

Abstract
Growth hormone secretagogue receptors (GHSRs) in the central nervous system (CNS) mediate hyperphagia and adiposity induced by acyl ghrelin (AG). Evidence suggests that des-AG (dAG) has biological activity through GHSR-independent mechanisms. We combined in vitro and in vivo approaches to test possible GHSR-mediated biological activity of dAG. Both AG (100 nmol/L) and dAG (100 nmol/L) significantly increased inositol triphosphate formation in human embryonic kidney-293 cells transfected with human GHSR. As expected, intracerebroventricular infusion of AG in mice increased fat mass (FM), in comparison with the saline-infused controls. Intracerebroventricular dAG also increased FM at the highest dose tested (5 nmol/day). Chronic intracerebroventricular infusion of AG or dAG increased glucose-stimulated insulin secretion (GSIS). Subcutaneously infused AG regulated FM and GSIS in comparison with saline-infused control mice, whereas dAG failed to regulate these parameters even with doses that were efficacious when delivered intracerebroventricularly. Furthermore, intracerebroventricular dAG failed to regulate FM and induce hyperinsulinemia in GHSR-deficient (Ghsr−/−) mice. In addition, a hyperinsulinemic-euglycemic clamp suggests that intracerebroventricular dAG impairs glucose clearance without affecting endogenous glucose production. Together, these data demonstrate that dAG is an agonist of GHSR and regulates body adiposity and peripheral glucose metabolism through a CNS GHSR-dependent mechanism.
Introduction
Ghrelin, a hormone predominately secreted from the stomach (1), regulates multiple aspects of energy metabolism, including feeding and adiposity (2,3), and is therefore a potential target for therapeutic strategies to prevent or cure obesity. Ghrelin acts directly on pancreatic islets to modulate glucose-stimulated insulin secretion (GSIS), which has made it a popular target for type 2 diabetes therapies (reviewed in 4). Ghrelin circulates in an acylated form (acyl ghrelin [AG]) and a des-acylated form (des-AG [dAG]) (1). Although the exact ratio of circulating AG to dAG varies depending on metabolic status (5,6), the majority of ghrelin circulates in the dAG form. There is currently only one known ghrelin receptor, the growth hormone secretagogue receptor (GHSR) 1a. This receptor is expressed in multiple areas of the central nervous system (CNS), where it mediates AG-induced feeding and adiposity. The presence of an acyl side chain (mainly n-octanoic acid) attached to the ghrelin peptide is required for full agonism of GHSR (1). Some in vitro evidence suggests that dAG interacts with GHSR, although this occurs at significantly lower levels compared with AG (1,7). Therefore, dAG was initially considered an inactive by-product of ghrelin secretion and degradation. Despite this, multiple reports have suggested that dAG acts in peripheral tissues and in the brain to regulate biological actions, including the control of feeding (8,9), body temperature (10), muscle atrophy (11), GSIS (12,13), and lipid metabolism (14,15). All of these actions are attributed to GHSR-independent mechanisms.
Many peripherally secreted hormones such as leptin, glucagon, or glucagon-like peptide 1 act in the CNS to regulate energy metabolism, and interestingly, a growing body of literature has highlighted that these hormones act in the CNS to regulate glucose homeostasis (16–19). Peripherally derived AG acts centrally to promote a positive energy balance (3) and acts directly on pancreatic islet cells to regulate insulin secretion (20,21). However, it is not well established whether AG regulates glucose metabolism through central mechanisms. Rats given chronic central administration of AG have elevated fasting insulin levels (22), which is independent of hyperphagia induced by ghrelin (23) and suggests that AG acts in the CNS to regulate circulating insulin levels.
In addition to the gut, ghrelin is expressed in the brain, where both AG and dAG can be detected (24–26), and therefore, understanding the central actions of these peptides is of interest. In contrast to AG, the central action of dAG in regulating energy and glucose metabolism remains poorly characterized. Some reports suggest that dAG action in the brain plays a role in the control of feeding (8,9). A role for dAG acting in peripheral tissues to regulate insulin secretion has been described (12,27,28), although the CNS control of glucose metabolism by dAG has not been explored. Previous reports demonstrate a lack of dAG-induced activation of GHSR (1), which has led researchers to assume that dAG-mediated effects are solely GHSR independent. However, a potential role for GHSR-mediated dAG action has yet to be directly tested in vivo. The aim of this study was to compare CNS-mediated action of AG and dAG on feeding, adiposity, and GSIS in mice. Furthermore, we aimed to determine whether GHSR mediates the biological actions of dAG using a combination of both in vitro and in vivo techniques.
Research Design and Methods
Peptide Synthesis
Rat AG and dAG were synthesized using in situ neutralization for Boc chemistry, purified by preparative chromatography, and characterized by high-performance liquid chromatography and mass spectral analysis, as described previously (3).
Cell Culture and Transfection
Human embryonic kidney (HEK)-293 cells were grown in Dulbecco’s modified Eagle’s medium. Dulbecco’s modified Eagle’s medium was supplemented with 10% FBS (PAA Laboratories GmbH), 100 units/mL penicillin, 100 µg/mL streptomycin (Biochrom AG), and 2 mmol/L l-glutamine (Invitrogen). For seeding of HEK-293 cells, 48-well plates were coated with poly-l-lysine (Biochrome); 83.3 ng plasmid DNA per well was transfected using 0.9 µL Metafectene (Biontex) per well.
Measurement of Intracellular Inositol Triphosphate Formation
Intracellular inositol triphosphate (IP3) levels were determined using a luciferase reporter assay (Promega). HEK-293 cells were seeded into 48-well plates (5 × 104 cells/well). Equal amounts of receptor-DNA (pcDps) and a reporter construct containing a response element and the firefly luciferase gene under control of the nuclear factor of activated T cells were cotransfected. Cells were stimulated 2 days after transfection for 6 h at 37°C with 5% CO2 with AG (10 nmol/L and 100 nmol/L) or with dAG (10 nmol/L and 100 nmol/L), and lysed with 100 µL 1× Passive Lysis Buffer (Promega). IP3 formation was determined by luciferase activity according to the manufacturer’s instructions.
Animals
Male C57BL/6 mice (12 weeks old; The Jackson Laboratory, Bar Harbor, ME) were maintained on a standard chow diet (Teklad; Harlan). After receiving surgery, animals were singly housed and maintained on a 12-h light/dark cycle at 22°C with free access to food and water unless noted otherwise. Ghsr−/− mice and wild-type (WT) control mice both on a C57BL/6 background were received from Regeneron Pharmaceuticals and bred in our facilities as described previously (29). The average body weight (BW) of the mice used in these studies was 26.8 g. All studies were approved by and performed according to the guidelines of the Institutional Animal Care and Use Committee of the University of Cincinnati.
Subcutaneous and Intracerebroventricular Infusions in Mice
For all surgical procedures, mice were anesthetized using 5% isoflurane in oxygen in an induction chamber and then maintained on 2.5% isoflurane delivered by a nose cone. Subcutaneous infusions of AG and dAG were delivered by subcutaneously implanted osmotic mini-pumps (1007D; Alzet, Cupertino, CA). For intracerebroventricular infusions, mice were stereotaxically implanted (David Kopf Instruments, Tujunga, CA) with a cannula (brain infusion kit #3; Alzet, Cupertino, CA) placed in the lateral cerebral ventricle as previously described (30). A polyethylene catheter attached the cannula to an osmotic mini-pump (1007D; Alzet, Cupertino, CA) that was subcutaneously implanted.
Indirect Calorimetry
For measurements of locomotor activity (LA), energy expenditure (EE), and respiratory exchange ratio (RER), animals were implanted with intracerebroventricular mini-pumps as described above and then placed in an indirect calorimetry system for 7 days (TSE Systems Gmbh, Bad Homburg, Germany).
Intraperitoneal Glucose Tolerance Test
Intraperitoneal glucose tolerance tests (IPGTTs) were performed by injection of glucose (2 g/kg, 20% w/v d-glucose in 0.9% w/v saline; Sigma) after a 16-h fast. Tail vein blood samples for blood glucose (BG) measurements were collected at 0, 10, 30, 60, and 120 min after the injection, and were measured using a handheld glucometer (Freestyle Lite). Tail vein blood samples (60 µL per time point) for insulin measurements were collected at 0, 10, and 60 min. Plasma insulin was determined using either a radioimmunoassay (Sensitive Rat Insulin RIA; Millipore) or an ELISA (Ultra Sensitive Mouse Insulin ELISA Kit; Crystal Chem) as indicated in the corresponding figure legends.
Body Composition Measurements
Whole-body composition (fat mass [FM] and lean mass [LM]) was measured using nuclear magnetic resonance technology (31) (EchoMRI-100; Echomedical Systems, Houston, TX). Initial body composition for all experiments was taken the day prior to surgery (d − 1). For dose-response intracerebroventricular infusion studies (Fig. 2) and indirect calorimetry studies (Fig. 3), final body composition was analyzed on day 7 after surgery. For studies involving analysis of glucose tolerance (Figs. 4, 5, and 6), final body composition was analyzed on day 6 immediately after the IPGTT (∼20 h fasted).
Figure 2.

Effect of chronic intracerebroventricular dAG or AG infusion on food intake, BW, and FM. C57/BL6 mice were given intracerebroventricular infusion of saline, dAG, or AG for 7 days. AG and dAG were infused using increasing doses (0.04, 0.2, 1.0, and 5.0 nmol/day). There was a significant effect of the treatment on 7-day cumulative food intake (P < 0.05 AG vs. dAG, two-way ANOVA). Consistently, a nonlinear regression analysis followed by an extra sum-of-squares F test detected a significant difference between the curves fitting AG and dAG effects (A; P = 0.0185). A two-way ANOVA detected a significant effect of the dose of both AG and dAG on BW (B; P < 0.05) and FM (C; P < 0.001). A nonlinear regression analysis followed by an extra sum-of-squares F test detected a significant difference (P = 0.0318) between the potency of AG and dAG on FM (C). n = 6–10 animals per group. Sal, saline.
Figure 3.

Effect of chronic intracerebroventricular dAG or AG infusion on RER, EE, and LA. C57BL/6 mice were given intracerebroventricular infusion of saline, dAG, or AG (5 nmol/day) for 7 days while being housed in an indirect calorimetry system. AG and dAG had no effect on 7-day cumulative food intake (A). Both AG and dAG increased BW (B; *P < 0.05 vs. saline, one-way ANOVA with Tukey post hoc test) and FM (C; **P < 0.01, ***P < 0.001 vs. saline, one-way ANOVA with Tukey post hoc test). D: Effect of AG and dAG on RER. AG significantly increased RER in the final 4 days of infusion (E; *P < 0.05, AG vs. saline; two-way ANOVA with Bonferroni post hoc test). Neither AG nor dAG altered EE throughout the 7-day infusion period (F and G). H: Effect of AG and dAG on LA. Only AG maintained the suppression of LA in the final 4 days of infusion (I; **P < 0.01, AG vs. saline; two-way ANOVA with Bonferroni post hoc test). n = 7–8 animals per group. Sal, saline.
Figure 4.

Effect of chronic intracerebroventricular dAG or AG infusion on energy and glucose metabolism. C57BL/6 mice were given intracerebroventricular infusion of saline, dAG, or AG for 6 days (5 nmol/day). AG significantly increased the 6-day cumulative food intake (A; *P < 0.05 vs. saline, one-way ANOVA with Tukey post hoc test). After the infusion period, mice that received intracerebroventricular AG or dAG lost less BW (B; **P < 0.01, ***P < 0.001 vs. saline, one-way ANOVA with Tukey post hoc test) and FM relative to saline-treated control animals (C; **P < 0.01, ***P < 0.001 vs. saline, one-way ANOVA with Tukey post hoc test). D: Neither AG nor dAG intracerebroventricular infusion altered intraperitoneal glucose tolerance. Plasma insulin levels were measured by radioimmunoassay and were elevated in mice treated with AG or dAG (E; *P < 0.05 dAG vs. saline; ##P < 0.01 AG vs. saline; two-way ANOVA with Bonferroni post hoc test). n = 8–9 animals per group.
Figure 5.

Effect of chronic subcutaneous dAG or AG infusion on energy and glucose metabolism. C57BL/6 mice were subcutaneously implanted with mini-pumps that infused AG or dAG for 6 days (120 nmol/day). A: Neither ghrelin compound had a significant effect on 6-day cumulative food intake. After the infusion period, mice treated with AG lost less BW (B) and FM relative to saline-treated controls (C), whereas dAG had no effect. Neither AG nor dAG infusion altered intraperitoneal glucose tolerance (D). Plasma insulin levels were measured by radioimmunoassay and were elevated in subcutaneous AG-treated mice at 60 min after glucose injection (E). *P < 0.05, ***P < 0.001 AG vs. saline, one-way ANOVA; ##P < 0.01 AG vs. saline; two-way ANOVA with Bonferroni post hoc test. n = 8–9 animals per group.
Figure 6.

Effect of chronic intracerebroventricular dAG infusion on energy and glucose metabolism in Ghsr−/− mice. Ghsr−/− and age-matched WT mice were given intracerebroventricular infusion of saline or dAG ghrelin for 6 days (5 nmol/day). A: Intracerebroventricular dAG infusion did not alter feeding in WT or Ghsr−/− animals. After the infusion period, WT mice treated with intracerebroventricular dAG lost less BW (B) and FM (C) relative to WT saline-treated controls. Intracerebroventricular dAG infusion had no effect on BW (B) or FM (C) in Ghsr−/− mice. D: Intracerebroventricular dAG did not alter intraperitoneal glucose tolerance in either WT or Ghsr−/− animals. Plasma insulin levels were measured by ELISA and were elevated in WT mice treated with intracerebroventricular dAG (E) compared with WT mice treated with intracerebroventricular saline. E: Intracerebroventricular dAG treatment did not alter glucose-stimulated plasma insulin levels in Ghsr−/− mice compared with intracerebroventricular saline-treated Ghsr−/− mice. *P < 0.05, **P < 0.01, ***P < 0.001, WT saline vs. WT dAG; two-way ANOVA with Bonferroni post hoc test. n = 7–8 animals per group.
Catheterization for Hyperinsulinemic-Euglycemic Clamps
The left common carotid artery and right jugular vein of the mice were catheterized for clamp studies as previously described (32). Immediately upon completion of catheterization, animals were implanted with an intracerebroventricular cannula attached to a subcutaneously placed osmotic mini-pump, as described above. After surgery, animals were given subcutaneous injections of buprenorphine (Buprenex; 0.28 mg/kg; Reckitt Benckiser Healthcare, Richmond, VA), meloxicam (0.25 mg/100 g BW; Metacam, St. Joseph, MO), and 1 mL warm saline.
Hyperinsulinemic-Euglycemic Clamps
On day 5 after surgery, hyperinsulinemic-euglycemic clamps (HECs) were performed as previously described (32). Mice were conscious during the entire experimental procedure. After a 5-h fast, catheter lines were exteriorized and connected to infusion pumps. A 5-µCi bolus of [3-3H]glucose (Perkin Elmer Life Sciences, Boston, MA) was given, followed by a 0.05 µCi/min infusion for 120 min before insulin infusion. At 100 min, a blood sample (50 µL) was taken to determine basal glucose and insulin levels as well as basal glucose turnover. The insulin clamp started at 120 min with a continuous infusion of insulin at a rate of 4 mU/kg/min. During the clamp, the [3-3H]glucose infusion rate was increased to (0.1 µCi/min) to maintain constant specific activity. Dextrose (50 g/100 mL) was infused as necessary to maintain euglycemia (∼130 mg/dL) on the basis of feedback from frequent arterial glucose measurements by handheld glucometers (Accu-check Aviva glucometer). Saline-washed erythrocytes previously collected from donor mice were infused throughout the experimental period to prevent a fall of hematocrit. A 12-µCi bolus of 2[14C]deoxyglucose was given at 198 min. Blood samples (20 µL) were taken every 10 min from t = 200–240 min and processed to determine plasma [3-3H]glucose and 2[14C]deoxyglucose. At the end of the clamp period, mice were killed with an injection of sodium pentobarbital. Tissues were collected and stored at −80°C for further analysis.
Tracer Calculations
Rates of glucose appearance, endogenous glucose production (EGP), and glucose utilization were calculated using steady-state equations, as previously described (33). Briefly, EGP was calculated by determining the total rate of glucose appearance (this comprises both glucose production and any exogenous glucose infused to maintain the desired glycemic levels) and subtracting the amount of exogenous glucose infused. Tissue-specific glucose uptake was calculated from tissue 2-d-glucose content, as previously described (33).
Statistical Analysis
Statistical analysis was performed using Prism version 5.0 (GraphPad Software, San Diego, CA). Statistical significance was determined either by unpaired Student t test, one-way ANOVA followed by Tukey multiple-comparison post hoc test or two-way ANOVA followed by Bonferroni multiple-comparison post hoc test. For nonlinear regression analysis, data sets were fitted using the least-squares method following the equation: [Y = bottom + (top − bottom)/(1 + 10^(LogEC50 − X)], where EC50 is half-maximal effective concentration, and were compared using the extra sum of squares F test. All results are given as the mean ± SEM. Results were considered statistically significant at P < 0.05.
Results
AG and dAG Activate GHSR in HEK-293 Cells
To determine whether dAG activates GHSR in vitro, we incubated GHSR-transfected HEK-293 cells with dAG. We found that 100 nmol/L dAG challenge resulted in a significant increase in IP3 turnover above the basal constitutive activity of the receptor (Fig. 1), which is similar to what other groups have found at this concentration of dAG in stable transfected CHO cells (34). The IP3 accumulation induced by AG was approximately twofold greater than that induced by dAG (Fig. 1).
Figure 1.

Effect of AG and dAG on IP3 formation in HEK-293 cells transfected with human GHSR. Functional in vitro assays were performed in transiently transfected HEK-293 cells to investigate Gq/11-mediated IP3 formation after stimulation with dAG (10 and 100 nmol/L) or AG (10 and 100 nmol/L) for 6 h. Stimulation with 100 nmol/L dAG or 10 and 100 nmol/L AG significantly increased IP3 formation (*P < 0.05; ***P < 0.001). IP3 formation was determined via a reporter construct containing a response element and the firefly luciferase gene under control of the nuclear factor of activated T cells. All data indicate a fold increase of the empty vector (pcDps)-transfected cells (MOCK) basal activity 3,961 ± 528 relative light units for IP3, which was set as 1. Data are the mean ± SEM of two experiments performed in triplicate for Gq/11 activation. Statistical significance was determined by Student unpaired two-tailed t test.
Food Intake and Body Composition After Chronic Intracerebroventricular Infusion of dAG and AG
Our in vitro data suggested that dAG is a weaker agonist of GHSR compared with AG, and, therefore, we hypothesized that infusion of dAG directly into the brain will have similar, but less potent, effects as AG on feeding, BW, and FM. To test this, we performed a 7-day chronic central infusion of either AG or dAG at increasing equimolar doses in mice (0, 0.04, 0.2, 1.0, and 5.0 nmol/day). A two-way ANOVA detected a significant effect of the treatment on cumulative 7-day food intake (P < 0.01 AG vs. dAG) and an extra sum-of-squares F test comparing the curve fits for both treatments (P < 0.05; calculated EC50 = 0.096 vs. 0.253 nmol/day, for AG and dAG, respectively; Fig. 2A). Both treatments increased BW in a dose-dependent manner (P < 0.01 for dose as the main effect, as calculated by two-way ANOVA), with a similar potency (Fig. 2B). Relative to vehicle-infused controls, AG and dAG increased FM to a similar extent with the highest dose tested (5.0 nmol/day). The difference between the dose-response curves of AG and dAG was statistically significant (P < 0.05 by extra sum-of-squares F test for the calculated curve fittings; calculated EC50 = 0.073 vs. dAG; EC50 = 0.902 nmol/day for AG and dAG, respectively) (Fig. 2C). LM did not differ among the groups (data not shown).
LA, EE, and RER During Chronic Intracerebroventricular Infusion of dAG and AG
Increased FM induced by AG can occur in the absence of hyperphagia, which has been demonstrated (3). This occurs, in part, through the suppression of LA (35) and decreased fat utilization as a fuel source (3). We did not detect hyperphagia in dAG-treated animals, and, therefore, we investigated whether dAG increases adiposity through similar alterations in LA and metabolic fuel preference. We gave mice chronic intracerebroventricular infusions of AG or dAG (5 nmol/day) and placed them into an indirect calorimetry system. We did not detect differences in cumulative food intake in either the AG- or dAG-treated animals (Fig. 3A). Both compounds increased BW and FM after 7 days of intracerebroventricular infusion (Fig. 3B and C). Figure 3D depicts the RER of the mice during the 7-day infusion period. When compared with vehicle-treated animals, dAG has a strong tendency to increase RER in the final 4 days of infusion (P = 0.054 saline vs. dAG with treatment as the main effect; two-way ANOVA). However, when comparing all groups, only AG causes a statistically significant increase in RER (Fig. 3E). We did not detect changes in EE in either dAG- or AG-treated animals over the course of the infusion (Fig. 3F) or during the last 4 days of treatment (Fig. 3G), which is consistent with other reports that have demonstrated a lack of effect of AG on EE (3). Figure 3H shows the LA of the animals during the 7-day infusion period. AG suppresses LA during the final 4 days of infusion, whereas dAG has no effect on LA during the final 4 days of infusion (Fig. 3I).
Effects on Glucose Metabolism After Chronic Intracerebroventricular Infusion of AG and dAG
To investigate the central effects on glucose metabolism, mice received chronic intracerebroventricular infusion of AG or dAG (5 nmol/day). Animals were then exposed to an IPGTT after an overnight (16-h) fast. All groups showed similar BG excursions during the IPGTT (Fig. 4D). Both AG and dAG significantly increased insulin levels in comparison with vehicle-treated controls, suggesting a higher GSIS (Fig. 4E). Animals in all groups experienced a decrease in BW and FM over the course of infusion, which was likely due to the combination of an overnight fast and surgical intervention. However, animals receiving either dAG or AG lost less BW (Fig. 4B) and FM (Fig. 4C) relative to vehicle-infused controls. Decreases in LM were similar among all groups (data not shown). During the infusion period, AG significantly increased food cumulative 6-day food intake, whereas dAG did not alter food intake (Fig. 4A).
Effects on Energy and Glucose Metabolism After Chronic Subcutaneous Infusion of AG and dAG
To clarify whether the increases in plasma insulin were due to a centrally mediated action or leakage into peripheral tissues, we chronically infused an equimolar amount of AG and dAG (120 nmol/day) to mice. Animals were exposed to an IPGTT after an overnight (16-h) fast. BG excursions were similar in all groups (Fig. 5D). Plasma insulin levels during the IPGTT were elevated in the AG-treated group 60 min after glucose injection (Fig. 5B). Unlike central administration, subcutaneous infusion of dAG had no effect on GSIS (Fig. 5E). After the infusion period, animals receiving peripheral treatment of AG lost less BW (Fig. 5B) and FM (Fig. 5C) relative to vehicle-treated controls. Decreases in LM after the infusion period were also significantly less in animals receiving chronic subcutaneously delivered AG relative to vehicle-treated animals (saline −7.182 ± 0.5196 g, dAG −5.816 ± 0.3766 g, AG −5.205 ± 0.2920 g; **P < 0.01 saline vs. AG, one-way ANOVA with Tukey post hoc test). In contrast to the intracerebroventricular infusion shown in Fig. 4, chronic subcutaneous administration of a larger dose (24 times the amount given centrally) of dAG did not affect BW or FM (Fig. 5B and C). Food intake was similar among all groups during the infusion period (Fig. 5A).
Energy and Glucose Metabolism in Ghsr−/− Mice Receiving Chronic Intracerebroventricular dAG
To determine whether the regulation of FM and GSIS by central administration of dAG depends on GHSR activation, we performed chronic central infusion of dAG in WT and age-matched Ghsr−/− mice. Administration of an intraperitoneal glucose bolus after an overnight (16-h) fast did not elicit differences in BG excursions regardless of the genotype or the treatment (Fig. 6D). Consistent with Fig. 4E, dAG increased plasma insulin levels in WT mice but not in Ghsr−/− mice during the IPGTT (Fig. 6E). Cumulative 6-day food intake was not altered as a result of intracerebroventricular dAG treatment in WT or Ghsr−/− animals (Fig. 6A). After the infusion period, dAG-treated WT mice lost less BW and FM relative to vehicle-treated mice; however, dAG did not affect BW or FM in Ghsr−/− mice compared with vehicle-treated Ghsr−/− mice (Fig. 6B and C). Changes in LM were similar among all groups (data not shown).
HEC Clamp in Mice Treated With Chronic Intracerebroventricular dAG
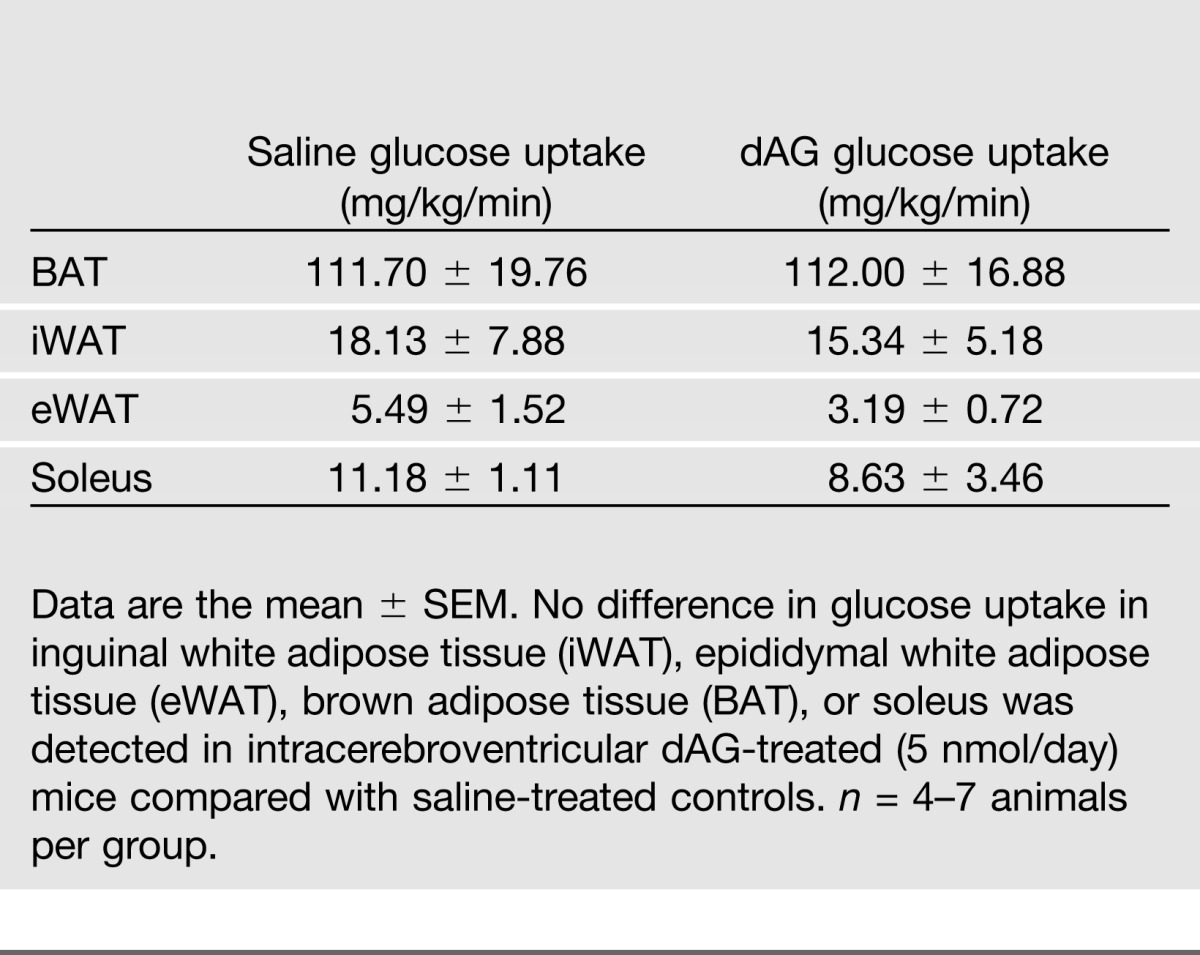
The glucose-stimulated hyperinsulinemia induced by intracerebroventricular AG or dAG treatment could be a result of the development of insulin resistance in these mice. To detect changes in hepatic and/or peripheral insulin sensitivity, we performed an HEC in mice that received chronic intracerebroventricular treatment of dAG (5 nmol/day). We used dAG in order to limit the confounding factor of hyperphagia that we commonly see with AG and also because we confirmed that the effects of dAG are mediated through the target of AG, GHSR. BG was clamped at similar levels between saline and dAG groups (Fig. 7A). The exogenous glucose infusion rate necessary to maintain these steady glucose levels was also similar between groups, indicating similar rates of whole-body insulin sensitivity (Fig. 7B). Saline- and dAG-treated animals had a similar basal EGP level, and insulin similarly suppressed EGP during the clamp period, indicating that central dAG treatment did not alter hepatic insulin sensitivity (Fig. 7C). However, whereas glucose clearance was significantly increased during the clamp in saline-treated animals (Fig. 7D), mice treated with intracerebroventricular dAG had a minor, but significant, impairment in their ability to increase peripheral glucose clearance during the clamp (Fig. 7D). Tissue-specific glucose uptake into soleus, inguinal white adipose tissue, epididymal white adipose tissue, and brown adipose tissue was similar between groups (Table 1).
Figure 7.

Effect of chronic intracerebroventricular dAG infusion on peripheral glucose homeostasis during an HEC. Mice received chronic intracerebroventricular infusion of dAG (5 nmol/day) for 5 days prior to an HEC. A: BG levels were clamped at similar levels in saline- and dAG-treated mice. B: Exogenous glucose infusion rate was similar in saline and dAG-treated mice. C: Basal EGP and suppression of EGP during the HEC were similar in saline- and dAG-treated mice. D: Animals treated with dAG had impaired insulin-mediated glucose disposal (*P < 0.05 saline vs. dAG; t test). n = 9–10 animals per group.
Table 1.
Effect of chronic intracerebroventricular dAG infusion on tissue-specific glucose uptake during an HEC
Discussion
Cell-based assays have demonstrated that ghrelin requires esterification with an acyl side chain on its Ser3 residue to activate GHSR at concentrations within the low-nanomolar range (1). This evidence led to the assumption that the biological effects of dAG are independent of GHSR activation. Here, we combined in vitro and in vivo approaches to demonstrate that dAG can activate GHSR, and activation at the level of the CNS leads to changes in the control of energy balance and glucose metabolism.
Several reports indicate that dAG interacts with GHSR at concentrations within the high-nanomolar or micromolar range. While some data indicate that dAG is a weak GHSR agonist (1,7), other data suggest that dAG is actually a full agonist of GHSR and induces similar maximal receptor stimulation as AG (34). Altogether, our in vitro results are consistent with the literature and demonstrate that dAG possesses GHSR agonism in vitro.
Our experiments consistently show that central infusion of dAG in the brain mimics the effect of AG on adiposity and hyperinsulinemia. Interestingly, the effect of dAG on adiposity is manifested at a higher potency than could be predicted based on functional assays in vitro. It should be noted that although central infusion of AG and dAG always led to relative increase in FM in comparison with the control group, we sometimes observe an absolute reduction in FM at the end of the experiment. We attribute that decrease to the impact of our experimental approach, which combines a major surgical procedure and IPGTT after overnight fasting in a relatively short period of time. This should be considered when comparing our results with others obtained in different conditions.
In contrast to the effect on adiposity and insulin secretion, we could not detect any significant effect of dAG on other parameters that are known to be regulated by central AG action including food intake (2), RER (3), or LA (35). It is noteworthy that the effect of AG on adiposity is more consistent across experiments than the effect on food intake. This difference emphasizes that, although AG commonly increases food intake, this increase is not necessary for the increase in FM induced by AG (3,30,35–37).
The reasons underlying the selectivity of dAG toward the control of adiposity over other AG-mediated effects as well as the relatively higher than expected potency in vivo will require further investigation. The reduced GHSR activation by dAG may explain part of the different outcomes observed between dAG and AG. Alternatively, intrinsic in vivo conditions may contribute to a relative increase in GHSR-mediated dAG biological activity, which may determine the selectivity over the control of different physiological effects. A growing area of interest is the ability of GHSR to heterodimerize with other G-protein-coupled receptors (38–41). Formation of these heteromers modifies GHSR basal activity as well as AG action (40). Furthermore, the amount of ghrelin present influences GHSR heterodimer formation as well as coupling to downstream intracellular signaling systems (39). Investigating whether dAG modulates GHSR heterodimerization with other G-protein-coupled receptors could help to elucidate the function and molecular mechanisms of endogenous dAG.
In addition to recapitulating central AG action on plasma insulin levels (23), we further expand on the role of both AG and dAG in the central regulation of glucose metabolism. We find that intracerebroventricular infusion of either ghrelin isoform causes hyperinsulinemia in response to a glucose bolus. The glucose-stimulated hyperinsulinemia induced by AG is blunted when AG is infused subcutaneously, whereas the glucose-stimulated hyperinsulinemia induced by dAG is completely ablated when dAG is delivered subcutaneously. Considering the increased efficacy of dAG when administered centrally in comparison with peripherally as well as the absence of effect when administered centrally in Ghsr−/− mice, our results demonstrate that CNS-GHSR plays a critical role in mediating the effect of dAG in the control of FM and insulin secretion. Intriguingly, ghrelin is produced in the brain in areas involved in the control of energy balance (24,26,42,43). Whether neurally derived dAG plays a physiological role in the control of GHSR activity requires additional investigation. The loss of effectiveness of dAG regulating FM in Ghsr−/− mice strongly suggests that dAG shares the mechanisms of action whereby AG controls adiposity, including the regulation of the sympathetic nervous system activity (37).
Our results also highlight the role of CNS-GHSR as a positive regulator of insulin secretion. This role is in contrast with the inhibitory action of GHSR in the periphery on GSIS (21,44). We find a stronger effect on GSIS after intracerebroventricular infusion of AG or dAG compared with their peripheral administration. The lack of a peripheral action of dAG is consistent with other groups reporting no effect of chronic peripheral dAG treatment on body composition, glucose tolerance, or GSIS in chow-fed mice (27). The inhibitory action of peripheral GHSR signaling on insulin release could contribute, at least in part, to counteract the CNS-GHSR-mediated hyperinsulinemic action of AG and dAG. Thus, GHSR may play distinct roles in the control of insulin secretion depending on the site of action of AG or dAG. The underpinnings of the control of insulin release by CNS-GHSR activity have not been completely investigated. The HEC suggests that central dAG impairs peripheral glucose clearance, which may contribute to the development of hyperinsulinemia. However, it is likely that other mechanisms, including a direct action in the pancreas, play a role in the hyperinsulinemia induced by the action of both AG and dAG in the brain.
Collectively, our data demonstrate that dAG regulates adiposity and plasma insulin levels through the interaction with GHSR in the CNS and suggest that dAG may be a functional endogenous agonist of GHSR. Furthermore, these data emphasize that adiposity and hyperinsulinemia induced by dAG are independent of changes in food intake. Our data highlight the plurality of the ghrelin system and implicate that further studies are necessary to fully understand each component.
Article Information
Acknowledgments. The authors thank S. Amburgy, D.M. Arble, E. Bartley, A.P. Chambers, J. Holland, J. Pressler, C. Raver, and J. Sorrell for their excellent technical assistance.
Funding. This work was supported by National Institutes of Health (NIH) grant 5R01DK-077975 (D.P.-T.), Deutsche Forschungsgemeinschaft grant DFG BI893/2-1 (H.B.), NIH grant R24DK-093434-01 (P.T.P. and M.H.T.), EurOCHIP grant FP7-HEALTH-2009-241592 (M.H.T.).
Duality of Interest. No potential conflicts of interest relevant to this article were reported.
Author Contributions. K.M.He. was responsible for data collection, study conception and design, data analysis and interpretation, and writing of the manuscript. C.L.P. and A.M. collected, analyzed, and interpreted data. N.O., S.S., and K.M.Ha. contributed to the design of the experimental approach and collected data. D.L.S. provided essential research tools. P.T.P. and M.H.T. advised on study concept and critical revision of the manuscript. R.D. provided essential research tools and contributed to the discussion of the manuscript. H.B. contributed to the study design, interpretation of the data, and discussion of the manuscript. D.A.S. contributed to the study design, and data analysis and interpretation, as well as the discussion and critical revision of the manuscript. D.P.-T. advised on the study conception and design of the experimental approach, and contributed to the writing and critical revision of the manuscript. D.P.-T. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
References
- 1.Kojima M, Hosoda H, Date Y, Nakazato M, Matsuo H, Kangawa K. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature 1999;402:656–660 [DOI] [PubMed] [Google Scholar]
- 2.Nakazato M, Murakami N, Date Y, et al. A role for ghrelin in the central regulation of feeding. Nature 2001;409:194–198 [DOI] [PubMed] [Google Scholar]
- 3.Tschöp M, Smiley DL, Heiman ML. Ghrelin induces adiposity in rodents. Nature 2000;407:908–913 [DOI] [PubMed] [Google Scholar]
- 4.Heppner KM, Tong J, Kirchner H, Nass R, Tschöp MH. The ghrelin O-acyltransferase-ghrelin system: a novel regulator of glucose metabolism. Curr Opin Endocrinol Diabetes Obes 2011;18:50–55 [DOI] [PubMed] [Google Scholar]
- 5.Kirchner H, Gutierrez JA, Solenberg PJ, et al. GOAT links dietary lipids with the endocrine control of energy balance. Nat Med 2009;15:741–745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu J, Prudom CE, Nass R, et al. Novel ghrelin assays provide evidence for independent regulation of ghrelin acylation and secretion in healthy young men. J Clin Endocrinol Metab 2008;93:1980–1987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bednarek MA, Feighner SD, Pong SS, et al. Structure-function studies on the new growth hormone-releasing peptide, ghrelin: minimal sequence of ghrelin necessary for activation of growth hormone secretagogue receptor 1a. J Med Chem 2000;43:4370–4376 [DOI] [PubMed] [Google Scholar]
- 8.Chen CY, Inui A, Asakawa A, et al. Des-acyl ghrelin acts by CRF type 2 receptors to disrupt fasted stomach motility in conscious rats. Gastroenterology 2005;129:8–25 [DOI] [PubMed] [Google Scholar]
- 9.Toshinai K, Yamaguchi H, Sun Y, et al. Des-acyl ghrelin induces food intake by a mechanism independent of the growth hormone secretagogue receptor. Endocrinology 2006;147:2306–2314 [DOI] [PubMed] [Google Scholar]
- 10.Inoue Y, Nakahara K, Maruyama K, et al. Central and peripheral des-acyl ghrelin regulates body temperature in rats. Biochem Biophys Res Commun 2013;430;278–283 [DOI] [PubMed] [Google Scholar]
- 11.Porporato PE, Filigheddu N, Reano S, et al. Acylated and unacylated ghrelin impair skeletal muscle atrophy in mice. J Clin Invest 2013;123:611–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Benso A, St-Pierre DH, Prodam F, et al. Metabolic effects of overnight continuous infusion of unacylated ghrelin in humans. Eur J Endocrinol 2012;166:911–916 [DOI] [PubMed] [Google Scholar]
- 13.Gauna C, Kiewiet RM, Janssen JA, et al. Unacylated ghrelin acts as a potent insulin secretagogue in glucose-stimulated conditions. Am J Physiol Endocrinol Metab 2007;293:E697–E704 [DOI] [PubMed] [Google Scholar]
- 14.Giovambattista A, Gaillard RC, Spinedi E. Ghrelin gene-related peptides modulate rat white adiposity. Vitam Horm 2008;77:171–205 [DOI] [PubMed] [Google Scholar]
- 15.Thompson NM, Gill DA, Davies R, et al. Ghrelin and des-octanoyl ghrelin promote adipogenesis directly in vivo by a mechanism independent of the type 1a growth hormone secretagogue receptor. Endocrinology 2004;145:234–242 [DOI] [PubMed] [Google Scholar]
- 16.Mighiu PI, Yue JT, Filippi BM, et al. Hypothalamic glucagon signaling inhibits hepatic glucose production. Nat Med 2013;19:766–772 [DOI] [PubMed] [Google Scholar]
- 17.Morton GJ, Schwartz MW. Leptin and the central nervous system control of glucose metabolism. Physiol Rev 2011;91:389–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Obici S, Zhang BB, Karkanias G, Rossetti L. Hypothalamic insulin signaling is required for inhibition of glucose production. Nat Med 2002;8:1376–1382 [DOI] [PubMed] [Google Scholar]
- 19.Sandoval DA, Bagnol D, Woods SC, D’Alessio DA, Seeley RJ. Arcuate glucagon-like peptide 1 receptors regulate glucose homeostasis but not food intake. Diabetes 2008;57:2046–2054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dezaki K, Hosoda H, Kakei M, et al. Endogenous ghrelin in pancreatic islets restricts insulin release by attenuating Ca2+ signaling in beta-cells: implication in the glycemic control in rodents. Diabetes 2004;53:3142–3151 [DOI] [PubMed] [Google Scholar]
- 21.Tong J, Prigeon RL, Davis HW, et al. Ghrelin suppresses glucose-stimulated insulin secretion and deteriorates glucose tolerance in healthy humans. Diabetes 2010;59:2145–2151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nesić DM, Stevanović DM, Ille T, Petricević S, Masirević-Drasković G, Starcević VP. Centrally applied ghrelin affects feeding dynamics in male rats. J Physiol Pharmacol 2008;59:489–500 [PubMed] [Google Scholar]
- 23.Kim MS, Namkoong C, Kim HS, et al. Chronic central administration of ghrelin reverses the effects of leptin. Int J Obes Relat Metab Disord 2004;28:1264–1271 [DOI] [PubMed] [Google Scholar]
- 24.Cowley MA, Smith RG, Diano S, et al. The distribution and mechanism of action of ghrelin in the CNS demonstrates a novel hypothalamic circuit regulating energy homeostasis. Neuron 2003;37:649–661 [DOI] [PubMed] [Google Scholar]
- 25.Mondal MS, Date Y, Yamaguchi H, et al. Identification of ghrelin and its receptor in neurons of the rat arcuate nucleus. Regul Pept 2005;126:55–59 [DOI] [PubMed] [Google Scholar]
- 26.Sato T, Fukue Y, Teranishi H, Yoshida Y, Kojima M. Molecular forms of hypothalamic ghrelin and its regulation by fasting and 2-deoxy-d-glucose administration. Endocrinology 2005;146:2510–2516 [DOI] [PubMed] [Google Scholar]
- 27.Delhanty PJ, Huisman M, Baldeon-Rojas LY, et al. Des-acyl ghrelin analogs prevent high-fat-diet-induced dysregulation of glucose homeostasis. FASEB J 2013;27:1690–1700 [DOI] [PubMed] [Google Scholar]
- 28.Gauna C, Delhanty PJ, van Aken MO, et al. Unacylated ghrelin is active on the INS-1E rat insulinoma cell line independently of the growth hormone secretagogue receptor type 1a and the corticotropin releasing factor 2 receptor. Mol Cell Endocrinol 2006;251:103–111 [DOI] [PubMed] [Google Scholar]
- 29.Pfluger PT, Kirchner H, Günnel S, et al. Simultaneous deletion of ghrelin and its receptor increases motor activity and energy expenditure. Am J Physiol Gastrointest Liver Physiol 2008;294:G610–G618 [DOI] [PubMed] [Google Scholar]
- 30.Heppner KM, Chaudhary N, Müller TD, et al. Acylation type determines ghrelin’s effects on energy homeostasis in rodents. Endocrinology 2012;153:4687–4695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tinsley FC, Taicher GZ, Heiman ML. Evaluation of a quantitative magnetic resonance method for mouse whole body composition analysis. Obes Res 2004;12:150–160 [DOI] [PubMed] [Google Scholar]
- 32.Kim DH, Sandoval D, Reed JA, et al. The role of GM-CSF in adipose tissue inflammation. Am J Physiol Endocrinol Metab 2008;295:E1038–E1046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ayala JE, Bracy DP, Julien BM, Rottman JN, Fueger PT, Wasserman DH. Chronic treatment with sildenafil improves energy balance and insulin action in high fat-fed conscious mice. Diabetes 2007;56:1025–1033 [DOI] [PubMed] [Google Scholar]
- 34.Gauna C, van de Zande B, van Kerkwijk A, Themmen AP, van der Lely AJ, Delhanty PJ. Unacylated ghrelin is not a functional antagonist but a full agonist of the type 1a growth hormone secretagogue receptor (GHS-R). Mol Cell Endocrinol 2007;274:30–34 [DOI] [PubMed] [Google Scholar]
- 35.Pfluger PT, Castañeda TR, Heppner KM, et al. Ghrelin, peptide YY and their hypothalamic targets differentially regulate spontaneous physical activity. Physiol Behav 2011;105:52–61 [DOI] [PubMed] [Google Scholar]
- 36.Perez-Tilve D, Heppner K, Kirchner H, et al. Ghrelin-induced adiposity is independent of orexigenic effects. FASEB J 2011;25:2814–2822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Theander-Carrillo C, Wiedmer P, Cettour-Rose P, et al. Ghrelin action in the brain controls adipocyte metabolism. J Clin Invest 2006;116:1983–1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kern A, Albarran-Zeckler R, Walsh HE, Smith RG. Apo-ghrelin receptor forms heteromers with DRD2 in hypothalamic neurons and is essential for anorexigenic effects of DRD2 agonism. Neuron 2012;73:317–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Park S, Jiang H, Zhang H, Smith RG. Modification of ghrelin receptor signaling by somatostatin receptor-5 regulates insulin release. Proc Natl Acad Sci USA 2012;109:19003–19008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rediger A, Piechowski CL, Yi CX, et al. Mutually opposite signal modulation by hypothalamic heterodimerization of ghrelin and melanocortin-3 receptors. J Biol Chem 2011;286:39623–39631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schellekens H, van Oeffelen WE, Dinan TG, Cryan JF. Promiscuous dimerization of the growth hormone secretagogue receptor (GHS-R1a) attenuates ghrelin-mediated signalling. J Biol Chem 2013;288:181–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kageyama H, Kitamura Y, Hosono T, et al. Visualization of ghrelin-producing neurons in the hypothalamic arcuate nucleus using ghrelin-EGFP transgenic mice. Regul Pept 2008;145:116–121 [DOI] [PubMed] [Google Scholar]
- 43.Menyhért J, Wittmann G, Hrabovszky E, et al. Distribution of ghrelin-immunoreactive neuronal networks in the human hypothalamus. Brain Res 2006;1125:31–36 [DOI] [PubMed] [Google Scholar]
- 44.Tong J, Prigeon RL, Davis HW, Bidlingmaier M, Tschöp MH, D’Alessio D. Physiologic concentrations of exogenously infused ghrelin reduces insulin secretion without affecting insulin sensitivity in healthy humans. J Clin Endocrinol Metab 2013;98:2536–2543 [DOI] [PMC free article] [PubMed] [Google Scholar]