

Abstract
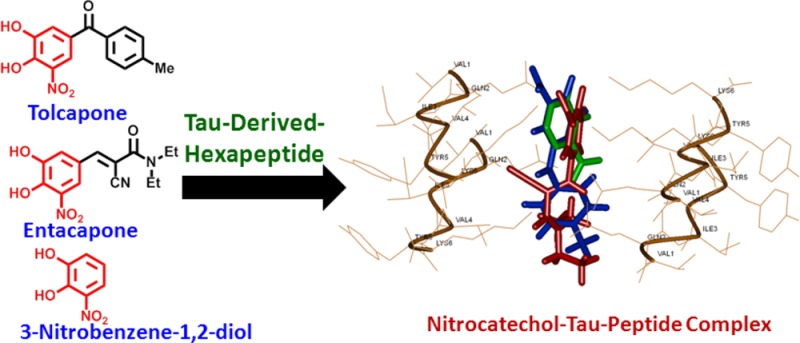
The nitrocatechol derivatives tolcapone (1) and entacapone (2), used as adjunctive therapy in the treatment of Parkinson’s disease, were investigated for their potential to inhibit the tau-derived-hexapeptide 306VQIVYK311. They were compared to small molecules that contain similar pharmacophores including the catechol derivatives (dopamine 3 and epinephrine 4), nitroderivatives (nifedipine 5 and chloramphenicol 6), nitrocatechol isomers (7 and 8), and a tolcapone derivative (13) lacking the nitrocatechol moiety. The aggregation kinetics by thioflavin S fluorescence assay indicates that both tolcapone (1) and entacapone (2) exhibit antiaggregation properties. These findings were supported by transmission electron microscopy (TEM) and circular dichroism (CD) spectroscopy measurements which suggest that the nitrocatechol (3,4-dihydroxy-5-nitrophenyl) moiety is a suitable pharmacophore in the design of new tau-aggregation inhibitors. Furthermore, tolcapone (1) was identified as most active compound with antiaggregation activity (46% inhibition of fluorescence intensity at 50 μM), which was supported by TEM data. The in silico steric zipper model of the tau-derived-hexapeptide 306VQIVYK311 indicates that the 3,4-dihydroxy-substituent present in tolcapone (1) and entacapone (2) underwent polar contacts with lysine side chains (VQIVYK), whereas the charged 5-nitrosubstituent was in close contact with lysine side chain present in the steric zipper region suggesting the critical role of a nitrocatechol (3,4-dihydroxy-5-nitrophenyl) pharmacophore present in tolcapone (1) and entacapone (2) in tau-hexapeptide binding and prevention of β-sheet assembly. Our results have significant implications in the design and development of tau-aggregation inhibitors.
Keywords: Alzheimer’s disease, tau aggregation, tau-derived-hexapeptide, nitrocatechols, tolcapone, entacapone
The mechanisms underlying protein misfolding and aggregation are of significant interest considering their role in the pathophysiology of numerous amyloid-based diseases.1−4 Neurodegenerative disorders such as Alzheimer’s disease (AD) and Parkinson’s disease (PD) are characterized by the accumulation of protein aggregates including amyloid-β (Aβ), tau (τ), and alpha-synuclein (α-syn) respectively.1,2,5−7 These amyloid deposits (plaques, neurofibrillary tangles or NFTs and Lewy bodies) form as a result of misfolding proteins and the progressive maturation of the soluble monomeric peptides into larger and insoluble cross-β-sheet assemblies, suggestive of shared aggregation dynamics.7−11 While these extra/intracellular proteinaceous deposits are mainly considered an end-result of the aggregation pathway, they do play a significant role in neurotoxicity, disease pathology, and diagnosis. These rolling toxicity cascades impair cellular metabolism, gene regulation, and, consequently, widespread neuronal cell death that leads to dementia.12−17
When examining the tauopathy disease mechanism in AD, it has been established that the downstream consequences of hyper-phosphorylated tau (hp-tau) lead to its dissociation from neuronal microtubule assemblies. This dissociation impacts the integrity of the neuroskeletal structure, and the hp-tau monomers initiate the rapid aggregation cascade to form intracellular NFTs resulting in metabolic impairment and cellular toxicity.18,19 Recently, tauopathy has gained significant research interest, as multiple trials on targeting Aβ alone as a potential therapeutic target seem to fail at the clinical level.20 Tau proteins are known to form aggregates known as paired helical filaments (PHFs) which can further grow to form NFTs. The molecular mechanisms of tau-protein aggregation and misfolding have been investigated by studying critical fragments of the protein sequence. One such sequence is the hexapeptide 306VQIVYK311 segment from the microtubule binding region of tau protein, which is known to promote nucleation dependent tau-aggregation. In this regard, the hexapeptide Ac-VQIVYK-NH2 (AcPHF6) is considered as a model peptide to investigate tau-aggregation. It is a well characterized hexapeptide that forms β-sheets and undergoes fibrillation efficiently in vitro to form cross-β-sheet structure similar to Aβ. This short peptide serves as a suitable model to study small molecule inhibitors of tau-aggregation.13,14,21−25
From a therapeutics research perspective, a number of drug classes have been discovered and proposed as suitable inhibitors of tau aggregation.26,27 A recent study showed that the small molecules, tolcapone (1) and entacapone (2) shown in Figure 1, exhibit antiaggregation properties against Aβ and α-synuclein.28 It should be noted that both tolcapone and entacapone are known to act as catechol-O-methyltransferase (COMT) inhibitors and are currently FDA approved as adjunctive therapies in PD.29 Both of these molecules contain a nitrocatechol (3,4-dihydroxy-5-nitrophenyl) pharmacophore. In our study, we investigated the ability of nitrocatechol, aromatic-nitro, and catechol containing small molecules such as tolcapone (1), entacapone (2), dopamine (3), epinephrine (4), chloramphenicol (5), nifedipine (6), and other small molecules 7–13 (Figure 1) to prevent the tau hexapeptide Ac-VQIVYK-NH2 (AcPHF6) aggregation by in vitro kinetics using thioflavin S (ThS) based fluorescence measurements, transmission electron microscopy (TEM), circular dichroism (CD) spectroscopy, and molecular modeling investigations. These studies demonstrate for the first time that the nitrocatechol moiety can be a useful pharmacophore in the development of tau-aggregation inhibitors.
Figure 1.

Chemical structures of small molecules used (1–13).
Results and Discussion
Test Compound Selection
In 2010, Lashuel and co-workers reported that two catechol-O-methyltransferase (COMT) inhibitors, tolcapone (Tasmar) and entacapone (Comtan), demonstrated promising activities against Aβ and α-syn fibril formation while eliciting neuroprotective effects against amyloid-induced toxicity.28 Either of these nitrocatechol-containing COMT inhibitors is typically prescribed in conjunction with levodopa as a primary treatment for PD patients. Interestingly, both of these drugs have a common feature which is the presence of a nitrocatechol pharmacophore. These findings prompted us to consider nitrocatechols as potential tau-aggregation inhibition pharmacophores in a tau-protein-derived hexapeptide AcPHF6 model. Test compound selection (Figure 1) was based on chemical and bioisosteric similarity to acquire structure–activity relationship (SAR) data. The nitrocatechol derivatives tolcapone (1) and entacapone (2); the catechol neurotransmitters dopamine (3) and epinephrine (4); marketed therapies containing an aromatic-nitro group such as chloramphenicol (5) and nifedipine (6); and small molecule building blocks including nitrocatechol isomers (7 and 8), catechol (9), aromatic nitro derivatives (10–12), and methylbenzophenone (13), a derivative of tolcapone that lacks the nitrocatechol pharmacophore, were investigated (Figure 1). The results were compared with known aggregation inhibitors curcumin and orange G.
AcPHF6 Aggregation and Inhibition Studies by ThS Fluorescence
The antiaggregation potential of the test compounds were measured by a ThS based kinetic assay. Compounds were evaluated at 0.1, 1, 10, and 50 μM concentrations in triplicates (two independent experiments) in the presence of AcPHF6 (100 μM). Figure 2A shows the effect of reference compound curcumin on tau-hexapeptide aggregation at various concentrations. In the absence of curcumin, AcPHF6 aggregates rapidly as monitored by fluorescence over a period of 120 min (black curve, Figure 2A). In the presence of curcumin, a significant decrease in AcPHF6 aggregation was seen in a concentration dependent manner. At a low concentration of 0.1 μM, curcumin did not decrease the fluorescence intensity (green curve, Figure 2A). However, at increasing concentration, there was a gradual decline in fluorescence intensity, indicating slow aggregation and inhibition. At the highest tested concentration (50 μM), curcumin displayed significant suppression of aggregation of AcPHF6 (orange curve, Figure 2A). Furthermore, in a recent study, Nowick and co-workers calculated the time at midpoint of fluorescence relative to the ThS control (indicated by tm value) as a useful indicator to monitor the time needed for the aggregation of AcPHF6 in the presence and absence of inhibitors.25 In this regard, our study shows that tm in the presence of 50 μM curcumin was about 23 min (23.0 ± 1.2 min) whereas tm was around 14 min (14.3 ± 1.2 min) in the absence of curcumin. This indicates the ability of curcumin to slow down AcPHF6 aggregation. Furthermore, it was able to delay aggregation process early on. A similar trend was observed for orange G where when incubated at 50 μM (tm = 19.5 ± 2.2 min), it was able to suppress and delay AcPHF6 aggregation similar to curcumin (Figure 2B). Furthermore, we investigated the effects of the nitrocatechols, tolcapone 1 and entacapone 2 (Figure 3). Increasing concentrations of tolcapone led to a decline in fluorescence intensity as a function of time (Figure 3A), suggesting the ability of tolcapone to decrease AcPHF6 aggregation (tm = 12.5 ± 1.0 min). Maximum drop was observed at a concentration of 50 μM, while monitoring ThS fluorescence at 120 min intervals. In comparison, entacapone also prevented AcPHF6 aggregation as monitored by ThS fluorescence which was directly proportional to its concentration (tm = 16.0 ± 0.6 min). Maximum reduction in fluorescence was observed at the highest concentration used (50 μM, 120 min interval, Figure 3B) reaching a plateau after 40 min. Interestingly, there appears to be a correlation between the lipophilicity parameters of tolcapone and entacapone and their ability to decrease ThS fluorescence intensity when monitored at 120 min. Tolcapone with an additional aromatic (phenyl) ring is much more lipophilic relative to entacapone (Table S1, Supporting Information) and is able to exhibit superior inhibition of AcPHF6 aggregation relative to entacapone (at 50 μM, 120 min interval).
Figure 2.
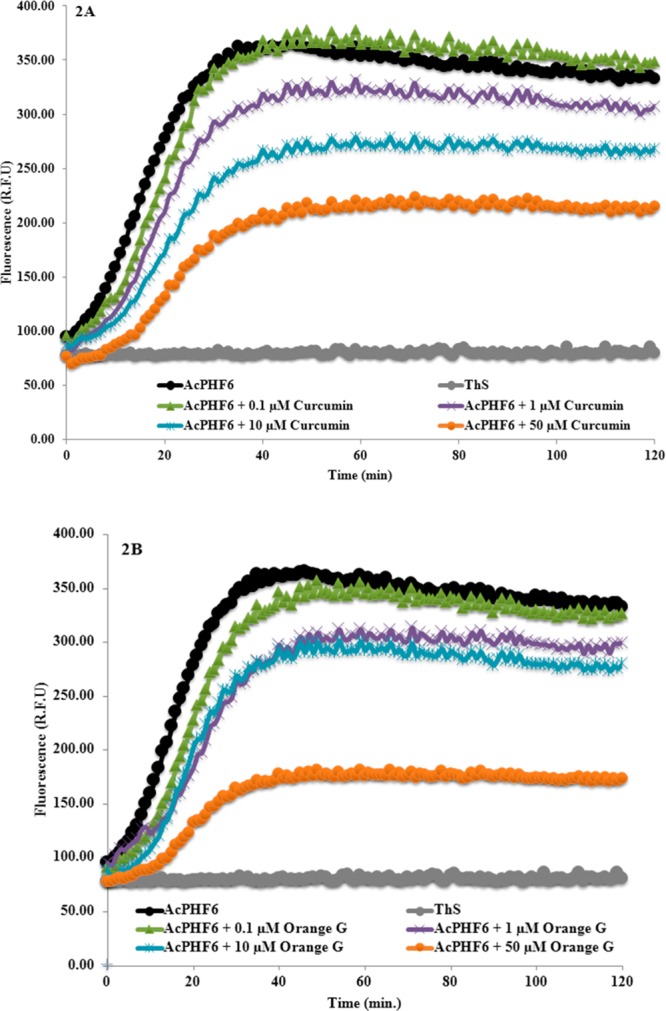
Effect of various concentrations of curcumin (A) and orange G (B) on AcPHF6 aggregation (in triplicates from two independent experiments).
Figure 3.
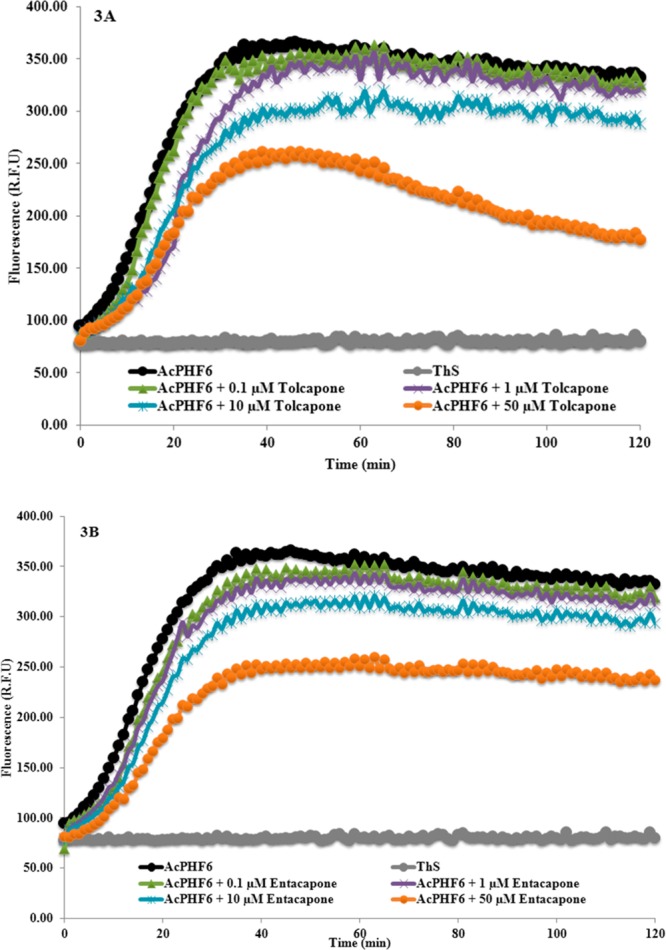
Effect of various concentrations of tolcapone 1 (A) and entacapone 2 (B) on AcPHF6 aggregation (in triplicates from two independent experiments).
In order to understand the role of the nitrocatechol moiety in preventing AcPHF6 aggregation, we investigated the ability of small building blocks such as the nitrocatechol isomers (7) and (8), and catechol (9, Figure 1). Strikingly, the nitrocatechol (7) exhibited a concentration dependent decline in the fluorescence intensity similar to the nitrocatechol containing agents, tolcapone and entacapone, with a tm of 16.0 ± 0.7 min as shown in Figure 4A. Maximum reduction was observed at the highest concentration tested (50 μM), whereas the nitrocatechol isomer (8) exhibited weaker inhibition of AcPHF6 aggregation (Figure S2A, Supporting Information) compared to nitrocatechol (7). Interestingly, the catechol (9) failed to exhibit any significant reduction in ThS fluorescence (Figure 4B). To further confirm the ability of the nitrocatechol moiety to prevent AcPHF6 aggregation, we screened catechol containing molecules (dopamine 3 and epinephrine 4), nitroaromatic molecules such as the antibacterial agent chloramphenicol (5), the calcium channel blocker nifedipine (6), and other nitroaromatics including nitrophenol (10), nitrobenzene (11), and nitroanthracene (12, Figure 1). Markedly, none of these small molecules were able to exhibit any significant inhibition of AcPHF6 aggregation (Figures S1–S4, Supporting Information). This further indicates that a nitrocatechol pharmacophore plays a major role in preventing AcPHF6 aggregation. Furthermore, a tolcapone derivative, methylbenzophenone (13), that lacks the critical nitrocatechol moiety failed to prevent AcPHF6 aggregation validating the critical nature of a nitrocatechol pharmacophore in preventing tau-hexapeptide aggregation (Figure S5, Supporting Information).
Figure 4.
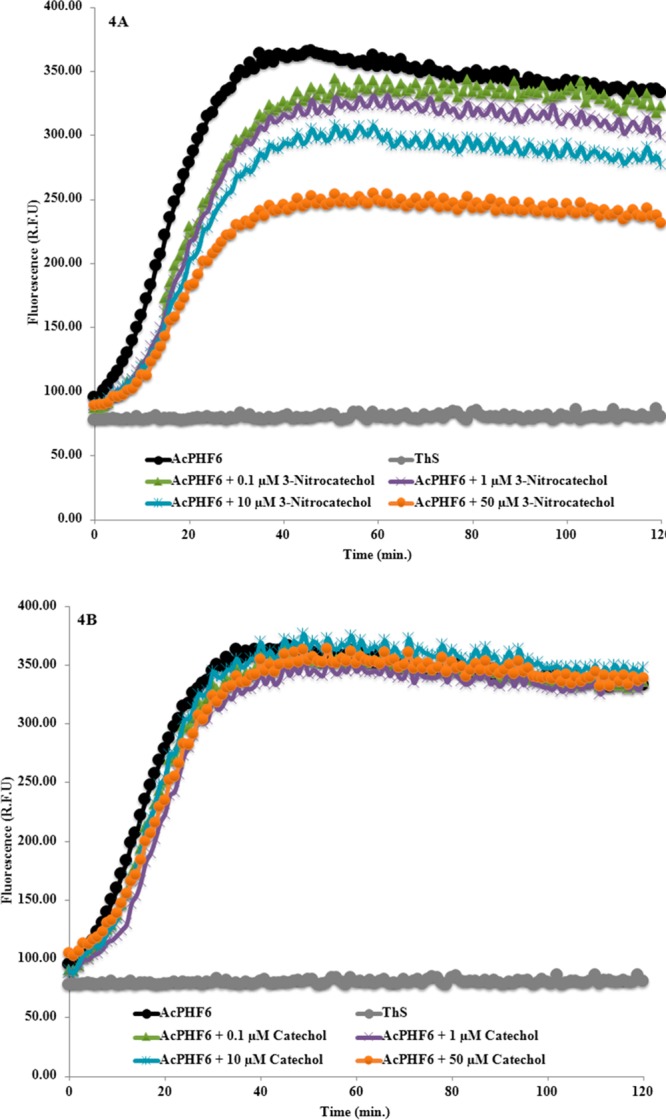
Effect of various concentrations of nitrocatechol isomer 7 (A) and catechol 9 (B) on AcPHF6 aggregation (in triplicates from two independent experiments).
A summary of the fluorescence intensity profiles at 120 min interval for various test compounds at 50 μM (curcumin, orange G, tolcapone 1, methylbenzophenone 13, entacapone 2, nitrocatechol 7, dopamine 3, and catechol 9) in the presence of AcPHF6 (100 μM) is given as a bar graph (Figure 5). The reduction in their fluorescence intensities, relative to AcPHF6 alone, was 46% (tolcapone), 29% (entacapone) and 30% (nitrocatechol), respectively, demonstrating their ability to prevent tau-hexapeptide aggregation. In contrast, dopamine (3), catechol (9), and the tolcapone derivative (13) were ineffective as indicated by almost no reduction in their fluorescence intensity (Figure 5). This observation can be attributed to the lack of a nitrocatechol pharmacophore. It should be noted that some reports have indicated that phenolic antioxidants can interfere with thioflavin-T fluorescence (ThT) while evaluating amyloid beta (Aβ)-42 fibril formation under basic conditions, suggesting that other methods such as TEM techniques should be used to confirm ThT fluorescence results.30 In order to assess this, we performed UV (400–700 nm) and fluorescence (excitation = 440 nm and emission = 490 nm) scans for the tested compounds. In addition, potential interference in fluorescence measurements due to the interaction between ThS and test compounds was also investigated. These studies indicated that, in our assay conditions, the test compounds do not exhibit significant absorption or fluorescence at the highest tested concentration (50 μM), ruling out their potential interference in the assay measurements (Figures S6–S8, Supporting Information). It should be noted that our assays were conducted at pH 7.2 whereas reported ThT based amyloid assays measure fluorescence at basic pH of 8.5.30 Basic pH is known to affect the degree of conjugation and wavelength of absorption of phenolic compounds. The highly conjugated reference agent, orange G, with two negatively charged sulfonate groups did exhibit absorption (UV λmax = 478 nm, Figure S6, Supporting Information) indicating its ability to interfere with the ThS assay. However, orange G is a known inhibitor of tau aggregation and its X-ray crystal structure with the tau-hexapeptide has been reported.21
Figure 5.

Effect of curcumin, orange G, tolcapone 1, methylbenzophenone 13, entacapone 2, nitrocatechol 7, dopamine 3, and catechol 9 on AcPHF6 aggregation at 120 min (in triplicates from two independent experiments).
The disaggregation potential of tolcapone (1), entacapone (2) and nitrocatechol isomers (7 and 8) was monitored over a period of 24 h (Figure S9, Supporting Information). Interestingly, these investigations demonstrate the ability of nitrocatechols 1, 2, 7, and 8 to cause disaggregation of tau-derived-hexapeptide. The disaggregation activity was linear and time dependent which further supports the antiaggregation properties of a nitrocatechol pharmacophore.
Transmission Electron Microscopy (TEM)
In order to confirm the role of the nitrocatechol pharmacophore in preventing AcPHF6 inhibition, TEM investigations were conducted to monitor the change in morphology of the tau-hexapeptide aggregates in the presence and absence of nitrocatechol containing agents, tolcapone (1), entacapone (2), and the nitrocatechol isomers 7 and 8. Both curcumin and orange G were used as reference compounds. The TEM images of AcPHF6 in the absence of test compounds, display strong aggregation after 24 h incubation. Images reveal the presence of both filaments (Figure 6A, highlighted by white arrows, width ∼ 6 nm) and fibrillar aggregates (Figure 6B, highlighted by black arrows, width ∼ 11 nm) species. In the presence of either orange G or curcumin (both at 10 μM), a reduction in the ability of AcPHF6 to form tau filaments was observed (Figure 6C and E). This effect was more pronounced with orange G. At a higher test concentration (50 μM), the scan revealed that both of them were able to reduce the formation of individual tau filaments as well as fibrils to a greater extent (Figure 6D and F) with orange G exhibiting almost no fibrillar aggregates.
Figure 6.

TEM images of reference compounds curcumin and orange G (10 and 50 μM) with AcPHF6 after 24 h incubation. (A, B) 100 μM AcPHF6 alone, white arrows represent individual filaments and black arrows represent fibrils; (C) 10 μM orange G and AcPHF6; (D) 50 μM orange G and AcPHF6; (E) 10 μM curcumin and AcPHF6; (F) 50 μM curcumin and AcPHF6. Bars represent 100 nm.
Furthermore, the effect of tolcapone and entacapone on AcPHF6 aggregation (Figure 7) shows that both of them were able to reduce the extent of fibril formation when incubated at 10 μM. Noticeably, at a higher concentration (50 μM), both of them markedly decrease the extent of filaments and aggregate formation (Figure 7B vs D) with tolcapone completely reducing the formation of filament and fibril species which was comparable to orange G (50 μM). This result supports our observation in the aggregation kinetics assay, where tolcapone exhibited greater propensity to prevent AcPHF6 aggregation (Figure 3A) compared to entacapone. The TEM images for the nitrocatechol isomers 7 (3-nitrocatechol) and 8 (2-nitrocatechol) are shown in Figure 8. At both 10 and 50 μM test concentrations, the nitrocatechol isomer 7 was able to suppress AcPHF6 aggregation which is consistent with our in vitro aggregation kinetics assay data (Figure 4A) although this was not as effective as either tolcapone or entacapone (50 μM). The nitrocatechol isomer 8 did inhibit aggregation; however, the effect was not as pronounced, even at 50 μM test concentration when compared to either tolcapone (1) or entacapone (2) or nitrocatechol 7 (Figure 8D). These studies show the ability of nitrocatechols (1, 2, and 7) to prevent AcPHF6 aggregation, further confirming the remarkable ability of a nitrocatechol (4-susbtituted-3-nitrobenzene-1,2-diol) pharmacophore as a tau-hexapeptide aggregation inhibitor.
Figure 7.

TEM images of test compounds (10 and 50 μM) with AcPHF6 after 24 h incubation. (A) 10 μM tolcapone 1 and AcPHF6; (B) 50 μM tolcapone 1 and AcPHF6; (C) 10 μM entacapone 2 and AcPHF6; (D) 50 μM entacapone 2 and AcPHF6. Bars represent 100 nm.
Figure 8.

TEM images of test compounds (10 and 50 μM) with AcPHF6 after 24 h incubation. (A) 10 μM nitrocatechol 7 and AcPHF6; (B) 50 μM nitrocatechol 7 and AcPHF6; (C) 10 μM nitrocatechol 8 and AcPHF6; (D) 50 μM nitrocatechol 8 and AcPHF6. Bars represent 100 nm.
Circular Dichroism (CD) Spectroscopy
In order to determine the ability of nitrocatechol-containing small molecules tolcapone (1), entacapone (2) and nitrocatechol (7) to prevent AcPHF6 aggregation and β-sheet assembly, conformational change of the peptide was monitored by far-UV CD spectroscopy. Initially, the conformation of tau hexapeptide AcPHF6 (2 mg/mL) was analyzed in water and 4-morpholinepropanesulfonic acid (MOPS) buffer. In agreement with a previous work, AcPHF6 in water displayed far-UV CD spectra with an intense negative minimum at ∼195 nm and a weak negative shoulder at ∼220 nm, which exhibits a dominantly disordered structure.31 In the presence of MOPS buffer (5 mM MOPS, 150 mM NaCl, pH 7.2), AcPHF6 polymerized rapidly and formed a β-sheet structure, characterized by the negative maximum ellipticity at ∼218 nm and a positive maximum at ∼195 nm. This β-sheet structure was stable when monitored over a period of 24 h. The effect of test compounds on the β-sheet structure of AcPHF6, was investigated by recording the far-UV CD spectra of the hexapeptide in the presence of test compounds at t = 0, 2, and 24 h, respectively. At each time point, the ellipticities of the CD spectra at 218 nm (characteristic of β-sheet) and at 205 nm (characteristic of random coil) were compared and their ratio was calculated. The AcPHF6 peptide underwent polymerization to form β-sheet in the absence of test compounds, as indicated by a negative maximum at 218 nm and positive ellipticity below 205 nm (Figure 9A). On the other hand, an inhibitor of AcPHF6 polymerization would impede the peptide aggregation process and formation of the β-sheet conformation. This results in an enhanced negative ellipticity in the 200–210 nm range, and a positive θ205/θ218 ratio, in the far-UV CD spectra of AcPHF6. A stronger inhibitor of tau aggregation may further enhance the negative ellipticity of tau peptide’s far-UV CD spectra around 205 nm and result in an even greater positive θ205/θ218 ratio. In these measurements, the aggregated β-sheet structure of AcPHF6 remained stable for at least 24 h in the absence of test compounds (Figure 9). Comparative θ205/θ218 ratio of AcPHF6 in the presence of test compounds at t = 24 h revealed that tolcapone (1), entacapone (2), nitrocatechol (7), and the reference agent orange G were able to inhibit β-sheet assembly (Figure 9B). The far-UV CD spectra of AcPHF6 in the presence of these test compounds exhibited a strong negative maximum ellipticity around 205 nm and a negative shoulder at ∼218 nm (Figure 9A). In addition, the θ205/θ218 ratios for AcPHF6 in the presence of test compounds remained around/above one during the 24 h experiment (Figure 9B). This study further demonstrates the ability of the nitrocatechol moiety to act as a tau-hexapeptide aggregation inhibiting pharmacophore.
Figure 9.
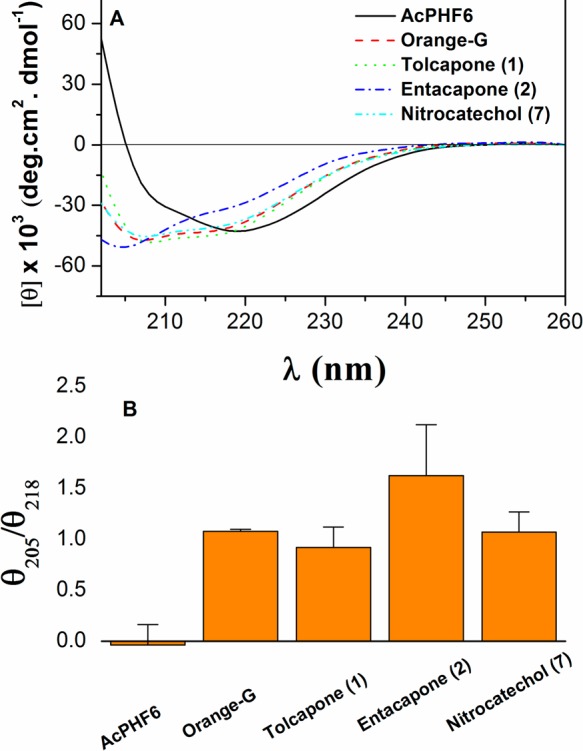
(A) Comparative far-UV CD spectra of AcPHF6 (2 mg/mL) in the presence and absence of test compounds after 24 h incubation. (B) Comparative θ205/θ218 ratios in the far UV-CD spectra of AcPHF6 in the absence and presence of test compounds at t = 24 h.
Computational Modeling
In order to understand the potential binding modes and antiaggregation properties of nitrocatechols, we investigated their binding interactions by molecular docking. In order to do this, we developed a β-sheet model of AcPHF6 tau hexapetide in silico for the first time. The X-ray coordinates of the tau-hexapeptide fragment was extracted from the solved structure to model two fiber layers consisting of parallel and antiparallel cross-β-strands.21 The molecular modeling experiments were conducted using the CDOCKER program to dock orange G, tolcapone (1), entacapone (2), dopamine (3), nitrocatechol (7), and catechol (9). Their binding modes were investigated and compared with the cocrystal structure of VQIVYK bound to orange G. It should be noted that orange G is known to bind in a space formed between pairs of steric zippers facing the lysine amino acid side chains protruding from the parallel β-strands which prevents fibril aggregation.21 Our modeling study is consistent with this observation where test compounds were bound in this region and underwent interactions with lysine residues lining the steric zipper motif.
Figure 10A shows the binding interactions of tolcapone 1. The molecule was oriented parallel to fiber axis. The catechol substituents of 3,4-dihydroxy-5-nitrophenyl (nitrocatechol) underwent polar contacts with three lysine side chains (distance < 2.80 Å) that line the steric zipper and the charged nitro-substituent formed a salt-bridge with another lysine side chain (distance = 2.80 Å). The ketone (C=O) that links the nitrocatechol moiety to a toluene substituent formed a salt-bridge with lysine side chain. Interestingly, the aromatic ring of toluene substituent underwent a cation−π interaction with lysine side chain (distance = 5.77 Å). Interestingly, entacapone 2 exhibited a similar binding mode in the steric zipper and underwent polar contacts with three lysine side chains protruding into the fibril junction (Figure 10B). However, the nitro-substituent was further away from the lysine side chain as compared to tolcapone (2.80 Å vs 3.02–3.17 Å). Moreover, the lack of an additional aromatic ring in entacapone takes away the stabilizing effect of a cation−π interaction seen with tolcapone which might explain the weaker inhibition of tau-aggregation seen with entacapone. Figure 11A shows the binding mode of the nitrocatechol isomer 7, in the tau-hexapeptide steric zipper. Compared to tolcapone 1 and entacapone 2, the nitrocatechol underwent interactions with fewer lysine side chains and formed fewer polar interactions. This suggests that the presence of additional bulk at C-1 would have a stabilizing effect on its binding ability. A comparison of binding modes of tolcapone 1, entacapone 2, and nitrocatechol 7 is shown in Figure 11B. Other tested compounds or building blocks that do not contain a nitrocatechol pharmacophore such as dopamine (3) and catechol (9) did not exhibit similar binding modes and had fewer polar contacts with lysine side chains (Figure S10, Supporting Information). This suggests that the addition of a 5-nitrogroup next to a 3,4-dihydroxyphenyl (catechol) substituent in tolcapone (1) and entacapone (2) provides a negatively charged center, which can undergo salt-bridge with a positively charged lysine group that could be critical in preventing tau-peptide aggregation. The molecular modeling studies are further supported by disaggregation kinetics (Figure S9, Supporting Information) indicating the ability of tolcapone (1), entacapone (2) and nitrocatechol isomer (7) to disrupt β-sheet assembly leading to disaggregation of preformed 306VQIVYK311 fibril assembly.
Figure 10.

(A) Binding mode of tolcapone (1, ball and stick) and (B) entacapone (2, ball and stick) in the steric zipper model of tau-derived-hexapeptide 306VQIVYK311. Two layers of fiber made up of individual β-strands are shown. The hydrogen atoms are removed for clarity. Distance parameters are shown in red (Å units).
Figure 11.

(A) binding mode of nitrocatechol (7, ball and stick) in the steric zipper model of tau-derived-hexapeptide 306VQIVYK311. Two layers of fiber made up of individual β-strands are shown. Distance parameters are shown in red (Å units). (B) Overlay of binding modes of tolcapone 1 (blue), entacapone 2 (red) and nitrocatechol 3 (green) in the tau-hexapeptide steric zipper β-strand assembly. The hydrogen atoms are removed for clarity.
Figure 12 shows the charge density maps for the nitrocatechols: tolcapone 1, entacapone 2, nitrocatechol isomer 7, nitrocatechol isomer 8, catechol 9, and orange G. This shows that the presence of a negatively charged nitro-substituent in 1, 2 and 7 increases polar surface area to interact with amino acids that line the steric zippers which is known to be 60% polar (Gln and Lys) and 40% nonpolar (Val and carbon side chain of Lys).21 It should be noted that the crystal structure of a tolcapone derivative in the COMT enzyme shows that the nitro-substituent undergoes polar contact with a charged lysine side chains along with the catechol moiety.32 This further confirms that a 4-susbtituted-3-nitrobenzene-1,2-diol (nitrocatechol) is a suitable pharmacophore to design small molecules as tau aggregation inhibitors.
Figure 12.
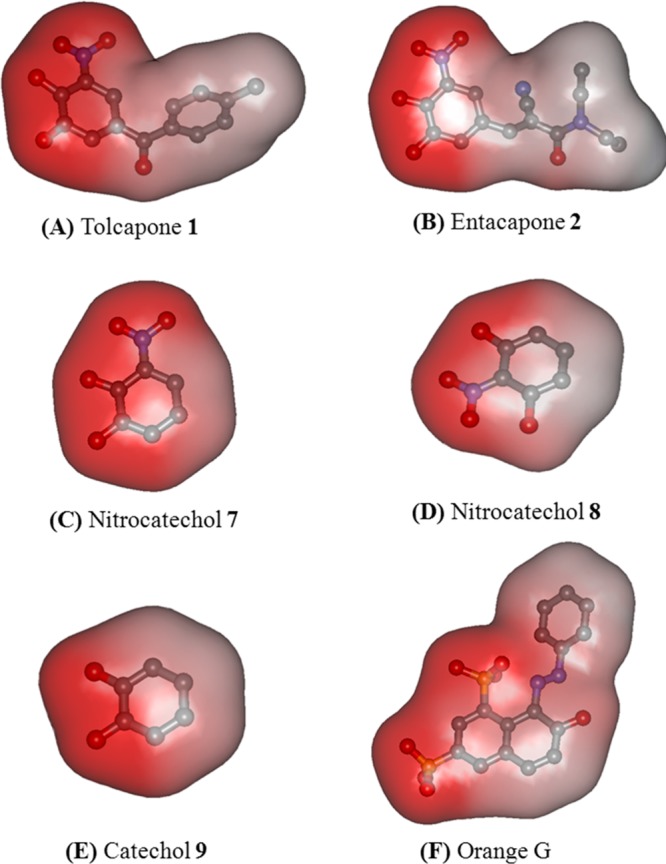
Charge density maps for (A) tolcapone 1, (B) entacapone 2, (C) nitrocatechol 7, (D) nitrocatechol 8, (E) catechol 9, and (F) Orange G. The hydrogen atoms are removed for clarity. Red color indicates polar region, and blue/gray color indicates nonpolar region in the molecule.
Conclusions
Our investigations reveal that the 3,4-dihydroxy-5-nitrophenyl (nitrocatechol) moiety present in the marketed the anti-Parkinson’s agents tolcapone and entacapone can prevent the aggregation of tau-derived hexapeptide. Tolcapone exhibited better inhibition of AcPHF6 aggregation as monitored by aggregation kinetics and TEM studies. Our results, coupled with previous report on tolcapone’s anti-amyloid aggregation properties, indicate potential “drug repurposing” of tolcapone to treat Alzheimer’s disease due to its super CNS penetration as compared to entacapone.28 Furthermore, the in silico β-sheet steric zipper modeling indicate that a catechol moiety coupled with a charged nitro-substituent (forming a 4-substituted-3-nitrobenzene-1,2-diol group) will assist in forming additional polar contacts with the lysine side chains lining the steric zipper cavity of tau-hexapeptide β-sheet. This study provides an opportunity to design novel small molecules possessing a nitrocatechol pharmacophore as chemical tools to study protein aggregation and to develop agents to treat the taupathy associated with Alzheimer’s disease.
Methods
Chemicals and Reagents
All chemicals and reagents (3–13, Figure 1, curcumin and orange G) were purchased from Sigma-Aldrich, Alfa Aesar, Acros Organics, or Fisher Scientific with a minimum purity of 97% and were used without further purification. Tolcapone and entacapone were purchased from Toronto Research Chemicals, Toronto, Ontario, Canada. Tau AcPHF6 peptide was purchased from Celtek Peptides, Nashville, TN.
AcPHF6 Aggregation Kinetics
The ability of test compounds to inhibit the tau-derived hexapeptide (AcPHF6) aggregation was examined using a ThS based assay according to a previously reported method.24 The aggregation assay was carried out with orange G and curcumin as reference agents. AcPHF6 was freshly prepared by dissolving in ultrapure water (Cayman Chemical Company, Ann Arbor, MI) to obtain a 1 mM stock solution. The ThS (Sigma-Aldrich, T1892) stock solution, 0.5 mg/mL was freshly prepared using a 20 mM MOPS buffer with 0.01% NaN3 and adjusted to pH 7.2, and the test compound stock solutions were also prepared with the same buffer and assay grade DMSO to achieve a maximum 1% (v/v) DMSO concentration in the assay run. At this concentration, DMSO did not interfere with the spectral measurements. The kinetic aggregation assay was carried out by incubating 20 μL of ThS, 140 μL of MOPS buffer, and 20 μL of test compound stock solutions at room temperature for 5 min before adding 20 μL of 1 mM AcPHF6 stock solution (final well concentration was 100 μM). Final concentration of test compounds ranged from 0.1, 1, 10, and 50 μM. Each 96-well plate (Costar, black clear bottom) was run with orange G and curcumin controls, MOPS blanks (140 μL of MOPS buffer and 60 μL of ultrapure water), and ThS blanks (140 μL of MOPS buffer, 20 μL of ThS and 20 μL of ultrapure water). Test compounds were used in triplicates and fluorescence was monitored at 440 nm (excitation) and 490 nm (emission) using a Molecular Devices SpectraMax spectrofluorometer. Readings were taken at every minute over a 2 h period with gentle shaking between each scan. Results were expressed as mean ± SD in triplicates of two independent experiments.
Transmission Electron Microscopy (TEM)
The morphology of AcPHF-6 was investigated in the presence of curcumin, orange G, tolcapone (1), entacapone (2), and nitrocatechol isomers 7 and 8. The phosphotungstic acid (PTA) hydrate was purchased from Sigma (cat. P4006). Formvar carbon-coated copper grids (400 mesh) were purchased from Canemco & Marivac, Quebec, Canada (Cat. 324-1). The AcPHF6 peptide was dissolved in ultrapure water to a 1 mM stock solution, and test compound stocks were prepared using assay grade DMSO and 20 mM MOPS buffer with 0.01% NaN3 and adjusted to pH 7.2 (max 1% v/v DMSO final concentration). Fibrillization assays were carried out in a 96-well plate by taking 160 μL of MOPS buffer, 20 μL of test compound aliquots, and 20 μL of AcPHF6 peptide and were incubated at room temperature for 24 h. The final AcPHF6 peptide concentration was 100 μM, whereas test compounds were used in two different concentrations (10 and 50 μM). Mesh grids were prepared by depressing the grid onto a piece of parafilm, followed by adding a 5 μL aliquot in the center of the grid. Grids were air-dried for 24 h at room temperature followed by negative staining with 5 μL of 2% PTA for 10 s, removing excess stain with filter paper wedges and further drying at room temperature for an additional 24 h. Grids were scanned using a Philips CM 10 transmission electron microscope at 60 kV and micrographs were obtained using a 14-megapixel AMT camera.
Circular Dichroism (CD) Spectroscopy
Far-UV CD spectra of AcPHF6 hexapeptide (2 mg/mL) in the absence and presence of test compounds were measured on an Aviv 215 spectropolarimeter (Aviv Biomedical, Lakewood Township, NJ). All spectra were measured in MOPS buffer (5 mM MOPS, 150 mM NaCl, 0.01% sodium azide, pH 7.2). A minimum concentration of DMSO (0.2–0.5% v/v) was used to solubilize test compounds. At these concentrations, DMSO did not interfere with the spectral measurements. All far-UV CD measurements were carried out in a 0.01 cm path length quartz cells, at 1 nm resolution (25 °C). Ellipticities of subtracted spectra are reported as mean residue ellipticity ± SD. The reported spectra were an average of either duplicate or triplicate (two independent experiments) measurements. The CD spectra of tau-derived hexapeptide in the absence and presence of test compounds were monitored over the time interval t = 0, 2, and 24 h respectively.
Computational Modeling
Discovery Studio (DS) Client v2.5.0.9164 (2005-09), Accelrys Software Inc. was used to perform docking experiments of tolcapone (1), entacapone (2), dopamine (3), nitrocatechol (7), and catechol (9) to a steric zipper β-sheet assembly of tau-derived-hexapeptide 306VQIVYK311 as per Eisenberg’s model.21,22 The cross-β-sheet structure was modeled by using the X-ray coordinates of VQIVYK segment cocrystallized with the inhibitor orange G (PDB code: 3OVL). The water molecules were removed and peptide was prepared using the protein preparation tool in DS. This hexapeptide was used to manually construct a steric zipper model of tau-hexapeptide-β-sheet made up of two parallel and two antiparallel β-strand assembly by maintaining an intrastrand distance of about 5 Å and an interstrand distance of about 10 Å.21,33 In order to remove any bad contacts or bumps, the β-sheet assembly was subjected to 1000 steps of steepest descent minimization (0.1 kcal/mol) followed by another 1000 steps of conjugate gradient minimization (0.1 kcal/mol) using the SHAKE constraints and CHARMM force field. Test compounds were built using the ligand preparation tool. The docking experiments were carried out by using CDOCKER in DS with CHARMM force field. The ligand binding site was identified by using the binding location of orange G from crystal structure, and a sphere of 12 Å radius was generated to define the binding site. The docking was performed after deleting orange G by simulated annealing with 2000 heating steps, 700 K heating target temperature and 500 cooling steps, 300 K cooling target temperature to generate 10 docked ligand poses. The β-sheet/ligand complex was ranked based on CDOCKER interaction energy in kcal/mol. The β-sheet/ligand complex was evaluated by measuring various polar and nonpolar interactions such as hydrogen bonding interactions, electrostatic interactions, van der Waal’s interactions, and hydrophobic interactions.
Glossary
Abbreviations
- Alzheimer’s disease
AD
- amyloid-beta
Aβ
- tau
τ
- alpha-synuclein
α-syn
- thioflavin-S
ThS
- morpholinepropanesulfonic acid
MOPS
- transmission electron microscopy
TEM
- phosphotungstic acid
PTA
- circular dichroism
CD
- neurofibrillary tangles
NFTs
Supporting Information Available
Supplementary Figures S1–S10 and Table S1. This materials is available free of charge via the Internet http://pubs.acs.org
Author Contributions
P.P.N.R and T.M. conceived the project and designed experiments. T.M. and T.H. performed experiments. T.M., T.H., and P.P.N.R. analyzed and interpreted results. T.M., T.H., M.J.-N. and P.P.N.R. wrote and revised the manuscript.
The authors would like to thank the Faculty of Science, Office of Research, the School of Pharmacy at the University of Waterloo, Ontario Mental Health Foundation (graduate scholarship for TM), Alexander Graham Bell Canada Graduate Scholarship (for TH), Canada Foundation for Innovation (CFI), Ontario Research Fund (ORF) and Natural Sciences and Engineering Research Council (NSERC), Canada for infrastructure and financial support.
The authors declare no competing financial interest.
Supplementary Material
References
- Soto C. (2003) Unfolding the role of protein misfolding in neurodegenerative diseases. Nat. Rev. Neurosci. 4, 49–60. [DOI] [PubMed] [Google Scholar]
- Cohen E. F.; Kelly J. W. (2003) Therapeutic approaches to protein-misfolding diseases. Nature 426, 905–909. [DOI] [PubMed] [Google Scholar]
- Chiti F.; Dobson C. M. (2006) Protein misfolding, functional amyloid and human disease. Annu. Rev. Biochem. 75, 333–366. [DOI] [PubMed] [Google Scholar]
- Dill K. A.; MacCallum J. L. (2012) The protein-folding problem, 50 years on. Science 338, 1042–1046. [DOI] [PubMed] [Google Scholar]
- Haydar S. N.; Yun H.; Staal R. G. W.; Hirst W. D. (2009) Small-molecule protein-protein interaction inhibitors as therapeutic agents for neurodegenerative diseases: recent progress and future directions. Annu. Rep. Med. Chem. 44, 51–69. [Google Scholar]
- Suh Y.; Checler F. (2002) Amyloid precursor protein, presenilins, and α-synuclein: molecular pathogenesis and pharmacological applications in Alzheimer’s disease. Pharmacol. Rev. 54, 469–525. [DOI] [PubMed] [Google Scholar]
- Cavalli A.; Bolognesi M. L.; Minarini A.; Rosini M.; Tumiatti V.; Recanatini M.; Melchiorre C. (2008) Multi-target-directed ligands to combat neurodegenerative diseases. J. Med. Chem. 54, 347–372. [DOI] [PubMed] [Google Scholar]
- Yam A. Y.; Wang X.; Gao C. M.; Connolly M. D.; Zuckermann R. N.; Bleu T.; Hall J.; Fedynyshyn J. P.; Allauzen S.; Peretz D.; Salisbury C. M. (2011) A universal method for detection of amyloidogenic misfolded proteins. Biochemistry 50, 4322–4329. [DOI] [PubMed] [Google Scholar]
- Sayre L. M.; Perry G.; Smith M. A. (2008) Oxidative stress and neurotoxicity. Chem. Res. Toxicol. 21, 172–188. [DOI] [PubMed] [Google Scholar]
- Ross C. A.; Poirier M. A. (2004) Protein aggregation and neurodegenerative disease. Nat. Med. 10, S10–S17. [DOI] [PubMed] [Google Scholar]
- Taylor J. P.; Hardy J.; Fischbeck K. H. (2002) Toxic proteins in neurodegenerative disease. Science 296, 1991–1995. [DOI] [PubMed] [Google Scholar]
- Glabe C. G.; Kayed R. (2006) Common structure and toxic function of amyloid oligomers implies a common mechanism of pathogenesis. Neurology 66, S74–S78. [DOI] [PubMed] [Google Scholar]
- Meraz-Rios M. A.; Lira-De Leon K. I.; Campos-Pena V.; De Anda-Hernandez M. A.; Mena-Lopez R. (2010) Tau oligomers and aggregation in Alzheimer’s disease. J. Neurochem. 112, 1353–1367. [DOI] [PubMed] [Google Scholar]
- Lasagna-Reeves C. A.; Castillo-Carranza D. L.; Guerrero-Munoz M. J.; Jackson G. R.; Kayed R. (2010) Preparation and characterization of neurotoxic tau oligomers. Biochemistry 49, 10039–10041. [DOI] [PubMed] [Google Scholar]
- Matharu B.; Gibson G.; Parsons R.; Huckerby T. N.; Moore S. A.; Cooper L. J.; Millichamp R.; Allsop D.; Austen B. (2009) Galantamine inhibits β-amyloid aggregation and neurotoxicity. J. Neurol. Sci. 280, 49–58. [DOI] [PubMed] [Google Scholar]
- Jan A.; Hartley D. M.; Lashuel H. A. (2010) Preparation and characterization of toxic abeta aggregates for structural and functional studies in Alzheimer’s disease research. Nat. Protoc. 5, 1186–1209. [DOI] [PubMed] [Google Scholar]
- Singh P. K.; Kotia V.; Ghosh D.; Mohite G. M.; Kumar A.; Maji S. K. (2013) Curcumin modulates α-synuclein aggregation and toxicity. ACS Chem. Neurosci. 4, 393–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gendron T. F.; Petrucelli L. (2009) The role of tau in neurodegeneration. Mol. Neurodegener. 4, 13–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felician O.; Sandson T. A. (1999) The neurobiology and pharmacotherapy of Alzheimer’s disease. J. Neuropsychiatry Clin. Neurosci. 11, 19–31. [DOI] [PubMed] [Google Scholar]
- Hopkins C. R. (2011) ACS chemical neuroscience molecule spotlight on ELND006: Another γ-secretase inhibitor fails in the clinic. ACS Chem. Neurosci. 2, 279–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landau M.; Sawaya M. R.; Faull K. F.; Laganowsky A.; Jiang L.; Sievers S. A.; Liu J.; Barrio J. R.; Eisenberg D. (2011) Towards a pharmacophore for amyloid. PLoS Biol. 9, e1001080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawaya M. R.; Sambashivan S.; Nelson R.; Ivanova M. I.; Sievers S. A.; Apostol M. I.; Thompson M. J.; Balbirnie M.; Wiltzius J. W.; McFarlane H. T.; Madsen A. O.; Riekel C.; Eisenberg D. (2007) Atomic structures of amyloid cross β-spines reveal varied steric zippers. Nature 447, 453–457. [DOI] [PubMed] [Google Scholar]
- Sievers S. A.; Karanicolas J.; Chang H. W.; Jiang L.; Zirafi O.; Stevens J. T.; Munch J.; Baker D.; Eisenberg D. (2011) Structure-based design of non-natural amino-acid inhibitors of amyloid fibril formation. Nature 475, 96–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng J.; Liu C.; Sawaya M. R.; Vadha B.; Khan S.; Woods R. J.; Eisenberg D.; Goux W. J.; Nowick J. S. (2011) Macrocyclic β-sheet peptides that inhibit the aggregation of a tau-protein-derived hexapeptide. J. Am. Chem. Soc. 133, 3144–3157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng J.; Baghkhanian A. M.; Nowick J. S. (2013) A hydrophobic surface is essential to inhibit the aggregation of a tau-protein derived hexapeptide. J. Am. Chem. Soc. 135, 6846–6852. [DOI] [PubMed] [Google Scholar]
- Bulic B.; Pickhardt M.; Mandelkow E. M.; Mandelkow E. (2010) Tau protein and tau aggregation inhibitors. Neuropharmacology 59, 276–289. [DOI] [PubMed] [Google Scholar]
- Brunden K. R.; Trojanowski J. Q.; Lee V. M. Y. (2009) Advances in tau-focused drug discovery for Alzheimer’s disease and related taupathies. Nat. Rev. Drug Discovery 8, 783–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giovanni S. D.; Eleuteri S.; Paleologou K. E.; Yin G.; Zweckstetter M.; Carrupt P. A.; Lashuel H. A. (2010) Entacapone and tolcapone, two catechol-o-methyltransferase inhibitors, block fibril formation of α-synuclein and β-amyloid and protect against amyloid-induced toxicity. J. Biol. Chem. 285, 14941–14954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsala S. Z.; Gioulis M.; Ceravolo R.; Tinazzi M. (2012) A systematic review of catechol-o-methyltransferase inhibitors: efficacy and safety in clinical practice. Clin. Neuropharmacol. 35, 185–190. [DOI] [PubMed] [Google Scholar]
- Hudson S. A.; Ecroyd H.; Kee T. W.; Carver J. A. (2009) The thioflavin T fluorescence assay for amyloid fibril detection can be biased by the presence of exogenous compounds. FEBS J. 276, 5960–5972. [DOI] [PubMed] [Google Scholar]
- Goux W. J.; Kopplin L.; Nguyen A. D.; Leak K.; Rutkofsky M.; Shanmuganandam V. D.; Sharma D.; Kirschner D. A. (2004) The formation of straight and twisted filaments from short tau peptides. J. Biol. Chem. 279, 26868–26875. [DOI] [PubMed] [Google Scholar]
- Palma P. N.; Rodrigues M. L.; Archer M.; Bonifacio M. J.; Loureiro A. I.; Learmonth D. A.; Carrondo M. A.; Soares-Da-Silva P. (2006) Comparative study of ortho- and meta-nitrated inhibitors of catechol-O-methyltransferase: interactions with the active site and regioselectivity of O-methylation. Mol. Pharmacol. 70, 143–153. [DOI] [PubMed] [Google Scholar]
- Ma B.; Nussinov R. (2002) Stabilities and conformations of Alzheimer’s amyloid peptide oligomers (A, A, and A): sequence effects. Proc. Natl. Acad. Sci. U.S.A. 99, 14126–14131. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.