

Abstract
Insulin regulates heart metabolism through the regulation of insulin-stimulated glucose uptake. Studies have indicated that insulin can also regulate mitochondrial function. Relevant to this idea, mitochondrial function is impaired in diabetic individuals. Furthermore, the expression of Opa-1 and mitofusins, proteins of the mitochondrial fusion machinery, is dramatically altered in obese and insulin-resistant patients. Given the role of insulin in the control of cardiac energetics, the goal of this study was to investigate whether insulin affects mitochondrial dynamics in cardiomyocytes. Confocal microscopy and the mitochondrial dye MitoTracker Green were used to obtain three-dimensional images of the mitochondrial network in cardiomyocytes and L6 skeletal muscle cells in culture. Three hours of insulin treatment increased Opa-1 protein levels, promoted mitochondrial fusion, increased mitochondrial membrane potential, and elevated both intracellular ATP levels and oxygen consumption in cardiomyocytes in vitro and in vivo. Consequently, the silencing of Opa-1 or Mfn2 prevented all the metabolic effects triggered by insulin. We also provide evidence indicating that insulin increases mitochondrial function in cardiomyocytes through the Akt-mTOR-NFκB signaling pathway. These data demonstrate for the first time in our knowledge that insulin acutely regulates mitochondrial metabolism in cardiomyocytes through a mechanism that depends on increased mitochondrial fusion, Opa-1, and the Akt-mTOR-NFκB pathway.
Introduction
The heart requires a constant supply of metabolic substrates and oxygen. Although in the adult heart nearly 70% of energy requirements are met by free fatty acid oxidation, cardiomyocytes have a high metabolic flexibility evidenced by their ability to use different molecules as energetic substrates (1). Although dependence on alternative substrates is well documented, under pathological conditions, such as cardiac hypertrophy and heart failure (2), there is accumulating evidence that glycolysis-derived ATP is also an important energy source that contributes to excitation–contraction coupling in healthy cardiomyocytes (3).
In the heart, insulin regulates glucose transport, glycolytic rate, glycogen synthesis, growth, cardiomyocyte contractility, and survival (4), acting mainly through the insulin receptor substrate family proteins (IRS-1/2), phosphoinositide 3-kinase (PI3K), Akt (5), and the metabolic signaling hub mammalian target of rapamycin (mTOR) (6). Epidemiological evidence associates diabetes with a higher risk of cardiovascular disease (7) that is largely attributable to insulin resistance, hyperlipidemia, and hyperglycemia (8). Although insulin-mediated glucose uptake is impaired in the heart in both obesity and diabetes, changes in insulin signaling to mTOR are less well understood (9). Of note, mitochondrial function is impaired in diabetic individuals (10); their skeletal muscle contains fewer mitochondria than age-matched healthy individuals (11), and the expression of optic atrophy protein-1 (Opa-1) and dynamin-related GTPase mitofusin-2 (Mfn2), two proteins of the mitochondrial fusion machinery, are dramatically altered in obese and insulin-resistant patients (12). Moreover, it has been suggested that mitochondrial dynamics are associated with distinct states of mitochondrial function (13). Thus, we hypothesized that insulin and its downstream signaling pathways regulate cardiomyocyte metabolism by controlling the morphology of the mitochondrial network. The present study demonstrates that insulin stimulates cardiomyocyte mitochondrial metabolism by promoting mitochondrial fusion through a mechanism involving the Akt-mTOR-NFκB signaling pathway and Opa-1.
Research Design and Methods
Reagents
Insulin was obtained from Novo Nordisk. Antibodies against Opa-1 (polyclonal and monoclonal), Mfn1, Mfn2, PGC1α, and oxidative phosphorylation (OXPHOS) subunits were purchased from Abcam. Phospho-mTOR, mTOR, phospho-Akt, Akt, and IκBα antibody were from Cell Signaling, and mtHsp70 antibody was from Affinity BioReagents. Tetramethylrhodamine methyl ester (TMRM), MitoTracker Green FM, and FBS were from Invitrogen. Anti-Drp-1 and cytochrome-c (Cyt-c) antibodies were from Becton Dickinson, whereas anti-Fis1 antibodies were from Enzo Life Sciences. Anti-β-tubulin antibody, carbonyl cyanide m-chlorophenylhydrazone (CCCP), Dulbecco’s modified Eagle’s medium, M199 medium, Bay 11-7085, and other reagents were from Sigma-Aldrich Co. All the inhibitors (rapamycin, Akti VIII, genistein, Y-294002, cycloheximide, and actinomycin D) were from Calbiochem. Protein assay reagents were from Bio-Rad. AsMfn2 and microOpa-1 (both Ad5 serotype) generation and use was as previously described (14). Cardiomyocytes were transduced with adenoviral vectors at a multiplicity of infection (MOI) of 1,000, except for AdIκBα (MOI 300), 48 h before insulin treatment. Empty adenovirus (mock), AdLacZ, or a scrambled microRNA were used as controls.
Culture of Cardiomyocytes
Cardiomyocytes were isolated from hearts of neonatal Sprague-Dawley rats as described previously (15). Rats were bred in the animal breeding facility of the University of Chile. All studies conform to the eighth edition of the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by our Institutional Ethics Review Committee. Primary cell cultures were incubated with or without insulin (10 nmol/L) for 0–24 h in Dulbecco’s modified Eagle’s medium/M199 (4:1) containing 10% FBS in the presence or absence of the different inhibitors.
Cardiomyocyte Transfection
Small interfering RNAs (siRNAs) for Oma-1 I and II and negative control (MISSION; Sigma-Aldrich Co.) were used according to manufacturer instructions. The siRNAs used for knockdown experiments were as follows: negative control, catalog number SIC001; Oma-1 I, sense (5′-GACCUAAACAUCACUGUCA-3′), antisense (5′-UGACAGUGAUGUUUAGGUC-3′); and Oma-1 II, sense (5′-GCCAUAAGAGAGGUCCGGA-3′), antisense (5′-UCCGGACCUCUCUUAUGGC-3′). Both siRNAs were tested, obtaining the best results with the siRNA that we called II, which was the one used for all the subsequent experiments (Supplementary Fig. 4).
Cell Lines Culture
L6 rat muscle cells were maintained in myoblast monolayer culture as previously described (16). For all experiments, cells were seeded in Minimum Essential Alpha Medium (α-MEM) containing 10% FBS for 2 days. After this period, α-MEM was changed to containing 2% FBS for the next 2 days when experiments were executed. Parallel HL-1 human cardiomyocytes were seeded in Claycomb medium supplemented with 10% FBS for 2 days (17), after which the medium was replaced and the experiments performed.
Mitochondrial Dynamics Analysis
Cells were incubated for 30 min with MitoTracker Green FM (400 nmol/L) and maintained in Krebs solution. Confocal image stacks were captured with a Zeiss LSM-5, Pascal 5 Axiovert 200 microscope using LSM 5 version 3.2 image capture and analysis software and a Plan-APOCHROMAT 63x/1.4 Oil DIC objective as previously described (15). Images were deconvolved with ImageJ software (National Institutes of Health), and volume reconstitution of Z-stacks of thresholded images was performed. The number and individual volume of each object (mitochondria) were quantified with the ImageJ 3D Object Counter plug-in. Each experiment was done at least four times, and 16–25 cells per condition were quantified. An increase in mitochondrial volume and a decrease in the number of mitochondria were considered as fusion criteria (15,18). The percentage of cells with a fusion pattern was also determined (18).
Immunofluorescence Studies, Colocalization Analysis, and Cyt-c Release
Cells were fixed, permeabilized, blocked, and incubated with primary antibodies (anti- mtHsp70, Drp-1, Fis1, or Cyt-c). Secondary antibodies were antimouse Alexa 456 for mtHsp70 and Drp-1, antimouse 488 for Cyt-c, and antirabbit IgG 488 for Fis1. For the colocalization analysis, only one focal plane was analyzed. Images obtained were deconvolved, and background was subtracted with the use of ImageJ software. Colocalization between proteins was quantified by the Mander coefficients as previously described (15,19). Cyt-c immunufluorescence and redistribution were evaluated by counting the cells with a diffuse pattern (15,20).
Fluorescence Recovery After Photobleaching
For fluorescence recovery after photobleaching (FRAP) measurements, cells were loaded with 200 nmol/L TMRM for 30 min at 37°C in Krebs solution. TMRM was excited at 561 nm, and fluorescence emission was detected with a 650/710 emission filter. Bleaching of TMRM fluorescence was applied in an ∼25 μm2 square at randomly chosen regions, and fluorescence intensity was normalized to the intensity levels before and after bleaching. The fluorescent images were collected every 0.4–2.0 s and analyzed frame by frame with ImageJ software.
ATP Measurements
ATP content in cells was determined with a luciferin/luciferase-based assay (CellTiter-Glo Kit; Promega).
Flow Cytometry Analysis of Mitochondrial Membrane Potential and Mitochondrial Mass
Mitochondrial membrane potential (Ψmt) and mitochondrial mass were measured after loading cardiomyocytes with TMRM (200 nmol/L) or MitoTracker Green FM (400 nmol/L) for 30 min. Afterward, cells underwent trypsinization, and fluorescence was assessed by flow cytometry (excitation/emission 543/560 and 488/530 for TMRM and MitoTracker Green FM, respectively) with a FACScan system (Becton Dickinson). CCCP (50 μmol/L) and oligomycin (10 μmol/L) for 30 min were used as positive and negative controls for the Ψmt measurements.
Mitochondrial DNA Quantification
Mitochondrial DNA (mtDNA) content was determined by quantitative real-time PCR (qPCR) as previously described (21). β-actin and mitochondrial cytochrome-b were used as nuclear and mtDNA markers, respectively.
qPCR for Opa-1 and Mfn2
Real-time PCR was performed with SYBR green (Applied Biosystems) as previously described (21). Data for each transcript were normalized to both GAPDH and 18S rRNA as internal controls with the 2-ΔΔCt method. Primers used were as follows: Opa-1 rat forward 5′ TGACAAACTTAAGGAGGCTGTG 3′; Opa-1 rat reverse 5′-CATTGTGCTGAATAACCCTCAA-3′; Mfn2 rat forward 5′-CAGCGTCCTCTCCCTCTGAC-3′; Mfn2 rat reverse 5′-GGTCCAGGTCAGTCGCTCAT-3′.
Live Cell Oxygen Consumption Determination
Oxygen consumption determinations in cells were assayed polarographically (22) with a Clark electrode 5331 (YSI Inc.).
Transmission Electron Microscopy
Cells were fixed in 2.5% glutaraldehyde in sodium cacodylate buffer, embedded in 2% agarose, postfixed in buffered 1% osmium tetroxide and stained in 2% uranyl acetate, dehydrated with an ethanol graded series, and embedded in EMbed-812 resin. Thin sections were cut on an ultramicrotome and stained with 2% uranyl acetate and lead citrate. Images were acquired on an FEI Tecnai G2 Spirit electron microscope equipped with an LaB6 source and operating at 120 kV. Measurements of mitochondrial area, circularity, and mitochondrial cristae–integrated density were made with the Multi Measure ROI tool of ImageJ.
Western Blot Analysis
Cell and tissue total protein extracts were analyzed as previously described (15). Protein content was normalized by β-tubulin or β-actin.
Hyperinsulinemic-Euglycemic Clamp Procedure in Mice
Wild-type C57BL6 mice were catheterized under avertin anesthesia, and after a 48-h recovery period, they were fasted overnight and infused with insulin at a constant flow rate and 50% dextrose at a variable rate to maintain a glucose level of 100–150 mg/dL as described by Huang et al. (23). Glucose was measured at 5-min intervals, and glycemia was considered at steady‐state after three successive equal glucometer readings (23). Mice were killed and their hearts harvested for fiber isolation and Western blot analysis.
Mitochondrial Respiration in Permeabilized Cardiac Fibers
Mitochondrial oxygen consumption and ATP production were measured in permeabilized cardiac fibers obtained from 5–7-week-old mice infused with saline (sham or control) or after hyperinsulinemic-euglycemic clamps by previously described protocols (24).
Transgenic Animals, Rapamycin Administration, and Diet
MyrAkt1 mice were previously generated by Walsh and colleagues (25,26). MyrAkt1 mice have two transgenes: tetracycline transactivator (tTA) driven by α-myosin heavy chain promoter and a myrAkt1 transgene under the tetO promoter. MyrAkt1 mice were fed with a doxycycline (DOX) chow 1 g/kg until the predetermined time (8 weeks of age) for myrAkt1 transgene induction. MyrAkt1 mice are on a mixed background. Rapamycin 2 mg/kg/day i.p. was administered 1 day before withdrawal of DOX chow and continued until 1 day before tissue harvest. For high-fat diet studies, mice were fed a diet containing 36% fat (60% calories from fat) (F3282; Bio-Serv, Frenchtown, NJ) starting at weaning (27). Body weight was measured weekly.
Statistical Analysis
Data are the mean ± SEM of the number of independent experiments indicated and represent experiments performed on at least three separate occasions with similar outcomes. Data were analyzed by one-way ANOVA, and comparisons between groups were performed with protected Tukey test. Statistical significance was defined as P < 0.05.
Results
Insulin Promotes Mitochondrial Fusion in Cardiomyocytes
Treatment of rat cardiomyocytes with insulin (10 nmol/L for 3 h) induced the appearance of large interconnected mitochondria mainly in the perinuclear area of cells (Fig. 1A) and significantly increased the percentage of cells displaying fused mitochondria from 19 ± 10% to 85 ± 7% (Fig. 1B, top panel). Mitochondrial network integrity was assessed by measuring the total number of mitochondria per cell and the volume of individual mitochondrion through three-dimensional reconstitution of confocal stacks (15). Insulin gradually increased the mean volume per individual mitochondrion (or detected element), showing a 32% and 129% increase after 0.5 and 3 h of incubation, respectively (Fig. 1B, middle panel), an effect that was reversed after 6 h. Conversely, the number of mitochondria per cell decreased significantly from 150 ± 38 (control) to 61 ± 8 at 3 h of insulin treatment (Fig. 1B, bottom panel). To further validate the assay of mitochondrion volume, we quantified the volume of individual mitochondrion along the different stacks of one micrograph (Fig. 1C, left panel). If the elements were connected during the stack, the software automatically assigned the same color pattern to the same mitochondrion. With this approach, we were able to calculate the volume for each mitochondrion and build histograms with this information (Fig. 1C, right panel). The treatment of cardiomyocytes with insulin (3 h) changed the distribution pattern of mitochondrion size, decreasing the abundance of smaller mitochondria and increasing the number of mitochondria with intermediate and higher values, which corroborated the results obtained with the quantification of the mean mitochondrion volume and total number of mitochondria per cell. Thus, in all subsequent experiments, we used these values as an index of mitochondrial fusion. A 30-min insulin exposure was both required and sufficient to achieve the subsequent fusion 3 h later (Supplementary Fig. 1A and B), but this effect dissipated by 6 h of insulin treatment (Supplementary Fig. 1C and D). To further support the findings with MitoTracker Green fluorescence, we performed the same analysis with indirect immunocytochemistry for mtHsp70 to detect mitochondria (Supplementary Fig. 1E and F). We found that insulin increased mitochondrial mean volume (21%) and decreased the number of mitochondria per cell (from 140 ± 16 to 98 ± 8). With this technique, the decreases in mitochondrial number where similar to those seen with MitoTracker Green; however, the increase in volume was less. We postulate that the difference may be a result of morphological changes during the fixation and permeabilization steps required for this method. Despite these limitations, indirect immunocytochemistry corroborated the trends documented in live cells with MitoTracker Green.
Figure 1.
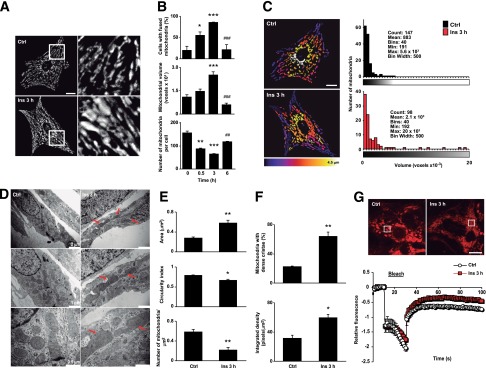
Insulin increases mitochondrial fusion in cultured cardiomyocytes. A: Time course of insulin on mitochondrial morphology. Cells were incubated with insulin (10 nmol/L) at indicated times and then loaded with MitoTracker Green. Multislice imaging reconstitution was obtained by confocal microscopy to show mitochondrial morphology. The scale bar is 10 μm. B: Percentage of cells with fused mitochondria. The individual mitochondrial volume and number of mitochondria per cell were determined. Data are mean ± SEM (n = 4). *P < 0.05, **P < 0.01, and ***P < 0.001 vs. 0 h; ##P < 0.01 and ###P < 0.001 vs. 3 h. C: Representative Z-stack reconstruction of individual mitochondrial structures and volume histograms of the representative images (n = 4). D: Representative transmission electron microscopy images from control and insulin-treated cells. Three different magnifications of the same cell are shown. Arrows indicate fused mitochondria. E and F: Mitochondrial area, circularity index, number of mitochondria per square millimeter, percentage of mitochondria with dense cristae, and cristae-integrated density were quantified from the images in D. Data are from 100 mitochondria from each control and insulin-treated cell examined from three separate experiments. Data are mean ± SEM (n = 3). *P < 0.05 and **P < 0.01 vs. control. G: FRAP analysis of the mitochondrial network (top). Bleaching of TMRM fluorescence was applied in an ∼25-μm2 square at randomly chosen regions where indicated (bleach), and fluorescence intensity was normalized to the intensity levels before and after bleaching. Data are from 15 cells examined from three separate experiments. Ctrl, control; Ins, insulin.
To examine detailed changes in mitochondrion morphology in response to insulin stimulation, we performed electron microscopy analysis. The data showed a significant insulin-dependent increase in the number of large mitochondria and organelle redistribution toward the perinuclear zone (Fig. 1D). In addition, insulin increased mitochondrion size (+107%) and decreased the circularity index (from 0.78 ± 0.02 to 0.66 ± 0.04) and mitochondrial density (36%). Furthermore, insulin increased the percentage of cells displaying a dense cristae structure (64%), which was quantified by the parameter of integrated density for this organelle (Fig. 1E and F).
To evaluate the functional connectivity of the mitochondrial network, we carried out FRAP experiments on cardiomyocytes labeled with TMRM. Figure 1G shows that after randomly bleaching chosen regions in control- or insulin-treated mitochondrial networks, the rate and magnitude of fluorescence recovery was greater in the insulin-treated mitochondria (slope for the first 10 s after the bleach: control 0.030 ± 0.007, insulin 1.647 ± 1.250, P < 0.001), indicating an increased connectivity of the mitochondrial network.
Insulin Increases Mitochondrial Protein Opa-1 Levels and Mitochondrial Function
In mammalian cells, Mfn and Opa-1 are the main regulators of mitochondrial fusion (28). The two isoforms of Mfn (Mfn1 and Mfn2) are localized to the outer mitochondrial membrane (29), whereas Opa-1 is localized to the inner mitochondrial membrane (IMM), where it is required for membrane tethering and fusion. Opa-1 also participates in cristae remodeling, an important determinant of mitochondrial metabolism. At least eight isoforms of Opa-1 exist, grouped as long (L) and short (S), whose distribution depends on the energetic state of the cell. All isoforms are capable of participating in normal IMM fusion (30,31). In the whole heart, five Opa-1 isoforms have been detected, although whether cell-type specificity exists is not known (32).
To elucidate the mechanism of insulin-dependent mitochondrial fusion, changes in Opa‑1 and Mfn2 levels were assessed as well as the levels and localization of the fission protein Drp-1. Western blot analysis showed that Drp1, Mfn1, and Mfn2 levels did not change after insulin treatment, whereas the total content of Opa-1 increased (Fig. 2A). Two different antibodies were used to probe for Opa-1: A polyclonal antibody recognized at least three Opa-1 isoforms in cardiomyocytes (fold increase with insulin 2.0 ± 0.2), and a monoclonal antibody recognized two bands corresponding to the L and S isoforms of the protein (fold increase with insulin 2.5 ± 0.4) (Fig. 2A and B). Of note, mitochondrial localization of the fission protein Drp-1 also decreased in the experimental condition, suggesting that insulin may decrease the rate of mitochondrial fission in addition to stimulating mitochondrial fusion (Supplementary Fig. 2A). Because mitochondrial uncoupling induces Opa-1 proteolysis (33), cells treated with either insulin or CCCP were analyzed with the polyclonal antibody. Whereas insulin promoted an increase in both L and S isoforms, CCCP decreased total Opa-1. This antibody also detected a nonspecific cross-reacting band that did not change with CCCP (Supplementary Fig. 2B). Because the monoclonal Opa-1 antibody was most effective at detecting early changes in Opa-1 protein levels, this antibody was used in all the subsequent in vitro experiments in cardiomyocytes (excepting those where the information of different isoforms was important) as well as in all the in vivo studies. The polyclonal Opa-1 antibody proved preferable for work with other cell types.
Figure 2.

Insulin increases the levels of the mitochondrial Opa-1 protein without changing mitochondrial total mass. A: Total protein extracts were prepared from cells incubated with insulin (10 nmol/L) for the indicated times. Opa-1, Drp-1, mtHsp70, Mfn1, Mfn2, and β-tubulin levels were determined by Western blot. For the determination of Opa-1 protein levels, two antibodies were used: polyclonal (first panel) and monoclonal (fourth panel). Representative Western blots are shown (n = 4). Protein content was normalized with anti-β-tubulin. B: Densitometric analysis of normalized Opa-1 levels from total extracts. Data are mean ± SEM (n = 4). *P < 0.05 and **P < 0.01 vs. 0 h. C: Mitochondrial OXPHOS and PGC1α protein levels were analyzed from total protein extracts from cells incubated with insulin (10 nmol/L) for the indicated times. Representative Western blot, one of four with similar outcomes (n = 4). D: Mitochondrial mass analysis using MitoTracker Green and flow cytometry and real-time PCR for mtDNA. Data are mean ± SEM (n = 5). E: Quantification of ∆Ψm in cells treated with insulin (10 nmol/L) for the times indicated. CCCP (50 μmol/L) and oligomycin (10 μmol/L) were used as negative and positive controls, respectively. Data are mean ± SEM (n = 5). *P < 0.05 and ***P < 0.001 vs. 0 h; #P < 0.05 vs. 3 h. F: Intracellular ATP levels and oxygen consumption determined in control or treated cells with 10 nmol/L insulin for 3 h. For the oxygen consumption assays, respiration was assayed under basal and uncoupled conditions (maximum respiration) with 200 nmol/L CCCP (n = 7 and 4 for ATP and oxygen measurements, respectively). *P < 0.05, **P < 0.01, and ***P < 0.001 vs. 0 h (basal); #P < 0.05 vs. 3 h (basal insulin respiration). Ab, antibody; nDNA, nuclear DNA; Oligo, oligomycin.
Gene expression analysis with qPCR indicated an increase in the Opa1 mRNA transcripts after insulin treatment (Supplementary Fig. 2C). Accordingly, preincubation of cardiomyocytes with either actinomycin D or cycloheximide promoted mitochondrial fission and abrogated all the insulin-dependent effects on mitochondrial morphology (Supplementary Fig. 2D). Cycloheximide also prevented the insulin-dependent increase in Opa-1 protein, supporting a model in which insulin acts by increasing Opa-1 synthesis rather than by inhibiting its degradation (Supplementary Fig. 2E).
To determine whether the changes observed in Opa-1 occurred in conjunction with an increase in mitochondrial biogenesis, we evaluated changes in the total mitochondrial mass by quantifying different constitutive mitochondrial proteins as mtHsp70 (Fig. 2A) and three core proteins of the OXPHOS machinery. Insulin did not change the levels of any of these mitochondrial components, and there was no change in the level of PGC1α, a central regulator of mitochondrial biogenesis (Fig. 2C). These results were further confirmed by flow cytometry in cells stained with MitoTracker Green and by mtDNA abundance quantification (Fig. 2D). These results suggest that insulin increases Opa-1 levels and mitochondrial fusion in cardiomyocytes without altering the overall mitochondrial mass.
Mitochondrial fusion has been associated with increases in mitochondrial OXPHOS (34,35). Therefore, we evaluated Ψmt and cellular ATP content as a measure of mitochondrial function after insulin treatment (Fig. 2E and F). Both Ψmt and ATP levels increased as early as 0.5 h after insulin addition and remained elevated until 3 h (21 and 19%, respectively). However, prolonged stimulation (6 h) resulted in a significant decrease in both parameters. To directly assess changes in mitochondrial OXPHOS, we measured cardiomyocyte oxygen consumption rates at baseline and under maximal uncoupling conditions. Insulin promoted a rise in both baseline and CCCP‑uncoupled cellular oxygen consumption, increasing the respiratory control ratio from 1.7 to 2.2 after 3 h of insulin treatment (Fig. 2F).
Insulin Stimulates Mitochondrial Fusion and Function in the Human Cardiomyocyte Cell Line HL-1 and in Rat Skeletal Muscle Myoblasts
We proceeded to further evaluate the mitochondrial morphology and oxygen consumption rates in the human cardiomyocyte HL-1 and rat skeletal muscle L6 cell lines. Under similar experimental conditions (3 h of insulin treatment [100 nmol/L]), we replicated the response of cardiomyocytes in both cell lines (Supplementary Fig. 3). Collectively, these results indicate that insulin controls mitochondrial function and morphology. Studies were then performed to determine whether these phenomena were mechanistically linked.
Knockdown of Opa-1 and Mfn2 Suppresses the Effects of Insulin on Cardiomyocyte Mitochondrial Morphology and Function
To determine whether mitochondrial fusion changes are directly related to the effects of insulin on mitochondrial function, we used a microRNA directed against Opa-1 (Supplementary Fig. 4A). Knockdown of Opa-1 in cardiomyocytes increased the percentage of cells with fragmented mitochondria (from 25 ± 8% to 80 ± 5%), decreased mitochondrion mean volume (from 1,067 ± 66 to 802 ± 91 voxels), and markedly increased the number of mitochondria per cell (from 150 ± 10 to 183 ± 13) relative to cells treated with a scrambled microRNA (Fig. 3A and B). Opa-1 knockdown prevented all the insulin-induced changes in mitochondrial function (Fig. 3C), suggesting that Opa-1 is necessary for the enhancement in mitochondrial function observed after insulin treatment.
Figure 3.

Downregulation of mitochondrial fusion proteins Opa-1 and Mfn2 alters mitochondrial morphology and the metabolic response to insulin in cardiomyocytes. A: Cells were transduced for 48 h with an adenovirus encoding a scrambled microRNA (miCtrl) or a microRNA against Opa-1 (miOpa-1). Representative confocal images of cells loaded with MitoTracker Green are shown. Cells were transduced with miCtrl (MOI 1,000) or with the miOpa‑1 (MOI 1,000) for 48 h before imaging. Scale bar represents 10 μm. B: Percentage of cells with fragmented mitochondria, individual mitochondrial volume, and number of mitochondria per cell were determined for the miCtrl- and miOpa-1–treated cells. Data are mean ± SEM (n = 4). *P < 0.05 and **P < 0.01 vs. miCtrl. C: After 48 h of transduction with respective adenoviruses, the cells were exposed to insulin (10 nmol/L) for the times indicated and Ψmt, intracellular ATP content, and oxygen consumption were quantified. Data are mean ± SEM (n = 4). *P < 0.05, **P < 0.01, and ***P < 0.001 vs. miCtrl 0 h; #P < 0.05 vs. miCtrl insulin 3 h; &P < 0.05 and &&P < 0.01 vs. respective time with miCtrl. D–F: Cells were transduced for 48 h with an adenovirus coding for an AsMfn2 or an empty adenovirus (Mock) and subjected to the same analysis as miOpa-1–treated cells. Analysis of mitochondrial morphology (E) and metabolism (F) was performed as described previously in Fig. 2. Cells were transduced with AsMfn2 (MOI 1,000) or Mock (MOI 1,000) for 48 h before insulin treatment. Data are mean ± SEM (n = 4). *P < 0.05 vs. Mock 0 h, #P < 0.05 vs. Mock insulin 3 h.
Because Opa-1 also controls mitochondrial cristae remodeling (31), which might have a direct effect on mitochondrial metabolism, independent of its role in fusion, we also increased mitochondrial fission with an antisense adenovirus against Mfn2 (AsMfn2) (Supplementary Fig. 4B) (15). AsMfn2 induced mitochondrial fragmentation, decreased the mitochondrial mean volume by 25%, and prevented the increase in mitochondrial metabolism in response to insulin (Fig. 3D–F), supporting our hypothesis that mitochondrial fusion is a critical element in the metabolic effect of insulin in cardiomyocytes.
To establish a direct relationship between the rise in the Opa-1 protein levels and increased mitochondrial fusion and function, we downregulated the mitochondrial protease Oma-1 in cardiomyocytes. We hypothesize that the downregulation of this protease, known as a regulator of the Opa-1 levels in mitochondria (33,36), could transiently increase Opa-1 levels, enhancing mitochondrial fusion and function. We used two different siRNAs, obtaining the best results with the second one in terms of Oma-1 knockdown; however, knockdown of Oma-1 with either led to an increase in Opa-1 (Supplementary Fig. 4C). Oma-1 knockdown itself stimulated mitochondrial fusion, measured as an increase in mitochondrial mean volume and a decrease in the number of mitochondria per cell (Supplementary Fig. 4D and E), as well as boosted mitochondrial respiration (Supplementary Fig. 4F). Pretreatment with rapamycin did not alter the outcome of Oma-1 knockdown.
Of note, Opa-1 can also help to preserve cardiomyocyte survival because siRNA against Opa-1 decreases cellular survival in cells treated with C2-ceramides, an apoptotic factor known to cause cellular death through mitochondrial fragmentation (15,20). Insulin treatment protected cardiomyocytes from C2-ceramides, whereas insulin protection was lost with knockdown of Opa-1 (Supplementary Fig. 4G).
Insulin Increases Opa-1 and Mitochondrial Function In Vivo
To evaluate the effects of insulin in vivo, C57BL6 mice were subjected to hyperinsulinemic-euglycemic clamps (23). After 2 h of treatment, hearts were harvested and used for protein analysis and oxygen consumption determination in permeabilized cardiac fibers. Akt Ser473 phosphorylation in hearts was significantly increased relative to sham controls, showing that insulin signaling was activated during the glucose clamp (Supplementary Fig. 5A). With use of the monoclonal Opa-1 antibody, the hyperinsulinemic-euglycemic clamp increased the total levels of Opa‑1 protein by 1.8 ± 0.1-fold without changing Mfn2 levels (Fig. 4A and B). Similar changes were observed when analyzing the mRNA levels for Opa-1 and Mfn2 (Fig. 4C). As shown in Fig. 4D, permeabilized fibers obtained from insulin-perfused mice also exhibited enhanced ADP-stimulated (state 3) mitochondrial respiration rate (38%), ATP synthesis (53%), and ATP/O ratio (33%) after incubation with palmitoylcarnitine. Furthermore, hearts from mice fed with a high-fat diet for 16 weeks showed decreased Opa-1 protein levels compared with regular diet controls (Supplementary Fig. 5B and C). These results provide evidence that insulin stimulation of mitochondrial function is correlated with an increase in Opa-1 protein levels in vivo and that loss of insulin sensitivity may lead to a decrease in Opa-1.
Figure 4.

Insulin regulates Opa-1 protein levels and mitochondrial function in vivo. A: Wild-type (WT) mice were subjected to hyperinsulinemic-euglycemic clamps for 2 h. Hearts of sham and operated animals were collected, and the proteins obtained were used for Western blot detection of Opa-1, Mfn2, and β-actin. B: Quantitative analysis of protein expression is shown. Data are mean ± SEM (n = 6 for each group). *P < 0.05 vs. sham. C: Opa-1 and Mfn2 gene expression in hearts isolated after 90 min of hyperinsulinemic-euglycemic clamp. mRNA was amplified by real-time PCR and normalized to β-actin. Data are mean ± SEM (n = 8). *P < 0.05 vs. control 0 h. D: Cardiac fibers obtained from sham (n = 6) and 2-h–clamped (n = 6) animals were saponin permeabilized, incubated with palmitoylcarnitine, and used for the determination of state 3 mitochondrial respiratory parameters, ATP synthesis rate, and ATP/O ratios. State 3 oxygen consumption refers to ADP (1 μmol/L)-stimulated respiration. Data are mean ± SEM. *P < 0.05 vs. sham. E: Opa-1, pAkt, Akt, pS6K, and S6K protein levels in myrAkt1 transgenic mice treated with DOX and rapamycin (Rapa) (2 mg/kg/day). Hearts of sham and treated animals were collected, and the proteins obtained were used for Western blot detection of Opa-1 and β-tubulin. F–H: Quantitative analysis of protein expression is shown. Data are mean ± SEM (n = 4 for each group). *P < 0.05, **P < 0.01, and ***P < 0.001 vs. WT vehicle; ##P < 0.01 and ###P < 0.001 vs. myrAkt1 vehicle; &&&P < 0.001 vs. WT Rapa.
Insulin Controls Mitochondrial Morphology and Function Through the Akt-mTOR-NFκB Signaling Pathway
Insulin controls metabolic homeostasis through the IRS-PI3K-Akt signaling cascade, acting in part through FOXO1 and mTOR (37). Whether this pathway plays a role in the regulation of mitochondrial morphology and function remains unknown. We used a transgenic model where inducible Akt activation in the heart was achieved in mice expressing a myrAkt1 transgene (25,26). Akt activation after DOX withdrawal triggered an increase in Opa-1 protein levels by 1.40 ± 0.02-fold relative to wild-type littermates. This change was inhibited by treating the mice with rapamycin (Fig. 4E and 4F). Moreover, rapamycin inhibited the phosphorylation of S6K (a downstream effector of mTOR) but had no effect on Akt phosphorylation (Fig. 4E, G, and H).
Similar to the in vivo findings, insulin treatment of cultured cardiomyocytes induced a significant increase in both Akt and mTOR phosphorylation at 0.5 and 3 h (Fig. 5A and B). Preincubation with rapamycin (100 nmol/L) inhibited insulin-induced mTOR phosphorylation (Fig. 5B). After 6 h of insulin treatment, phosphorylation of both proteins decreased in agreement with prior evidence suggesting that prolonged insulin stimulation leads to Akt desensitization (38,39). Consistent with this, a second insulin pulse (10 nmol/L for 15 min) after a prolonged insulin exposure (6–24 h) showed a lower Akt phosphorylation compared with cells not previously treated with the hormone (Fig. 5C). To correlate the results obtained in mice with those obtained in rat cardiomyocytes, we used pharmacological inhibitors to determine whether Akt and mTOR signaling were involved in the increase in mitochondrial fusion after insulin treatment. Both Akti (a specific inhibitor of Akt1/2) and rapamycin fully suppressed the insulin-dependent increase in mitochondrion mean volume as well as the decrease in the number of mitochondria per cell (Fig. 5D). The general inhibition of the insulin receptor and PI3K, with genistein and LY‑294002, respectively, showed this same inhibitory effect (Supplementary Fig. 6A). These results confirm the involvement of the IRS-PI3K-Akt-mTOR signaling pathway in regulating mitochondrial morphology. Combined use of Akti and rapamycin had no synergistic effect (Supplementary Fig. 6B), consistent with mTOR acting downstream of Akt in cardiomyocytes (40,41). Preincubation of cardiomyocytes with cytochalasin B (a Glut transporter inhibitor) diminished mitochondrial fusion response to insulin (Supplementary Fig. 6C). This result supports previous studies, demonstrating the need for glucose uptake for full mTOR activation (41). Inhibition of Akt and mTOR with Akti or rapamycin completely abolished the insulin-dependent Opa-1 increase (Fig. 5E).
Figure 5.

Signaling through the IR-PI3K-Akt-mTOR-NFκB pathway mediates the changes observed in mitochondrial morphology, Opa-1 protein levels, and cardiomyocyte mitochondrial metabolism following insulin stimulation. A–C: Time course of insulin-mediated Akt and mTOR phosphorylation. Cardiomyocytes were preincubated with insulin (10 nmol/L) and rapamycin (Rapa) (100 nmol/L) for the times indicated. p-Akt (Ser473), total Akt, p-mTOR (Ser2448), and total mTOR levels were determined by Western blot. Densitometric analysis for normalized p-Akt (A and C) and p-mTOR (B) levels are shown. Data are mean ± SEM (n = 3). *P < 0.05 and **P < 0.01 vs. 0 h. D and E: Effect of preincubation with Akti VIII (Akti) (10 μmol/L) or Rapa (100 nmol/L) on the modulation of mitochondrial fusion (n = 4) (D) or Opa-1 levels induced by insulin (n = 3) (E). *P < 0.05 and **P < 0.01 vs. control and #P < 0.05 vs. insulin 3 h. F: mTOR inhibition abolishes the metabolic boost induced by insulin treatment. Ψmt, intracellular ATP levels, and oxygen consumption rate were determined in cells treated with insulin with or without Rapa preincubation (n = 4). *P < 0.05 and ***P < 0.001 vs. 0 h; #P < 0.05 and ###P < 0.001 vs. insulin 3 h. G and H: Effect of preincubation with Bay 11-7085 (10 μmol/L) on the modulation of mitochondrial fusion (n = 4) (G) and Opa-1 levels induced by insulin (n = 3) (H). **P < 0.01 and ***P < 0.001 vs. control; #P < 0.05, ##P < 0.01, and ###P < 0.001 vs. insulin 3 h. I: Blunted insulin-mediated Opa-1 protein induction by insulin in cardiomyocytes transduced with an AdIκBα adenovirus. Representative Western blots are shown (n = 3). Protein content was normalized using anti-β-tubulin. Data are mean ± SEM. **P < 0.01 vs. AdLacZ without insulin treatment and ##P < 0.01 vs. AdLacZ with insulin 3 h. Ctrl, control; Ins, insulin.
Because mTOR appeared to be required for insulin to influence mitochondrial morphology and metabolism, we assayed the effect of rapamycin treatment on Ψmt, intracellular ATP levels, and oxygen consumption. Fig. 5F shows that the ability of insulin to increase mitochondrial function was completely abrogated after rapamycin preincubation. Collectively, these data strongly suggest that insulin stimulation enhances mitochondrial function in cardiomyocytes through a mechanism that is mediated in part by an mTOR-dependent increase in Opa-1–regulated mitochondrial fusion.
Currently, transcriptional factors involved in the regulation of Opa-1 expression remain unknown. In light of previous reports describing Akt-mTOR–dependent regulation of the transcription factor NFκB (42) and its demonstrated ability to influence insulin responses and mitochondrial function (43), we investigated the relationship between NFκB and mitochondrial morphology. As shown in the Supplementary Figs. 2D and 7A and B, treatment with actinomycin D to inhibit transcription inhibited the insulin-dependent increase in Opa-1 protein levels and mitochondrial fusion. In silico analysis indicated the presence of an evolutionarily conserved NFκB binding site in the Opa-1 promoter (Supplementary Fig. 7C). Moreover, insulin decreased the protein levels of the NFκB repressor IκBα, consistent with an increase in NFκB activity (Supplementary Fig. 7D). Both mitochondrial fusion (Fig. 5G) and increase in Opa-1 protein level induced by insulin (Fig. 5H) were abolished by the NFκB inhibitor Bay 11-7085. Expression of an NFκB super-repressor also cancelled the Opa-1 increase induced by insulin (Fig. 5I). Together, these data suggest that insulin-mediated changes in Opa-1 protein levels and mitochondrial fusion are influenced by NFκB acting downstream of the Akt-mTOR pathway.
Discussion
In cardiomyocytes, mitochondrial dynamics are emerging as fundamental biological processes important for controlling not only mitochondrion shape, but also its function, which can play a key role to determining cell survival. A full understanding of the contribution of mitochondrial dynamics to cardiac physiology and function is still lacking. Our findings are the first to our knowledge to show that insulin controls mitochondrial dynamics in cultured cardiomyocytes by stimulation of mitochondrial fusion without changing mitochondrial biogenesis. This process is controlled by an increase in Opa-1 protein level, leading to an enhanced mitochondrial function. These changes depend, at least in part, on the Akt-mTOR-NFκB pathway.
One of the most interesting findings of the present study is the increase in the levels of the Opa‑1 protein in response to insulin. Mammalian Opa-1 has been studied in in vitro cell culture systems, and beyond its role in mitochondrial fusion, it has been suggested to play roles in preventing apoptosis and maintaining mtDNA (44). Opa-1 also controls cristae structure of the IMM and, thereby, the release of Cyt-c during apoptosis (44). However, studies focusing on the role of Opa-1 in mammalian tissues have been limited because homozygous mutations of Opa-1 lead to embryonic lethality in mice. Mice with heterozygous mutations of Opa-1 show normal phenotypes except for age-dependent degradation of optic nerves (45,46). Recent studies showed that dysregulation of Opa-1 in Oma1 knockout mice induced insulin resistance and impaired glucose homeostasis and thermogenesis. Oma–/– mice develop metabolic defects similar to those seen with high-fat feeding, illustrating the importance of a proper balance between the L and S forms of Opa-1 for maintaining mitochondrial function (47). In Drosophila, an Opa-1 heterozygous mutation is associated with decreased heart rate and cardiac arrhythmia (48). In adult mice with decreased Opa-1 expression (Opa-1–/–), oxidative and respiratory capacity changes only after an important hemodynamic stress. These changes likely result from incompletely understood compensatory adaptations that help to maintain a fused mitochondrial morphology in response to stress (49). The results presented here establish a role for the Opa-1 protein in insulin-dependent control of muscle cell metabolism. These data provide novel insights that may link the changes in Opa-1 levels observed in obesity and diabetes with the pathogenesis and progression of insulin resistance. The results complement the studies of Keller et al. (50), who showed that islet Opa-1 levels decrease before the onset of diabetes in ob/ob mice, and Zhang et al. (51), who used the Cre-loxP system to delete mouse Opa-1 in pancreatic β-cells. In the model by Zhang et al., β-cells lacking Opa-1 maintained normal mtDNA copy numbers; however, the amount and activity of electron transport chain complex IV were significantly decreased, leading to impaired glucose-stimulated ATP production and insulin secretion. These findings in β-cells are consistent with the data from cardiomyocytes presented in the current study.
Today, strong evidence implicates mitochondrial dysfunction in diseases such as diabetes and insulin resistance; however, the relationship among mitochondrial dynamics, mitochondrial biogenesis, and mitophagy in the pathophysiology of these disorders is incompletely understood. In cardiomyocytes, these important aspects of mitochondrial biology have only recently begun to be addressed. Progress may have been limited by the general perception that the highly structured organization of adult ventricular cardiomyocytes prevents mitochondrial dynamics from playing a relevant role in cellular physiology (52). However, new studies are rapidly changing this assumption. Cardiac tissues contain higher levels of proteins involved in mitochondrial dynamics than other tissues (14,15,53), and in vitro data suggest that hyperglycemia induces mitochondrial fragmentation in cardiac cells, resulting in cell death (18). Moreover, Makino et al. (54) demonstrated that coronary endothelial cells from murine diabetic hearts displayed mitochondrial fragmentation associated with reduced Opa-1 protein levels. However, a full understanding of the contribution of mitochondrial dynamics to cardiac physiology and function is still incomplete.
Thus, the present study is significant in that we define an important mechanism linking mitochondrial dynamics in cardiomyocytes with the regulation of mitochondrial energy metabolism by insulin. We propose a novel regulatory pathway involved in the control of Opa-1 protein levels that is mediated by Akt, mTOR, and NFκB. This signaling module has been implicated in the onset of a variety of metabolic diseases. The data suggest an important relationship not only among Opa-1, mitochondrial morphology, and metabolism, but also with insulin signaling, and potentially with the onset of insulin resistance, by revealing the existence of a novel link between mitochondrial morphology and insulin signaling in cardiac and skeletal muscle cells.
Article Information
Acknowledgments. The authors thank Fidel Albornoz (Facultad de Ciencias Químicas y Farmacéuticas, Universidad de Chile) for excellent technical assistance.
Funding. V.P. held a travel PhD fellowship from Bicentennial Program, Comisión Nacional de Investigación Científica y Tecnológica (CONICYT). CONICYT provided fellowships to V.P., H.E.V., A.d.C., J.K., C.P., C.L.-C., and F.J. as well as postdoctoral funding 3110114 to R.T. This work was supported by CONICYT (grant Anillo ACT 1111 to M.C. and S.L.), the American Heart Association (10POST4340064 to M.I., 0640084N to J.A.H., and 06552024 to B.A.R.), the American Heart Association-Jon Holden DeHaan Foundation (to J.A.H.), the National Institutes of Health (HL-075173, HL-080144, and HL-090842 to J.A.H.; HL-072016 and HL-097768 to B.A.R.; and HL-087947 and DK-092065 to E.D.A.), Fondo Nacional de Desarrollo Científico y Tecnológico (grant 1120212 to S.L.), and Fondo de Financiamiento de Centros de Investigación en Áreas Prioritarias (grant 1501006 and 15130011 to S.L.). Additional funding was provided by grant 2009SGR915 from the Generalitat de Catalunya, CIBERDEM (Instituto de Salud Carlos III) to the laboratory of A.Z. and by Ministerio de Educación y Ciencia (SAF2008-03803).
Duality of Interest. No potential conflicts of interest relevant to this article were reported.
Author Contributions. V.P. carried out the project design and all experiments, except those stated separately, and wrote the manuscript. H.E.V. contributed to the project design and manuscript writing. M.I. conducted the qPCR and mtDNA analysis. A.d.C. contributed to the L6 experiments. R.T. carried out the mTOR Western blot analysis. D.J. and Y.Z. contributed to the in vivo glucose clamps studies in mice and the processing of the cardiac samples from those experiments. J.K. contributed to the Akt Western blot analyses, high-fat diet studies, and data discussion. C.P. and C.L.-C. carried out the adenovirus purification and contributed to their standardization. F.J. and J.F. contributed to the normalization of oxygen consumption measures and data discussion. E.N. built the Opa-1 microRNA adenovirus. M.C. guided the adenovirus purification and contributed to the preparation of figures. D.A.B. contributed to the high-fat diet studies. A.K. contributed to the data discussion and manuscript editing. J.A.H. and B.A.R. contributed to the electron microscopy studies, FRAP experiments, and data discussion. E.D.A. conceived the design and discussion of the in vivo experiments with transgenic animals and rapamycin administration. A.Z. provided all the adenoviruses and contributed to the data discussion and project conception. S.L. conceived and supervised the whole project, discussed the data, and edited the final manuscript. S.L. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db13-0340/-/DC1.
References
- 1.Lopaschuk GD, Ussher JR, Folmes CDL, Jaswal JS, Stanley WC. Myocardial fatty acid metabolism in health and disease. Physiol Rev 2010;90:207–258 [DOI] [PubMed] [Google Scholar]
- 2.Neubauer S. The failing heart—an engine out of fuel. N Engl J Med 2007;356:1140–1151 [DOI] [PubMed] [Google Scholar]
- 3.Saks VA, Kuznetsov AV, Vendelin M, Guerrero K, Kay L, Seppet EK. Functional coupling as a basic mechanism of feedback regulation of cardiac energy metabolism. Mol Cell Biochem 2004;256:185–199 [DOI] [PubMed] [Google Scholar]
- 4.Abel ED. Myocardial insulin resistance and cardiac complications of diabetes. Curr Drug Targets Immune Endocr Metabol Disord 2005;5:219–226 [DOI] [PubMed] [Google Scholar]
- 5.Muniyappa R, Montagnani M, Koh KK, Quon MJ. Cardiovascular actions of insulin. Endocr Rev 2007;28:463–491 [DOI] [PubMed] [Google Scholar]
- 6.Abel ED. Insulin signaling in heart muscle: lessons from genetically engineered mouse models. Curr Hypertens Rep 2004;6:416–423 [DOI] [PubMed] [Google Scholar]
- 7.Huxley R, Barzi F, Woodward M. Excess risk of fatal coronary heart disease associated with diabetes in men and women: meta-analysis of 37 prospective cohort studies. BMJ 2006;332:73–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Laakso M. Cardiovascular disease in type 2 diabetes from population to man to mechanisms: the Kelly West Award Lecture 2008. Diabetes Care 2010;33:442–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Abel ED, O’Shea KM, Ramasamy R. Insulin resistance: metabolic mechanisms and consequences in the heart. Arterioscler Thromb Vasc Biol 2012;32:2068–2076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lowell BB, Shulman GI. Mitochondrial dysfunction and type 2 diabetes. Science 2005;307:384–387 [DOI] [PubMed] [Google Scholar]
- 11.Kelley DE, He J, Menshikova EV, Ritov VB. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes 2002;51:2944–2950 [DOI] [PubMed] [Google Scholar]
- 12.Zorzano A, Liesa M, Palacín M. Role of mitochondrial dynamics proteins in the pathophysiology of obesity and type 2 diabetes. Int J Biochem Cell Biol 2009;41:1846–1854 [DOI] [PubMed] [Google Scholar]
- 13.Benard G, Bellance N, Jose C, Melser S, Nouette-Gaulain K, Rossignol R. Multi-site control and regulation of mitochondrial energy production. Biochim Biophys Acta 2010;1797:698–709 [DOI] [PubMed]
- 14.Bach D, Pich S, Soriano FX, et al. Mitofusin-2 determines mitochondrial network architecture and mitochondrial metabolism. A novel regulatory mechanism altered in obesity. J Biol Chem 2003;278:17190–17197 [DOI] [PubMed] [Google Scholar]
- 15.Parra V, Eisner V, Chiong M, et al. Changes in mitochondrial dynamics during ceramide-induced cardiomyocyte early apoptosis. Cardiovasc Res 2008;77:387–397 [DOI] [PubMed] [Google Scholar]
- 16.JeBailey L, Wanono O, Niu W, Roessler J, Rudich A, Klip A. Ceramide- and oxidant-induced insulin resistance involve loss of insulin-dependent Rac-activation and actin remodeling in muscle cells. Diabetes 2007;56:394–403 [DOI] [PubMed] [Google Scholar]
- 17.Claycomb WC, Lanson NA, Jr, Stallworth BS, et al. HL-1 cells: a cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc Natl Acad Sci U S A 1998;95:2979–2984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yu T, Robotham JL, Yoon Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc Natl Acad Sci U S A 2006;103:2653–2658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Manders E, Verbeek FJ, Aten JA. Measurement of co‐localization of objects in dual‐colour confocal images. J Microsc 1993;169:375–382 [DOI] [PubMed] [Google Scholar]
- 20.Parra V, Moraga F, Kuzmicic J, et al. Calcium and mitochondrial metabolism in ceramide-induced cardiomyocyte death. Biochim Biophys Acta 2013;1832:1334–1344 [DOI] [PMC free article] [PubMed]
- 21.Boudina S, Sena S, Theobald H, et al. Mitochondrial energetics in the heart in obesity-related diabetes: direct evidence for increased uncoupled respiration and activation of uncoupling proteins. Diabetes 2007;56:2457–2466 [DOI] [PubMed] [Google Scholar]
- 22.Troncoso R, Vicencio JM, Parra V, et al. Energy-preserving effects of IGF-1 antagonize starvation-induced cardiac autophagy. Cardiovasc Res 2012;93:320–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang J, Gabrielsen JS, Cooksey RC, et al. Increased glucose disposal and AMP-dependent kinase signaling in a mouse model of hemochromatosis. J Biol Chem 2007;282:37501–37507 [DOI] [PubMed] [Google Scholar]
- 24.Boudina S, Sena S, O’Neill BT, Tathireddy P, Young ME, Abel ED. Reduced mitochondrial oxidative capacity and increased mitochondrial uncoupling impair myocardial energetics in obesity. Circulation 2005;112:2686–2695 [DOI] [PubMed] [Google Scholar]
- 25.Shiojima I, Sato K, Izumiya Y, et al. Disruption of coordinated cardiac hypertrophy and angiogenesis contributes to the transition to heart failure. J Clin Invest 2005;115:2108–2118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhu Y, Pereira RO, O’Neill BT, et al. Cardiac PI3K-Akt impairs insulin-stimulated glucose uptake independent of mTORC1 and GLUT4 translocation. Mol Endocrinol 2013;27:172–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Curtis JM, Grimsrud PA, Wright WS, et al. Downregulation of adipose glutathione S-transferase A4 leads to increased protein carbonylation, oxidative stress, and mitochondrial dysfunction. Diabetes 2010;59:1132–1142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Song Z, Ghochani M, McCaffery JM, Frey TG, Chan DC. Mitofusins and OPA1 mediate sequential steps in mitochondrial membrane fusion. Mol Biol Cell 2009;20:3525–3532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol 2003;160:189–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Olichon A, Emorine LJ, Descoins E, et al. The human dynamin-related protein OPA1 is anchored to the mitochondrial inner membrane facing the inter-membrane space. FEBS Lett 2002;523:171–176 [DOI] [PubMed] [Google Scholar]
- 31.Meeusen S, DeVay R, Block J, et al. Mitochondrial inner-membrane fusion and crista maintenance requires the dynamin-related GTPase Mgm1. Cell 2006;127:383–395 [DOI] [PubMed] [Google Scholar]
- 32.Akepati VR, Müller E-C, Otto A, Strauss HM, Portwich M, Alexander C. Characterization of OPA1 isoforms isolated from mouse tissues. J Neurochem 2008;106:372–383 [DOI] [PubMed] [Google Scholar]
- 33.Guillery O, Malka F, Landes T, et al. Metalloprotease-mediated OPA1 processing is modulated by the mitochondrial membrane potential. Biol Cell 2008;100:315–325 [DOI] [PubMed] [Google Scholar]
- 34.Pich S, Bach D, Briones P, et al. The Charcot-Marie-Tooth type 2A gene product, Mfn2, up-regulates fuel oxidation through expression of OXPHOS system. Hum Mol Genet 2005;14:1405–1415 [DOI] [PubMed] [Google Scholar]
- 35.Zanna C, Ghelli A, Porcelli AM, et al. OPA1 mutations associated with dominant optic atrophy impair oxidative phosphorylation and mitochondrial fusion. Brain 2008;131:352–367 [DOI] [PubMed] [Google Scholar]
- 36.McBride H, Soubannier V. Mitochondrial function: OMA1 and OPA1, the grandmasters of mitochondrial health. Curr Biol 2010;20:R274–R276 [DOI] [PubMed] [Google Scholar]
- 37.Cheng Z, Tseng Y, White MF. Insulin signaling meets mitochondria in metabolism. Trends Endocrinol Metab 2010;21:589–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shanik MH, Xu Y, Skrha J, Dankner R, Zick Y, Roth J. Insulin resistance and hyperinsulinemia: is hyperinsulinemia the cart or the horse? Diabetes Care 2008;31(Suppl. 2):S262–S268 [DOI] [PubMed] [Google Scholar]
- 39.Marshall S, Olefsky JM. Effects of insulin incubation on insulin binding, glucose transport, and insulin degradation by isolated rat adipocytes. Evidence for hormone-induced desensitization at the receptor and postreceptor level. J Clin Invest 1980;66:763–772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moschella PC, Rao VU, McDermott PJ, Kuppuswamy D. Regulation of mTOR and S6K1 activation by the nPKC isoforms, PKCepsilon and PKCdelta, in adult cardiac muscle cells. J Mol Cell Cardiol 2007;43:754–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sharma S, Guthrie PH, Chan SS, Haq S, Taegtmeyer H. Glucose phosphorylation is required for insulin-dependent mTOR signalling in the heart. Cardiovasc Res 2007;76:71–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dan HC, Cooper MJ, Cogswell PC, Duncan JA, Ting JP-Y, Baldwin AS. Akt-dependent regulation of NF-kappaB is controlled by mTOR and Raptor in association with IKK. Genes Dev 2008;22:1490–1500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mauro C, Leow SC, Anso E, et al. NF-κB controls energy homeostasis and metabolic adaptation by upregulating mitochondrial respiration. Nat Cell Biol 2011;13:1272–1279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Olichon A, Guillou E, Delettre C, et al. Mitochondrial dynamics and disease, OPA1. Biochim Biophys Acta 2006;1763:500–509 [DOI] [PubMed]
- 45.Davies VJ, Hollins AJ, Piechota MJ, et al. Opa1 deficiency in a mouse model of autosomal dominant optic atrophy impairs mitochondrial morphology, optic nerve structure and visual function. Hum Mol Genet 2007;16:1307–1318 [DOI] [PubMed] [Google Scholar]
- 46.Alavi MV, Bette S, Schimpf S, et al. A splice site mutation in the murine Opa1 gene features pathology of autosomal dominant optic atrophy. Brain 2007;130:1029–1042 [DOI] [PubMed] [Google Scholar]
- 47.Quirós PM, Ramsay AJ, Sala D, et al. Loss of mitochondrial protease OMA1 alters processing of the GTPase OPA1 and causes obesity and defective thermogenesis in mice. EMBO J 2012;31:2117–2133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dorn GW, 2nd, Clark CF, Eschenbacher WH, et al. MARF and Opa1 control mitochondrial and cardiac function in Drosophila. Circ Res 2011;108:12–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Piquereau J, Caffin F, Novotova M, et al. Down-regulation of OPA1 alters mouse mitochondrial morphology, PTP function, and cardiac adaptation to pressure overload. Cardiovasc Res 2012;94:408–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Keller MP, Choi Y, Wang P, et al. A gene expression network model of type 2 diabetes links cell cycle regulation in islets with diabetes susceptibility. Genome Res 2008;18:706–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang Z, Wakabayashi N, Wakabayashi J, et al. The dynamin-related GTPase Opa1 is required for glucose-stimulated ATP production in pancreatic beta cells. Mol Biol Cell 2011;22:2235–2245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hom J, Sheu S-S. Morphological dynamics of mitochondria—a special emphasis on cardiac muscle cells. J Mol Cell Cardiol 2009;46:811–820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Imoto M, Tachibana I, Urrutia R. Identification and functional characterization of a novel human protein highly related to the yeast dynamin-like GTPase Vps1p. J Cell Sci 1998;111:1341–1349 [DOI] [PubMed] [Google Scholar]
- 54.Makino A, Scott BT, Dillmann WH. Mitochondrial fragmentation and superoxide anion production in coronary endothelial cells from a mouse model of type 1 diabetes. Diabetologia 2010;53:1783–1794 [DOI] [PMC free article] [PubMed] [Google Scholar]