trifluoroacetates,6b halodifluoroacetates,7 fluorosulfonyldifluoroacetates,6f,7a or xanthates,8 which can be trifluoromethylated in the presence of stoichiometric quantities of transition metals (eq 2). However, for economic and environmental considerations, catalytic methods are desirable. To this end, a Cu-based catalyst recently converted allylic bromides or chlorides (one step from allylic alcohols) to trifluoromethanes using the Ruppert-Prakash reagent (eq 3).9
In contrast, we envisioned an attractive approach might involve the conversion of an alcohol to a halodifluoroacetic ester, followed by a catalytic decarboxylative trifluoromethylation (eq 4). Although trifluoromethylation reactions of halodifluoroacetic esters have been conducted using stoichiometric CuI,6d–e,7,10 catalytic reactions have proven elusive over many years. Herein, we report a Cu-catalyzed conversion of allyl bromodifluoroacetic ester into trifluoromethanes, and preliminary mechanistic findings that distinguish this reaction from analogous Cu-mediated reactions.
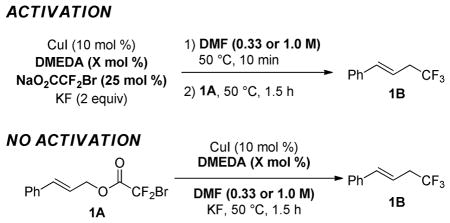
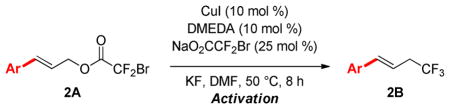
Initial screening of catalysts and conditions revealed that the Cu-mediated reaction of cinnamyl bromodifluoroacetate (1A) could be adapted to provide a Cu-catalyzed procedure. Several CuI salts and ligands provided catalytic activity, and CuI and N,N′-dimethylethylenediamine (DMEDA) were selected as an inexpensive and efficient system that is already available in many synthetic chemistry laboratories and stockrooms worldwide.11 The optimized catalyst included the use of 10 mol % CuI, 10 mol % DMEDA as a ligand, 25 mol % NaO2CCF2Br, KF (2 equiv), in DMF (1.0 M) at 50 °C. Importantly, a beneficial activation procedure was identified that involved the heating of the catalyst and reagents in DMF at 50 °C prior to addition of the substrate (vide infra).
Three parameters, including reaction concentration, ligand, and activation of the catalyst, proved most critical for obtaining a high yield of product (Table 1). Compared with the optimized reaction conditions (entry 1), reactions run at lower concentration (entry 2), or without employing the activation procedure (entry 3) provided less efficient catalyst systems as defined by the ratio of yield/conversion. Employing the activation procedure, the use of CuI/DMEDA seemed to provide a less-active catalyst than that derived from CuI alone at the 1.5 h time point (entries 1, 4); however, when the reactions were allowed to proceed to full conversion, a higher yield of product was reproducibly obtained using CuI/DMEDA (entries 5–6). Thus, DMEDA could serve to stabilize the active catalyst against decomposition near the end of the reaction.
Table 1.
Sensitivity of Cu-Catalyzed Trifluoromethylation to Concentration, Ligand, and Activation of Catalyst.a
| ||||||
|---|---|---|---|---|---|---|
| entry | activation | [1A] (M) | DMEDA (mol %) | yieldb (%) | convb (%) | yield/conv |
| 1 | √ | 1.0 | 10 | 52 | 64 | 0.82 |
| 2 | √ | 0.33 | 10 | 63 | 85 | 0.74 |
| 3 | – | 1.0 | 10 | 26 | 54 | 0.49 |
| 4 | √ | 1.0 | 0 | 66 | 79 | 0.83 |
| 5c | √ | 1.0 | 10 | 83 | 100 | 0.83 |
| 6c | √ | 1.0 | 0 | 72 | 100 | 0.72 |
| 7 | – | 1.0 | 0 | 50 | 66 | 0.76 |
| 8 | – | 0.33 | 0 | 52 | 78 | 0.67 |
Reactions were performed with 0.20 mmol substrate, 0.050 mmol NaO2CCF2Br, 0.40 mmol KF in 0.20 mL DMF at 50 °C for 1.5 h.
Conversion and yield data were determined by GC/FID analysis using dodecane as an internal standard. Each data point represents an average of 2–4 experiments.
The reactions were run for 8 h instead of 1.5 h.
A variety of 2-, 3- and 4-substituted cinnamyl bromodifluoroacetates (2A) were compatible with the present reaction (Table 4). Electron-deficient (entries 1–5) and electron-neutral cinnamyl systems (entries 7–8) reacted in good yields, although an electron rich substrate provided the product in slightly lower yield (entry 9). In general, substrates capable of affording resonance stabilized allyl cations (e.g., 4-NMe2) were unstable to both acidic and basic conditions, which limited purification and storage of this class of substrates. Aryl (pseudo)halides were well tolerated, and did not undergo aromatic trifluoromethylation (entries 1–3, 6). In addition, compounds bearing heterocycles were tolerated (entries 9–10). Finally, on an 8 mmol scale, over 1.9 g of material was obtained in high yield (entry 2), which suggests that the present reaction could be amenable to larger scale processes.
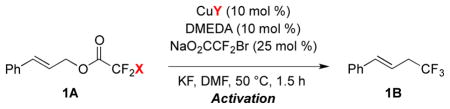
Table 4.
Unique Reactivity of Allyl Bromodifluoroacetes.a
| ||||||
|---|---|---|---|---|---|---|
| entry | X | Y | other | conv (%)b | yield (%)b | yield/conv |
| 1 | F | I | - | 26 | 0 | 0 |
| 2 | Cl | I | - | 31 | 1 | 0.03 |
| 3 | Br | I | - | 52 | 46 | 0.88 |
| 4 | I | I | - | 53 | 25 | 0.47 |
| 5 | Br | I | +100% KI | 68 | 38 | 0.56 |
| 6 | Br | (MeCN)4PF6 | 8 h | >99 | 79 | 0.79 |
Reactions were performed with 0.20 mmol substrate, 0.050 mmol NaO2CCF2Br, 0.40 mmol KF in 0.20 mL DMF at 50 °C for 1.5 h.
Conversion and yield data were determined by GC/FID analysis using dodecane as an internal standard.
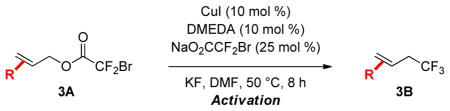
Disubstituted and non-conjugated allylic esters (3A) containing a diverse array of functional groups provided moderate to good yields of E alkene products (3B, Table 3). Substituents at the α and β positions of the styrene were tolerated (entries 1–3), and non-conjugated allylic systems displayed good reactivity (entries 4–7). Several aliphatic functional groups were compatible with the reaction, including esters, imides, and benzyl ethers (entries 5–7). In entries 5–6, disastereomeric mixtures of substrates (3A, E/Z = 4:1) provided thermodynamically-stable E-allyl trifluoromethane products 3B in excellent selectivities (E/Z > 19:1). Further, the reaction of a pure Z-alkene substrate afforded the E product in excellent diastereoselectivity (entry 7). When monitoring the reaction by both GC/FID and 19F NMR, slow isomerization of the substrate was observed, while the E-product was formed in greater than 15:1 dr throughout the course of the reaction. In control reactions, the Z-substrate was stable when treated with KF in DMF at 50 °C. These data could implicate the existence of a π-allyl intermediate that reacts to generate the more stable E-product.6b,9 However at present, other explanations for this isomerization phenomenon cannot be excluded.
Table 3.
Disubstituted and Non-Conjugated Allylic Bromodifluoroacetates Undergo Decarboxylative Trifluoromethylation.a
| ||||
|---|---|---|---|---|
| entry | E/Z of 3Ab | product | E/Z of 3Bc | yield (%)d |
| 1 | >98:2 |
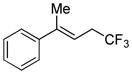
|
>98:2 | 55 (60) |
| 2 | >98:2 |
|
>98:2 | 74 (80) |
| 3 | – |
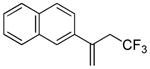
|
– | 56 (51) |
| 4 | >98:2 |
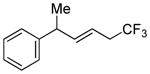
|
>98:2 | 78 (75) |
| 5e | 78:22 |
|
97:3 | 76 (75) |
| 6e | 79:21 |
|
98:2 | 85 (82) |
| 7 | <2:98 |
|
96:4 | 81 (79) |
Reactions were performed with 0.20 mmol substrate, 0.020 mmol CuI, 0.020 mmol DMEDA, 0.050 mmol NaO2CCF2Br, 0.40 mmol KF in 0.20 mL DMF at 50 °C for 8 h following 10 min activation.
Determined by 1H NMR.
Determined by 19F NMR.
Isolated yield, number in parentheses indicates 19F NMR yield using α,α,α-trifluorotoluene as an internal standard.
18 h.
Using the Cu/DMEDA-based catalyst system, bromodifluoroacetic esters provided unique reactivity (Table 4). A trend of increasing reactivity was observed for cinnamyl trifluoroacetate < chlorodifluoroacetate < bromodifluoroacetate (entries 1–3);12 however, the reaction of cinnamyl difluoroiodoacetate provided a low yield of product (entry 4). It was hypothesized that I−, generated as a byproduct of the reaction, inhibited catalysis. In support of this theory, the addition of exogenous KI to the reaction of cinnamyl bromodifluoroacetate decreased the yield of product (entry 5). Combined, these two findings suggest that I− does not participate in the catalytic reaction. In fact, the Cu-catalyzed reaction could be conducted in the complete absence of I− (entry 6), a key feature that distinguishes the present Cu-catalyzed reaction from previously reported Cu-mediated reactions.7
Although thorough mechanistic studies have not been conducted, the present Cu-catalyzed reaction likely involves a mechanism distinct from CuI-mediated reactions of allyl halodifluoroacetates. For the Cu-catalyzed reaction, the activation procedure presumably serves to convert the precatalytic combination of CuI/DMEDA/NaO2CCF2Br/KF into the active catalyst, LnCu–CF3 (Scheme 2A). LnCu–CF3 can then promote direct trifluoromethylation of the substrate without participation of I−. The substitution reaction potentially involves a π-allyl intermediate, which has been proposed in other allylic substitution reactions using LnCu–CF3 in both stoichiometric6b and catalytic9 systems. In contrast, the Cu-mediated reaction invokes I− as a key feature of the mechanism (Scheme 2B).7a In this case, I− participated by converting the bromodifluoroacetic ester to an allyl iodide, which then reacted with Cu–CF3. Studies to thoroughly explore the mechanism of the present transformation, including the activation procedure, are ongoing, and results will be presented in due time. Further, ongoing work aims to expand the scope of this process to alternate classes of substrates are currently underway.
Scheme 2.

Proposed Catalytic Cycle Involving a Reactive Cu–CF3 Species.
In conclusion, a catalytic method for the conversion of allylic alcohols to trifluoromethanes via bromodifluoroacetic esters has been developed. Conjugated and non-conjugated substrates bearing a variety of functional groups afford α-substituted trifluoromethylated products in moderate to good yield and excellent diastereoselectivity for the E-stereoisomer. Beneficial aspects of this transformation include: 1) employment of a mild, inexpensive and atom-economical source of CF3 in near-stoichiometric quantity; 2) development of a shortened strategy for converting readily available allylic alcohols into trifluoromethyl analogs; 3) the ability to conduct trifluoromethylation reactions using only a catalytic quantity of metal. Finally, functionalization of the allyl trifluoromethane-based product should be useful for accessing more complex fluorinated compounds.7b,13
Supplementary Material
Scheme 1.

An Economical Approach for the Conversion of Allylic Alcohols to Trifluoromethanes
Table 2.
Substituted Cinnamyl Bromodifluoroacetates Undergo Decarboxylative Trifluoromethylation.a
| ||
|---|---|---|
| entry | Ar | Yield (%)b |
| 1 |
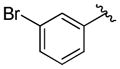
|
81 (82) |
| 2c | 89 (87) | |
| 3 |
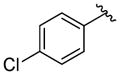
|
72 (76) |
| 4 |
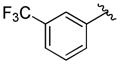
|
71 (75) |
| 5 |
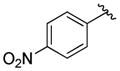
|
83 (80) |
| 6 |
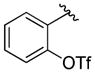
|
75 (75) |
| 7 |
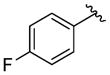
|
65 (79) |
| 8 |
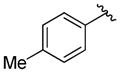
|
71 (79) |
| 9 |
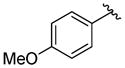
|
57 (60) |
| 10 |
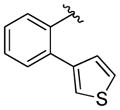
|
80 (78) |
| 11 |
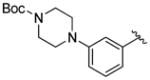
|
58 (57) |
Reactions were performed with 0.20 mmol substrate, 0.020 mmol CuI, 0.020 mmol DMEDA, 0.050 mmol NaO2CCF2Br, 0.40 mmol KF in 0.20 mL DMF at 50 °C for 8 h following 10 min activation.
Isolated yield, number in parentheses indicates 19F NMR yield using α,α,α-trifluorotoluene as an internal standard.
Reaction conducted on an 8 mmol scale.
Acknowledgments
We thank the donors of the Herman Frasch Foundation for Chemical Research (701-HF12) for support of this project, and the NIGMS Training Grant on Dynamic Aspects of Chemical Biology (T32 GM08545) for a graduate traineeship (B. R. A.). We also gratefully acknowledge support of from the University of Kansas Office of the Provost, Department of Medicinal Chemistry, and New Faculty General Research Fund (2302264).
Footnotes
Supporting Information Available. Experimental procedures and compound characterization data are available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Theodoridis G. Fluorine-Containing Agrochemicals: An Overview of Recent Developments. In: Tressaud A, editor. Fluorine and the Environment: Agrochemicals, Archaeology, Green Chemistry & Water. Elsevier B. V; Amsterdam: 2006. [Google Scholar]
- 2.(a) Bégué J-P, Bonnet-Delpon D. Bioorganic and Medicinal Chemistry of Fluorine. John Wiley & Sons; Hoboken: 2008. [Google Scholar]; (b) Ojima I, editor. Fluorine in Medicinal Chemistry and Chemical Biology. Blackwell Publishing Ltd; West Sussex: 2009. [Google Scholar]
- 3.(a) Patil Y, Ameduri B. Prog Polym Sci. 2013;38:703. [Google Scholar]; (b) Dhara MG, Banerjee S. Prog Polym Sci. 2010;35:1022. [Google Scholar]
- 4.For some recent reviews, see: Liang T, Neumann CN, Ritter T. Angew Chem, Int Ed. 2013;52:8214. doi: 10.1002/anie.201206566.Liu G. Org Biomol Chem. 2012;10:6243. doi: 10.1039/c2ob25702e.Hollingworth C, Gouverneur V. Chem Commun. 2012;48:2929. doi: 10.1039/c2cc16158c.Tomashenko OA, Grushin VV. 2011;111:4475. doi: 10.1021/cr1004293.
- 5.(a) Fustero S, Román R, Sanz-Cervera JF, Simón-Fuentes A, Bueno J, Villanova S. J Org Chem. 2008;73:8545. doi: 10.1021/jo801729p. [DOI] [PubMed] [Google Scholar]; (b) Del Valle JR, Goodman M. Angew Chem, Int Ed. 2002;41:1600. doi: 10.1002/1521-3773(20020503)41:9<1600::aid-anie1600>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]; (c) Jeannot F, Gosselin G, Mathe C. Org Biomol Chem. 2003;1:2096. doi: 10.1039/b303093h. [DOI] [PubMed] [Google Scholar]; (d) Qiu XL, Qing FL. J Org Chem. 2002;67:7162. doi: 10.1021/jo0257400. [DOI] [PubMed] [Google Scholar]; (e) Serafinowski PJ, Brown CA. Tetrahedron. 2000;56:333. [Google Scholar]; (f) Chang J, Liu K, McEachem EJ, Mu C, Selenick HG, Shi F, Vocaldo DJ, Wang Y, Wei Z, Zhou Y, Zhu Y. Alectos Therapeutics Inc., Can., Merck Sharp & Dohme Corp; WO 2012062157 A1. 2012 May 18;; (g) Tam TF, Leung-Toung R, Wang Y, Zhao Y. Apotex Technologies Inc., Can; WO 2008116301 A1. 2008 Oct 2;
- 6.(a) Kobayashi Y, Yamamoto K, Kumadaki I. Tetrahedron Lett. 1979;20:4071. [Google Scholar]; (b) Larsson JM, Pathipati SR, Szabó KJ. J Org Chem. 2013;78:7330. doi: 10.1021/jo4010074. [DOI] [PubMed] [Google Scholar]; (c) Bouillon J-P, Maliverney C, Merenyi R, Viehe HG. J Chem Soc, Perkin Trans. 1991;1:2147. [Google Scholar]; (d) Su DB, Duan JX, Chen QY. Tetrahedron Lett. 1991;32:7689. [Google Scholar]; (e) Duan JX, Su DB, Chen QY. J Fluorine Chem. 1993;61:279. [Google Scholar]; (f) Chen QY. J Fluorine Chem. 1995;72:241. [Google Scholar]
- 7.(a) Duan J-X, Chen Q-Y. J Chem Soc, Perkin Trans. 1994;1:725. [Google Scholar]; (b) Tan L, Chen C-y, Larsen RD, Verhoeven TR, Reider PJ. Tetrahedron Lett. 1998;39:3961. [Google Scholar]
- 8.Zhu L, Liu S, Douglas JT, Altman RA. Chem Eur J. 2013;19:12800. doi: 10.1002/chem.201302328. [DOI] [PubMed] [Google Scholar]
- 9.Miyake Y, Ota S-i, Nishibayashi Y. Chem—Eur J. 2012;18:13255. doi: 10.1002/chem.201202853. [DOI] [PubMed] [Google Scholar]
- 10.(a) MacNeil JG, Jr, Burton DJ. J Fluorine Chem. 1991;55:225. [Google Scholar]; (b) Duan JX, Su DB, Wu JP, Chen QY. J Fluorine Chem. 1994;66:167. [Google Scholar]; (c) Langlois BR, Roques N. J Fluorine Chem. 2007;128:1318. [Google Scholar]; (d) McReynolds KA, Lewis RS, Ackerman LKG, Dubinina GG, Brennessel WW, Vicic DA. J Fluorine Chem. 2010;131:1108. [Google Scholar]; (e) Li Y, Chen T, Wang H, Zhang R, Jin K, Wang X, Duan C. Synlett. 2011;12:1713. [Google Scholar]; (f) Schareina T, Wu XF, Zapf A, Cotté A, Gotta M, Beller M. Top Catal. 2012;55:426. [Google Scholar]
- 11.Surry DS, Buchwald SL. Chem Sci. 2010;1:13–31. doi: 10.1039/C0SC00107D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.At higher temperatures, the chlorodifluoroacetic ester provided mild activity, while the trifluoroacetic ester provided no reactivity.
- 13.(a) Shimizu R, Egami H, Hamashima Y, Sodeoka M. Angew Chem, Int Ed. 2012;51:4577. doi: 10.1002/anie.201201095. [DOI] [PubMed] [Google Scholar]; (b) Wang CJ, Sun X, Zhang X. Angew Chem, Int Ed. 2005;44:4933. doi: 10.1002/anie.200501332. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.