

Abstract
Amyloid oligomers represent the primary pathological species for neurodegenerative diseases such as Alzheimer’s and Parkinson’s diseases. Toxic oligomers are formed by many different proteins and peptides, but their polydispersity makes them highly dynamic and heterogeneous. One way to stabilize these structures is to prepare constrained peptides that can be used to study amyloid intermediates, to identify oligomer-specific drugs, and to generate conformational antibodies. These conformational antibodies have demonstrated that oligomers share a common epitope. In this research, we used a 40-amino acid unstructured segment of prion protein (Prp) 109–148 with substitutions of methionine for glycine (Prp-G) residues to prepare a stable and homogeneous population of β-sheet oligomer mimics. These structures were characterized by multiple biophysical and biochemical techniques that show characteristic features of oligomers. Finally, this preparation was not detected by three different sequence dependent prion antibodies.
Keywords: Amyloid oligomer, constrained peptide, prion
Of the protein misfolding pathologies, amyloid diseases are the most clinically relevant due to their high prevalence in the population, the most notable being Alzheimer’s and Parkinson’s diseases. For many years, it was assumed that fibrillar Aβ amyloid plaques were responsible for most of the neurodegenerative changes in AD.1 However, neuronal loss correlates poorly with the distribution and amount of Aβ in the form of plaques.2−4 In addition, some transgenic animals display cognitive deficits prior to the onset of Aβ plaque accumulation.5,6 Dementia correlates better with soluble Aβ levels than with insoluble, fibrillar deposits,7,8 suggesting that oligomeric forms of Aβ may represent the primary toxic species in AD. Indeed, soluble oligomers have been implicated as primary causative agents in many different degenerative diseases where the accumulation of large fibrillar deposits may be either inert or protective.9
A key issue in the investigation of amyloid structures is the description of both the growth mechanism from monomeric precursors and the structural features of toxic oligomers. However, the intrinsically disordered nature of these assemblies makes it very difficult to get solid data on their structural features. Our approach achieved stabilization of these structures by preparing conformationally constrained peptides and using them to generate different epitopes that are common to β-sheet oligomer conformation. Antioligomer antibodies have provided a more rational means of classifying these structures based on their underlying structural organization rather than on differences in size or sample preparation.10−13 However, it is well described that these two peptides share many features,14,15 and perhaps the generation of amyloid peptides with higher sequence variability can generate antibodies that recognize novel epitopes common to amyloid oligomers.
Results and Discussion
We used a peptide that includes 109–148 amino acids from human prion protein (hPrp) and substituted M for G residues. These substitutions were made to avoid disulfide bonds in oligomer mimic preparations. This mutant peptide was called Prp-G. Then we used CSSP2 to predict amyloidogenic regions that are indicated in boxes. This software predicted the loss of the GSAMS amyloid region in mutant peptide16 (Figure 1) . Then, we covalently coupled this peptide to colloidal gold nanoparticles with an average diameter of 5.3 nm. Coupling was done via carboxy terminal thiol according to the protocol described to prepare mimic.12
Figure 1.

Alignment of hPrp wild-type and mutant peptide made with ClustalW.17 Amino acid characteristics are designated by color, with acidic in blue, basic in purple, small and hydrophobic in red, and hydroxyl, histidine, and glycine in green. Asterisks indicate positions which have a single, fully conserved residue. Regions indicated in boxes are predicted as stretches more prone to aggregate by CSSP2 software.
In order to characterize the conformation of Prp mimic, we measured the fluorescence emitted by Thioflavin T (ThT) and Bis-ANS using Aβ40 fibrils and oligomers as controls in both assays (Figure 2a and b, respectively). ThT fluorescence assay has been widely used to characterize the kinetics of fibril formation. This probe exhibits an increase in fluorescence intensity at 490 nm when bound to protofibrils and fibrils.18 Prp mimic showed low intensity values of fluorescence similarly to Aβ40 oligomers, while there was an increase for Aβ40 fibrils. This supports the previous observation that pure amyloid oligomers, though β-sheet rich, have lower affinity to ThT compared to amyloid fibrils.12 Bis-ANS is a probe that exhibits low fluorescence in aqueous solutions and high fluorescence with proteins containing exposed hydrophobic patches19 mimic.12 The mimic in the presence of this probe showed intensity values of fluorescence similar to Aβ40 oligomers and low fluorescence was detected with fibrils.
Figure 2.
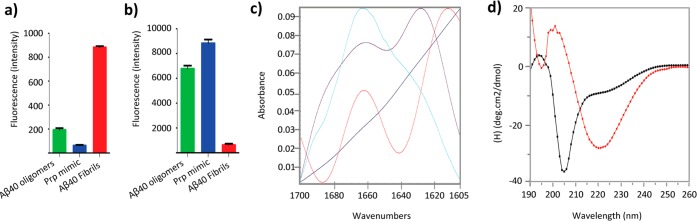
(a) Thioflavin T and (b) Bis-ANS assay for oligomers and fibrils of Aβ40 and Prp mimic. Fluorescence intensity values in the graphs were obtained by the subtraction of their respective blanks. (c) FT-IR absorption spectra in the amide I region of Aβ40 monomer (aqua), oligomer (purple), Prp mimic (red), and gold nanoparticles (blue). (d) CD spectra of Prp-G without gold nanoparticles (black) and Prp mimic (red).
Another feature of oligomers is that they exhibit β-sheet conformation. To determine the presence of β-sheet conformation in this mimic, we used Fourier transform Infrared (FT-IR) and circular dichroism (CD) spectroscopy. FT-IR spectra showed two components at 1612 and 1662 cm–1 as can be seen in Figure 2c. These bands can be assigned to β-sheet secondary structure and turns because they are in the region of 1630–1610 cm–1 and 1660–1670 cm–1, respectively.20 We compared these results to Aβ40 monomer and oligomers, and we could see a band at 1630 cm–1 indicating β-sheet structure and another at 1662 cm–1 indicating turn content, while monomer only displayed a band at 1662 cm–1, corresponding to turn content only. CD spectra of denatured Prp-G showed a negative peak at 205 nm, suggesting a random coil conformation, while Prp mimic showed a negative peak at 221 nm, suggesting β-sheet-rich structure (Figure 2d). It has been shown that ThT fluorescence and β-sheet content analyzed by CD spectra increase during the Aβ aggregation process, showing their maxima when fibrils are formed.21 Indeed, in this instance, Prp mimics showed reduced fluorescence with ThT and β-sheet secondary structure evaluated by FT-IR and CD, when compared to fibrils. Then, we determined particle homogeneity by size exclusion chromatography (SEC), atomic force microscopy (AFM), and dynamic light scattering (DLS). When the same sample was analyzed by SEC, we detected that 76.2% of the species had a molecular weight (MW) of 69.7 kDa and 11.8% had 184.5 kDa and no Prp-G monomer was detected at 3.8 kDa (Figure 3a). These results show a more heterogeneous preparation. Perhaps this was caused by the long run-time inherent to SEC analysis, as increased run-time allows for a higher degree of dissociation, aggregation, and equilibrium shift down the multistep kinetic pathway.22
Figure 3.
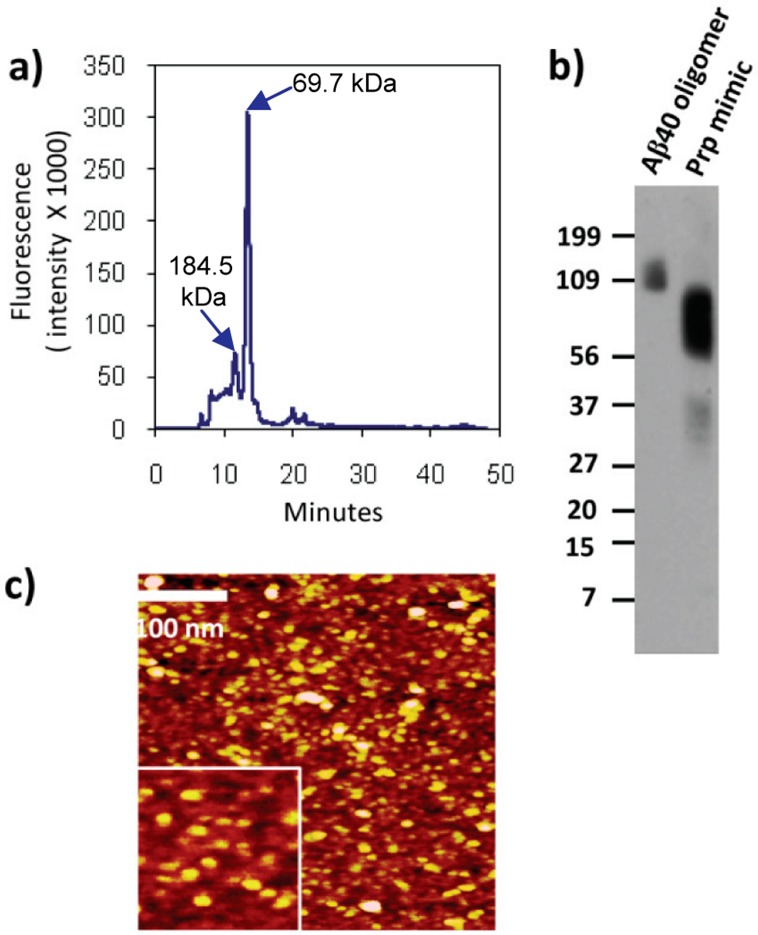
Homogeneity of Prp mimics was evaluated by (a) size exclusion chromatography. Detection of Prp-mimic was performed by fluorescence using 280 and 350 nm corresponding to excitation and emission wavelengths, respectively. (b) Western blot with A11 and (c) AFM of Prp mimic.
Then, we probed Prp mimic against A11 antioligomer antibody by Western blot and found positive signal for SDS resistant species ranging from 109 to 160 kDa for Aβ40 oligomers while Prp 109–148 mimic showed positive signal for species ranging from 31 to 40 kDa and 56 to 100 kDa (Figure 3b). Atomic force microscopy (AFM) clearly distinguishes the morphologies of different species on the aggregation pathway.23 AFM image analysis revealed that 90.82% of this preparation had an average diameter of 10.691 ± 3.63 nm and those remaining had an average diameter of 26 ± 5.6 nm (Figure 3c), while DLS showed that 96.8% mass of the particles had a radius of 5.81 ± 0.96 nm while the remaining had 22.42 ± 6.11 nm. If we model oligomers as spherical particles, AFM and DLS show that most of the particles have similar average sizes.
Finally, we tested the accessibility of Prp mimic to sequence dependent antibodies by dot blot analysis using 3F4, Pri308, and F89/160.1.5 antibodies that recognize the 109–114, 115–135, and 139–142 regions, respectively (Figure 4a). This preparation was not detected by any sequence-dependent antibody used in this test (Figure 4b). Then we probed Prp mimic with antioligomer antibody A11 and antifibrillar antibody OC using Aβ monomer, oligomer, and fibrils as controls (Figure 4c). Prp mimic showed positive signal only with A11 antibody, indicating that this mimic displays oligomer conformation with no contamination of monomers or fibrils;10,12,13 moreover, mimic did not aggregate further to form fibrils even after 2 months at 4 °C. After conformational transition of Prpc to the PrPSc state, region 111–135 becomes inaccessible to antibodies.24 Consistent with this view, this region has an intrinsic tendency to adopt β-sheet-rich structure reminiscent of PrPSc.25−27 Conversely, PrP molecules that are deleted in the hydrophobic region have greater conformational stability than WT (wild-type) PrP28 and are refractory to PrPSc-induced conversion.29,30 From the antibodies used in this research, only Pri 308 has been reported to block Prp oligomer toxicity, (31) but in our case this antibody could not detect Prp mimics. One common problem with amyloid oligomers is that they are unstable and disappear as mature fibrils and amorphous aggregates in solution; our results clearly show a preparation that represents a stable oligomeric transitional aggregation state that is not recognized by any sequence dependent antibody. This method can be used for the preparation of mimic from peptides and protein fragments, which is invaluable for studying aggregation pathways, and the screening for antioligomer small molecules. Moreover, stable mimic can be used as antigen to generate and screen polyclonal and monoclonal antibodies and to identify toxic conformations displayed by diverse sequences. In conclusion, this preparation showed the interaction between Prp-G peptides because any sequence-specific antibody was able to detect epitopes in this region and showed an aggregation state with high hydrophobicity and β sheet conformation that amyloid oligomers showed too.
Figure 4.

(a) Different peptide stretches recognized by Prp sequence antibodies. (b) Prp-G without gold nanoparticles and Prp mimic probed with different anti-Prp sequence antibodies. It is clear that the conformationally constrained mimics are not recognized by the sequence antibodies, and (c) only antioligomer specific antibody recognized the mimic by dot blot.
Acknowledgments
We are grateful to Dr. Luis Holthauzen the director of Solution Biophysics Laboratory for valuable assistance, and Drs. George Jackson and Yogesh Wairkar for their useful suggestions.
Supporting Information Available
Experimental details. This material is available free of charge via the Internet at http://pubs.acs.org.
This project was supported by the Mitchell Center for Neurodegenerative Diseases and the Cullen Family Trust for Health Care.
The authors declare the following competing financial interest(s): Rakez Kayed has patent applications on the compositions and methods related to amyloid oligomers and antibodies. This did not play a role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Supplementary Material
References
- Terry R. D. (1996) The pathogenesis of Alzheimer disease: an alternative to the amyloid hypothesis. J. Neuropathol. Exp. Neurol. 55, 1023–1025. [PubMed] [Google Scholar]
- Crystal H.; Dickson D.; Fuld P.; Masur D.; Scott R.; Mehler M.; Masdeu J.; Kawas C.; Aronson M.; Wolfson L. (1988) Clinico-pathologic studies in dementia: nondemented subjects with pathologically confirmed Alzheimer’s disease. Neurology 38, 1682–1687. [DOI] [PubMed] [Google Scholar]
- Davies L.; Wolska B.; Hilbich C.; Multhaup G.; Martins R.; Simms G.; Beyreuther K.; Masters C. L. (1988) A4 amyloid protein deposition and the diagnosis of Alzheimer’s disease: prevalence in aged brains determined by immunocytochemistry compared with conventional neuropathologic techniques. Neurology 38, 1688–1693. [DOI] [PubMed] [Google Scholar]
- Price J. L.; Morris J. C. (1999) Tangles and plaques in nondemented aging and ″preclinical″ Alzheimer’s disease. Ann. Neurol. 45, 358–368. [DOI] [PubMed] [Google Scholar]
- Billings L. M.; Oddo S.; Green K. N.; McGaugh J. L.; Laferla F. M. (2005) Intraneuronal Abeta causes the onset of early Alzheimer’s disease-related cognitive deficits in transgenic mice. Neuron 45, 675–688. [DOI] [PubMed] [Google Scholar]
- Westerman M. A.; Cooper-Blacketer D.; Mariash A.; Kotilinek L.; Kawarabayashi T.; Younkin L. H.; Carlson G. A.; Younkin S. G.; Ashe K. H. (2002) The relationship between Abeta and memory in the Tg2576 mouse model of Alzheimer’s disease. J. Neurosci. 22, 1858–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean C. A.; Cherny R. A.; Fraser F. W.; Fuller S. J.; Smith M. J.; Beyreuther K.; Bush A. I.; Masters C. L. (1999) Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer’s disease. Ann. Neurol. 46, 860–866. [DOI] [PubMed] [Google Scholar]
- Lue L. F.; Kuo Y. M.; Roher A. E.; Brachova L.; Shen Y.; Sue L.; Beach T.; Kurth J. H.; Rydel R. E.; Rogers J. (1999) Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer’s disease. Am. J. Pathol. 155, 853–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haass C.; Selkoe D. J. (2007) Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nat. Rev. Mol. Cell Biol. 8, 101–112. [DOI] [PubMed] [Google Scholar]
- Kayed R.; Head E.; Sarsoza F.; Saing T.; Cotman C. W.; Necula M.; Margol L.; Wu J.; Breydo L.; Thompson J. L.; Rasool S.; Gurlo T.; Butler P.; Glabe C. G. (2007) Fibril specific, conformation dependent antibodies recognize a generic epitope common to amyloid fibrils and fibrillar oligomers that is absent in prefibrillar oligomers. Mol. Neurodegener. 2, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayed R.; Pensalfini A.; Margol L.; Sokolov Y.; Sarsoza F.; Head E.; Hall J.; Glabe C. (2009) Annular protofibrils are a structurally and functionally distinct type of amyloid oligomer. J. Biol. Chem. 284, 4230–4237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayed R.; Head E.; Thompson J. L.; McIntire T. M.; Milton S. C.; Cotman C. W.; Glabe C. G. (2003) Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 300, 486–489. [DOI] [PubMed] [Google Scholar]
- Wu J. W.; Breydo L.; Isas J. M.; Lee J.; Kuznetsov Y. G.; Langen R.; Glabe C. (2010) Fibrillar Oligomers Nucleate the Oligomerization of Monomeric Amyloid {beta} but Do Not Seed Fibril Formation. J. Biol. Chem. 285, 6071–6079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butterfield S. M.; Lashuel H. A. (2010) Amyloidogenic protein-membrane interactions: mechanistic insight from model systems. Angew. Chem., Int. Ed. Engl. 49, 5628–5654. [DOI] [PubMed] [Google Scholar]
- Andreetto E.; Yan L. M.; Tatarek-Nossol M.; Velkova A.; Frank R.; Kapurniotu A. (2010) Identification of hot regions of the Aβ-IAPP interaction interface as high-affinity binding sites in both cross- and self-association. Angew. Chem., Int. Ed. Engl. 49, 3081–3085. [DOI] [PubMed] [Google Scholar]
- Yoon S.; Welsh W. J.; Jung H.; Yoo Y. D. (2007) CSSP2: an improved method for predicting contact-dependent secondary structure propensity. Comput. Biol. Chem. 31, 373–377. [DOI] [PubMed] [Google Scholar]
- Larkin M. A.; Blackshields G.; Brown N. P.; Chenna R.; McGettigan P. A.; McWilliam H.; Valentin F.; Wallace I. M.; Wilm A.; Lopez R.; Thompson J. D.; Gibson T. J.; Higgins D. G. (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948. [DOI] [PubMed] [Google Scholar]
- Groenning M. (2010) Binding mode of Thioflavin T and other molecular probes in the context of amyloid fibrils-current status. J. Chem. Biol. 3, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawe A.; Sutter M.; Jiskoot W. (2008) Extrinsic fluorescent dyes as tools for protein characterization. Pharm. Res. 25, 1487–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson M.; Mantsch H. H. (1995) The use and misuse of FTIR spectroscopy in the determination of protein structure. Crit. Rev. Biochem. Mol. Biol. 30, 95–120. [DOI] [PubMed] [Google Scholar]
- Prangkio P.; Yusko E. C.; Sept D.; Yang J.; Mayer M. (2012) Multivariate analyses of amyloid-beta oligomer populations indicate a connection between pore formation and cytotoxicity. PLoS One 7, e47261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philo J. S. (2006) Is any measurement method optimal for all aggregate sizes and types?. AAPS J. 8, E564–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper J. D.; Wong S. S.; Lieber C. M.; Lansbury P. T. Jr. (1999) Assembly of A beta amyloid protofibrils: an in vitro model for a possible early event in Alzheimer’s disease. Biochemistry 38, 8972–8980. [DOI] [PubMed] [Google Scholar]
- Peretz D.; Williamson R. A.; Matsunaga Y.; Serban H.; Pinilla C.; Bastidas R. B.; Rozenshteyn R.; James T. L.; Houghten R. A.; Cohen F. E.; Prusiner S. B.; Burton D. R. (1997) A conformational transition at the N terminus of the prion protein features in formation of the scrapie isoform. J. Mol. Biol. 273, 614–622. [DOI] [PubMed] [Google Scholar]
- Gasset M.; Baldwin M. A.; Lloyd D. H.; Gabriel J. M.; Holtzman D. M.; Cohen F.; Fletterick R.; Prusiner S. B. (1992) Predicted alpha-helical regions of the prion protein when synthesized as peptides form amyloid. Proc. Natl. Acad. Sci. U.S.A. 89, 10940–10944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagliavini F.; Prelli F.; Verga L.; Giaccone G.; Sarma R.; Gorevic P.; Ghetti B.; Passerini F.; Ghibaudi E.; Forloni G.; et al. (1993) Synthetic peptides homologous to prion protein residues 106–147 form amyloid-like fibrils in vitro. Proc. Natl. Acad. Sci. U.S.A. 90, 9678–9682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmona M.; Morbin M.; Massignan T.; Colombo L.; Mazzoleni G.; Capobianco R.; Diomede L.; Thaler F.; Mollica L.; Musco G.; Kourie J. J.; Bugiani O.; Sharma D.; Inouye H.; Kirschner D. A.; Forloni G.; Tagliavini F. (2003) Structural properties of Gerstmann-Straussler-Scheinker disease amyloid protein. J. Biol. Chem. 278, 48146–48153. [DOI] [PubMed] [Google Scholar]
- Thaa B.; Zahn R.; Matthey U.; Kroneck P. M.; Burkle A.; Fritz G. (2008) The deletion of amino acids 114–121 in the TM1 domain of mouse prion protein stabilizes its conformation but does not affect the overall structure. Biochim. Biophys. Acta 1783, 1076–1084. [DOI] [PubMed] [Google Scholar]
- Holscher C.; Delius H.; Burkle A. (1998) Overexpression of nonconvertible PrPc delta114–121 in scrapie-infected mouse neuroblastoma cells leads to trans-dominant inhibition of wild-type PrP(Sc) accumulation. J. Virol. 72, 1153–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norstrom E. M.; Mastrianni J. A. (2005) The AGAAAAGA palindrome in PrP is required to generate a productive PrPSc-PrPC complex that leads to prion propagation. J. Biol. Chem. 280, 27236–27243. [DOI] [PubMed] [Google Scholar]
- Simoneau S.; Rezaei H.; Sales N.; Kaiser-Schulz G.; Lefebvre-Roque M.; Vidal C.; Fournier J. G.; Comte J.; Wopfner F.; Grosclaude J.; Schatzl H.; Lasmezas C. I. (2007) In vitro and in vivo neurotoxicity of prion protein oligomers. PLoS Pathog. 3, e125. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.