

Abstract
Motivated by the promises of gene therapy, there is a large interest in developing non-viral lipid-based vectors for therapeutic applications due to their nonimmunogenicity, low toxicity, ease of production, and the potential of transferring large pieces of DNA into cells. In fact, cationic lipid (CL) based vectors are among the prevalent synthetic carriers of nucleic acids (NAs) currently used in human clinical gene therapy trials worldwide. These vectors are studied both for gene delivery with CL–DNA complexes and gene silencing with CL–siRNA (short-interfering RNA) complexes. However, their transfection efficiencies and silencing efficiencies remain low compared to those of engineered viral vectors. This reflects the currently poor understanding of transfection-related mechanisms at the molecular and self-assembled levels, including a lack of knowledge about interactions between membranes and double stranded NAs and between CL–NA complexes and cellular components. In this review, we describe our recent efforts to improve the mechanistic understanding of transfection by CL–NA complexes, which will help to design optimal lipid-based carriers of DNA and siRNA for therapeutic gene delivery and gene silencing.
Keywords: multivalent cationic lipid, cholesterol, siRNA, small angle x-ray scattering, gene delivery
1 Introduction
In this chapter, we provide an overview of our recent efforts to develop a fundamental science base for the design and preparation of optimal lipid-carriers of DNA and siRNA for gene therapy and gene silencing. We employ synthesis of custom multivalent lipids, synchrotron x-ray diffraction (XRD) techniques, optical and cryo-electron microscopy as well as biological assays in order to correlate the structures, chemical and biophysical properties of cationic lipid (CL)–NA complexes to their biological activity and to clarify the interactions between CL–NA complexes and cellular components. Earlier work has been reviewed elsewhere [1-7] and will not be covered exhaustively here.
Motivation
Gene therapy—addressing disease at the level of the genetic cause, typically with nucleic acid (NA) “drugs”—holds great promise for future medical applications. In fact, numerous clinical trials are currently ongoing, targeting cancers, inherited diseases, and many other disorders with this novel medical approach [9,10]. Concurrently, substantial research efforts are directed towards developing and fundamentally understanding NA carriers (vectors). These include engineered viruses as well as synthetic vectors, where the negatively charged NA is complexed with cationic liposomes [2,4,11-15] or cationic polyelectrolytes [16-18]. Synthetic (non-viral) vectors have garnered much interest due to their nonimmunogenicity and their ability to transfer very large DNA pieces into cells (which is not feasible with viral vectors) [19]. To improve their efficiencies by rational design, significant ongoing research efforts are aimed at elucidating the mechanisms of action of non-viral vectors intended for therapeutic applications. Cationic lipid-based vectors are studied both for gene delivery (as CL–DNA complexes) and gene silencing (as CL–siRNA (short interfering RNA) complexes). Their transfection efficiency (TE; a measure of the expression of an exogenous gene that is transferred) and silencing efficiency (SE: a measure of specific post-transcriptional silencing of the gene targeted by the transferred siRNA), however, remain low compared to viral vectors. Understanding the pathways and mechanisms governing the interaction of CL–DNA complexes and cells is crucial to make lipid-mediated gene delivery therapeutically viable. The complexity of the transfection process—from initial attachment of a CL–DNA complex to the plasma membrane to internalization of the complex via endocytosis, its release from the endosome followed by the dissociation of the CLs from the NA and (in the case of DNA transfection) finally the transport of the NA into the nucleus followed by successful gene expression—suggests that an interplay of many critically important parameters needs to be considered in order to achieve successful NA delivery.
Gene Delivery Barriers and Complex Properties
The many barriers to successful gene delivery range from serum stability to endosomal release and delivery to the nucleus [20]. Physico-chemical parameters of CL–DNA complexes often strongly affect their ability to overcome these barriers. Two examples of key parameters which impact the TE of CL–DNA complexes are the membrane charge density (σM, average charge per unit area of the membrane) of the cationic lipid membranes and the cationic lipid to DNA charge ratio, ρchg [4,7,21]. These parameters are directly affected by complex composition, since the membrane charge density is defined by the ratio and nature of cationic and neutral lipid in the membrane. Another important parameter is the nanoscopic internal structure of the complexes, which is affected by the choice of lipids and complex composition.
Structures of CL–DNA Complexes
CL–DNA complexes form spontaneously when solutions of cationic liposomes (typically containing both a cationic lipid and a neutral “helper” lipid) are combined. We have discovered several distinct nanoscale structures of CL–DNA complexes by synchrotron x-ray diffraction, three of which are schematically shown in Fig. 1. These are the prevalent lamellar phase with DNA sandwiched between cationic membranes (LαC) [22], the inverted hexagonal phase with DNA encapsulated within inverse lipid tubes (HIIC) [23], and the more recently discovered HIC phase, with hexagonally arranged rod-like micelles surrounded by DNA chains forming a continuous substructure with honeycomb symmetry [24]. Both the neutral lipid and the cationic lipid can drive the formation of specific structures of CL–DNA complexes. The inverse cone shape of DOPE favors formation of the HIIC phase, while the formation of micelles in the HIC phase is driven by a highly charged (16+), cone-shaped multivalent cationic lipid.
Fig. 1.

Mixing DNA and cationic liposomes results in the spontaneous formation of CL–DNA complexes with equilibrium self-assembled structures. The schematics show the local (nanoscale) interior structure of CL–DNA complexes as derived from synchrotron x-ray diffraction data. (A) The lamellar LαC phase of CL–DNA complexes with alternating lipid bilayers and DNA monolayers [22]. (B) The inverted hexagonal HIIC phase of CL–DNA complexes, comprised of DNA inserted within inverse lipid tubules, which are arranged on a hexagonal lattice [23]. (C) The more recently discovered hexagonal HICI phase of CL–DNA complexes, where a cationic lipid with a large dendritic headgroup leads to the formation of rod-like lipid micelles arranged on a hexagonal lattice with DNA inserted within the interstices with honeycomb symmetry [24]. Reprinted in part from [23] and [24] with permission. LaC and HIIC phase images Copyright 1998 American Association for the Advancement of Science. HIC phase image Copyright 2006 American Chemical Society
The Effect of Complex Structure on Transfection Mechanism and Efficiency
The internal structure of the complexes can directly determine the mechanism of transfection [4,23,25]. We have established that for LαC CL–DNA complexes, the membrane charge density (σM) is a predictive parameter for transfection efficiency [21] (see Sect. 2), i.e., the data for monovalent and multiva-lent CLs are described by a simple bell-curve. In contrast, for inverted hexagonal HIIC CL–DNA complexes, TE is independent of σM, suggesting a distinctly different mechanism of transfection. Consistent with the TE data, confocal microscopy revealed distinctly different CL–DNA complex pathways and interactions with cells, which depended on both the structure (HIIC versus LαC) and, for LαC complexes, on σM [25]. Thus, the mechanism of transfection by CL–DNA complexes is dependent both on their structure and, for a given structure, on chemical and physical parameters of the complexes.
For lamellar CL–DNA complexes, endosomal escape via activated fusion limits TE and strongly depends on σM, whereas the inverted hexagonal phase promotes fusion of the CL–DNA complex membranes with cellular membranes independent of σM (see Fig. 3). For HIIC CL–DNA complexes, a model was proposed that recognizes the importance of the outer (water-facing) layer of positive curvature around the inverted hexagonal CL–DNA complex [25]. The lipids in the outer layer have a negative spontaneous curvature and thus are energetically frustrated, which favors fusion of the complexes’ membranes with extra-cellular and endosomal membranes encountered along the gene transfer pathway.
Fig. 3.

(A) Transfection efficiency (TE) as a function of mol % DOPC for DNA complexes prepared with MVL2 (diamonds), MVL3 (squares), MVL5 (triangles), TMVL5 (inverted-triangles), and DOTAP (open circles). All data was taken at ρchg = 2.8. (B) The same TE data plotted against the membrane charge density, σσ shows that TE of the lamellar LαC complexes describes a universal, bell-shaped curve as a function of σM (the solid line is a Gaussian fit to the data). Data for DOTAP/DOPE complexes (open circles, HIIC phase) deviate from the universal curve, indicative of a distinctly different transfection mechanism for the inverted hexagonal phase. Three regimes of transfection efficiency are labeled. Reproduced with permission from [21]. Copyright 2005 John Wiley & Sons Limited
2 A Universal Curve for Transfection Efficiency versus Membrane Charge Density
In this section, we describe some of our efforts focused on clarifying the role of the membrane charge density (σM) as a key chemical parameter for transfection by LαC CL–DNA complexes. In a previous study, we had tentatively identified σM as a universal parameter for transfection by lamellar complexes [25]. However, that study was limited by the range of charge densities accessible with commercially available lipids.
To more thoroughly study the dependence of TE on σM and evaluate a broad range of higher charge densities, we synthesized a series of new multivalent lipids (MVLs) with headgroup valencies ranging from +2 to +5 which allowed systematic variation of headgroup size and charge [21, 26]. Fig. 2 shows the chemical structures of the lipids DOTAP, DOPC, DOPE and MVL5. DOTAP is a commercially available, commonly used univalent lipid (UVL). X-ray diffraction showed that the new MVLs form CL–DNA complexes that exhibit the lamellar LαC phase (Fig. 1A). Fig. 3 shows TE results for complexes transfecting mouse fibroblast cells at various MVL/DOPC ratios [21]. Also included is data for the monovalent lipid DOTAP mixed with DOPC, as a control system. The complexes were prepared at the optimum cationic lipid/DNA charge ratio σchg = 2.8, and the amount of DNA and cationic lipid per sample was kept constant. Thus, only the amount of neutral lipid varies between data points.
Fig. 2.
Chemical structures of the zwitterionic neutral lipids DOPC (1,2-dioleoyl-sn-glycero-3-phosphatidylcholine) and DOPE (1,2-dioleoyl-sn-glycero-3-phosphatidylethanolamine) and the cationic lipids DOTAP (1,2-dioleoyl-3-trimethylammonium-propane, a UVL) and MVL5 (a custom-synthesized MVL)
Fig. 3A shows the TE data as a function of the mole fraction of cationic lipid. For all cationic lipids, a maximum in TE as a function of lipid composition is observed: at 65 mol % for MVL2, 70 mol % for MVL3, 50 mol % for MVL5, 55 mol % for TMVL5, and 90 mol % for DOTAP. The optimal molar ratio results in a TE that is close to three decades higher than that of the lowest transfecting complexes in these systems, and each data set fits a skewed bell-shaped curve. Fig. 3B shows the data of Fig. 3A plotted versus the membrane charge density, σM (σM = [1-Φnl/(Φnl + rΦcl)σcl), where r = Acl/Anl is the ratio of the headgroup areas of the cationic and the neutral lipid; σcl = eZ/Acl is the charge density of the cationic lipid with valence Z (measured experimentally [21]); Φnl and Φcl are the mole fractions of the neutral and cationic lipids, respectively. We used Anl = 72 Å2, rDOTAP = 1, rMVL2 = 1.05 ± 0.05, rMVL3 = 1.30 ± 0.05, rMVL5 = 2.3 ± 0.1, rTMVL5 = 2.5 ± 0.1, ZDOTAP = 1, ZMVL2 = 2.0 ± 0.1, ZMVL3 = 2.5 ± 0.1 ZMVL5 = ZTMVL5 = 4.5 ± 0.1). The resulting solid curve going through the data describes a Gaussian TE = TE0 + A exp-[(σM-σ0)/w]2, with optimal charge density σ0 = 17.0 ± 0.1 ×10–3 e/Å2, TE0 = −(2.4 ± 0.4) ×107 RLU/mg protein A = 9.4 ± 0.6 ×108 RLU/mg protein, w = 5.8 ± 0.5 ×10–3 e/Å2.
Remarkably, all the data points for cationic lipids with different valence merge onto a single bell-shaped curve. This identifies σσ, rather than the charge of the lipid, as a universal parameter for transfection by lamellar LαC CL–DNA complexes (i.e. a predictor of transfection efficiency). The bell curve of Fig. 3B identifies three distinct regimes related to interactions between complexes and cells: at low σM (Regime I), TE increases with increasing σM; at intermediate σM (Regime II), TE exhibits saturated behavior; and unexpectedly, at high σM, TE decreases with increasing σM [21].
The TE data, combined with our confocal microscopy data for low and high TE LαC complexes interacting with cells [25] suggests a model of cellular uptake of LαC complexes depicted schematically in Fig. 4 [21]. The initial attachment of CL–DNA complexes to cells is mediated by electrostatics (Fig. 4a) and followed by cellular uptake via endocytosis (Fig. 4b). At low σM < σM * (Regime I, Fig. 3B), transfection is limited by endosomal escape (Fig. 4c and Fig. 4d). As σM increases towards an optimal value σM ≈ σM * (near the boundary between Regimes I and II shown in Fig. 3B), TE increases exponentially with σM over three orders of magnitude as the complexes are able to overcome this barrier by fusing with the endosomal membrane and releasing smaller complexes into the cytoplasm (Fig. 4e and Fig. 4f). In the regime of high σM > σM * (Regime III, Fig. 3B), accessible to us for the first time with the custom synthesized multivalent cationic lipids [21,26], complexes are able to escape the endosome, yet they exhibit a decreasing level of efficiency as σM further increases, presumably due to the DNA's inability to dissociate from the highly charged membranes of complexes in the cytosol (Fig. 4e and Fig. g). The optimal TE in Regime II reflects the compromise between opposing requirements (Fig. 4f): escape from endosomes requires high σM, but dissociation of complexes in the cytoplasm requires low σM. Future optimization of TE requires decoupling these requirements. The following two sections show how specific neutral or cationic lipid components are able to force deviations from the universal curve.
Fig. 4.

Model of cellular uptake of LαC complexes. Complexes adhere to cells due to electrostatics (a) and enter through endocytosis (b and c). Low σM complexes remain trapped in the endo-some (d). High σM complexes escape the endosome (e) where released DNA may form aggregates with cationic biomolecules (f) or the complexes are less able to dissociate and less DNA is available (g). Reproduced with permission from [21]. Copyright 2005 John Wiley & Sons Limited
3 The Role of Cholesterol and Structurally Related Molecules in Enhancing Transfection by Cationic Liposome–DNA Complexes
Motivated by its important role in gene delivery, we have studied the effect of cholesterol (chol) and several analogs on the transfection efficiency of lamellar CL–DNA complexes in vitro [27]. As evident from the results on DOPC/DOTAP and DOPE/DOTAP vectors, the nature of the neutral lipid component is an important parameter that is worth further exploration. Conveniently, a number of neutral lipids are commercially available. In addition, modifiying the neutral lipid component has the potential to improve TE in a regime (at low σM) where DNA dissociation from the complex in the cytosol is not yet a barrier to transfection.
Several reports in the literature state that DOPE, while successfully used for in vitro gene delivery, is a poor helper lipid for in vivo applications [28-32]. Instead, for reasons that are not understood, lipid mixtures for successful transfection in vivo seem to require cholesterol [33]. In fact, an equimolar mixture of cholesterol and DOTAP is widely used for in vivo experiments and clinical trials. Cholesterol has also been included in liposomes along with cationic DOTAP and fusogenic DOPE to form a potent mixture used to study the treatment of ovarian cancer by delivery of the p53 tumor suppressor gene [34,35].
We recently discovered an unexpectedly large enhancement in TE of poorly transfecting lamellar CL–DNA complexes upon incorporation of cholesterol. To elucidate the cause of this enhancement, we studied the effect of added cholesterol and structurally related molecules in the low membrane charge density regime (low ΦDOTAP) of the DOTAP/DOPC–DNA system. In addition to cholesterol, we investigated sterols (ergosterol, the plant version of cholesterol, and β-estradiol, an estrogen), other steroids (progesterone, a progestin hormone, and dihydroisoandrosterone, a testosterone precursor) and ergocalciferol (which derives from a sterol precursor by opening of a central ring) [27]. Thus, we were able to correlate the biophysical properties of membranes, inter-membrane interactions and changes in TE to structural properties of the steroid molecules. While keeping the membrane charge (i.e. the lipid/DNA charge ratio and the molar fraction of DOTAP) constant, we gradually replaced DOPC molecules by cholesterol or its analogs. TE of low-transfecting DOTAP/DOPC–DNA complexes (ΦDOTAP = 0.3) increases by a factor of ten with the inclusion of only 15 mol% cholesterol, and further inclusion of cholesterol continues to exponentially increase TE.
3.1 Structure and Membrane Charge Density of CL–DNA Complexes Containing Cholesterol and Analogs
X-ray diffraction showed that DOTAP/DOPC–DNA complexes containing added cholesterol or structurally related molecules form a single lamellar phase. The only exception is ergocalciferol, where two lamellar phases coexist for Φergocalciferol ≥ 0.2.
At Φchol ≥ 0.4, phase coexistence of CL–DNA complexes and cholesterol monohydrate crystals is observed, which means that the lipid composition (and most notably of ΦDOTAP) in the complex differs from that of the lipid mixture used for preparation of the complexes. This is in agreement with previous reports in the literature, which also observe membrane saturation with cholesterol at about 40 mol% [36,37]. The phase behavior of DOTAP/DOPC–DNA complexes containing cholesterol analogs is similar: they exhibit membrane saturation at high analog content, coexisting with phase-separated cholesterol analog for Φergosterol ≥ 0.4, Φergocalciferol ≥ 0.4, and for Φsteroid ≥ 0.3 in the case of β-estradiol, progesterone and dihydroisoandrosterone.
Thus, the structural features of β-estradiol, progesterone and dihydroisoandrosterone, i.e., absence of an alkyl tail and the presence of a second polar group (see Fig. 6), seem to favor membrane saturation at lower molar fractions of steroid compared to cholesterol. In addition, XRD shows that the lamellar repeat distance d (see Fig. 1A, d = δw + δm) of complexes containing these steroids is about 5 Å shorter than that of corresponding complexes containing cholesterol or ergosterol. This suggests a different packing of these groups of molecules within the lipid bilayer.
Fig. 6.

(A) TEs of DOTAP/DOPC/steroid–DNA complexes. The TE data for ergosterol 2 and ergocalciferol 3 follows a similar dependence on the steroid content in the membrane as that of cholesterol 1: TE rapidly increases with Φsteroid. In contrast, addition of β-estradiol 4, progesterone 5 and dehydroisoandrosterone 6 only modestly enhances TE until high steroid contents (35 mol% and higher) are reached, where phase separation occurs and TE suddenly increases to values comparable with TE of cholesterol-containing complexes. The major structural differences between these two groups of molecules are the absence of the terminal alkyl chain and the presence of a second polar moiety in case of 4–6. (B) Chemical structure of the investigated steroid molecules. (C) TEs of DOTAP/DOPC/steroid–DNA complexes plotted as a function of experimentally obtained σM. The data for cholesterol (dark circles) and ergosterol (dark triangles) deviate significantly from the universal TE curve (black solid line), whereas the TE data for progesterone (grey triangles) and dehydroisoandrosterone (grey circles) nearly follow the universal behavior. Reprinted with permission from [27]. Copyright 2009 American Chemical Society
At the isoelectric point, the membrane charge density of the lipid bilayer in lamellar complexes can be calculated from the observed DNA spacing dDNA (see Fig. 1A), because the negative charge of DNA has to neutralize the positive charge on the adjacent lipid bilayers [22,38,39]. A simple geometrical calculation, taking the lamellar geometry of the CL–DNA complex into account, yields [39]:
| (1) |
where e is the elementary charge and 3.4 Å corresponds to the bare distance between two charges along a DNA molecule.
As expected due to the small headgroup area of cholesterol (Achol = 40 Å2, while ADOPC = 72 Å2) [40,41], the membrane charge density of DOTAP/DOPC/Chol–DNA complexes increases with cholesterol content. Exchanging DOPC for cholesterol reduces the total membrane area while the membrane charge, given by ΦDOTAP = 0.3, remains constant; thus, σM increases. A particularly strong increase in σM occurs for Φchol ≥ 0.4, where part of the cholesterol is not incorporated in the complex. This results in an increased ΦDOTAP and thus σM.
3.2 Transfection Efficiency of CL–DNA Complexes Containing Cholesterol and Analogs
Fig. 5A shows the transfection efficiency of (lamellar) DOTAP/DOPC–DNA complexes (circles) as a function of ΦDOTAP. TE increases over several orders of magnitude with the molar fraction of cationic DOTAP. Also evident from this plot is the dramatic increase in TE upon partially replacing DOPC with cholesterol at ΦDOTAP = 30% (markers of different shapes correspond to different Φchol; see legend). TE increases by a factor of ten with the addition of only 15 mol% cholesterol, and further addition of cholesterol continues to exponentially increase TE. Of note, the amount of DOTAP and DNA is constant for all data points.
Fig. 5.

(A) TE of DNA complexes of binary DOTAP/DOPC lipid mixtures (black circles). Their TE increases over several orders of magnitude with increasing molar fraction of monovalent DOTAP (ΦDOTAP). Grey symbols represent TE of DNA complexes of ternary DOTAP/DOPC/Chol lipid mixtures with constant ΦDOTAP = 0.3. Different symbol shapes correspond to different Φchol (cf. legend). (B) The TE of the DNA complexes of ternary DOTAP/DOPC/Chol lipid mixtures (empty circles) plotted against σM significantly deviates from the universal bell shaped curve observed for binary systems [21]. Reprinted with permission from [27]. Copyright 2009 American Chemical Society
Transfection Efficiency and Membrane Charge Density
Using the experimentally obtained values of σM (calculated using Equation 1), Fig. 5B plots TE of DOTAP/DOPC/Chol–DNA complexes (empty circles) as a function of membrane charge density, together with the universal curve and the TE data used for its derivation [21].
TE of the DOTAP/DOPC/Chol–DNA complexes strongly deviates from the universal bell-shaped curve observed for binary systems. The TE of cholesterol-containing complexes increases more rapidly with increasing cholesterol content than the increase in membrane charge density predicts, for 0 < Φchol ≤ 0.4. No further TE increase is seen for Φchol > 0.4 (where the membrane is saturated with cholesterol: Φchol, membrane = 0.4 = const.).
The Effect of Cholesterol Analogs
Fig. 6A shows the TEs of DOTAP/DOPC–DNA complexes containing the structural analogs of cholesterol. Two distinct trends can be observed. The data for ergosterol and ergocalciferol are similar to that of cholesterol: TE rapidly increases with the increasing molar fraction of steroid, dramatically improving TE. In contrast, when β-estradiol, progesterone and dihydroisoandrosterone are incorporated into CL–DNA complexes, TE rises less rapidly until membrane saturation occurs at high steroid contents (35 mol% and higher). At this point, TE suddenly increases to values comparable with those obtained for cholesterol. The major structural differences between the two groups of molecules are the absence of the short alkyl chain attached to the hydro-phobic core and the presence of a second polar moiety in its place in case of β-estradiol, progesterone and dihydroisoandrosterone (see the chemical structures in Fig. 6B).
Fig. 6C shows the TEs of the DOTAP/DOPC/steroid–DNA complexes plotted as a function of σM. The membrane charge densities were obtained from X-ray diffraction data using Eq. 1. The data for cholesterol (dark circles) and ergosterol (dark triangles) deviate significantly from the universal TE curve (black solid line), whereas the TE data for progesterone (grey triangles) and dehydroisoandrosterone (grey circles) nearly follow the universal behavior.
Reduced Hydration-Repulsion Causes Enhanced Transfection Efficiency
The results described above show that adding cholesterol and certain analogs increases TE more than the resulting increase in membrane charge density would predict. Previous work has demonstrated that CL–DNA complexes at low σM transfect poorly due to inefficient endosomal escape (which involves fusion) [21,25]. Thus, our findings suggest that cholesterol and certain analogs facilitate fusion of the membranes of the complex and the endosome, independent of their effect on σM. A possible explanation for this is the overall reduction of the hydration repulsion layer of the membrane.
As two lipid membrane surfaces approach each other, short-range hydration and steric repulsions set in at distances between 1 nm to 3 nm and exponentially increase with decay lengths between 0.08 nm to 0.64 nm for the range of lipids and surfactants that have been studied to date [42]. Hydration repulsion forces result from the presence of water molecules strongly bound to hydrophilic membrane lipids, because of the energy required to dehydrate the lipids as the membranes approach each other [42]. The term steric interaction refers to excluded volume effects, which include the effects of thermal height fluctuations of the lipid membranes. The adhesion energy for oppositely charged membranes (at given positive and negative charge densities), will be optimized as the hydration/steric repulsive forces are decreased, allowing the membranes to approach more closely. For oppositely charged membranes, increased adhesion will facilitate fusion [42,43], which is favored by electrostatics. That is, membranes comprised of cationic/anionic lipids (after fusion) have a lower electrostatic energy compared to two approaching membranes with cationic and anionic lipids in different membranes (before fusion). Furthermore, the entropy of mixing is increased when oppositely charged membranes fuse.
It is known that the hydration repulsion layer of cholesterol is much smaller than that of DOPC [42,44]. Therefore, exchanging DOPC for cholesterol enhances fusion [42]. For CL–DNA complexes, this enhanced fusion of the membranes of the complex with the endosomal membrane facilitates endosomal release and increases TE. Ergosterol and ergocalciferol show the same effect. On the other hand, CL–DNA complexes containing progesterone, estradiol and dehydroisoandrosterone show different behavior, even though they likely reduce the average hydration repulsion layer of the membrane in the same way as cholesterol. A possible explanation for this phenomenon is that these steroid molecules also enhance the repulsion of the membranes because of increased protrusion forces. Progesterone, estradiol and dehydroisoandrosterone possess two polar groups, one at each end of the polycyclic framework, which dictate a positioning of the molecules close to the water interface (due to their increased hydrophilicity). The resulting protrusion forces appear to cancel the benefits of the reduced hydration repulsion layer with respect to the activated fusion with the endosomal membrane.
To test the hypothesis that the reduction of the hydration repulsion layer by cholesterol is responsible for the enhancement of TE, we have performed transfection experiments with DNA complexes of a ternary mixture of DOTAP, DOPC and PC-cholesterol (cholesteryl-phosphatidylcholine), a cholesterol derivative in which the hydroxyl group of cholesterol has been replaced by a phosphatidylcholine group. The chemical structure of PC-cholesterol is shown in Fig. 7B. The headgroups of PC-cholesterol and DOPC are essentially identical, thus having a similar (if not identical) hydration repulsion layer. Fig. 7A compares the TE of lamellar DNA-complexes of DOTAP/DOPC/Chol (black squares) and DOTAP/DOPC/PC-cholesterol lipid mixtures (grey bowties), again at constant ΦDOTAP = 0.3. The data show the large increase in transfection efficiency by ≈ two decades as DOPC lipids are replaced by cholesterol (black squares). In contrast, if instead DOPC is replaced by cholesterol-PC (grey bowties) no increase in TE is observed. This is strong evidence that the reduction of the hydration repulsion layer is, indeed, crucial for the TE enhancement.
Fig. 7.

(A) A comparison of the TE of DOTAP/DOPC/Chol–DNA complexes (black squares) and DOTAP/DOPC/PC-cholesterol–DNA complexes (grey bowties). The replacement of DOPC with PC-cholesterol, which has a similarly hydrated headgroup, fails to increase TE. (B) The chemical structures of cholesterol and PC-cholesterol. Reprinted with permission from [27]. Copyright 2009 American Chemical Society
In summary, our findings suggest that cholesterol and certain analogs are a highly valuable neutral lipid component (“helper lipid”) for CL–DNA complexes because they facilitate endosomal escape by reducing the repulsive hydration and protrusion forces. They are thus able to lower the kinetic barrier for fusion of the cationic membranes of CL–DNA complexes with the anionic membrane of the endosome and increase TE, in addition to their beneficial effect on σM.
4 Highly Charged Multivalent Cationic Lipids with Dendritic Headgroups (DLs) Promote Novel Structures and Mechanisms
The cationic lipids exhibiting universal behavior in our earlier studies (see Sect. 2) ranged in their headgroup valency from 2+ to 5+ [21]. The corresponding upper limit of σM, for membranes of pure pentavalent MVL5, was 27.17×10−3 e/Å2. To study the transfection behavior of CL–DNA complexes at even higher membrane charge densities σM, we synthesized a series of highly charged lipids with dendritic headgroups (DLs) and studied their DNA complexes [24,45,46].
4.1 Synthesis of DLs
Dendrimers are monodisperse, highly branched spherical molecules [47]. They are typically assembled by adding AB2 building blocks to a central core, thus yielding sequential “generations” of increasing size and endgroup number. Employing a building block approach for lipid design and synthesis, we have prepared a series of multivalent DLs based on ornithine as the AB2 building block [24,45]. Fig. 8 shows the chemical structures, molecular models and valencies at full protonation for the studied DLs. Branching ornithine groups (highlighted in by rectancles) double the number of end groups with cationic charges in each generation. Using both zeta potential measurements and an ethidium bromide displacement assay [24,45,48], the charges of the lipid headgroups effective in DNA complexation were determined as 4.0 ± 0.2 for MVLG2, 7.9 ± 0.3 for MVLG3, 8.0 ± 0.1 for MVLBisG1, and 14.6 ± 0.4 for MVLBisG2 independent of ΦDOPC. Thus, the headgroup charges of the DLs are very close to their charge at full protonation. To date, only very few other lipids with a similar number of charges in the headgroup have been reported [49]. Mixing of these lipids with neutral DOPC results in liposomes having σM of up to 40×10−3 e/Å2.
Fig. 8.

Chemical structures, maximum charge, and molecular models of the DLs MVLG2, MVLG3, MVLBisG1, and MVLBisG2. Branching ornithine spacer groups (highlighted by rectangles) double the number of end groups in each generation. The lipid tails are underlaid with a rounded rectangle, and the cationic end groups (carboxyspermine (4+) or ornithine (2+)) and their charged moieties are also highlighted
The synthesis of the branched core of the lipid headgroups [24,45] proceeds in the same manner as that of multiple antigenic peptides (MAPs) [50,51] or polyethylene glycol-dendritic oligo-lysine block copolymers [52]. It starts from ornithine methyl ester, which is acylated with Boc-protected ornithine. After deprotection of the amino groups, this process is repeated to increase the headgroup generation. By aminolysis of the methyl ester of the headgroup building block with an excess of ethylene diamine, a spacer with a distal amino group is introduced in a single step. In the synthesis of the headgroup moieties of MVLBisG1 and MVLBisG2, the final acylation is performed using Boc-protected carboxyspermine, which yields four charges after deprotection. This building block was prepared starting from ornithine through Michael-addition of acrylonitrile, followed by reduction of the cyano-groups using Raney-Nickel [53-55] and Boc-protection of the resulting amino groups [26]. To finish the synthesis of the DLs, the headgroup-spacer moiety is coupled with our lipid building block [56]. The product is purified extensively and finally deprotected using TFA.
4.2 Novel Structures of DL/DOPC–DNA Complexes
MVLG2(4+)/DOPC–DNA self-assemblies exhibit the lamellar Lαc phase for all ΦDOPC. XRD indicates a very tight packing of DNA molecules within MVLG2/DOPC–DNA complexes even at low σM. This is consistent with previous findings for the multivalent lipids MVL3(3+) and MVL5(5+) by Farago et al., who attributed the tight packing found even at low σM to a unique DNA locking mechanism involving the multivalent headgroups [57].
MVLBisG2(16+) bears the largest headgroup of the studied DLs, the size of which results in a conical molecular shape, favoring positive spontaneous membrane curvature. When mixed with cylindrically shaped DOPC, MVLBisG2 exhibits a rich phase diagram [58]. Cryo-TEM revealed that micelles coexist with vesicles at 0.5 ≤ ΦMVLBisG2 < 0.75. At ΦMVLBisG2 ≥ 0.75, the MVLBisG2/DOPC lipid mixture forms only micelles.
X-ray diffraction of MVLBisG2/DOPC–DNA complexes reveals the lamellar LαC phase for ΦMVLBisG2 < 0.2. In a narrow interval around ΦMVLBisG2 ≈ 0.25, the novel hexagonal CL–DNA complex phase (HIC, see also Fig. 1C) is found [24], with coexistence of the two phases at ΦMVLBisG2 = 0.2. At ΦMVLBisG2 = 0.4, a phase transition to a distorted hexagonal lattice occurs, persisting up to ΦMVLBisG2 = 1. This phase is characterized by broad diffraction peaks with the ratio of peak positions q2/q1 = 1.6. Similarly, the phase transition from lamellar to hexagonal can be clearly identified by the change in q2/q1 from 2 to 1.7. The phase transition from the hexagonal phase to the distorted hexagonal phase coincides with the appearance of micelles in the MVLBisG2/DOPC lipid mixture at ΦMVLBisG2 ≈ 0.5, suggesting a direct impact of the presence of micelles on the assembly of MVLBisG2/DOPC–DNA complexes. At higher ΦMVLBisG2 (0.6 ≤ ΦMVLBisG2 ≤ 1), XRD experiments further revealed a coexisting phase of tightly packed DNA bundles [46].
Fig. 9 shows schematic depictions of the two newly discovered structures described above. A cross section of a distorted hexagonal lattice is shown in Fig. 9A, displaying lipid micelles of an elliptical cross section and DNA molecules localized in the interstitial space. The distortion of the lattice is likely caused by the asymmetry in the micellar shape. X-ray diffraction data shows that the distortion increases with ΦMVLBisG2.
Fig. 9.

(A) Schematics of the molecular structure of DL–DNA complexes assembled in slightly disordered HIC; (B) DNA bundles surrounded by a cloud of micelles. The depletion–attraction force caused by micelles and the screening of the electrostatic interaction in the system enables the formation of the DNA bundles. Reprinted with permission from [46]. Copyright 2009 American Chemical Society
Fig. 9B shows a schematic of the DNA bundle phase observed at ΦMVLBisG2 > 0.5. The bundling phase requires the presence of salt (as found in the cell culture medium used for all our experiments) and is formed by the interplay of the salt-induced screening of the electrostatic interactions and the depletion–attraction [59,60] caused by the lipid micelles. While depletion–attraction has previously been reported for like-charged or neutral objects, the screening of the electrostatic interactions enables this effect to be observed also between DNA and the oppositely charged lipid micelles, for which the electrostatic interactions are attractive. The presence of salt not only facilitates bundling of DNA by reducing the electro-static repulsion between DNA molecules. It also reduces the electrostatic attraction between positively charged micelles and negatively charged DNA (Fig. 9A) to a level where it is less than the entropy gained by the micelles upon confining the DNA into bundles (Fig. 9B).
As expected from their intermediate headgroup size and charge, MVLG3(8+) or MVLBisG1(8+) form DL/DOPC–DNA complexes which occupy a middle ground in their phase behavior. Fig. 10A,B shows X-ray diffraction data for MVLG3/DOPC–DNA complexes and MVLBisG1/DOPC–DNA complexes, respectively, at three different compositions: ΦDL = 0.2, 0.4, and 1. DL/DOPC–DNA complexes of both lipids form a lamellar Lαc phase for ΦDL ≤ 0.5. Fig. 10C,D shows plots of the ratios of the peak positions q2/q1 and q3/q1 versus ΦDL which signify the nature of the DL–DNA self-assembly. For the lamellar phase, q2 = 2q1 and q3 = 3q1, which is clearly satisfied for ΦDL ≤ 0.5. For 0.5 < ΦDL < 0.8, the ratio between the first and the second order peaks q2/q1 is 1.7 (√3), while q2/q1 = 1.6 for ΦDL ≥ 0.8. This suggests a sequence of phases similar to that observed for MVLBisG2/DOPC–DNA complexes, from Lαc to HIC to a distorted hexagonal phase. An indication of a DNA bundle phase is only seen for ΦMVLG3 = 1 (a characteristic DNA bundle peak at q = 0.241 Å−1).
Fig. 10.

X-ray diffraction data for (A) MVLG3/DOPC–DNA complexes and (B) MVLBisG1/ DOPC–DNA complexes at ΦDL = 0.2, 0.4, and 1. (C) Ratio of the first and second order diffraction peaks, q2/q1, and (D) ratio of the first and third order diffraction peaks, q3/q1, plotted as a function of ΦDL. (E) The spacing d = 2π/q1 as a function of ΦDL. (F) Plot of dDNA as a function of increasing ΦDL in lamellar complexes. Reprinted with permission from [46]. Copyright 2009 American Chemical Society
4.3 Transfection Efficiency of DL–DNA Complexes
We have mapped the transfection efficiency of DL/DOPC–DNA complexes as a function of molar fraction of DL (ΦDL) and the cationic lipid/DNA charge ratio (ρchg). As observed for DOTAP and multivalent lipids with valencies up to +5, TE at the optimal ΦDL increases with ρchg up to a saturation value. Interestingly, this value is higher for the DLs (ρchg ≈ 4.5) then for previously investigated lipids (ρchg ≈ 3). [45]
The TE data of DL/DOPC–DNA complexes at ρchg = 4.5 and ρchg = 8 as a function of σM is plotted in Fig. 11. Also shown are fits representing the universal TE curves at those values of ρchg (black solid lines) [21,46]. TE of MVLG2(4+)/DOPC–DNA complexes exhibits the previously observed dependence on σM and closely follows the universal curve. However, the data for both MVLBisG1/DOPC–DNA complexes as well as MVLBisG2/DOPC–DNA complexes deviate strongly from the universal TE curve for σM ≥ 18×10−3 e/Å2, which is close to the maximum of the universal TE curve. Instead of dropping, TE of these DL–DNA complexes remains high beyond this value of σM. This behavior is reminiscent of the TE of DOTAP/DOPE–DNA complexes, which is independent of σM, albeit at low membrane charge densities. DOTAP/DOPE–DNA complexes exhibit the inverted hexagonal phase at low σM, and their constant, high TE reflects their different mechanism of action (see also Sect. 1) [25].
Fig. 11.

TE of DL/DOPC–DNA complexes containing MVLG2, MVLBisG1 or MVLBisG2 plotted as a function of σM for two different values of ρchg. (A) TE at ρchg = 4.5 and (B) TE at ρchg = 8. The solid line represents the universal TE curve. [21] The solid symbols mark data for DL/DOPC–DNA complexes in the lamellar phase, while empty symbols correspond to DL/DOPC–DNA complexes in hexagonal phases. Reprinted with permission from [46]. Copyright 2009 American Chemical Society
Correlations between Structure and TE of DL–DNA Complexes
As evident from Fig. 11, where different symbols are used to distinguish TE data for lamellar (filled symbols) and non-lamellar phases (open symbols), DL/DOPC–DNA complexes in the LαC phase closely follow the universal behavior. These are MVLG2/DOPC–DNA complexes at all ΦDL and DNA complexes of the other DLs at low ΦDL. In contrast, TE of the HIC phase and the new distorted hexagonal and DNA bundle phases is not only high but independent of ΦDL and thus σM. The appearance of non-lamellar phases therefore coincides with the deviation from the universal TE curve, suggesting a different mechanism of action for the different structures of DNA complexes.
The non-lamellar DL–DNA complexes exhibit enhanced TE (over lamellar complexes of the same σM) in the σM regime where release of DNA from the complex is thought to be limiting TE. The structure of the HIC complexes gives a clue as to its possible role in the transfection mechanism and high TE. In contrast to the LαC phase, both the HIC phase and the distorted hexagonal phase exhibit a continuous sub-structure of DNA within the complexes. The DNA bundle phase observed with MVLBisG2 even seems to allow the delivery of a lipid-free subphase of DNA. The existence of a continuous DNA substructure likely facilitates release of DNA, because all DNA is accessible as soon as a part of it is exposed to the cell interior. Interestingly, the TE of these complexes does not exceed that of the optimized lamellar complexes, which may hint at the presence of another barrier for complexes of high σM.
MVLBisG2 Efficiently Transfects MEFs, a Hard-to-Transfect Cell Line
Another important discovery was made when comparing MVLBisG2 and DOTAP in a number of different cell lines. As shown in Fig. 12, complexes of MVLBisG2 efficiently transfect a variety of mouse and human cells in culture [24]. Their TE reaches or surpasses that of optimized complexes prepared from commercially available DOTAP. Most importantly, complexes containing MVLBisG2 are significantly more transfectant over the entire composition range in mouse embryonic fibroblasts (MEFs). MEFs are important as feeder cells for embryonic stem cells and are a cell line that is empirically known to be hard to transfect.
Fig. 12.

Transfection efficiencies for DOTAP/DOPC and MVLBisG2/DOPC complexes in four different cell lines, plotted against the mole fraction of cationic lipid. The data points were obtained at a constant σchg (7 for HeLa cells, 4.5 for all others), corresponding to a constant amount of DNA applied to the cells for each data point in a plot. Remarkably, MVLBisG2 complexes are significantly more transfectant in mouse embryonic fibroblasts, a cell line empirically know to be hard to transfect and of large practical importance as feeder cells for embryonic stem cells. Reprinted with permission from [24]. Copyright 2006 American Chemical Society
5 CL–siRNA Complexes for Gene Silencing
A novel direction in CL-based nucleic acid delivery research worldwide, including our laboratory, has arisen out of the recent major discovery of RNA interference (RNAi) as an evolutionary conserved post-transcriptional gene-silencing pathway, in the nematode worm C. elegans in 1998 [61], in plants [62,63], and in filamentous fungi [64,65]. The finding that short interfering RNA (siRNA) (19-27 bp strands of dsRNA, with 2nt 3′–overhangs) lead to sequence specific gene silencing via RNAi when introduced into mammalian cells [66,67], without evoking the interferon pathway [68,69], has led to a surge in research activity aiming to utilize the pathway more broadly in functional genomics studies [70,71] and therapeutic applications [72-76]. The specificity of the RNAi machinery has been demonstrated by its ability to discriminate between mRNA targets with only one base pair difference [72]. Thus, in principle, siRNAs may be designed that selectively knock down the expression of any given gene product for which the sequence of the gene is known.
The therapeutic applications of RNAi are currently being explored, with potential targets including cancers and viral infections [74-78]. However, the utility of RNAi is limited by the efficiency and toxicity of the available siRNA delivery vehicles. To improve cationic lipid-based vectors, it is important to gain a better understanding of the relationship between the chemical-physical parameters and the biological, gene silencing activity of cationic lipid–siRNA (CL–siRNA) complexes. Understanding the mechanism of action of CL–siRNA complexes in vitro will allow for the optimization of lipid carriers for siRNA molecules, thereby making them a viable alternative to virus-based delivery methods which avoid their safety, immunogenicity and production issues.
When investigating CL–siRNA complexes with the objective of optimizing their silencing efficiency (SE), it is of particular interest to reveal similarities and important differences with the process of optimizing transfection efficiency of CL–DNA complexes, which involves delivery of long dsDNA. Two key differences from the outset are the fact that siRNA complexes transport a much shorter cargo, and that they only need to deliver it to the cytoplasm, where the RNAi machinery is located. The shorter length of the siRNA duplex is expected to result in a weaker electrostatic stabilization of the complexes with CLs, and may, for some membrane compositions, lead to different structures.
We have found that efficient delivery of siRNAs to cells in culture requires a molar charge ratio (σchg, cationic lipid/nucleic acid) nearly an order of magnitude larger than that optimal for CL–DNA complexes. This larger σchg needed for efficient silencing results in a larger amount of cationic lipid per cell. Thus, toxicity becomes an important issue to consider in some composition regimes. This implies that cationic multivalent lipids (MVLs) should be better vectors compared to univalent lipids, because a smaller number of MVLs is required for a given σchg of the complex. We have compared the silencing efficiency and toxicity of CL–siRNA complexes in mammalian cells, using monovalent DOTAP and custom synthesized pentavalent MVL5 [21,26] as cationic lipids [79].
MVL5(5+) exhibits superior silencing efficiency over a large range in the composition and σchg phase diagram compared to monovalent DOTAP and was significantly less toxic. In fact, MVL5-based vectors achieved near-complete, specific silencing, a result that could not be attained using DOTAP-based vectors. In addition, the experiments showed that DOPE is not a viable neutral lipid for siRNA delivery due to its toxicity.
5.1 Structures of CL–siRNA Complexes
DOTAP/DOPC–siRNA complexes exhibit the lamellar (LαsiRNA) structure at 0 < ΦDOPC< 0.9 (mole fraction of DOPC) at σchg =10 (Fig. 13A). The LαsiRNA structure is similar to the LαC structure shown in Fig. 1A, with the layer of DNA replaced by a layer of siRNA. An XRD scan displaying the [00L] layering peaks is shown in Fig. 13B. Fig. 13C shows an XRD pattern of lamellar MVL5/DOPC–siRNA complexes at ΦDOPC = 0.6 and ρchg = 10. The lamellar structure was observed for all ΦDOPC > 0.3. For ΦDOPC ≤ 0.3 a distinct, new phase was observed, the structure of which remains to be determined. This is in contrast to MVL5/DOPC–DNA complexes, which exhibit the lamellar structure for all values of ΦDOPC > 0.1 [21]. An important observation is the absence of NA–NA correlation peaks that are typically seen with CL–DNA complexes [22] in the DOTAP/DOPC–siRNA complexes . The short length of siRNA molecules disfavors 2D nematic liquid crystal ordering of siRNA rods (with orientational order). However, broad correlation peaks are observed for MVL5/DOPC–siRNA complexes. XRD showed that DOTAP/DOPE–siRNA complexes exhibit the lamellar structure for ΦDOPE < 0.4 and the inverted hexagonal structure (HIIsiRNA) for ΦDOPE > 0.6, with a coexistence region in between [79]. The HIIsiRNA structure is similar to the HIIC structure shown in Fig. 1B, with siRNA inserted in the inverse tubular micelles.
Fig. 13.

(A) Schematic of a lamellar (LαsiRNA) DOTAP/DOPC–siRNA complex. Partial bilayers have been removed, exposing 19 bp siRNAs in the isotropic phase. (B,C) Synchrotron x-ray data of CL–siRNA complexes reveal lamellar (LαsiRNA) patterns for DOTAP/DOPC–siRNA complexes (B) and MVL5 DOPC complexes (C). Note the broad siRNA–siRNA correlation peak in (C), between q002 and q003. Reprinted with permission from [79]. Copyright 2007 American Chemical Society
5.2 Gene Silencing Activities of CL–siRNA Complexes
In order to quantify and effectively compare the gene silencing activity of CL–siRNA complexes, we measured the effect of lipid composition and ρchg on both the target gene knockdown and non-specific gene silencing (with the latter correlating to cytotoxicity). We prepared CL–siRNA complexes with monovalent (DOTAP) or pentavalent (MVL5) cationic lipid [26] combined with one of two commonly used neutral lipids (NLs), DOPC or DOPE. The delivered siRNA targeted the firefly luciferase mRNA and consisted of a 21 bp long siRNA. Varying the neutral lipid enabled us to elucidate its contribution to structure and SE for the CL–siRNA complexes.
To enable meaningful comparisons of gene silencing efficiencies, we developed a dual luciferase assay which allows us to distinguish the contributions from specific and non-specific gene silencing for a given vector. Mouse L-cells where first co-transfected with plasmids encoding the Firefly (FF) and Renilla (RL) luciferases. The cells were then either transfected with CL–siRNA complexes (at a given ΦNL and ρchg) with siRNA targeting the mRNA for FF luciferase, or used as controls. A dual luciferase assay was used to measure the expression of FF (denoted FF(ΦNL,ρchg)) and RL (denoted RL(ΦNL,ρchg)) luciferase genes. For each measurement of FF and RL, expression levels were also measured in corresponding control cells (on volumes containing the same number of cells), yielding the controls FFcont and RLcont. Thus, by measuring FF(ΦNL,ρchg), FFcont, RL(ΦNL,ρchg), and RLcont, one readily obtains the total normalized target gene knockdown KT = 1 −FF(ΦNL,ρchg)/FFcont, and the normalized non-specific gene knockdown KNS = 1 −RL(ΦNL,ρchg)/RLcont.
The total knockdown KT includes silencing resulting from two separate contributions: one due to sequence-specific silencing of the target FF by the siRNA, and another, from the non-specific suppression of protein production. The non-specific knockdown KNS measures this sequence-independent global suppression protein production by CL–siRNA complexes due to cytotoxicity, and is determined by measuring the silencing of the off-target RL gene in cells transfected with CL–siRNA complexes containing siRNA which targets the distinctly different FF luciferase mRNA. As outlined below, cytotoxicity and thus KNS appears to be dominated by the cationic liposome component. Optimal gene silencing would correspond to KT approaching 1 and KNS approaching zero, where silencing is both complete and sequence-specific.
Fig. 14 (left to right) shows plots of the total knockdown KT and non-specific knockdown KNS as a function of ρchg at ΦNL= 0.4 for MVL5/DOPC–siRNA, DOTAP/DOPC–siRNA, and DOTAP/DOPE–siRNA complexes, respectively. Similar behavior was observed at ΦNL = 0.1. The data show that, for the lamellar MVL5/DOPC–siRNA complexes, the non-specific knockdown remains nearly constant and low with KNS < 0.1 for 2.8 ≤ ρchg ≤ 20, while KT exhibits a rapid nonlinear growth to KT ≈ 0.9 (for ρchg between 10 and 15), indicative of significant sequence-specific gene silencing (Fig. 14, left). In contrast, such a region of relatively high KT and low KNS was not observed for the lamellar phases of monova-lent DOTAP/DOPC–siRNA complexes at ΦNL= 0.4 (or ΦNL= 0.1), where KT increased slowly from 0.4 to 0.55 with KNS ≈ 0.2 (Fig. 14, middle). Furthermore, for DOTAP/DOPE–siRNA complexes (for which XRD indicates co-existence of the lamellar and inverted hexagonal structure), substantial non-specific knockdown KNS (related to cell toxicity) is observed even at low ρchg ≈ 5 (Fig. 14, right). This data is in striking contrast to DOTAP/DOPE–DNA inverted hexagonal complexes, which exhibit high TE in cell culture with low toxicity [25], albeit at lower ρchg.
Fig. 14.
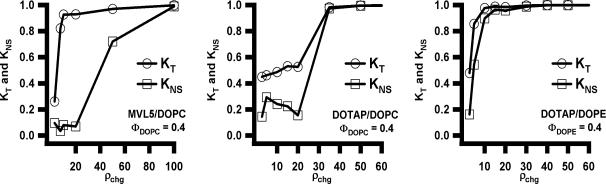
Total (KT, open circles) and non-specific (KNS, open squares) gene knockdown vs. cationic lipid/siRNA molar charge ratio (ρchg) at ΦNL= 0.4 for MVL5/ DOPC–siRNA (Left), DOTAP/DOPC–siRNA (Middle), and DOTAP/DOPE–siRNA (Right) complexes targeting luciferase mRNA in transfected L-cells. Reprinted with permission from [79]. Copyright 2007 American Chemical Society
Fig. 15A shows total gene knockdown (KT) data comparing the silencing efficiency of MVL5/DOPC–siRNA and DOTAP/DOPC–siRNA complexes at ρchg=15 (i.e. in the regime where the non-specific knockdown shown in Fig. 14 is relatively low) as a function of ΦNL. Complexes containing pentavalent MLV5 show high silencing efficiency over a broad range with KT ≈ 0.9 for 0 < ΦNL < 0.5. In contrast, KT of DOTAP-containing complexes remains relatively low and drops from 0.6 to ≈ 0.5 in the same range.
Fig. 15.

Total gene knockdown (KT) with siRNA complexes targeting the luciferase mRNA in transfected mouse L-cells as a function of mole fraction of neutral lipid ΦNL at ρchg = 15 (A) and ρchg = 2.8 (B). Reprinted with permission from [79]. Copyright 2007 American Chemical Society
For a comparison at a charge ratio typically employed in DNA transfection, Fig. 15B shows the total gene knockdown for MVL5/DOPC–siRNA, DOTAP/DOPC–siRNA, and DOTAP/DOPE–siRNA complexes at ρchg = 2.8, which exhibits optimal transfection efficiency for DOTAP containing CL–DNA complexes with very low cell toxicity [21,25]. At this lower charge ratio, CL–siRNA complexes are generally inefficient at gene silencing. While the silencing efficiency of MVL5/DOPC–siRNA complexes greatly increases as ρchg approaches 10, there was only a modest increase for the DOTAP containing complexes (Fig. 14).
5.3 Cytotoxicity of CL–siRNA Complexes and Liposomes
Cytotoxicity data for CL–siRNA complexes (MVL5/DOPC–siRNA, DOTAP/DOPC–siRNA, DOTAP/DOPE–siRNA) as a function of ΦNL are shown in Fig. 16. The filled triangles (ρchg= 10) and filled circles (ρchg= 50) represent toxicity data for cells incubated with complexes. Also plotted are the toxicities measured when cells were incubated with corresponding equivalent amounts of cationic liposomes without siRNA (open triangle and open circle). At ρchg= 10, only the DOTAP/DOPE–siRNA complexes and DOTAP/DOPE liposomes showed toxicity. For these systems, the toxicity exhibits a marked broad peak as a function of ΦDOPE. This is in contrast to the DOTAP/DOPC–siRNA and MVL5/DOPC–siRNA complexes and the corresponding liposomes, where toxicity is low for all ΦDOPC. For ρchg = 50, siRNA complexes and CLs showed significant toxicity for all lipid combinations. Thus, the toxicity data correlate well with the measured non-specific knockdown values KNS (Fig. 14) and confirm the use of KNS as an indicator for cell viability. Because the degree of cytotoxicity is qualitatively similar for cells incubated with either CL–siRNA complexes or cationic liposomes alone, its origin appears to be the lipid component of the complex.
Fig. 16.
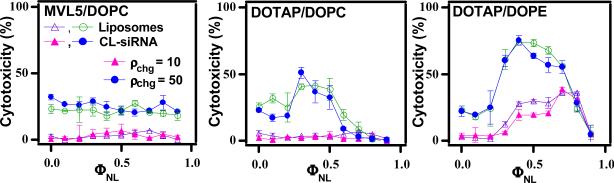
Cytotoxicity of CL–siRNA complexes (MVL5/DOPC–siRNA, DOTAP/DOPC–siRNA, DOTAP/DOPE–siRNA) targeting the FF luciferase mRNA in mouse L-cells and the corresponding cationic liposomes (without siRNA) as a function of ΦNL (mole fraction of neutral lipid). The filled triangles (ρchg= 10) and filled circles (ρchg= 50) represent toxicity data for cells incubated with complexes. Also plotted are the toxicities measured when cells were incubated with corresponding equivalent amounts of cationic liposomes without siRNA (open triangle (ρchg= 10) and open circle (ρchg= 50)). Cytotoxicity was measured by quantifying the amount of released lactate dehydrogenase from cells with damaged membranes. Reprinted with permission from [79]. Copyright 2007 American Chemical Society
In summary, the data on total knockdown KT, non-specific knockdown KNS, and cell cytotoxicity show that MVL5/DOPC–siRNA complexes have a significantly higher silencing efficiency (with KT ≈ 0.9 and KNS < 0.1) and lower cell toxicity over a broader range of ρchg and ΦNL than monovalent DOTAP/DOPC–siRNA complexes, with the latter not showing a regime with KT approaching 1 at low KNS [79]. This means that MVL5/DOPC–siRNA complexes are the only viable siRNA vector out of those tested, since a high total silencing simply amounts to a global suppression of protein production if KNS is also high, as in the case of DOTAP vectors.
6 Similarities and Differences in the Performance of Multivalent Lipids (MVLs) and Univalent Lipids (UVLs)
Since the synthetic effort required to prepare the lipid is generally greater for MVLs than for UVLs, with a view towards applications it is prudent to compare the overall performance of UVLs and MVLs, in particular considering the results shown in Sect. 2. Our work shows that while optimized formulations of MVLs and UVLs perform similarly in some cases, there are two important applications in which MVLs are far superior.
Systems where MVLs and UVLs have comparable performance
As we have elaborated in Sect. 2 and Sect. 4.3, the TE of lamellar DNA complexes of MVLs and UVLs shows universal behavior when plotted against the membrane charge density, implying that optimized complexes of MVLs and UVLs transfect equally well. Importantly, the fact that the universal curve is bell-shaped (where TE is plotted logarithmically) implies that optimization of the lipid composition is crucial for objectively comparing lipid performance. Interestingly, while complexes in the HIC and HIIC phase do not follow this universal curve, their TE no more than equals that of optimized lamellar complexes.
Systems where MVLs are superior to UVLs
We have identified two scenarios of high relevance in nucleic acid delivery for which MVLs are clearly superior to UVLs. These are plasmid delivery to hard to transfect cell lines and gene silencing with siRNA. In these applications, MVLs yield results that simply cannot be achieved by UVLs. As described in Sect. 4.3.2, a very highly charged MVL (MVLBisG2, 16+) has proven to be superior to the UVL DOTAP in MEFs, a hard to transfect cell line. We attribute this superiority of MVLBisG2 to its ability to induce the recently discovered hexagonal HIC structure, which is not found with UVLs. This unexpected finding is a very significant result, since hard to transfect cell lines are one of the most important current topics of basic research on DNA delivery. The second highly relevant difference between UVLs and MVLs is seen in the delivery of siRNA, where our studies show that MVLs are far superior (see Sect. 5.2). MVL5-based vectors showed near-complete and specific silencing, a result that could not be obtained with UVL vectors. MVLs permit using the large lipid/siRNA charge ratios required for high specific gene silencing without significant lipid-induced toxicity.
7 Future Directions
The ultimate goal of non-viral vector development is to rationally design and optimize vectors that are viable for in vivo applications. Non-viral vectors that can be successful at a task as complex as the delivery of nucleic acids in vivo likely will be sophisticated multi-component systems. En route to such systems, lipids specifically designed to lower or overcome known barriers to nucleic acid delivery will allow detailed investigations on the relevance of these barriers, ultimately leading to improved and “virus-like” lipid vectors. If these strategies are successful, they will benefit the development of more efficient non-viral vectors for research and therapeutic applications. At the very least, the improved understanding of barriers to successful delivery gained from this work will point the way to further improvement. Insights obtained on intracellular barriers and toxicity issues are relevant for both in vitro and in vivo applications since both share these barriers. Targeted vectors, which make use of cell specific attachment and internalization capabilities, will benefit not only in vivo applications but also in vitro work with hard to transfect cell lines. The same is true for lipids that facilitate endosomal escape, which may serve to recover efficiency that is lost by sterical stabilization.
Many of the known barriers to transfection may be addressed by custom synthesized lipids. For example, easily biodegradable cationic lipids should (i) enhance TE of CL–DNA complexes in the high membrane charge density regime, where dissociation and release of DNA from the cationic lipid membrane of the complex in the cytoplasm appears to be a barrier to TE, and (ii) reduce toxicity in gene silencing applications with CL–siRNA complexes.
In addition, for CL–siRNA complexes, further exploration of the relationships between cationic lipid valence, complex stability, silencing efficiency, and cytotoxicity also with a series of multivalent lipids such as the MVLS or DLs is a logical next step.
7.1 Non-viral Vectors for In Vivo Gene Delivery
In vivo transfection by CL–DNA complexes poses a number of additional requirements when compared with transfection in vitro. This most notably shows in the fact that formulations which are optimal for transfection in vitro are not the best or even suitable for in vivo applications [29,80,81]. Addition of high amounts of serum to the transfection medium has a similar effect.
While the causes of this phenomenon are still the subject of active research, it is clear that CL–DNA complexes for systemic administration need to be stable in the circulatory system long enough to at least reach the target organ. Sterical stabilization by PEGylation, i.e. addition of PEG-lipids (PEG = Poly(ethylene glycol), can achieve this goal by preventing the attachment of opsonins and by limiting the activation of the complement system [82]. PEGylation also confers colloidal stability, high solubility and a small, well-defined size to CL–DNA complexes [83-85]. A stable, consistent particle size of about 100 nm diameter, as achieved by PEGylation, is most advantageous for cancer therapy in vivo, being too large for fast renal excretion and too small for rapid clearance by the reticuloendothelial system (RES) [85]. Particles of this size have the added advantage that they accumulate in tumors and sites of inflammation – the so-called enhanced permeation and retention (EPR) effect [86].
However, complexes that have been stabilized against aggregation and degradation by PEGylation exhibit much lower TE than their un-shielded counterparts [83,85,87,88]. PEGylation reduces the ability of CL–DNA complexes to attach to cells via electrostatic interactions and inhibits escape from the endosomes. These properties need to be regained by adding lipids that perform specific functions. Thus, to counter the undesired effects of a PEG coating, PEG-lipids with targeting ligands and degradable PEG-lipids, which shed their PEG chains after endocytosis, may prove valuable.
To regain efficient cell attachment capabilities, the PEGylated complex must be decorated with ligands that bind to receptors on the surface of the target cells. Fortuitously for in vivo applications, screening of the charge-mediated attraction between CL–DNA complexes and sulfated cell surface proteoglycans by the PEG shell offers the opportunity to replace this unspecific attraction with specific, ligand mediated interactions. Thus, PEGylation indirectly enables targeting which allows for delivery to a specific cell or tissue type after systemic injection. Of the many potential targeting ligands, peptides are of particular interest. Recently, in vivo phage display methods have revealed a system of “vascular zip codes” which vary not only from tissue to tissue but also from healthy to diseased tissue [89-91].
Similar to cell attachment, endosomal escape of simple lamellar complexes is a process driven by electrostatics [21,25,83] (see Sect. 2) and therefore inhibited by PEGylation. A strategy to recover efficient endosomal escape is to prepare PEG-lipids in which the PEG chains are attached via bonds that are quickly cleaved as the endosomal pH is lowered in the course of maturation. This practically converts the shielded complex back into an unshielded complex. Several acid-labile moieties have been investigated for similar purposes, e.g., hydrazones [92], vinyl ethers [93] and orthoesters [87,94].
7.2 Novel Liposome Structures: Block Liposomes
The landmark discovery of liposomes by A. D. Bangham in the early 1970s [95] sparked intense interest in them by the scientific community. Because of their similarities to biological membranes, they are used in model studies of cell-cell interactions as well as interactions between membranes of eucaryotic organelles. Furthermore, their ability to stably encapsulate liquid solutions has enabled their use as chemical carriers, and liposomes will continue to have a major impact in the medical field as drug and gene delivery systems.
Using optical microscopy and cryo-TEM, we have recently discovered block liposomes, which are liposomes consisting of connected, but distinctly shaped nanoscale liposome blocks: spheres or pears connected to tubes or rods [58,96,97]. The key to this discovery is the curvature stabilizing ability of our new, highly charged DL MVLBisG2 (see Sect. 4) [24]. dendritic lipid.
Fig. 17A-D shows cryo-TEM images of diblock (sphere-rod) liposomes, which consist of micellar nanorods (arrows) attached to spherical vesicles. Their diameter equals the thickness of the bilayer (~ 4 nm) and their lengths can reach up to several micrometer. A lower magnification image (Fig. 17D) shows a collection of these novel block liposomes with different spherical vesicle sizes. Fig. 17E shows a schematic depiction of this remarkable liposome structure. The large charge and persistence lengths of the rods provide ideal conditions for templating of nanostructures (e.g. wires or needles).
Fig. 17.

(A-D) Cryo-TEM images of diblock (sphere-rod) liposomes comprised of liquid-phase lipid nanorods (white arrows) connected to spherical vesicles. The lipid nanorods are stiff cylindrical micelles with an aspect ratio ≈1000. Their diameter equals the thickness of a lipid bilayer (≈4 nm) and their length reaches up to several micrometers, with a persistence length on the order of millimeters. (C) An inset of B, demonstrating the thickness of the nanorod: white arrow heads point out a thickness of≈4 nm (approximate bilayer thickness, identical for the spherical vesicle and the nanorods). (E) Schematic of a MVLBisG2/DOPC sphere-rod diblock liposome. Reprinted with permission from [58]. Copyright 2008 American Chemical Society
Two asymmetric triblock (pear-tube-pear) liposomes (with vesicles of differing size capping the nanotube) are shown in Fig. 18A. The high-magnification inset shows that the tubular section has an inner lumen diameter of ~ 10 nm (Fig. 18B). A diblock (pear-tube) liposome with inner diameter of order 50 nm is seen in Fig 2C. Fig. 18E shows a diblock liposome (lower arrow) and several block liposomes (second, third, and fourth arrows from bottom), which are either di- or triblocks. Schematic depictions of block liposomes containing tubular sections are shown in Fig. 18F. These lipid nanotubes and nanorods may find applications in biotechnology and drug/gene delivery. Importantly, their membranes are in the liquid (chain-melted) phase, which is mandatory for the functional incorporation of membrane proteins.
Fig. 18.

(A-E) Cryo-TEM images of triblock (pear-tube-pear) and diblock (pear-tube) liposomes comprised of liquid-phase lipid nanotube segments capped by spherical vesicle. The tubule blocks (arrows) are the first examples of liquid-phase (chain-melted) tubes with diameter on the nanometer scale (between 10 nm and 50 nm). (A) Triblock liposomes (pear-tube-pear). (B) An inset of panel A, disclosing the hollow tubular structure (white arrowheads and white bar point out the bilayer thickness of 4 nm). (C) A diblock liposome. (D) One block liposome encapsulated within another one. (E) A group of block liposomes. (F) Schematics of the MVLBisG2/DOPC tri- and diblock liposomes, manifesting the symmetry breaking between outer and inner mono-layer. Reprinted with permission from [58]. Copyright 2008 American Chemical Society
New theory will be required to describe the phase diagram of these block liposomes. In particular, theories have to break new ground in explaining why nanorods and tubes stay attached to spherical vesicles. All current theories of lipid self-assemblies (based on Helfrich's theory of membranes [98]), in contrast, predict spherical, tubular, and micellar shaped liposomes but only as separate objects. In our experiments, not a single instance of an isolated rod- or tube-shaped liposome (i.e., not connected to a sphere- or pear-shaped vesicle) was found.
Acknowledgments
KKE, AZ, AA, NFB, HME and CRS were supported primarily by NIH GM-59288-11, and in part by DOE-BES grant DE-FG02-06ER46314 (lipid microstructure) and NSF-DMR 0803103 (lipid phase behavior). CSM and CES were supported by NIH AI-12520 and AI-20611. Cryo-TEM experiments were conducted at the National Resource for Automated Molecular Microscopy which is supported by the National Institutes of Health though the National Center for Research Resources’ P41 program (RR17573). The X-ray diffraction work was carried out at the Stanford Synchrotron Radiation Laboratory which is supported by the Department of Energy.
Abbreviations
- Chol
cholesterol
- CL
cationic lipid
- DL
lipid with dendritic headgroup
- DOPC
1,2-dioleoyl-sn-glycero-3-phosphatidylcholine
- DOPE
1,2-dioleoyl-sn-glycero-3-phosphatidylethanolamine
- DOTAP
1,2-dioleoyl-3-trimethylammonium-propane
- FF
Firefly
- MEF
Mouse embryonic fibroblast
- MVL
multivalent lipid
- NA
nucleic acid
- NL
neutral lipid
- PEG
Poly(ethylene glycol)
- RL
Renilla
- RNAi
RNA interference
- SE
silencing efficiency
- siRNA
small interfering RNA
- TE
transfection efficiency
- UVL
univalent lipid
- XRD
X-ray diffraction
References
- 1.Safinya CR, Koltover I. Self Assembled Structures of Lipid–DNA Nonviral Gene Delivery Systems from Synchrotron X-Ray Diffraction. In: Huang L, Hung M-C, Wagner E, editors. Non-Viral Vectors for Gene Therapy. Academic Press; San Diego: 1999. [Google Scholar]
- 2.Ewert KK, Slack NL, Ahmad A, Evans HM, Lin A, Samuel CE, Safinya CR. Cationic Lipid–DNA Complexes for Gene Therapy: Understanding the Relationship between Complex Structures and Gene Delivery Pathways at the Molecular Level. Curr Med Chem. 2004;11:1241–1253. doi: 10.2174/0929867043456160. [DOI] [PubMed] [Google Scholar]
- 3.Ewert K, Evans H, Ahmad A, Slack L, Lin A, Martin-Herranz A, Safinya CR. Lipoplex Structures and their Distinct Cellular Pathways. In: Huang L, Hung M-C, Wagner E, editors. Non-Viral Vectors for Gene Therapy. 2nd edn. Elsevier; San Diego: 2005. Part I (Advances in Genetics 53) [Google Scholar]
- 4.Ewert K, Ahmad A, Evans H, Safinya CR. Cationic lipid–DNA complexes for non-viral gene therapy: relating supramolecular structures to cellular pathways. Expert Opin Biol Ther. 2005;5:33–53. doi: 10.1517/14712598.5.1.33. [DOI] [PubMed] [Google Scholar]
- 5.Safinya CR, Ewert KK, Ahmad A, Evans HM, Raviv U, Needleman DJ, Lin AJ, Slack NL, George CX, Samuel CE. Cationic Liposome–DNA Complexes: From Liquid Crystal Science to Gene Delivery Applications. Phil Transact Royal Soc A. 2006;364:2573–2596. doi: 10.1098/rsta.2006.1841. [DOI] [PubMed] [Google Scholar]
- 6.Ewert KK, Samuel CE, Safinya CR. Lipid–DNA Interactions: Structure-Function Studies of Nanomaterials for Gene Delivery. In: Dias R, Lindman B, editors. Interaction of DNA with Surfactant and Polymers. Blackwell; Boston, MA: 2008. [Google Scholar]
- 7.Ewert KK, Ahmad A, Bouxsein NF, Evans HM, Safinya CR. Non-Viral Gene Delivery with Cationic Liposome–DNA Complexes. In: Le Doux J, editor. Gene Therapy Protocols. 3rd edn. Humana Press; Totowa, NJ: 2008. [DOI] [PubMed] [Google Scholar]
- 8.Edelstein ML, Abedi MR, Wixon J, Edelstein RM. Gene therapy clinical trials worldwide 1989–2004 - an overview. J Gene Med. 2004;6:597–602. doi: 10.1002/jgm.619. [DOI] [PubMed] [Google Scholar]
- 9.Edelstein ML, Abedi MR, Wixon JJ. Gene therapy clinical trials worldwide to 2007: An update. Gene Med. 2007;9:833–842. doi: 10.1002/jgm.1100. [DOI] [PubMed] [Google Scholar]
- 10. [5 Nov 2009];The Journal of Gene Medicine Clinical Trial site. http://www.wiley.co.uk/genetherapy/clinical.
- 11.Huang L, Hung M-C, Wagner E, editors. Nonviral Vectors for Gene Therapy. Academic Press; San Diego: 1999. [Google Scholar]
- 12.Huang L, Hung M-C, Wagner E, editors. Non-Viral Vectors for Gene Therapy. 2nd edn. Elsevier; San Diego: 2005. Part I Advances in Genetics, Vol 53. [Google Scholar]
- 13.Mahato RI, Kim SW, editors. Pharmaceutical Perspectives of Nucleic Acid-Based Therapeutics. Taylor & Francis; London and New York: 2002. [Google Scholar]
- 14.Chesnoy S, Huang L. Structure and function of lipid–DNA complexes for gene delivery. Annu Rev Biophys Biomol Struct. 2000;29:27–47. doi: 10.1146/annurev.biophys.29.1.27. [DOI] [PubMed] [Google Scholar]
- 15.Byk G, Dubertret C, Escriou V, Frederic M, Jaslin G, Rangara R, Pitard B, Crouzet J, Wils P, Schartz B, Scherman D. Synthesis, Activity, and Structure-Activity Relationship Studies of Novel Cationic Lipids for DNA Transfer. J Med Chem. 1998;41:224–235. doi: 10.1021/jm9704964. [DOI] [PubMed] [Google Scholar]
- 16.Boussif O, Lezoualc'h F, Zanta MA, Mergny MD, Scherman D, Demeniex B, Behr J-P. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: Polyethylenimine. Proc Natl Acad Sci USA. 1995;92:7297–7301. doi: 10.1073/pnas.92.16.7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haensler J, Szoka FC. Polyamidoamine Cascade Polymers Mediate Efficient Transfection of Cells in Culture. Bioconjugate Chem. 1993;4:372–379. doi: 10.1021/bc00023a012. [DOI] [PubMed] [Google Scholar]
- 18.Tang MX, Redemann CT, Szoka FC., Jr In Vitro Gene Delivery by Degraded Polyamidoamine Dendrimers. Bioconjugate Chem. 1996;7:703–714. doi: 10.1021/bc9600630. [DOI] [PubMed] [Google Scholar]
- 19.Thomas CE, Ehrhardt A, Kay MA. Progress and problems with the use of viral vectors for gene therapy. Nature Rev Genet. 2003;4:346–358. doi: 10.1038/nrg1066. [DOI] [PubMed] [Google Scholar]
- 20.Tranchant I, Thompson B, Nicolazzi C, Mignet N, Scherman D. Physicochemical optimisation of plasmid delivery with cationic lipids. J Gene Med. 2004;6:S24–S35. doi: 10.1002/jgm.509. [DOI] [PubMed] [Google Scholar]
- 21.Ahmad A, Evans HM, Ewert K, George CX, Samuel CE, Safinya CR. New Multivalent Lipids Reveal Bell-Curve for Transfection versus Membrane Charge Density: Nonviral Lipid–DNA Complexes for Gene Delivery. J Gene Med. 2005;7:739–748. doi: 10.1002/jgm.717. [DOI] [PubMed] [Google Scholar]
- 22.Rädler JO, Koltover I, Salditt T, Safinya CR. Structure of DNA-Cationic Liposome Complexes: DNA Intercalation in Multilamellar Membranes in Distinct Interhelical Packing Regimes. Science. 1997;275:810–814. doi: 10.1126/science.275.5301.810. [DOI] [PubMed] [Google Scholar]
- 23.Koltover I, Salditt T, Rädler JO, Safinya CR. An Inverted Hexagonal Phase of Cationic Liposome–DNA Complexes Related to DNA Release and Delivery. Science. 1998;281:78–81. doi: 10.1126/science.281.5373.78. [DOI] [PubMed] [Google Scholar]
- 24.Ewert KK, Evans HM, Zidovska A, Bouxsein NF, Ahmad A, Safinya CR. A columnar phase of dendritic lipid-based cationic liposome–DNA complexes for gene delivery: hexagonally ordered cylindrical micelles embedded in a DNA honeycomb lattice. J Am Chem Soc. 2006;128:3998–4006. doi: 10.1021/ja055907h. [DOI] [PubMed] [Google Scholar]
- 25.Lin AJ, Slack NL, Ahmad A, George CX, Samuel CE, Safinya CR. Three-Dimensional Imaging of Lipid Gene-Carriers: Membrane Charge Density Controls Universal Transfection Behavior in Lamellar Cationic Liposome–DNA Complexes. Biophys J. 2003;84:3307–3316. doi: 10.1016/S0006-3495(03)70055-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ewert K, Ahmad A, Evans HM, Schmidt H-W, Safinya CR. Efficient Synthesis and Cell-Transfection Properties of a New Multivalent Cationic Lipid for Non-Viral Gene Delivery. J Med Chem. 2002;45:5023–5029. doi: 10.1021/jm020233w. [DOI] [PubMed] [Google Scholar]
- 27.Zidovska A, Evans HM, Ahmad A, Ewert KK, Safinya CR. The Role of Cholesterol and Structurally Related Molecules in Enhancing Transfection by Cationic Liposome–DNA Complexes. J Phys Chem. 2009;113:5208–5216. doi: 10.1021/jp809000e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Song YK, Liu F, Chu S, Liu D. Characterization of cationic liposome-mediated gene transfer in vivo by intravenous administration. Hum Gene Ther. 1997;8:1585–1594. doi: 10.1089/hum.1997.8.13-1585. [DOI] [PubMed] [Google Scholar]
- 29.Liu Y, Mounkes LC, Liggitt HD, Brown CS, Solodin I, Heath TD, Debs RJ. Factors influencing the efficiency of cationic liposome-mediated intravenous gene delivery. Nature Biotechnol. 1997;15:167–173. doi: 10.1038/nbt0297-167. [DOI] [PubMed] [Google Scholar]
- 30.Audouy S, Molema G, de Leij L, Hoekstra D. Serum as a modulator of lipoplex-mediated gene transfection: dependence of amphiphile, cell type and complex stability. J Gene Med. 2000;2:465–476. doi: 10.1002/1521-2254(200011/12)2:6<465::AID-JGM141>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 31.Crook K, Stevenson BJ, Dubouchet M, Poreous DJ. Inclusion of cholesterol in DOTAP transfection complexes increases the delivery of DNA to cells in vitro in the presence of serum. Gene Ther. 1998;5:137–143. doi: 10.1038/sj.gt.3300554. [DOI] [PubMed] [Google Scholar]
- 32.Li S, Tseng WC, Stolz DB, Wu SP, Watkins SC, Huang L. Dynamic changes in the characteristics of cationic lipidic vectors after exposure to mouse serum: implications for intravenous lipofection. Gene Ther. 1999;6:585–594. doi: 10.1038/sj.gt.3300865. [DOI] [PubMed] [Google Scholar]
- 33.Ilies MA, Johnson BH, Makori F, Miller A, Seitz WA, Thompson EB, Balaban AT. Pyridinium cationic lipids in gene delivery: an in vitro and in vivo comparison of transfection efficiency versus a tetraalkylammonium congener. Arch Biochem Biophys. 2005;435:217–226. doi: 10.1016/j.abb.2004.12.010. [DOI] [PubMed] [Google Scholar]
- 34.Kim CK, Haider KH, Choi SH, Choi EJ, Ahn WS, Kim YB. Nonviral Vector for Efficient Gene Transfer to Human Ovarian Adenocarcinoma Cells. Gynecol Oncol. 2002;84:85–93. doi: 10.1006/gyno.2001.6480. [DOI] [PubMed] [Google Scholar]
- 35.Kim CK, Choi EJ, Choi SH, Park JS, Haider KH, Ahn WS. Enhanced p53 gene transfer to human ovarian cancer cells using the cationic nonviral vector, DDC. Gynecol Oncol. 2003;90:265–272. doi: 10.1016/s0090-8258(03)00248-8. [DOI] [PubMed] [Google Scholar]
- 36.Epand RM, Hughes DW, Sayer BG, Borochov N, Bach D, Wachtel E. Novel properties of cholesterol-dioleoylphosphatidylcholine mixtures. Biochim Biophys Acta–Biomembranes. 2003;1616:196–208. doi: 10.1016/j.bbamem.2003.08.006. [DOI] [PubMed] [Google Scholar]
- 37.Huang JY, Buboltz JT, Feigenson GW. Maximum solubility of cholesterol in phosphatidylcholine and phosphatidylethanolamine bilayers. Biochim Biophys Acta–Biomembranes. 1999;1417:89–100. doi: 10.1016/s0005-2736(98)00260-0. [DOI] [PubMed] [Google Scholar]
- 38.Koltover I, Salditt T, Safinya CR. Phase diagram, stability, and overcharging of lamellar cationic lipid–DNA self-assembled complexes. Biophys J. 1999;77:915–924. doi: 10.1016/S0006-3495(99)76942-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rädler JO, Koltover I, Jamieson A, Salditt T, Safinya CR. Structure and interfacial aspects of self-assembled cationic lipid–DNA gene carrier complexes. Langmuir. 1998;14:4272–4283. [Google Scholar]
- 40.Slotte JP, Jungner M, Vilcheze C, Bittman R. Effect of sterol side-chain structure on sterol-phosphatidylcholine interactions in monolayers and small unilamellar vesicles. Biochim Biophys Acta–Biomembranes. 1994;1190:435–443. doi: 10.1016/0005-2736(94)90105-8. [DOI] [PubMed] [Google Scholar]
- 41.Tristram–Nagle S, Petrache HI, Nagle JF. Structure and Interactions of Fully Hydrated Dioleoylphosphatidylcholine Bilayers. Biophys J. 1998;75:917–925. doi: 10.1016/S0006-3495(98)77580-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Israelachvili JN. Intermolecular and Surface Forces. 2nd ed. Academic Press; London: 1992. [Google Scholar]
- 43.Hed G, Safran SA. Attractive instability of oppositely charged membranes induced by charge density fluctuations. Phys Rev Lett. 2004;93(1–4):138101. doi: 10.1103/PhysRevLett.93.138101. [DOI] [PubMed] [Google Scholar]
- 44.Petrache HI, Harries D, Parsegian VA. Alteration of Lipid Membrane Rigidity by Cholesterol and Its Metabolic Precursors. Macromol Symp. 2004;219:39–50. [Google Scholar]
- 45.Ewert KK, Evans HM, Zidovska A, Bouxsein NF, Ahmad A, Safinya CR. Dendritic Cationic Lipids with Highly Charged Headgroups for Efficient Gene Delivery. Bioconjugate Chem. 2006;17:877–888. doi: 10.1021/bc050310c. [DOI] [PubMed] [Google Scholar]
- 46.Zidovska A, Evans HM, Ewert KK, Quispe J, Carragher B, Potter CS, Safinya CR. Liquid Crystalline Phases of Dendritic Lipid–DNA Self-Assemblies: Lamellar, Hexagonal and DNA Bundles. J Phys Chem B. 2009;113:3694–3703. doi: 10.1021/jp806863z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bosman AW, Janssen HM, Meijer EW. About Dendrimers: Structure, Physical Properties, and Applications. Chem Rev. 1999;99:1665–1688. doi: 10.1021/cr970069y. [DOI] [PubMed] [Google Scholar]
- 48.Boger DL, Fink BE, Brunette SR, Tse WC, Hedrick MP. A Simple, High-Resolution Method for Establishing DNA Binding Affinity and Sequence Selectivity. J Am Chem Soc. 2001;123:5878–5891. doi: 10.1021/ja010041a. [DOI] [PubMed] [Google Scholar]
- 49.Takahashi T, Harada A, Emi N, Kono K. Preparation of Efficient Gene Carriers Using a Polyamidoamine Dendron-Bearing Lipid: Improvement of Serum Resistance. Bioconjugate Chem. 2005;16:1160–1165. doi: 10.1021/bc050012f. [DOI] [PubMed] [Google Scholar]
- 50.Veprek P, Jezek J. Peptide and glycopeptide dendrimers. Part I. J Pept Sci. 1999;5:5–23. doi: 10.1002/(SICI)1099-1387(199905)5:5<203::AID-PSC181>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 51.Veprek P, Jezek J. Peptide and glycopeptide dendrimers. Part II. J Pept Sci. 1999;5:203–220. doi: 10.1002/(SICI)1099-1387(199905)5:5<203::AID-PSC181>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 52.Choi JS, Lee EJ, Choi YH, Jeong YJ, Park JS. Poly(ethylene glycol)-block-poly(L-lysine) dendrimer: Novel linear polymer/dendrimer block copolymer forming a spherical water-soluble polyionic complex with DNA. Bioconjugate Chem. 1999;10:62–65. doi: 10.1021/bc9800668. [DOI] [PubMed] [Google Scholar]
- 53.Brabender-van den Berg EMMD, Meijer EW. Poly(propylene imine) dendrimers - large-scale synthesis by heterogeneously catalyzed hydrogenations. Angew Chem Int Ed Engl. 1993;32:1308–1311. [Google Scholar]
- 54.Wörner C, Mülhaupt R. Polynitrile-functional and polyamine-functional poly(trimethylene imine) dendrimers. Angew Chem Int Ed Engl. 1993;32:1306–1308. [Google Scholar]
- 55.Behr J-P. Photohydrolysis of DNA by Polyaminobenzenediazonium Salt. Chem Commun. 1989;101:101–103. [Google Scholar]
- 56.Schulze U, Schmidt HW, Safinya CR. Synthesis of novel cationic poly(ethylene glycol) containing lipids. Bioconjugate Chem. 1999;10:548–552. doi: 10.1021/bc9801068. [DOI] [PubMed] [Google Scholar]
- 57.Farago O, Ewert KK, Ahmad A, Evans HM, Grønbech-Jensen N, Safinya CR. Transitions between Distinct compaction regimes in complexes of multivalent cationic lipids and DNA. Biophys J. 2008;95:836–846. doi: 10.1529/biophysj.107.124669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zidovska A, Ewert KK, Quispe J, Carragher B, Potter CS, Safinya CR. Block Liposomes from Curvature-Stabilizing Lipids: Connected Nanotubes, –rods or –spheres. Langmuir. 2009;25:2979–2985. doi: 10.1021/la8022375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vroege GJ, Lekkerkerker HNW. Phase transitions in lyotropic colloidal and polymer liquid crystals. Rep Prog Phys. 1992;55:1241–1309. [Google Scholar]
- 60.Vasilevskaya VV, Khokhlov AR, Matsuzawa Y, Yoshikawa K. Collapse of single DNA molecule in poly(ethylene glycol) solutions. J Chem Phys. 1995;102:6595–6602. [Google Scholar]
- 61.Fire A, Xu SQ, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–811. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- 62.Napoli CA, Lemieux C, Jorgensen R. Introduction of a chimeric chalcone synthase gene into petunia results in reversible co-suppression of homologous genes in trans. Plant Cell. 1990;2:279–289. doi: 10.1105/tpc.2.4.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jorgensen RA, Cluster PD, English J, Que Q, Napoli CA. Chalcone synthase cosuppression phenotypes in petunia flowers: comparison of sense vs antisense constructs and single-copy vs complex T-DNA sequences. Plant Mol Biol. 1996;31:957–973. doi: 10.1007/BF00040715. [DOI] [PubMed] [Google Scholar]
- 64.Cogoni C, Macino G. Post-transcriptional gene silencing across kingdoms. Curr Opin Genet Dev. 2000;10:638–643. doi: 10.1016/s0959-437x(00)00134-9. [DOI] [PubMed] [Google Scholar]
- 65.Hammond SM, Caudy AA, Hannon GJ. Post-transcriptional gene silencing by double-stranded RNA. Nature Rev Genet. 2001;2:110–119. doi: 10.1038/35052556. [DOI] [PubMed] [Google Scholar]
- 66.Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411:494–498. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- 67.Caplen NJ, Parrish S, Imani F, Fire A, Morgan RA. Specific inhibition of gene expression by small double-stranded RNAs in invertebrate and vertebrate systems. Proc Natl Acad Sci USA. 2001;98:9742–9747. doi: 10.1073/pnas.171251798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Samuel CE. Knockdown by RNAi—proceed with caution. Nature Biotechnol. 2004;22:280–282. doi: 10.1038/nbt0304-280. [DOI] [PubMed] [Google Scholar]
- 69.McAllister CS, Samuel CE. The RNA-activated Protein Kinase Enhances the Induction of Interferon-beta and Apoptosis Mediated by Cytoplasmic RNA Sensors. J Biol Chem. 2009;284:1644–1651. doi: 10.1074/jbc.M807888200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.McManus MT, Sharp PA. Gene silencing in mammals by small interfering RNAs. Nature Rev Genet. 2002;3:737–747. doi: 10.1038/nrg908. [DOI] [PubMed] [Google Scholar]
- 71.Elbashir SM, Harborth J, Weber K, Tuschl T. Analysis of gene function in somatic mammalian cells using small interfering RNAs. Methods. 2002;26:199–213. doi: 10.1016/S1046-2023(02)00023-3. [DOI] [PubMed] [Google Scholar]
- 72.Martinez LA, Naguibneva I, Lehrmann H, Vervisch A, Tchenio T, Lozano G, Harel-Bellan A. Synthetic small inhibiting RNAs: Efficient tools to inactivate oncogenic mutations and restore p53 pathways. Proc Natl Acad Sci USA. 2002;99:14849–14854. doi: 10.1073/pnas.222406899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hannon GJ, Rossi JJ. Unlocking the potential of the human genome with RNA interference. Nature. 2004;431:371–378. doi: 10.1038/nature02870. [DOI] [PubMed] [Google Scholar]
- 74.Karagiannis TC, El-Osta A. RNA interference and potential therapeutic applications of short interfering RNAs. Cancer Gene Ther. 2005;12:787–795. doi: 10.1038/sj.cgt.7700857. [DOI] [PubMed] [Google Scholar]
- 75.Sioud M. Therapeutic siRNAs. Trends Pharm Sci. 2004;25:22–28. doi: 10.1016/j.tips.2003.11.006. [DOI] [PubMed] [Google Scholar]
- 76.Caplen NJ. RNAi as a gene therapy approach. Expert Opin Biol Ther. 2003;3:575–586. doi: 10.1517/14712598.3.4.575. [DOI] [PubMed] [Google Scholar]
- 77.Spagnou S, Miller AD, Keller M. Lipidic carriers of siRNA: differences in the formulation, cellular uptake, and delivery with plasmid DNA. Biochemistry. 2004;43:13348–56. doi: 10.1021/bi048950a. [DOI] [PubMed] [Google Scholar]
- 78.Yin JY, Ma ZY, Selliah N, Shivers DK, Cron RQ, Finkel TH. Effective gene suppression using small interfering RNA in hard-to-transfect human T cells. J Immunol Meth. 2006;312:1–11. doi: 10.1016/j.jim.2006.01.023. [DOI] [PubMed] [Google Scholar]
- 79.Bouxsein NF, McAllister CS, Ewert KK, Samuel CE, Safinya CR. Structure and gene silencing activities of monovalent and pentavalent cationic lipid vectors complexed with siRNA. Biochemistry. 2007;46:4785–4792. doi: 10.1021/bi062138l. [DOI] [PubMed] [Google Scholar]
- 80.Faneca H, Simoes S, Pedroso de Lima MC. Association of albumin or protamine to lipoplexes: enhancement of transfection and resistance to serum. J Gene Med. 2004;6:681–692. doi: 10.1002/jgm.550. [DOI] [PubMed] [Google Scholar]
- 81.Yang J-P, Huang L. Overcoming the inhibitory effect of serum on lipofection by increasing the charge ratio of cationic liposome to DNA. Gene Ther. 1997;4:950–960. doi: 10.1038/sj.gt.3300485. [DOI] [PubMed] [Google Scholar]
- 82.Plank C, Mechtler K, Szoka FC, Wagner E. Activation of the complement system by synthetic DNA complexes: A potential barrier for intravenous gene delivery. Hum Gene Ther. 1996;7:1437–1446. doi: 10.1089/hum.1996.7.12-1437. [DOI] [PubMed] [Google Scholar]
- 83.Martin-Herranz A, Ahmad A, Evans HM, Ewert K, Schulze U, Safinya CR. Surface functionalized cationic lipid–DNA complexes for gene delivery: PEGylated lamellar complexes exhibit distinct DNA–DNA interaction regimes. Biophys J. 2004;86:1160–1168. doi: 10.1016/S0006-3495(04)74190-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hong K, Zheng W, Baker A, Papahadjopoulos D. Stabilization of cationic liposome-plasmid DNA complexes by polyamines and poly(ethylene glycol)-phospholipid conjugates for efficient in vivo gene delivery. FEBS Lett. 1997;400:233–237. doi: 10.1016/s0014-5793(96)01397-x. [DOI] [PubMed] [Google Scholar]
- 85.Kostarelos K, Miller AD. Synthetic, self-assembly ABCD nanoparticles; a structural paradigm for viable synthetic non-viral vectors. Chem Soc Rev. 2005;34:970–994. doi: 10.1039/b307062j. [DOI] [PubMed] [Google Scholar]
- 86.Maeda H, Greish K, Fang J. The EPR Effect and Polymeric Drugs: A Paradigm Shift for Cancer Chemotherapy in the 21st Century. Adv Polym Sci. 2006;193:103–121. [Google Scholar]
- 87.Choi JS, MacKay A, Szoka FC., Jr Low-pH-Sensitive PEG-Stabilized Plasmid-Lipid Nanoparticles: Preparation and Characterization. Bioconjugate Chem. 2003;14:420–429. doi: 10.1021/bc025625w. [DOI] [PubMed] [Google Scholar]
- 88.Wheeler JJ, Palmer L, Ossanlou M, MacLachlan I, Graham RW, Zhang YP, Hope MJ, Scherrer P, Cullis PR. Stabilized plasmid-lipid particles: construction and characterization. Gene Ther. 1999;6:271–281. doi: 10.1038/sj.gt.3300821. [DOI] [PubMed] [Google Scholar]
- 89.Ruoslahti E, Duza T, Zhang L. Vascular homing peptides with cell-penetrating properties. Curr Pharm Design. 2005;11:3655–3660. doi: 10.2174/138161205774580787. [DOI] [PubMed] [Google Scholar]
- 90.Ruoslahti E. Vascular zip codes in angiogenesis and metastasis. Biochem Soc Transact. 2004;32:397–402. doi: 10.1042/BST0320397. [DOI] [PubMed] [Google Scholar]
- 91.Laakkonen P, Åkerman ME, Biliran H, Yang M, Ferrer F, Karpanen T, Hoffman RM, Ruoslahti E. Antitumor activity of a homing peptide that targets tumor lymphatics and tumor cells. Proc Natl Acad USA. 2004;101:9381–9386. doi: 10.1073/pnas.0403317101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Walker GF, Fella C, Pelisek J, Fahrmeir J, Boeckle S, Ogris M, Wagner E. Toward Synthetic Viruses: Endosomal pH-Triggered Deshielding of Targeted Polyplexes Greatly Enhances Gene Transfer in Vitro and in Vivo. Mol Ther. 2005;11:418–425. doi: 10.1016/j.ymthe.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 93.Boomer JA, Thompson DH. Synthesis of acid-labile diplasmenyl lipids for drug and gene delivery applications. Chem Phys Lipids. 1999;99:145–153. doi: 10.1016/s0009-3084(99)00033-x. [DOI] [PubMed] [Google Scholar]
- 94.Zhu J, Munn RJ, Nantz MH. Self-cleaving ortho ester lipids: a new class of pH-vulnerable amphiphiles. J Am Chem Soc. 2000;122:2645–2646. [Google Scholar]
- 95.Bangham AD, Hill MW, Miller NGA. Preparation and Use of Liposomes as Models of Biological Membranes. Methods Membr Biol. 1973;1:1–68. [Google Scholar]
- 96.Zidovska A, Ewert KK, Quispe J, Carragher B, Potter CS, Safinya CR. The effect of salt and pH on block liposomes studied by cryogenic transmission electron microscopy. Biochim Biophys Acta - Biomembranes. 2009;1788:1869–1876. doi: 10.1016/j.bbamem.2009.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zidovska A, Ewert KK, Quispe J, Carragher B, Potter CS, Safinya CR. Block Liposomes: Vesicles of Charged Lipids with Distinctly Shaped Nanoscale Sphere-, Pear-, Tube-, or Rod-Segments. Meth Enzymol. 2009 doi: 10.1016/S0076-6879(09)65006-0. (accepted) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Helfrich W. Elastic Properties of Lipid Bilayers - Theory and Possible Experiments. Z Naturforsch C. 1973;28:693–703. doi: 10.1515/znc-1973-11-1209. [DOI] [PubMed] [Google Scholar]