

Abstract
Autism spectrum disorders (ASD) are neurodevelopmental disorders which are currently diagnosed solely on the basis of abnormal stereotyped behavior as well as observable deficits in communication and social functioning. Although a variety of candidate genes have been identified on the basis of genetic analyses and up to 20% of ASD cases can be collectively associated with a genetic abnormality, no single gene or genetic variant is applicable to more than 1–2 percent of the general ASD population. In this report, we apply class prediction algorithms to gene expression profiles of lymphoblastoid cell lines (LCL) from several phenotypic subgroups of idiopathic autism defined by cluster analyses of behavioral severity scores on the Autism Diagnostic Interview-Revised diagnostic instrument for ASD. We further demonstrate that individuals from these ASD subgroups can be distinguished from nonautistic controls on the basis of limited sets of differentially expressed genes with a predicted classification accuracy of up to 94% and sensitivities and specificities of ~90% or better, based on support vector machine analyses with leave-one-out validation. Validation of a subset of the “classifier” genes by high-throughput quantitative nuclease protection assays with a new set of LCL samples derived from individuals in one of the phenotypic subgroups and from a new set of controls resulted in an overall class prediction accuracy of ~82%, with ~90% sensitivity and 75% specificity. Although additional validation with a larger cohort is needed, and effective clinical translation must include confirmation of the differentially expressed genes in primary cells from cases earlier in development, we suggest that such panels of genes, based on expression analyses of phenotypically more homogeneous subgroups of individuals with ASD, may be useful biomarkers for diagnosis of subtypes of idiopathic autism.
Keywords: Autism, subphenotypes, gene expression, class prediction, blood biomarkers
Introduction
Autism spectrum disorders (ASD) are pervasive neurodevelopmental disorders that affect a broad spectrum of functions, but are diagnosed primarily on the basis of deficits in pragmatic language and communication, impaired ability to engage in reciprocal social interactions, as well as by stereotyped and repetitive behaviors often coupled with restricted interests. 1 Although many genetic studies have provided evidence for high heritability, 2–7 there are still no genetic markers that are unequivocally diagnostic for idiopathic ASD. This is in distinct contrast to the genetically-defined syndromic disorders, such as Fragile X,8, 9 tuberous sclerosis,10 Smith-Lemli-Opitz disease, 11 and Rett Syndrome, 12, 13 in which a fraction of affected individuals are also diagnosed with ASD. The difficulty in identifying genetic variants for idiopathic ASD is often attributed to the heterogeneity within the ASD population, which is manifested by the broad symptomatic profile of individuals on the ASD spectrum. Another problem associated with the majority of genetic analyses is that the individual single nucleotide polymorphisms (SNPs) and copy number variants (CNVs) identified as candidate biomarkers, even if replicated in a separate cohort, are each associated with a small percentage (typically <1–2%) of the general ASD population.
Although the majority of studies directed towards biomarker identification for ASD have focused on genetic variants, both common and rare, 4 we hypothesized that gene expression signatures may also be informative with respect to identification of ASD. However, realizing the challenges presented by the heterogeneity of ASD, we first divided the ASD population into subgroups sharing similar symptomatic profiles. 14 As shown in our earlier studies, dividing the ASD population into subgroups on the basis of cluster analyses of 123 scores from the Autism Diagnostic Interview-Revised (ADI-R) diagnostic instrument 15 resulted in the identification of distinct but overlapping gene expression profiles that characterized each of three ASD subgroups analyzed in comparison to nonautistic controls.16 In the current study, we conducted meta-analyses of our published gene expression profiles of lymphoblastoid cell lines (LCL) from these 3 phenotypic subgroups of ASD to identify differentially expressed genes that were robust in separating cases from controls. Here, we applied class prediction algorithms to identify and test differentially expressed genes that provide high sensitivity and specificity to separate cases and controls. A fraction of the differentially expressed genes for one of the subtypes was confirmed by high-throughput quantitative nuclease protection assays (qNPA), and then validated in part with a new set of samples, demonstrating the potential of this approach for developing a biomarker screen that can detect subtypes of ASD.
Materials and Methods
Analysis of data from ADI-R questionnaires to identify phenotypic subgroups
As previously described,14 ADI-R score sheets were downloaded for 1954 individuals with autism from the Autism Genetic Research Exchange (AGRE) phenotype database. A total of 63 items that were identical or comparable on both 1995 and 2003 versions of the ADI-R were included in our cluster analyses. “Current” and “ever” scores were used for most of these items, thus giving rise to the 123 scores used to phenotype individuals with ASD. Only items scored numerically from 0 – 3 (0 = normal; 3 = most severe) were analyzed. Cluster analyses of the item scores were used to divide the cases into 4 subgroups that were phenotypically distinguishable from each other on the basis of severity of symptoms across the 63 items, using “current” and “ever” scores for most items. Gene expression data of LCL from 3 of these ASD subgroups were used in this study as described below.
Selection of samples for large-scale gene expression analyses
In our previously published study16, lymphoblastoid cell lines (LCL) were selected from 3 of the 4 phenotypic groups for gene expression analyses to test “proof-of-concept” that expression profiles could separate samples from individuals with ASD from that of controls as well as to distinguish subtypes of ASD based on gene expression signatures. These groups included individuals with severe language impairment (L), those with milder symptoms across all domains (M), and those defined by notable presence of savant skills (S). Additional selection criteria were applied to exclude all female subjects (inasmuch as the male:female ratio in ASD is ~4:1), individuals with cognitive impairment (Raven’s scores < 70), those with reported genetic or chromosomal abnormalities (e.g., Fragile X, Retts, tuberous sclerosis, chromosome 15q11-q13 duplication), those born prematurely (< 35 weeks gestation), and those with diagnosed comorbid psychiatric disorders (e.g., bipolar disorder, obsessive compulsive disorder, severe anxiety), as reported in AGRE’s phenotypic database. In addition, a score < 80 on the Peabody Picture Vocabulary Test (PPVT) was used to confirm language deficits for those in the ASD subgroup identified by cluster analysis as having severe language impairment. For this study, 22 cases with severe language impairment and 22 controls from the original study were selected to validate differential expression of the putative classifier genes by quantitative nuclease protection assays.
Cell culture
LCL for the validation study were cultured as previously described 17 according to the protocol specified by the Rutgers University Cell and DNA Repository, which maintains the AGRE collection of biological materials from autistic individuals and relatives. Briefly, cells are cultured in RPMI 1640 (MediaTech) supplemented with 15% fetal bovine serum (Atlanta Biologicals), and 1% penicillin/streptomycin (Invitrogen). Cultures are split 1:2 every 3–4 days and cells are typically harvested for RNA isolation 3 days after a split while the cultures are in logarithmic growth phase. The RNA was analyzed for purity and integrity using a NanoDrop spectrophotometer and an Agilent 2100 Bioanalyzer.
Gene expression analyses using DNA microarrays
Gene expression profiling was accomplished using custom-printed TIGR 40K human arrays as previously described.17 Total RNA was isolated from LCL using the TRIzol (Invitrogen) isolation method according to the manufacturer’s protocols, and cDNA was synthesized, labeled, and hybridized to the microarrays as described in our earlier study, with the exception that cDNA from each sample was labeled with Cy-3 dye (Molecular Probes) and hybridized against Cy-5 labeled reference cDNA prepared from Universal human RNA (Stratagene). This “reference” design allows the flexibility to perform different comparisons among the samples since all expression values are measured against a common reference. After hybridization, washing of the arrays, and laser scanning to elicit dye intensities for each element on the array, the intensity data was normalized and filtered using Midas and analyzed using MeV, which are open-access software programs for DNA microarray analyses. 18 The raw and normalized gene expression data for these samples were deposited into the Gene Expression Omnibus (GEO), accession number GSE15402. All analyses for this study were performed with a 100% data filter which means that each gene included in the analyses must have an expression value in 100% of the samples. Unpaired t-tests on the normalized data from cases (either combined or subtyped by ASD) and controls were used to obtain significant differentially expressed genes (nominal p-value ≤ 0.01). These t-tests resulted in a total of 1197, 343, and 320 genes for the language-impaired (L), mild (M), and savant (S) subgroups, respectively, and 130 genes for the combined cases (A). The 100 most significant genes from each list were then subjected to class prediction and validation methods to select the most robust genes for predicting cases and controls.
Class prediction and validation methods
Two supervised machine learning methods were employed to identify highly predictive differentially expressed genes for ASD. Program modules for Uncorrelated Shrunken Centroids (USC) and Support Vector Machine (SVM) analyses were both contained within MeV software developed for microarray analyses.18 These methods were applied to discriminate each of the members of the ASD subgroups from controls as well as to discriminate members of the combined group of individuals with ASD from controls. The 100 most significant differentially expressed genes derived from the unpaired t-tests (p ≤ 0.01) were subsequently analyzed using USC analysis with 10-fold cross-validation to identify a reduced set of genes by removing highly correlated genes.19 SVM analyses20 with leave-one-out (LOO) cross-validation were used to determine the accuracy, sensitivity, and specificity of correctly assigning samples to case or control groups, using the genes from the respective USC analyses. The flow chart in Fig. 1 describes the workflow used in this study.
Fig. 1.

Workflow for class prediction analyses
Validation of putative “classifier” genes
High-throughput quantitative nuclease protection assays (qNPA) were used to confirm differential expression of a subset of predictive classifier genes derived for the severely language-impaired subtype of ASD as well as to validate differential expression of these genes in a completely new set of LCL derived from cases and controls. Although the complete lists of potentially predictive genes for this subtype ranged from 24 (Supplemental Table 5) to 29 (Supplemental Table 1), we restricted our assay to 14 genes because of the 16 gene/microtiter well format of the qNPA platform at the time of these studies and the need to include both positive and negative expression controls for the qNPA, the exploratory nature of this validation assay, and fiscal constraints. Gene selection criteria for the qNPA included presence on both gene lists, which included genes with adjusted Bonferroni p-values ≤ 0.01 and, in one case, high level of differential expression relative to control samples (FGFR1). The selected classifier genes/transcripts were: ALS2CL, BZRP, C12ORF30, CASP7, DDX26, FGFR1, FLJ11021, ITGAM, JAK1, MYLE, PTPN1, SFRS10, UPF1, and a transcript with GenBank# AI187812. Based upon SVM analysis using the microarray expression values, the predicted accuracy, sensitivity, and specificity for this set of 14 genes in differentiating cases from controls exceeded 93% (see last row of Table 1). Probes for these transcripts were designed by HighThroughputGenomics, Inc. (HTG) as a contract service which included the qNPA of RNA samples provided by our laboratory. The method of qNPA is described by Roberts et al.21 Two sets of RNA samples were provided for qNPA. The first set of samples included RNA from LCL of 22 male cases (severely language-impaired phenotype) and 22 male controls who were originally included in the large-scale gene expression study.16 The second set of samples analyzed by qNPA included RNA from LCL that were derived from 13 new cases of language-impaired male individuals with ASD identified by our previously described phenotyping method14 and 10 new age-matched male controls. The qNPA (performed by HTG) were conducted in triplicate for each sample and values that exceeded 3 standard deviations from the mean for other values in the series were discarded. Data for ALS2CL was low or non-existent for the majority of samples and were thus eliminated from the analyses. The coefficient of variance (CV) for all samples was typically <20%. Inasmuch as the expression values for different genes included in the qNPA covered more than 2 orders of magnitude, relative gene expression values (obtained by dividing the mean expression value for each sample by the highest mean expression for the respective gene) were used in order to bring expression data for all genes to within the numerical range of 0 to1. The resulting data was then used for SVM analyses as described above.
Table 1. Summary of accuracies, sensitivities, and specificities for class predictors based upon gene expression profiles.
Panels of predictor genes were selected by USC analyses of differentially expressed genes from DNA microarray analyses of LCL from a total of 87 individuals with ASD and 29 controls. The individuals with ASD were either divided into phenotypic subgroups based upon cluster analyses of ADI-R scores, as previously described14 or combined into one case group. SVM analyses with Leave-one-out (LOO) validation were used to determine the sensitivities and specificities associated with each set of genes in discriminating individuals from each ASD subtype (L, M, or S) from controls (C ) as well as the combined case group (A) from the controls. The performance of the 24 member gene set (shown in supplemental table 5) derived from adjusted Bonferroni analysis of the differentially expressed genes for the severely language-impaired phenotype (L) was also tested for comparison with the 29 gene set derived from USC analysis. Also shown are the class prediction data derived from the original microarray expression values of the 14 genes selected for qNPA analyses for individuals with the “L” phenotype vs. controls.
| Case-control | Accuracy of class predictor | Sensitivity | Specificity | ||
|---|---|---|---|---|---|
| Comparison | USC→SVM | [correct assignment] | (# genes) | (%) | (%) |
| L vs C | 93.3% | [56/60] | (29) | 96.6 | 90.3 |
| M vs C | 94.5% | [52/55] | (26) | 96 | 93.3 |
| S vs C | 94% | [47/50] | (18) | 96.6 | 90.5 |
| A vs C | 81.8% | [95/116] | (74) | 91.2 | 61.1 |
| L vs C (adj. Bonf.) | 90.0% | [54/60] | (24) | 90.3 | 89.6 |
| L vs C | 93.3% | [56/60] | (14)* | 93.5 | 93.1 |
14 genes used for qNPA
Pathway analysis of predictive classifier genes
Pathway analysis was accomplished using two different network prediction software packages, Pathway Studio 7 and Ingenuity Pathway Analysis (IPA). Where provided, the p-values, calculated on the basis of the Fisher Exact Test as implement by IPA which uses the complete set of annotated genes as the reference set, represent the probability that the indicated functions or disorders are not associated with the given set of genes.
Results and Discussion
A major goal of this study was to identify sets of genes that may be used to discriminate individuals with ASD from unaffected controls on the basis of gene expression profiles that may ultimately be used as biomarkers to develop a diagnostic screen for autism. Towards this goal, we performed DNA microarray analyses to obtain the gene expression profiles of LCL of 87 autistic male individuals who were divided into 3 phenotypic subgroups based on cluster analyses of scores on the ADI-R questionnaire 14, 16. Here, we applied gene classification and validation software in a meta-analysis of the data derived from the expression analyses to identify sets of genes that have a high statistical probability of predicting cases and controls for each of the 3 ASD subtypes that we studied. To establish proof-of-concept that small sets of differentially expressed genes may be used to distinguish cases from controls, we used high-throughput qNPA to first confirm that a subset of “classifier” genes for the severely language-impaired subtype of ASD could replicate the separation of cases and controls achieved by cDNA microarray analyses, and then tested the performance of this limited set of genes in classifying new samples of cases and controls on the basis of qNPA.
Identification of classifier genes for 3 phenotypic variants of ASD
The phenotypic subgroups of ASD that were studied included one group with severe language impairment (n = 31), another of moderate severity with noticeable savant skills (n = 30), and a subgroup with an overall mild phenotype (n = 26), as previously described.14 Gene expression data on LCL from these 3 phenotypic subgroups were obtained using a 40K TIGR human cDNA array with 39,936 probe elements.16 Using MeV microarray analysis software,18 the resulting data were subjected to a 100% data filter that eliminated genes that were undetectable in any one of the samples under study. Unpaired t-tests were performed on the filtered data from each of the ASD subgroups and from the nonautistic controls to identify significantly differentiated genes (nominal p ≤ 0.01) between each subgroup and the group of controls (n = 29). An unpaired t-test was also used to identify differentially expressed genes (nominal p ≤ 0.01) between the combined cases (n = 87) and the 29 controls. Two different supervised learning methods were used to select and validate genes from each of the resulting sets of differentially expressed genes for our predictive models. Uncorrelated Shrunken Centroids (USC) with 10-fold cross-validation 19 as implemented in MeV software18 was first used to select the most robust classifier genes from the lists of significant genes (Supplemental Tables 1–4). The limited sets of subtype-dependent classifier genes from the USC analyses (ranging from 18–29) were then entered into the support vector machine (SVM)20 software program using leave-one-out (LOO) cross-validation to test the gene classifier for each of the phenotypic variants. As shown in Figures 2A–C and Table 1, the SVM analyses suggest that gene classifiers based upon a relatively small number of differentially expressed genes can discriminate between each of the ASD phenotypic variants with an overall accuracy of ~93%, with the number and identity of classifier genes dependent on the phenotype. As shown in Table 1, the sensitivity of the predictive gene panels was ~96% for all 3 ASD subtypes, while the specificity ranged from 90–93%. As an alternative to the USC method of identifying highly predictive genes described above, we also employed a t-test with an adjusted Bonferroni correction for multiple testing (corrected p ≤ 0.01) to identify significantly differentially expressed genes between the severely language-impaired ASD subgroup and controls. The resultant set of 24 genes (Supplemental Table 5) could also correctly distinguish ASD from controls with 90% accuracy as indicated by SVM analysis (Table 1, row 5). Six of these genes overlapped with those identified by the USC algorithm. By comparison, if the combined autistic samples (n = 87) are tested against the nonautistic controls (n = 29) using the USC and SVM procedures described earlier, the accuracy of correct assignment to case or control groups is 81% with a sensitivity of ~91% and a specificity of 61%, based upon 74 differentially expressed genes (Table 1, Fig. 2D, and Supplemental Table 4), thus demonstrating the value of subphenotyping of cases to identify genes for improved classifier performance. Despite the low overall specificity, it is interesting to note that the classifier based on 74 genes shows the best performance in separating the most severely affected individuals with language impairment from the control group, with only one out of 31 ASD samples incorrectly scored as “negative”.
Fig. 2. Performance of “classifier” genes for ASD subtypes vs. control samples.

The differentially expressed genes used for class prediction were selected by USC with 10-fold cross-validation. SVM analyses of microarray expression data for the selected genes show: A) Separation of severely language-impaired ASD (L, red) from controls (C, turquoise) based on 29 genes; B) Separation of mild ASD (M, blue) from controls (C ) based on 27 genes; C) Separation of ASD with savant skills (S, yellow) from controls (C ) based on 18 genes; D) Separation of combined ASD samples (each subtype represented by its respective color) from controls.
Partial replication and validation of classifier gene expression differences using high-throughput quantitative nuclease protection assays
To test the ability of the proposed classifier genes to discriminate between ASD cases and controls, another highly sensitive method of detecting gene expression, high-throughput quantitative nuclease protection assay (qNPA), was used: 1) to confirm differential expression of putative classifier genes using LCL derived from individuals with severe language impairment and nonautistic controls that were used previously for DNA microarray analyses; and 2) to validate this same set of classifier genes with completely new LCL from cases and controls. The normalized qNPA data from each of these studies are provided in Supplemental Tables 6 and 7, respectively. Support Vector Machine analyses were used to assess the performance of the selected classifier genes in discriminating cases from controls in both studies. Table 2 summarizes the results of the SVM analysis (with LOO cross-validation) based on the qNPA expression data obtained using 22 cases and 22 controls from the original samples that were previously analyzed by DNA microarray analyses.16 As shown, the sensitivity and specificity of the test genes for assignment of samples to the correct groups (cases vs. controls) were 78 and 80%, respectively. This is in general agreement with the separation of individual samples based on unsupervised principal components analysis of the qNPA data (Fig. 3A), which captures >72% of the gene expression variation among the samples within the first 3 principal components.
Table 2.
Validation of selected classifier genes on original samples from severe language subtype by quantitative nuclease protection assay
| Sample Description | Number | Sensitivity (%) | Specificity (%) |
|---|---|---|---|
| Positive Cases | 22 | ||
| Classified as positive | 23 | ||
| True positives | 18 | 78.2 | |
| False negatives | 5 | ||
| Controls | 22 | ||
| Classified as negative | 21 | ||
| True negatives | 17 | 80.9 | |
| False positives | 4 |
Results are from SVM analysis of the qNPA data.
Fig. 3.
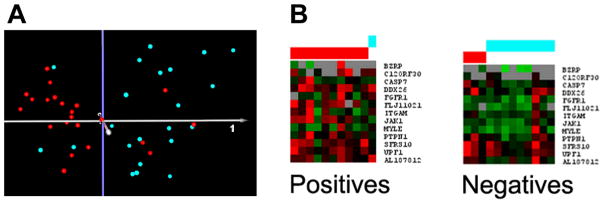
A) Principal components analysis of the qNPA data for cases and controls. Quantitative nuclease protection assays were performed on LCL [22 cases of the severely language-impaired subtype (red) and 22 controls (turquoise)] that were previously analyzed by DNA microarray analyses. Principal components analysis of the qNPA data shows good separation of the samples based on 14 differentially expressed genes, with 72% of the variance represented within the first 3 principal components. B) SVM analysis of the qNPA data for a new set of 13 cases (red) and 10 controls (turquoise). For the SVM analyses, cases were initially identified as positives and controls as negatives for training the classifier.
Table 3 and Fig. 3B show the results of SVM analysis using the qNPA data obtained with completely new LCL from the subgroup of severely language-impaired individuals with ASD and nonautistic controls. As shown, the sensitivity and specificity of the classifier genes when applied to this new set of samples were 90.9% and 75%, respectively. While the specificity is less than desired, it is notable that the sensitivity for identifying cases, which is highly desirable for screening purposes, exceeded 90%. It is also important to note that, due to the limited number of genes that could be tested per sample well in the qNPA, the number of potential classifier genes tested in the qNPA was restricted to less than half (14) of the 29 genes previously identified by class prediction analyses of the DNA microarray data. Furthermore, the controls used in the qNPA studies are siblings of individuals with ASD (but not of the cases used in this study) who may share an overlapping gene expression profile with their autistic sibling, but who do not exhibit behaviors or ASD characteristics that meet the diagnostic criteria for ASD. Both of these factors, coupled with the limited number of tested samples, may account for the lower than predicted sensitivity and specificity (~93%) for this set of genes based on DNA microarray data (Table 1, last row).
Table 3.
Class prediction performance of selected classifier genes on new samples from severe language subtype by quantitative nuclease protection assay
| Sample Description | Number | Sensitivity (%) | Specificity (%) |
|---|---|---|---|
| Positive Cases | 13 | ||
| Classified as positive | 11 | ||
| True positives | 10 | 90.9 | |
| False negatives | 1 | ||
| Controls | 10 | ||
| Classified as negative | 12 | ||
| True negatives | 9 | 75 | |
| False positives | 3 |
Results are from SVM analysis of the qNPA data.
Pathway analysis of predictive classifier genes
Although the usefulness of classifier genes as biomarkers of ASD need not depend explicitly on their functions relative to the disorder, we undertook pathway analyses to determine whether the identified genes were relevant to functions associated with ASD. Figure 4 shows the network generated using the 29 transcripts (Supplemental Table 1) that are predictive for identifying ASD individuals with severe language disorder. The functions associated with 15 annotated genes from this list include neurite outgrowth, embryonic development, cell proliferation and translation, which are all known to be impacted by ASD. Disorders associated with some of these genes include absence and myoclonic seizure, Huntington’s disease, major depression, and schizophrenia, demonstrating overlapping genes and pathways impacted by these different neurological disorders (Supplementary Table 8). The gene network constructed with the classifier genes associated with the “mild” phenotype (Supplemental Table 2) revealed functions associated with chromatin remodeling, muscle function, cell proliferation and differentiation, survival, and apoptosis (Figure 5), with two genes identified by Ingenuity Pathway Analysis software as being associated with neurological disease. MARCKS is involved in mania22 and microglial activation23 and an isoform of TRIO is implicated in Purkinje cell degeneration.24 The “savant” phenotype, classified according to only 18 transcripts (Supplemental Table 3), revealed a network that included dendrite morphogenesis, synapse maturation and transmission in addition to cell proliferation and apoptosis among the gene-associated biological functions (Figure 6). These results suggest that the genes identified by class prediction analyses are functionally meaningful with respect to what is known about the pathophysiology of autism.
Fig. 4.

Interactive gene network of classifier genes associated with the severely language-impaired subtype of ASD.
Fig. 5.

Interactive gene network of classifier genes associated with the “mild” subtype of ASD.
Fig. 6.

Interactive gene network of classifier genes associated with the “savant” subtype of ASD.
Study limitations and future directions
This study was undertaken in order to assess the feasibility of identifying a small set of genes capable of distinguishing individuals with ASD from unaffected, unrelated controls. However, as mentioned earlier, the unrelated controls from the AGRE repository are siblings of probands with ASD and may bear some gene expression similarities with that of individuals with ASD, which would have the effect of attenuating expression differences between the cases and controls in our study. In fact, a recent study on the gene expression profiles of case-control siblings and unrelated, unaffected individuals without a family history of ASD suggests that the gene expression pattern of some of the undiagnosed sibling controls resembled that of their affected sibling while the expression profile of other siblings resembled that of the unrelated controls.25 Another possible confounder might be that of population structure which has been reported in a meta-analysis26 of a genetics study27 that identified risk alleles for ASD where the cases and controls were reportedly from different ancestral populations. However, both cases and controls used in this study are from the AGRE collection of white Americans of various European ancestries which may reduce the effect, if any, of population structure on gene expression differences. Other limitations, related to the qNPA platform and cost per assay, were the use of less than the full set of classifier genes in the qNPA studies and the relatively small number of samples. Thus, the class predictor based on gene expression still requires optimization with regard to the number and selection of genes for each subtype of ASD. Nevertheless, the results from this pilot study still reveal the potential for developing a predictive and subtype-dependent gene classifier for ASD based on a limited set of genes. For clinical translation, these gene panels should be further investigated as potential biomarker screens for idiopathic autism using primary blood cells. Two recent studies demonstrate that transcriptomic signatures derived from both primary lymphocytes and mononuclear cells have a predictive value for identifying cases with accuracies of 68% and 91%, respectively,28, 29 thus reinforcing the idea that a diagnostic screen for ASD might be developed using peripheral tissues. Finally, since the mean age of the individuals with ASD represented in this study was 9.6 (range 4.5–17) years for the qNPA and 12.5 (range 5–37) years for the microarray analyses, longitudinal studies are needed in order to determine the earliest times of development for which expression differences can be reliably detected and used diagnostically.
Conclusions
This study is the first to report class prediction methods for identifying potential biomarkers of idiopathic autism based upon gene expression profiling of LCL. In particular, we establish proof-of-concept that individuals with idiopathic autism can be segregated from nonautistic controls with a moderate to high degree of accuracy, good sensitivity, and reasonable specificity based upon gene panels comprised of a relatively small number of differentially expressed genes, which are specific for different phenotypic variants of ASD. Although additional validation studies with a larger cohort of cases and controls are needed, and effective clinical translation must include confirmation of the differentially expressed genes in primary cells from cases obtained at younger ages, we suggest that the strategy demonstrated here of reducing clinical heterogeneity for class prediction analyses will aid in the identification of robust biomarkers for not only diagnosis of ASD, but also as pharmacogenomic indicators of the subphenotype of ASD which may be uniquely amenable to subtype-targeted therapies. Early identification of autism based on objective gene screening is a major first step towards early intervention and effective treatment of affected individuals.
Supplementary Material
Acknowledgments
VWH thanks Ms. Mara Steinberg for assistance in cell culturing and RNA preparation from the new set of LCL for the qNPA analysis. This study was supported by a supplement to NIH grant # R21 MH073393 (VWH). The funding agency had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript. Intellectual property (GWU and VWH) arising from this study has been licensed by SynapDx Corp. (Boston, MA) which played no role in any part of this study nor in the decision to publish this manuscript.
References
- 1.American Psychological Association. Diagnostic and Statistical Manual of Mental Disorders. 4. Washington, DC: American Psychological Association; 1994. [Google Scholar]
- 2.Freitag CM, Staal W, Klauck SM, Duketis E, Waltes R. Genetics of autistic disorders: Review and clinical implications. European Child and Adolescent Psychiatry. 2010;19(3):169–178. doi: 10.1007/s00787-009-0076-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Geschwind DH. Autism: Many genes, common pathways? Cell. 2008;135(3):391–395. doi: 10.1016/j.cell.2008.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.State MW, Levitt P. The conundrums of understanding genetic risks for autism spectrum disorders. Nat Neurosci. 2011;14(12):1499–1506. doi: 10.1038/nn.2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hallmayer J, Cleveland S, Torres A, et al. Genetic heritability and shared environmental factors among twin pairs with autism. Arch Gen Psychiatry. 2011;68(11):1095–1102. doi: 10.1001/archgenpsychiatry.2011.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bailey A, Le Couteur A, Gottesman I, et al. Autism as a strongly genetic disorder: Evidence from a british twin study. Psychol Med. 1995;25(1):63–77. doi: 10.1017/s0033291700028099. [DOI] [PubMed] [Google Scholar]
- 7.Folstein S, Rutter M. Infantile autism: A genetic study of 21 twin pairs. J Child Psychol Psychiatry. 1977;18(4):297–321. doi: 10.1111/j.1469-7610.1977.tb00443.x. [DOI] [PubMed] [Google Scholar]
- 8.Verkerk AJMH, Pieretti M, Sutcliffe JS, et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell. 1991;65(5):905–914. doi: 10.1016/0092-8674(91)90397-h. [DOI] [PubMed] [Google Scholar]
- 9.Sutcliffe JS, Nelson DL, Zhang F, et al. DNA methylation represses FMR-1 transcription in fragile X syndrome. Hum Mol Genet. 1992;1(6):397–400. doi: 10.1093/hmg/1.6.397. [DOI] [PubMed] [Google Scholar]
- 10.Povey S, Burley MW, Attwood J, et al. Two loci for tuberous sclerosis: One on 9q34 and one on 16p13. Ann Hum Genet. 1994;58(2):107–127. doi: 10.1111/j.1469-1809.1994.tb01881.x. [DOI] [PubMed] [Google Scholar]
- 11.Jira PE, Waterham HR, Wanders RJA, Smeitink JAM, Sengers RCA, Wevers RA. Smith-lemli-opitz syndrome and the DHCR7 gene. Ann Hum Genet. 2003;67(3):269–280. doi: 10.1046/j.1469-1809.2003.00034.x. [DOI] [PubMed] [Google Scholar]
- 12.Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23(2):185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- 13.Van den Veyver IB, Zoghbi HY. Methyl-CpG-binding protein 2 mutations in rett syndrome. Curr Opin Genet Dev. 2000;10(3):275–279. doi: 10.1016/s0959-437x(00)00083-6. [DOI] [PubMed] [Google Scholar]
- 14.Hu VW, Steinberg ME. Novel clustering of items from the autism diagnostic interview-revised to define phenotypes within autism spectrum disorders. Autism Res. 2009;2(2):67–77. doi: 10.1002/aur.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lord C, Rutter M, Couteur AL. Autism diagnostic interview-revised: A revised version of a diagnostic interview for caregivers of individuals with possible pervasive developmental disorders. Journal of Autism and Developmental Disorders. 1994;24(5):659–685. doi: 10.1007/BF02172145. [DOI] [PubMed] [Google Scholar]
- 16.Hu VW, Sarachana T, Kim KS, et al. Gene expression profiling differentiates autism case-controls and phenotypic variants of autism spectrum disorders: Evidence for circadian rhythm dysfunction in severe autism. Autism Res. 2009;2(2):78–97. doi: 10.1002/aur.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hu VW, Frank BC, Heine S, Lee NH, Quackenbush J. Gene expression profiling of lymphoblastoid cell lines from monozygotic twins discordant in severity of autism reveals differential regulation of neurologically relevant genes. BMC Genomics. 2006;7:118. doi: 10.1186/1471-2164-7-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Saeed AI, Sharov V, White J, et al. TM4: A free, open-source system for microarray data management and analysis. BioTechniques. 2003;34(2):374–378. doi: 10.2144/03342mt01. [DOI] [PubMed] [Google Scholar]
- 19.Yeung KY, Bumgarner RE. Multiclass classification of microarray data with repeated measurements: Application to cancer. Genome Biol. 2003;4(12):R83. doi: 10.1186/gb-2003-4-12-r83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brown MPS, Grundy WN, Lin D, et al. Knowledge-based analysis of microarray gene expression data by using support vector machines. Proc Natl Acad Sci U S A. 2000;97(1):262–267. doi: 10.1073/pnas.97.1.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roberts RA, Sabalos CM, LeBlanc ML, et al. Quantitative nuclease protection assay in paraffin-embedded tissue replicates prognostic microarray gene expression in diffuse large-B-cell lymphoma. Laboratory Investigation. 2007;87(10):979–997. doi: 10.1038/labinvest.3700665. [DOI] [PubMed] [Google Scholar]
- 22.Szabo ST, Machado-Vieira R, Yuan P, et al. Glutamate receptors as targets of protein kinase C in the pathophysiology and treatment of animal models of mania. Neuropharmacology. 2009;56(1):47–55. doi: 10.1016/j.neuropharm.2008.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hasegawa H, Nakai M, Tanimukai S, et al. Microglial signaling by amyloid beta protein through mitogen-activated protein kinase mediating phosphorylation of MARCKS. Neuroreport. 2001;12(11):2567–2571. doi: 10.1097/00001756-200108080-00055. [DOI] [PubMed] [Google Scholar]
- 24.Sun YJ, Nishikawa K, Yuda H, et al. Solo/Trio8, a membrane-associated short isoform of trio, modulates endosome dynamics and neurite elongation. Mol Cell Biol. 2006;26(18):6923–6935. doi: 10.1128/MCB.02474-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kong SW, Shimizu-Motohashi Y, Campbell MG, et al. Peripheral blood gene expression signature differentiates children with autism from unaffected siblings. Neurogenetics. 2013;14(2):143–152. doi: 10.1007/s10048-013-0363-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Belgard TG, Jankovic I, Lowe JK, Geschwind DH. Population structure confounds autism genetic classifier. Mol Psychiatry. 2013:1–3. doi: 10.1038/mp.2013.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Skafidas E, Testa R, Zantomio D, Chana G, Everall IP, Pantelis C. Predicting the diagnosis of autism spectrum disorder using gene pathway analysis. Mol Psychiatry. 2012:1–7. doi: 10.1038/mp.2012.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kong SW, Collins CD, Shimizu-Motohashi Y, et al. Characteristics and predictive value of blood transcriptome signature in males with autism spectrum disorders. PLoS ONE. 2012;7(12):e49475. doi: 10.1371/journal.pone.0049475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Glatt SJ, Tsuang MT, Winn M, et al. Blood-based gene expression signatures of infants and toddlers with autism. J Am Acad Child Adolesc Psychiatry. 2012;51(9):934–944.e2. doi: 10.1016/j.jaac.2012.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.