

Abstract

Recent studies present that the single arginine (R) residue in the sequence of Aβ42 adopts abundant β-sheet structure and forms stable salt bridges with various residues. Furthermore, experiments proposed that R stimulates the Aβ assembly and arginine (R) to alanine (A) mutation (R5A) decreases both aggregate formation tendency and the degree of its toxicity. However, the exact roles of R and R5A mutation in the structures of Aβ42 are poorly understood. Extensive molecular dynamics simulations along with thermodynamic calculations present that R5A mutation impacts the structures and free energy landscapes of the aqueous Aβ42 peptide. The β-sheet structure almost disappears in the Ala21–Ala30 region but is more abundant in parts of the central hydrophobic core and C-terminal regions of Aβ42 upon R5A mutation. More abundant α-helix is adopted in parts of the N-terminal and mid-domain regions and less prominent α-helix formation occurs in the central hydrophobic core region of Aβ42 upon R5A mutation. Interestingly, intramolecular interactions between N- and C-terminal or mid-domain regions disappear upon R5A mutation. The structures of Aβ42 are thermodynamically less stable and retain reduced compactness upon R5A mutation. R5A mutant-type structure stability increases with more prominent central hydrophobic core and mid-domain or C-terminal region interactions. Based on our results reported in this work, small organic molecules and antibodies that avoid β-sheet formation in the Ala21–Ala30 region and hinder the intramolecular interactions occurring between the N-terminal and mid-domain or C-terminal regions of Aβ42 may help to reduce Aβ42 toxicity in Alzheimer’s disease.
Keywords: Disordered protein, amyloid-β, mutation, toxicity, Alzheimer’s disease
Disordered proteins, that is, unstructured proteins, are considered to lack stable structures yet are at the center of severe diseases.1−4 One of these highly dynamic disordered proteins that has been directly associated with various severe diseases, including Alzheimer’s and cerebral amyloid angiopathy diseases, is amyloid-β (Aβ).5−100 The 42 amino acid residue Aβ alloform (Aβ42) is reported to be the toxic protein in Alzheimer’s disease due to its increased tendency toward aggregation in comparison to the more dominant 40 amino acid residue Aβ alloform (Aβ40).12−15 The sequence of Aβ42 is given as follows with the single arginine (R) residue in the sequence of Aβ42 presented in bold: D A E F R H D S G Y E V H H Q K L V F F A E D V G S N K G A I I G L M V G G V V I A.
Most recently, several groups have shown that Aβ alloforms possess random coil conformation along with distinct specific structuring characteristics.5,6,17−23 Relating such specific structural properties to the thermodynamic properties and biological characteristics is not a straightforward process. In fact, such research activities that aim to study the structures of monomeric and oligomeric disordered fibrillogenic proteins are challenging due to fast aggregation, rapid conformational changes, and solvent effects. Several groups have successfully reported distinct structuring characteristics in the N-terminal (D1–L16), central hydrophobic core (CHC; L17–A21), and C-terminal regions (A30–A42) of the wild-type Aβ42.5,6,17−20 Aβ peptides are capable of forming fibrils in the absence of other biospecies, indicating that the potential to form these fibrils resides mainly in the structures of Aβ.54,55 The many various conformations adopted by Aβ are directly linked to aggregate formation: Aβ peptide structures with α-helical and random coil conformations have been shown to aggregate more slowly, and Aβ peptide structures that possess β-sheet conformation are known to aggregate more rapidly.54,55 It seems likely that various factors including β-sheet structure formation and intramolecular peptide interactions facilitate monomeric Aβ interactions and that these structural characteristics drive the formation of toxic oligomers and amyloid fibrils.54,55 Regarding Aβ42, α-helix formation with high abundance has been reported in the central hydrophobic core region (L17–A21; CHC), and β-sheet structure has been detected in the N-terminal and C-terminal regions, with the latter being more prominent5,6,17−19,23,25,44 in the structures of the wild-type Aβ42 peptide. In addition, the abundant β-sheet structure formation in the C-terminal region of the wild-type Aβ42 peptide has been linked to the aggregation mechanism.5,6,16,18 Furthermore, the stabilization of the turn conformation at A21–A30 in the wild-type Aβ42 peptide has been associated with the formation of salt bridges and hydrophobic interactions.5,6,18 Interestingly, prominent β-sheet structure is formed at R5 located in the N-terminal region of Aβ42, and this residue forms also various stable intramolecular interactions with other residues of the peptide located in the N- and C-terminal or mid-domain (L17–G29) regions in an aqueous solution environment.5,6
Experimental R5A mutation studies showed a decrease both in the tendency toward Aβ aggregate formation and a reduced toxicity related to Alzheimer’s disease.28 Further experimental studies presented that R5A mutation depresses the interactions between Aβ and sphingomyelin, which also have been related to the degree of Aβ toxicity in Alzheimer’s disease.29 In addition, blocking the E3–F4–R5–H6 region in which the R5 residue is located with targeted antibodies was shown to inhibit the aggregation of Aβ.32−30 These foregoing studies presented that R5 stimulates the assembly of Aβ. However, the exact role of R5 and R5A mutation in the structures and free energy landscapes of Aβ42 in an aqueous medium at the atomic level with dynamics has not been studied before. In general, theoretical studies complement experiments and provide knowledge about the structural properties of proteins in solution. In fact, important information has been gained about the structural characteristics of disordered monomeric and oligomeric Aβ peptides in solution from theoretical and computational studies.4−6,17−19,20,23,27−50 For example, α-helix and β-sheet structure formations, which have been directly related to the toxicity of Aβ, have been detected in various regions of the disordered Aβ42 peptide at the atomic level using computational and theoretical tools.5,6,17−19,23 We recently developed a theoretical strategy that calculates the secondary structure transition stabilities per residue in the structures of disordered proteins, which has been successfully applied on wild- and genetic mutant-type disordered proteins at the center of Alzheimer’s and Parkinson’s diseases as well as type II diabetes.6−8 For gaining insights into the role of R and for providing detailed knowledge about the impact of R5A mutation on the structures and thermodynamic properties of the wild-type Aβ42 peptide, we performed extensive parallel tempering molecular dynamics simulations along with special sampling techniques. As shown in earlier studies including our own, the potential energy surfaces of large-size disordered proteins have been recognized to be rugged, and traditional MD simulations without applying special sampling techniques hinder conformational transitions between various local minima.4−8,100,18,19,20,27−50 Sampling problems can preclude success in the structural and thermodynamic property studies using MD simulations.4−8,100,53−51 Therefore, various groups have worked actively on the development of special sampling techniques for enabling more efficient simulation strategies that locate low-energy minima for biological macromolecules, such as disordered proteins. One approach that has been widely used for the simulations of disordered proteins is the parallel tempering method.39,40 Such simulations help to increase the conformational sampling in comparison to classical MD simulations without the application of special sampling techniques, which can be trapped in one specific potential minimum.4−8,100,18,19,20,27−50 Using parallel tempering MD simulations, we studied the structural properties including the secondary and tertiary structures as well as the free energy landscapes of the wild- and R5A mutant-type Aβ42 peptides in an aqueous solution medium and compared these characteristics to one another. The thermodynamic properties, including the impact of the enthalpic and entropic contributions are studied using both harmonic and quasi-harmonic methods and potential of mean force surfaces.
Results and Discussion
The calculated secondary structure components per residue show that the β-sheet formation at L17, E22, and S26–G29 that occurs in the structures of the wild-type Aβ42 peptide almost disappears upon R5A mutation in an aqueous solution environment (Figure 1). Residues V18–A21 located in the CHC region, as well as I31 and the C-terminal region except G33, G37, and I41 adopt more abundant β-sheet structure upon R5A mutation. A significant difference in the N-terminal region is noticed at E3–H6 and V12–K16 since these adopt more abundant 310-helix upon R5A mutation of the wild-type Aβ42 peptide. Furthermore, more prominent α-helical structure formation occurs at Y10–K16 and at E22–I32 of the wild-type peptide via R5A mutation. Less abundant α-helix and 310-helical structure are formed at L17–A21 and G33–V36 and at Y10–V12, A21–D23, and G33–V36 via R5A mutation, respectively. Knowledge about secondary structure interconversion stabilities that lead to specific helical and β-sheet structure formations can aid in the understanding of the role of dynamic changes in the aggregation mechanism and toxicity. In fact, experimental studies have aimed to obtain such knowledge for several decades. However, such detailed information has not been gained until most recently due to the lack of available theoretical and experimental tools. Using our new theoretical strategy, obtained results show that turn or helix to coil structure transitions overall are preferred in the conformations of the wild-type Aβ42 peptide (Figure 2A). This result supports the experimentally known disordered structure of Aβ42. In addition, distinct structuring transition trends, for example, turn to helix conversion per residue, are more stable than the coil or β-sheet to helix transitions at F4–K16 in the N-terminal region of Aβ42 (Figure 2A). Turn to helix is the most thermodynamically stable transition but coil to β-sheet conversion is also preferred over the helix or turn to β-sheet conversions in the CHC region. Moreover, the transition from helix to β-sheet is slightly more stable than the β-sheet to helix conversion in the CHC region (Figure 2A). Interestingly, the transitions from helix or coil to turn are the most stable ones for the A21–A30 decapeptide region of Aβ42 (Figure 2A). This finding supports previous experimental and theoretical studies that detected turn structure formation in this decapeptide region. Overall the most stable transitions resulting in β-sheet formation occur initially from a coil structure at R5, L17, F20, and I31. These are thermodynamically followed by coil to β-sheet conversions at H6, Y10, H13, V18, F19, A21, E22, S26–A30, G33, and G37–I41. Additionally, turn or helix to β-sheet conversions are thermodynamically preferred at R5, L17, and I32. Furthermore, residues V18–F20 prefer to convert from helix to β-sheet structure (Figure 2A). Results obtained from our simulations and calculations (Figures 1 and 2) for the structural characteristics of Aβ42 are in agreement with NMR measurements that present turn and bend-like formations at D7–E11 and F20–S26 (ref (56) and references therein). Moreover, the turn structure formation, which we detect with high abundance, at A21–A30 of Aβ42 was also reported by experiments (ref (56) and references therein).
Figure 1.
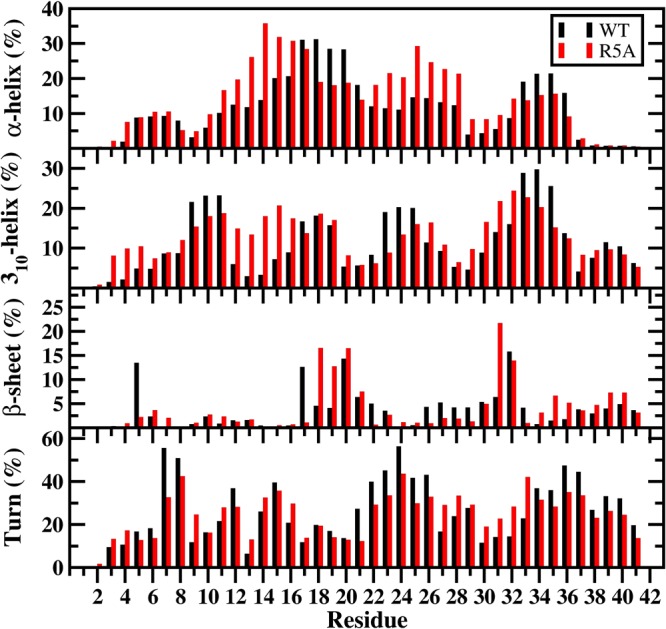
Wild-type and R5A mutant-type Aβ42 secondary structure components. Secondary structures along with their abundances per residue for the wild-type (black) and R5A mutant-type (red) Aβ42 peptides per residue. The abundances for the π-helix and coil structures are not displayed.
Figure 2.
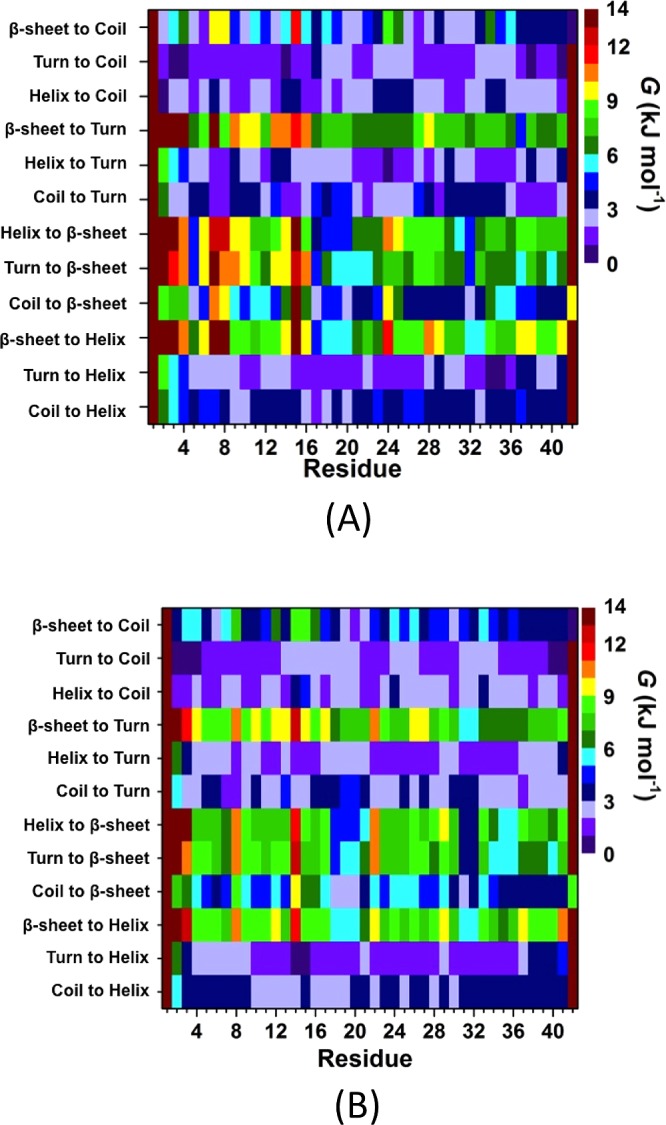
Secondary structure transition stabilities per residue of the wild-type and R5A mutant-type Aβ42 peptides. The stability of secondary structure transitions between two specific secondary structure components per residue for the wild-type (A) and R5A mutant-type (B) Aβ42 peptides in aqueous solution. The color scale corresponds to the free energy value associated with specific transitions between two secondary structure components for a specific residue.
Different than the wild-type Aβ42 peptide, turn or coil to helix transition is more stable at E3–Q15 located in the N-terminal region of the R5A mutant-type peptide (Figure 2B). Moreover, the coil to β-sheet conversion is more stable at H6, D7, and G9 upon R5A mutation. The stability of turn formation from helix or coil decreases slightly at A5–K16 upon R5A mutation of Aβ42. Interestingly, coil to β-sheet conversion becomes thermodynamically more preferred in the CHC region of the wild-type Aβ42 peptide upon R5A mutation. Helix or coil to turn transitions are slightly less preferred in the A21–A30 region of the R5A mutant-type peptide in comparison to the same region in the structures of the wild-type Aβ42 peptide. Instead, coil or turn to helix are thermodynamically more preferred and β-sheet to coil as well as β-sheet to turn conversions are less stable in the A21–A30 region upon R5A mutation. Coil to β-sheet transition becomes less stable at S26–A30 and L34–G38 upon R5A mutation. Moreover, the mid-domain region adopts more stable β-sheet structure initially resulting from turn or helix conformations in the wild-type in comparison to its mutant-form. In other words, R5A mutation results in less stable secondary structure conversions resulting in the formation of β-sheet in the mid-domain region of the wild-type peptide, which in turn might be directly associated with the depressed toxicity or decreased tendency toward aggregation via R5A mutation or blocking of the R residue utilizing antibodies. We also note that R5A mutation yields more structuring in the disordered nature of the wild-type Aβ42, for example, coil to helix conversion is more preferred in the CHC and in parts of the A21–A30 decapeptide regions, coil to β-sheet transition is more stable in parts of the CHC region, and coil to turn transition is thermodynamically more preferred at G33–V36 upon R5A mutation. Overall, helix or turn to β-sheet transition stability decreases in parts of the mid-domain and C-terminal regions of the R5A mutant-type peptide in comparison to the wild-type Aβ42 peptide. On the other hand, β-sheet to helix conversion becomes more preferred in the N-terminal region except D1–E3, S8, and H14 upon R5A mutation. These findings might further present that the decreased helix or turn to β-sheet structure conversion stabilities in parts of the mid-domain and C-terminal regions can be associated with the decreased toxicity observed in R5A mutant-type Aβ42 species. Based on these results, we find that helix to β-sheet transition is preferred over β-sheet to helix transition at Q15–F20, V24, K28–I31, and L34–I41 in the structures of the wild-type Aβ42. β-sheet to helix transition is more stable than the helix to β-sheet transition at G9, Y10, G25, and G33 in the structures of the wild-type peptide. For the R5A mutant-type, helix to β-sheet transition is preferred over β-sheet to helix transition at F4–D7, E11–H14, K16, V18–A21, V24, I31, I32, and L34–I41, and the β-sheet to helix transition is predominantly preferred over helix to β-sheet transition at E3 and E22.
The impact of R5A mutation on the structural stability of the wild-type Aβ42 peptide in an aqueous solution environment was studied using both harmonic and quasi-harmonic methods (Table 1). A comparison of the thermodynamic values reveals that the R5A mutation increases the conformational enthalpy (H) value by ∼840 kJ mol–1. Although a drastic change in the conformational entropy value (−TS) upon R5A mutation is not observed utilizing the harmonic method, the conformational entropy values calculated using the quasi-harmonic method show a decrease by 370 kJ mol–1 upon R5A mutation (Table 1). Nevertheless, the R5A mutation results in less stable structures of Aβ42 regardless of the chosen thermodynamic method. These results present that R5A mutation destabilizes the structures of Aβ42 in an aqueous solution environment. The impact of R5A mutation on the conformational preferences of the wild-type Aβ42 peptide based on specific structural characteristics is also assessed by calculating the potential of mean force (PMF) surfaces of the wild-type and R5A mutant-type Aβ42 peptides along the coordinates of radius of gyration (Rg) and end-to-end distance (RE-E) (Figure 3). This type of free energy landscape determines the most favorable protein structures based on the Rg and RE-E values, which are located in the regions with the lowest PMF values. The localized regions of these preferred structures are termed basins.
Table 1. The Calculated Average Enthalpy (H), Solvation Free Energy (Gsol), H – Gsol, Entropy (S), and Gibbs Free Energy (G) Values for the Wild-Type and R5A Mutant-Type Aβ42 Peptidesa.
| peptide | ⟨H – Gsol⟩ (kJ mol–1) | ⟨H⟩ (kJ mol–1) | –T⟨SNMA⟩ (kJ mol–1) | ⟨GNMA⟩ (kJ mol–1) | –T⟨SQH⟩ (kJ mol–1) | ⟨GQH⟩ (kJ mol–1) |
|---|---|---|---|---|---|---|
| WT | –188.0 (±281.4) | –2612.9 (±127.4) | 2196.4 (±39.3) | –4809.3 (±126.3) | –5895.3 (±18.0) | –8508.2 (±128.7) |
| R5A | 1022.1 (±298.6) | –1736.9 (±127.9) | 2196.8 (±43.5) | –3933.6 (±125.0) | –6151.3 (±44.3) | –7888.2 (±135.4) |
The subscripts NMA and QH represent the normal mode analysis and quasi-harmonic methods, respectively.
Figure 3.

WT and R5A mutant-type Aβ42 PMF surfaces. Change in the potential of mean force (ΔPMF) of the wild-type (A) and R5A mutant-type (B) Aβ42 peptides along the coordinates of radius of gyrations (Rg) and end-to-end distance (RE-E) in units of kJ mol–1. Representative structures of the most preferred PMF basins (basin IA and basin IB for the WT and basin I for the R5A mutant-type Aβ peptides) are displayed next to each PMF surface. The secondary structure components per residue for each structure are correlated with the color of the peptide backbone as follows: α-helix (blue), 310-helix (gray), π-helix (purple), β-sheet (red), β-bridge (black), turn (yellow), and coil (white).
Before evaluating the PMF surfaces, the probability distributions of the Rg were determined (see Supporting Information). The probability of Aβ42 conformations with Rg values greater than 12 Å significantly increases upon R5A mutation, yielding an ∼2 Å larger average Rg value in comparison to the structures of the wild-type peptide. This finding indicates that the presence of R results in a higher probability for more compact structures of the Aβ42 peptide than when R is replaced by A. The free energy landscape of Aβ42 is significantly altered by the R5A mutation. The PMF surface of the wild-type Aβ42 peptide presents two favorable PMF basins. One of these favorable PMF basins (basin IA) is located at Rg values between 9.8 and 11.0 Å and RE-E values between 9.9 and 17.0 Å, while the second favorable PMF basin (basin IB) is located at Rg values varying between 9.7 and 11.1 Å and RE-E values varying between 19.5 and 42.0 Å. Furthermore, the calculated PMF surfaces indicate that transitions between structures located in these two basins require the overriding of large energy barriers (>1kBT). The R5A mutation impacts this trend and results in the formation of only one most favorable PMF basin at Rg values varying between 10.0 and 15.0 Å and RE-E values varying between 16.0 and 50.0 Å (basin I). This finding indicates that the presence of R in the wild-type Aβ42 peptide plays a significant role in the appearance of two different preferred PMF basins along the coordinates of Rg and RE-E. In addition, the lowest PMF value of the wild-type is 12% smaller than the most favorable PMF value obtained for the R5A mutant-type peptide structures, which also shows that the wild-type peptide adopts more preferred structures than the R5A mutant-type Aβ42. This result is in accord with the thermodynamic results utilizing both the harmonic and quasi-harmonic methods (see above). Altogether, these results show that R5A mutation of the wild-type Aβ42 peptide results in thermodynamically less preferred structures due to conformational differences between the wild-type and R5A mutant-type Aβ42 peptides. In other words, the so-called “disorder promoting” R residue promotes thermodynamic stability in the structures of the wild-type Aβ42 peptide in comparison to its R5A mutant form structures in an aqueous solution medium.
Figure 4 depicts the probabilities of the intramolecular peptide interactions in the structures of the wild-type and R5A mutant-type Aβ42 peptides in an aqueous solution environment. In addition to the highly abundant interactions between adjacent residues (within ±5 residues of a specific residue) throughout the wild-type Aβ42, we also observe prominent intramolecular interactions between the CHC (L17–A21) and mid-domain/C-terminal (N27–G33) regions (Figure 4A). The high abundance of β-sheet formation in these regions and the prominent turn structure formation at E22–S26 indicates that these abundant interactions might be related to β-sheet formation in these regions of Aβ42 (see above). The N- and C-terminal regions interact with the mid-domain region of Aβ42 (Figure 4A). In addition, prominent interactions between E3–S8 and Y10–H14, CHC region and S8–V12 or G25–A30 occur in the structures of Aβ42 in an aqueous medium. We note that long-range intramolecular peptide interactions, which we detected for Aβ42, disappear or become less abundant upon R5A mutation (Figure 4B). Specifically, N- or C-terminal interactions with the mid-domain region of the wild-type Aβ42 disappear upon R5A mutation (Figure 4B), which explains why the R5A mutant-type adopts less compact and thermodynamically less stable structures (see above). On the other hand, shorter-range intramolecular interactions between N27–A30 and D23–N27 as well as between I32–V36 and G29–I31 are more abundant (up to 30%) in the structures of the R5A mutant-type Aβ42 peptide (Figure 4B) in comparison to the wild-type Aβ42 peptide (Figure 4A). Furthermore, interactions between the CHC region (L17–A21) and the residues adjacent to the CHC region (A21–V24) decrease by up to 26% upon R5A mutation. Overall, these results indicate that R plays a crucial role in the formation of long-range intramolecular interactions in the structures of the wild-type Aβ42 peptide, and once R is replaced by A, the peptide becomes less compact and less stable in an aqueous solution medium. In other terms, R5A mutation causes a disorder in terms of the tertiary structure properties of Aβ even though the same trend cannot be detected for the secondary structures (see above).
Figure 4.
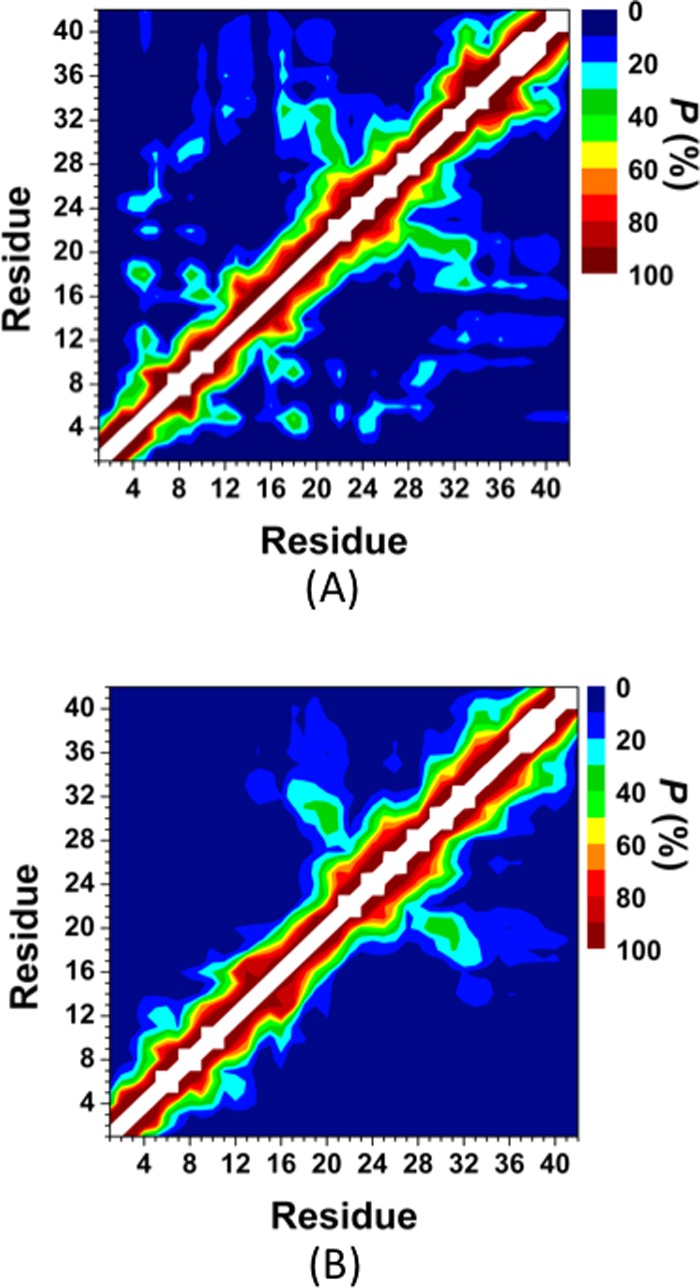
Wild-type and R5A mutant-type Aβ42 tertiary structures. Intramolecular interactions and abundances in the structures of the wild-type (A) and the R5A mutant-type (B) Aβ42 peptides. The color scale corresponds to the probability (P) of the distance between the centers of mass between two residues being ≤9 Å from each other.
The intramolecular peptide interactions for the structures along the PMF surface reveal that the interactions between the CHC and mid-domain/C-terminal regions occur in the most preferred PMF basin (Figure 5A). Furthermore, these interactions disappear with increasing PMF (Figure 5). This finding indicates that the CHC region and mid-domain/C-terminal region interactions increase the stability of the peptide. It is also interesting to note that the tertiary structures of the R5A mutant-type peptide in the most favorable PMF basin (Figure 5A) more closely resemble those of the wild-type Aβ42 structures of the least favorable PMF basin.5,6 Overall, these results indicate that the presence of R increases the formation of intramolecular interactions that result in thermodynamically more preferred structures. In other words, R, which is a residue generally known as a disorder promoting polar residue, largely causes specific structuring in the disordered peptide conformations in terms of intramolecular long-range interactions. The salt bridge formations between K28 and E22 or D23 were associated with the turn structure formation in the A21–A30 region of the wild-type Aβ42 peptide.5,6 The calculated salt bridge formation stabilities between K28 and E22 or D23 were analyzed by calculating the distribution of the distance between the Cγ and Nζ atoms of these residues are shown in Figure 6A,B, respectively. As shown in Figure 6A,B, the D23 and K28 salt bridge is more preferred than the E22 and K28 salt bridge at a distance of 4.0 Å (red curves, Figure 6). A vice versa trend is observed in the structures of the wild-type Aβ42 peptide (black curves, Figure 6). The salt bridge between D23 and K28 is about 60% more preferred in the structures of the R5A mutant-type rather than in those of the wild-type Aβ42 peptide (Figure 6B), which might be associated with the more abundant turn structure formation at K28 upon R5A mutation (Figure 1). In addition, we calculated the abundance of other possible salt bridges that are formed in the structures of the wild- and R5A mutant-type Aβ42 in an aqueous medium (Table 2). Based on these additional calculations, we find that the K16 and E11 as well as K16 and D7 salt bridges are formed with a slightly higher abundance in the structures of the R5A mutant-type Aβ42 (at 4.0 Å). In addition, the probability of the N-terminus and E3 salt bridge increase upon R5A mutation (Table 2). These findings indicate that R impacts the stability of the salt bridges formed between K28 and D23 in the structures of the wild-type Aβ42 monomer. Moreover, the probability of salt bridge formation for the R5A mutant-type Aβ42 peptide also shows specific trends according to the PMF values of the structures (Table 3). The formations of salt bridges between K16 and D7 or E11 as well as the C-terminus and K16 or K28 increase with decreasing PMF values (basins I to III).
Figure 5.

Intramolecular interactions in the structures of the wild-type Aβ42 peptide located in basin I (A.1), basin II (A.2), and basin III (A.3) and in the structures of the R5A mutant-type Aβ42 located in basin I (B.1), basin II (B.2), and basin III (B.3) of the PMF surfaces. The color scale corresponds to the probability (P) of the distance between the centers of mass between two residues being ≤9 Å from each other.
Figure 6.
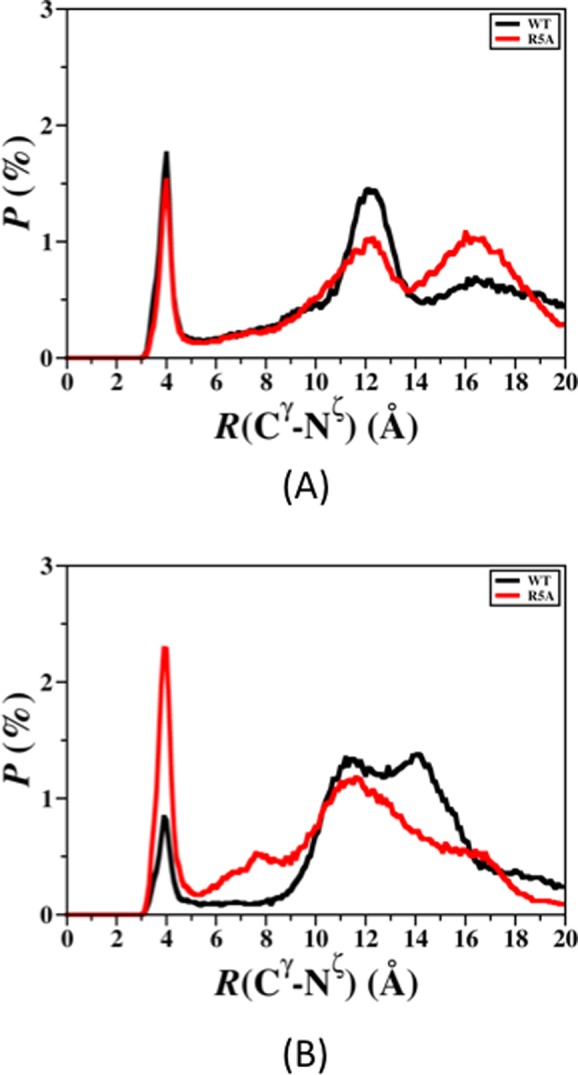
The calculated probability distribution of the distance between the Cγ atom of the E22 (A) or D23 (B) residues and the Nζ atom of the K28 residue for all converged structures of the wild-type (black) and R5A mutant-type (red) Aβ42 peptides.
Table 2. The Formed Salt Bridges of the Wild-Type and R5A Mutant-Type Aβ42 Peptidesa.
|
R(Cγ–Nζ) (%) |
|||||||
|---|---|---|---|---|---|---|---|
| WT |
R5A |
||||||
| donor | acceptor | ≤4 Å | ≤5 Å | ≤6 Å | ≤4 Å | ≤5 Å | ≤6 Å |
| Lys16 | Glu11 | 3.8 | 6.7 | 8.0 | 10.5 | 17.7 | 20.1 |
| Asp1 (−NH3+) | Glu3 | 2.8 | 4.5 | 5.3 | 8.0 | 12.7 | 14.3 |
| Lys16 | Asp7 | 3.8 | 8.3 | 11.1 | 5.6 | 7.6 | 8.1 |
| Lys28 | Ala42 (−COO–) | 1.8 | 2.6 | 2.9 | 3.0 | 4.4 | 4.9 |
| Lys16 | Glu3 | 0.9 | 1.6 | 1.8 | 1.4 | 2.2 | 2.4 |
| Lys16 | Ala42 (−COO–) | 5.6 | 8.1 | 9.4 | 1.1 | 1.7 | 1.9 |
| Asp1 (−NH3+) | Asp7 | 0.0 | 0.0 | 0.0 | 0.6 | 1.6 | 2.0 |
| Lys28 | Glu11 | 0.0 | 0.0 | 0.0 | 0.8 | 1.3 | 1.4 |
| Lys16 | Glu22 | 0.0 | 0.0 | 0.0 | 0.6 | 1.0 | 1.2 |
| R5 | Glu3 | 57.7 | 59.2 | 59.6 | |||
| R5 | Glu22 | 26.4 | 26.7 | 26.7 | |||
| R5 | Ala42 (−COO–) | 20.2 | 21.0 | 21.3 | |||
| R5 | Glu11 | 14.1 | 15.9 | 16.7 | |||
| R5 | Asp1 | 13.9 | 14.8 | 15.0 | |||
| R5 | Asp23 | 8.2 | 8.4 | 8.4 | |||
R(Cγ–Nζ) is the distance between carboxylate C atom (Cγ) and the side-chain or N-terminal N atom (Nζ) of the residues involved in the formed salt bridges. The values presented are the abundances of the R(Cγ–Nζ) being less than or equal to the specified distance in the table for the converged structures of the wild-type and R5A mutant-type Aβ42 peptides.
Table 3. The Formed Salt Bridges of the R5A Mutant-Type Peptides for the Structures Located in Each of the PMF Basinsa.
|
R(Cγ–Nζ) ≤ 4 Å |
||||
|---|---|---|---|---|
| donor | acceptor | I | II | III |
| Lys28 | Asp23 | 5.4 | 5.7 | 5.0 |
| Lys16 | Glu11 | 5.3 | 5.0 | 3.7 |
| Asp1 (−NH3+) | Glu3 | 4.1 | 3.8 | 4.8 |
| Lys28 | Glu22 | 3.0 | 2.0 | 2.0 |
| Lys16 | Asp7 | 2.9 | 2.3 | 1.5 |
| Lys28 | Ala42 (−COO–) | 1.7 | 0.7 | 0.9 |
| Lys16 | Glu3 | 0.8 | 0.4 | 0.2 |
| Lys16 | Ala42 (−COO–) | 0.6 | 0.3 | 0.1 |
| Asp1 (−NH3+) | Asp7 | 0.3 | 0.3 | 0.4 |
| Lys28 | Glu11 | 0.4 | 0.3 | 0.4 |
| Lys16 | Glu22 | 0.3 | 0.2 | 0.2 |
R(Cγ–Nζ) is the distance between carboxylate C atom (Cγ) and the side-chain or N-terminus N atom (Nζ) of the residues involved in the formed salt bridges. The values presented are the abundances of the R(Cγ–Nζ) being less than or equal to the specified distance in the table for the converged structures of the wild-type and R5A mutant-type Aβ42 peptides.
Overall, the reported structural and thermodynamic properties present that R5A mutation greatly impacts the structural and thermodynamic properties of the wild-type Aβ42 peptide in an aqueous solution medium. R promotes both order and disorder in the secondary structures of Aβ42. Besides, R5A mutation strengthens the disordered nature of Aβ42 regarding its tertiary structures. R5A mutation also causes less stable and less compact Aβ42 structures. Interestingly, β-sheet structure abundance increases in the C-terminal region, and it almost disappears in the A21–A30 decapeptide region of Aβ42 upon R5A mutation. We should mention here again that abundant β-sheet structure formation in the C-terminal region of monomeric and oligomeric Aβ has been related to the aggregation mechanism and toxicity in Alzheimer’s disease. However, experiments reported a decreased tendency toward aggregation and a depressed toxicity for the R5A mutant-type Aβ in comparison to its wild-type form. These findings show that the β-sheet structure formation in the C-terminal region of R5A mutant-type is more prominent than in the same region of its wild-type form but the synthetic mutant-type is less toxic. Therefore, we can conclude that the decreased toxicity in the R5A mutant-type Aβ is not related to β-sheet formation in the C-terminal region. Instead, R5A mutation reduces β-sheet formation in the A21–A30 decapeptide region of Aβ42. Additionally, intramolecular interactions between the N- and C-terminal, as well as N-terminal and mid-domain regions almost disappear upon R5A mutation of Aβ42 in an aqueous solution environment.
Methods
Separate REMD simulations of the wild-type and R5A mutant-type Aβ42 peptides were performed. The NMR structure (PDBID 1Z0Q) of the wild-type Aβ42 peptide was used as the initial geometry,52 and the R5A mutant-type structure was made by replacing the R residue with an A residue in the initial wild-type Aβ42 structure. The Amber11 software package37 utilizing the Amber ff99SB potential functions200 for proteins was used to perform the REMD simulations. The Onufriev–Bashford–Case implicit solvent model was chosen to represent the solution environment around the protein in order to avoid possible confined aqueous volume effects and specific heat errors on the simulated structures.5,6,39,40 Particle mesh Ewald method was used to treat the long-range interactions using a cutoff value of 25 Å.41,42 The temperature was maintained by employing Langevin dynamics with a collision frequency of 2 ps–1.41 Each REMD simulation utilized 24 different replicas with temperatures exponentially distributed between 280 and 400 K, as shown in our previous studies of the Aβ peptide.5,6,43 The integration time step was set to 2 fs with trajectories saved every 500 steps. The initial conformations of both peptides were equilibrated for 200 ps at each replica temperature. Then, both peptide structures were simulated for 300 ns per replica (for each peptide) with exchanges between replicas attempted every 5 ps, yielding a total simulation time of 7.2 μs and an exchange probability of 0.74 for both the wild-type and R5A mutant-type Aβ42 peptides. The convergence of both simulations occurred at 150 ns as determined via time-dependent secondary structure component abundance and the conformational internal energy values (see Supporting Information). All calculations of the structural and thermodynamic properties of both Aβ42 peptides were performed on the 150 000 structures obtained after convergence for each peptide from the replica closest to physiological temperature (∼310 K). The conformational stabilities of the wild-type and R5A mutant-type Aβ42 peptides were calculated utilizing the molecular mechanics/generalized Born surface area (MM/GBSA) method.44,45,46 The MM/GBSA method determines the conformational Gibbs free energy (G) from the potential energy (EMM), the solvation free energy (Gsol), and the entropy (S) at a specific temperature (T) via eq 1.
| 1 |
The Gsol is composed of electrostatic and nonpolar contributions, using the generalized Born and molecular surface methods for the estimation of these values, respectively. The entropic contribution to the conformational Gibbs free energy was estimated using both harmonic and quasi-harmonic methods via normal-mode analysis and Schlitter calculations, respectively.47,48 In addition, we calculated the potential of mean force (PMF) surfaces along the coordinates of radius of gyration (Rg) and end-to-end distance (RE-E), which we have previously shown to yield insights into the conformational preferences of the Aβ peptide.5,6,49 Following our recent studies, intramolecular peptide interactions were considered to occur when the centers of mass of two residues were within 9 Å of each other.5,6,49 Salt bridges are considered to occur when two hydrogen bonded atoms have opposite electrostatic charges. A hydrogen bond exists if the distance between donor hydrogen atom and the acceptor atom is ≤2.5 Å, and the hydrogen bond angle is larger than 113°.5,6,50 To gain detailed insights into the secondary structure properties, we applied the DSSP algorithm33 as well as our recently developed theoretical strategy that enables the predictions of secondary structure transition stabilities per residue with dynamics.6−8 This strategy, which we implemented in our molecular simulation program ProtMet, utilizes potential of mean force calculations from a conditional probability point of view to evaluate the stability of transitions between two different secondary structure components.6−8
Acknowledgments
This research was supported by an allocation and computing resources provided by the National Science Foundation (Grant No. TG-CHE110044). The calculations and simulations were performed on Kraken at the National Institute for Computational Sciences. The authors thank Ulrich Hansmann (University of Oklahoma), Rudolph Magyar (SNRL), and Emily A. Jarvis (Loyola University) for support and helpful discussions.
Glossary
Abbreviations:
- Aβ
amyloid-β
- CHC
central hydrophobic core
- MD
molecular dynamics
- EMD
replica exchange molecular dynamics
- PMF
potential of mean force
- NMA
normal-mode analysis
- QH
quasi-harmonic
- R
arginine
- R5A
arginine to alanine mutation
- Rg
radius of gyration
- RE-E
end-to-end distance
- MM/PBSA
molecular mechanics/Poisson–Boltzmann surface area
Supporting Information Available
Convergence figures for the wild-type and R5A mutant-type Aβ peptides, convergence figures for the PMF surfaces of both peptides, probability distribution of the Rg values for both peptides, convergence figures for the secondary structure transition stability calculations of the wild-type and R5A mutant-type Aβ peptides, secondary structure component abundances per residue for the structures located in different PMF basins of the wild-type and R5A mutant-type Aβ peptides, and intramolecular interaction maps and salt bridge abundances for the wild-type Aβ protein structures located in the different PMF basins on the PMF surface. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Uversky V. N.; Oldfield C. J.; Dunker A. K. (2008) Intrinsically disordered proteins in human diseases: Introducing the D(2) concept. Annu. Rev. Biophys. 37, 215–246. [DOI] [PubMed] [Google Scholar]
- Babu M. M.; van der Lee R.; de Groot N. S.; Gsponer J. (2011) Intrinsically disordered proteins: Regulation and disease. Curr. Opin. Struct. Biol. 21, 432–440. [DOI] [PubMed] [Google Scholar]
- Dyson H. J.; Wright P. E. (2005) Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 6, 197–208. [DOI] [PubMed] [Google Scholar]
- Uversky V. N.; Oldfield C. J.; Midic U.; Xie H. B.; Xue B.; Vucetic S.; Iakoucheva L. M.; Obradovic Z.; Dunker A. K. (2009) Unfoldomics of human diseases: Linking protein intrinsic disorder with diseases. BMC Genomics 10, S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise-Scira O.; Xu L.; Kitahara T.; Perry G.; Coskuner O. (2011) Amyloid-β peptide structure in aqueous solution varies with fragment size. J. Chem. Phys. 135, 205101. [DOI] [PubMed] [Google Scholar]
- Coskuner O.; Wise-Scira O.; Perry G.; Kitahara T. (2012) The Structures of the E22Δ Mutant-Type Amyloid-β Alloforms and the Impact of E22Δ Mutation on the Structures of the Wild-Type Amyloid-β Alloforms. ACS Chem. Neurosci. 4, 310–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise-Scira O.; Dunn A.; Aloglu A. K.; Sakallioglu I. T.; Coskuner O. (2013) Structures of the E46K Mutant-Type α-Synuclein Protein and Impact of E46K Mutation on the Structures of the Wild-Type α-Synuclein Protein. ACS Chem. Neurosci. 4, 498–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise-Scira O.; Aloglu A. K.; Dunn A.; Sakallioglu I. T.; Coskuner O. (2013) Structures and Free Energy Landscapes of the Wild-Type and A30P Mutant-Type α-Synuclein Proteins with Dynamics. ACS Chem. Neurosci. 4, 486–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson K. A.; Gregas M.; Becker J. A.; Kinnecom C.; Salat D. H.; Moran E. K.; Smith E. E.; Rosand J.; Rentz D. M.; Klunk W. E.; Mathis C. A.; Price J. C.; DeKosky S. T.; Fischman A. J.; Greenberg S. M. (2007) Imaging of amyloid burden and distribution in cerebral amyloid angiopathy. Ann. Neurol. 62, 229–234. [DOI] [PubMed] [Google Scholar]
- Vanbroeckhoven C.; Haan J.; Bakker E.; Hardy J. A.; Vanhul W.; Wehnert A.; Vegtervandervlis M.; Roos R. A. C. (1990) Amyloid-Beta Protein-Precursor Gene and Hereditary Cerebral-Hemorrhage with Amyloidosis (Dutch). Science 248, 1120–1122. [DOI] [PubMed] [Google Scholar]
- Atwood C. S.; Martins R. N.; Smith M. A.; Perry G. (2002) Senile plaque composition and posttranslational modification of amyloid-beta peptide and associated proteins. Peptides 23, 1343–1350. [DOI] [PubMed] [Google Scholar]
- Coskuner O.; Wise-Scira O. (2013) Structures and Free Energy Landscapes of the A53T Mutant-Type α-Synuclein Protein and Impact of A53T Mutation on the Structures of the Wild-Type α-Synuclein Protein with Dynamics. ACS Chem Neurosci 4, 1101–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann T.; Bieger S. C.; Bruhl B.; Tienari P. J.; Ida N.; Allsop D.; Roberts G. W.; Masters C. L.; Dotti C. G.; Unsicker K.; Beyreuther K. (1997) Distinct sites of intracellular production for Alzheimer’s disease A beta 40/42 amyloid peptides. Nat. Med. 3, 1016–1020. [DOI] [PubMed] [Google Scholar]
- Dahlgren K. N.; Manelli A. M.; Stine W. B.; Baker L. K.; Krafft G. A.; LaDu M. J. (2002) Oligomeric and fibrillar species of amyloid-beta peptides differentially affect neuronal viability. J. Biol. Chem. 277, 32046–32053. [DOI] [PubMed] [Google Scholar]
- Selkoe D. J. (1999) Translating cell biology into therapeutic advances in Alzheimer’s disease. Nature 399, A23–A31. [DOI] [PubMed] [Google Scholar]
- Younkin S. G. (1995) Evidence That a-Beta-42 Is the Real Culprit in Alzheimers-Disease. Ann. Neurol. 37, 287–288. [DOI] [PubMed] [Google Scholar]
- Sgourakis N. G.; Merced-Serrano M.; Boutsidis C.; Drineas P.; Du Z. M.; Wang C. Y.; Garcia A. E. (2011) Atomic-Level Characterization of the Ensemble of the A beta(1-42) Monomer in Water Using Unbiased Molecular Dynamics Simulations and Spectral Algorithms. J. Mol. Biol. 405, 570–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sgourakis N. G.; Yan Y. L.; McCallum S. A.; Wang C. Y.; Garcia A. E. (2007) The Alzheimer’s peptides A beta 40 and 42 adopt distinct conformations in water: A combined MD/NMR study. J. Mol. Biol. 368, 1448–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M. F.; Teplow D. B. (2008) Amyloid beta-Protein Monomer Folding: Free-Energy Surfaces Reveal Alloform-Specific Differences. J. Mol. Biol. 384, 450–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luttmann E.; Fels G. (2006) All-atom molecular dynamics studies of the full-length beta-amyloid peptides. Chem. Phys. 323, 138–147. [Google Scholar]
- Hou L. M.; Shao H. Y.; Zhang Y. B.; Li H.; Menon N. K.; Neuhaus E. B.; Brewer J. M.; Byeon I. J. L.; Ray D. G.; Vitek M. P.; Iwashita T.; Makula R. A.; Przybyla A. B.; Zagorski M. G. (2004) Solution NMR studies of the A beta(1–40) and A beta(1–42) peptides establish that the met35 oxidation state affects the mechanism of amyloid formation. J. Am. Chem. Soc. 126, 1992–2005. [DOI] [PubMed] [Google Scholar]
- Mitternacht S.; Staneva I.; Hard T.; Irback A. (2010) Comparing the folding free-energy landscapes of A beta 42 variants with different aggregation properties. Proteins 78, 2600–2608. [DOI] [PubMed] [Google Scholar]
- Lim K. H.; Collver H. H.; Le Y. T. H.; Nagchowdhuri P.; Kenney J. M. (2007) Characterizations of distinct amyloidogenic conformations of the A beta (1–40) and (1–42) peptides. Biochem. Biophys. Res. Commun. 353, 443–449. [DOI] [PubMed] [Google Scholar]
- Velez-Vega C.; Escobedo F. A. (2011) Characterizing the Structural Behavior of Selected A beta-42 Monomers with Different Solubilities. J. Phys. Chem. B 115, 4900–4910. [DOI] [PubMed] [Google Scholar]
- Burdick D.; Soreghan B.; Kwon M.; Kosmoski J.; Knauer M.; Henschen A.; Yates J.; Cotman C.; Glabe C. (1992) Assembly and aggregation properties of synthetic Alzheimer’s A4/beta amyloid peptide analogs. J. Biol. Chem. 267, 546–554. [PubMed] [Google Scholar]
- Zagorski M. G.; Barrow C. J. (1992) NMR studies of amyloid β-peptides: proton assignments, secondary structure, and mechanism of an α-helix → β-sheet conversion for a homologous, 28-residue, N-terminal fragment. Biochemistry 31, 5621–5631. [DOI] [PubMed] [Google Scholar]
- Yoshiike Y.; Akagi T.; Takashima A. (2007) Surface structure of amyloid-beta fibrils contributes to cytotoxicity. Biochemistry 46, 9805–9812. [DOI] [PubMed] [Google Scholar]
- Hebbar S.; Lee E.; Manna M.; Steinert S.; Kumar G. S.; Wenk M.; Wohland T.; Kraut R. (2008) A fluorescent sphingolipid binding domain peptide probe interacts with sphingolipids and cholesterol-dependent raft domains. J. Lipid Res. 49, 1077–1089. [DOI] [PubMed] [Google Scholar]
- Frenkel D.; Balass M.; Katchalski-Katzir E.; Solomon B. (1999) High affinity binding of monoclonal antibodies to the sequential epitope EFRH of beta-amyloid peptide is essential for modulation of fibrillar aggregation. J. Neuroimmunol. 95, 136–142. [DOI] [PubMed] [Google Scholar]
- Frenkel D.; Balass M.; Solomon B. (1998) N-terminal EFRH sequence of Alzheimer’s beta-amyloid peptide represents the epitope of its anti-aggregating antibodies. Journal of Neuroimmunology 88, 85–90. [DOI] [PubMed] [Google Scholar]
- Solomon B. (2003) Immunological approach for the treatment of Alzheimer’s disease. J. Mol. Neurosci. 20, 283–286. [DOI] [PubMed] [Google Scholar]
- Baumketner A.; Shea J. E. (2007) The structure of the Alzheimer amyloid beta 10–35 peptide probed through replica-exchange molecular dynamics simulations in explicit solvent. J. Mol. Biol. 366, 275–285. [DOI] [PubMed] [Google Scholar]
- Reddy G.; Straub J. E.; Thirumalai D. (2009) Influence of Preformed Asp23-Lys28 Salt Bridge on the Conformational Fluctuations of Monomers and Dimers of A beta Peptides with Implications for Rates of Fibril Formation. J. Phys. Chem. B 113, 1162–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarus B.; Straub J. E.; Thirumalai D. (2006) Dynamics of Asp23-Lys28 salt-bridge formation in A beta(10–35) monomers. J. Am. Chem. Soc. 128, 16159–16168. [DOI] [PubMed] [Google Scholar]
- Dupuis N. F.; Wu C.; Shea J.-E.; Bowers M. T. (2009) Human Islet Amyloid Polypeptide Monomers Form Ordered β-hairpins: A Possible Direct Amyloidogenic Precursor. J. Am. Chem. Soc. 131, 18283–18292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise-Scira O.; Xu L.; Perry G.; Coskuner O. (2012) Structures and free energy landscapes of aqueous zinc(II)-bound amyloid-beta(1–40) and zinc(II)-bound amyloid-beta(1–42) with dynamics. J. Biol. Inorg. Chem. 17, 927–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massi F.; Peng J. W.; Lee J. P.; Straub J. E. (2001) Simulation study of the structure and dynamics of the Alzheimer’s amyloid peptide congener in solution. Biophys. J. 80, 31–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergeron D. E.; Coskuner O.; Hudgens J. W.; Gonzalez C. A. (2008) Ligand exchange reactions in the formation of diphosphine-protected gold clusters. J. Phys. Chem. C 112, 12808–12814. [Google Scholar]
- Coskuner O. (2010) Single ion and dimerization studies of the Al(III) ion in aqueous solution. J. Phys. Chem. A 114, 10981–10987. [DOI] [PubMed] [Google Scholar]
- Coskuner O.; Jarvis E. A. A.; Allison T. C. (2007) Water dissociation in the presence of metal ions. Angew. Chem., Int.Ed. 46, 7853–7855. [DOI] [PubMed] [Google Scholar]
- Onufriev A.; Bashford D.; Case D. A. (2004) Exploring protein native states and large-scale conformational changes with a modified generalized born model. Proteins 55, 383–394. [DOI] [PubMed] [Google Scholar]
- Prakash M. K.; Barducci A.; Parrinello M. (2011) Replica Temperatures for Uniform Exchange and Efficient Roundtrip Times in Explicit Solvent Parallel Tempering Simulations. J. Chem. Theory Comput. 7, 2025–2027. [DOI] [PubMed] [Google Scholar]
- Lazo N. D.; Grant M.; Condron M. C.; Rigby A. C.; Teplow D. B. (2005) On the nucleation of amyloid β-protein monomer folding. Protein Sci. 14, 1581–1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case D. A., Darden T. A., Cheatham T. E., Simmerling C. L.; Wang J., Duke R. E., Luo R., Crowley M., Walker R. C., Zhang W., Merz K. M., Wang B., Hayik S., Roitberg A., Seabra G., Kolossvary L., Wong K. F., Paesani F., Vanicek J., Wu X., Brozell S. R., Steinbrecher T., Gohlke H., Yang L., Tan C., Mongon J., Hornak V., Gui C., Mathews D. H., Seetin M. J., Sagui C., Babin V., and Kollman P. A. (2008) Amber 10, University of California, San Francisco, CA.
- Hornak V.; Abel R.; Okur A.; Strockbine B.; Roitberg A.; Simmerling C. (2006) Comparison of multiple amber force fields and development of improved protein backbone parameters. Proteins 65, 712–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen M. P., and Tildesley D. J. (1999) Computer Simulation of Liquids, Clarendon Press, Oxford, U.K. [Google Scholar]
- Frenkel D., and Smit B. (2002) Understanding Molecular Simulation: From Algorithms to Applications, Vol. 4, Academic Press, San Diego, CA. [Google Scholar]
- van der Spoel D.; Patriksson A. (2008) A temperature predictor for parallel tempering simulations. Phys. Chem. Chem. Phys. 10, 2073–2077. [DOI] [PubMed] [Google Scholar]
- Kollman P. A.; Massova I.; Reyes C.; Kuhn B.; Huo S. H.; Chong L.; Lee M.; Lee T.; Duan Y.; Wang W.; Donini O.; Cieplak P.; Srinivasan J.; Case D. A.; Cheatham T. E. (2000) Calculating structures and free energies of complex molecules: Combining molecular mechanics and continuum models. Acc. Chem. Res. 33, 889–897. [DOI] [PubMed] [Google Scholar]
- Lee M. R.; Duan Y.; Kollman P. A. (2000) Use of MM-PB/SA in estimating the free energies of proteins: Application to native, intermediates, and unfolded villin headpiece. Proteins: Struct., Funct., Genet. 39, 309–316. [PubMed] [Google Scholar]
- Schlitter J. (1993) Estimation of Absolute and Relative Entropies of Macromolecules Using the Covariance-Matrix. Chem. Phys. Lett. 215, 617–621. [Google Scholar]
- Case D. A. (1994) Normal-Mode Analysis of Protein Dynamics. Curr. Opin. Struct. Biol. 4, 285–290. [Google Scholar]
- Kabsch W.; Sander C. (1983) Dictionary of Protein Secondary Structure - Pattern-Recognition of Hydrogen-Bonded and Geometrical Features. Biopolymers 22, 2577–2637. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.