

Transcriptional mechanisms responsible for low DAPK2 levels in particular AML subtypes.
Keywords: APL, neutrophil, acute myeloid leukemia, DRP-1
Abstract
DAPK2 is a proapoptotic protein that is mostly expressed in the hematopoietic tissue. A detailed DAPK2 expression analysis in two large AML patient cohorts revealed particularly low DAPK2 mRNA levels in APL. DAPK2 levels were restored in APL patients undergoing ATRA therapy. PML-RARA is the predominant lesion in APL causing transcriptional repression of genes important for neutrophil differentiation. We found binding of PML-RARA and PU.1, a myeloid master regulator, to RARA and PU.1 binding sites in the DAPK2 promoter. Ectopic expression of PML-RARA in non-APL, as well as knocking down PU.1 in APL cells, resulted in a significant reduction of DAPK2 expression. Restoring DAPK2 expression in PU.1 knockdown APL cells partially rescued neutrophil differentiation, thereby identifying DAPK2 as a relevant PU.1 downstream effector. Moreover, low DAPK2 expression is also associated with C/EBPα-mutated AML patients, and we found C/EBPα-dependent regulation of DAPK2 during APL differentiation. In conclusion, we identified first inhibitory mechanisms responsible for the low DAPK2 expression in particular AML subtypes, and the regulation of DAPK2 by two myeloid transcription factors underlines its importance in neutrophil development.
Introduction
Hematopoiesis is orchestrated by transcription factors that regulate, for example, differentiation, cell-cycle arrest, or hematopoietic growth factor genes. In neutrophil differentiation, the two myeloid transcription factors, PU.1 and C/EBPα, directly regulate genes important in myeloid differentiation, such as GM-CSFR [1], G-CSFR [2, 3] and M-CSFR [4] or CD11b [5, 6]. Attenuated expression or functions of these two transcription factors lead to a block in myeloid differentiation, which is a hallmark of AML. The AML subtype, APL, is characterized by the occurrence of the balanced translocation t(15;17) that leads to the expression of the oncogenic fusion protein PML-RARα. PML-RARα confers a differentiation arrest at the promyelocytic stage and contributes to increased self-renewal of leukemic blast cells [7]. In the clinics, APL patients are treated successfully with a combination of differentiation therapy using pharmacological doses of ATRA and chemotherapy [8]. ATRA treatment resolves the differentiation block in APL cells via the proteasomal degradation of PML-RARα [9], and PML-RARα also inhibits transcription of PU.1, and significant low PU.1 levels were found in this particular subtype of AML [10]. In addition, Wang et al. [11] showed that PML-RARα binds to and represses PU.1-mediated transactivation of myeloid genes in APL, describing a new mechanism of PML-RARα repression. On the other hand, C/EBPα is dysregulated frequently in AML by a different mechanism. Approximately 10% of AML patients show dominant-negative mutations in the C/EBPα coding region [12], and 3–15% of AML patients are positive for C/EBPα promoter hypermethylation [13–16]. In addition, C/EBPα expression is suppressed by the leukemogenic fusion proteins AML1-ETO, AML1-MDS-associated protein 1-ecotropic virus integration site 1 protein homolog, or core-binding factor β–smooth muscle myosin heavy chain [17–19]. Moreover, re-expressing PU.1 or C/EBPα in AML cells can restore differentiation of the leukemic blasts [20, 21].
DAPK2, also known as DRP-1, belongs to a family of proapoptotic Ca2+/calmodulin-regulated serine/threonine kinases and activates programmed cell death in different tissues [22]. DAPK1, its closest homolog, is inactivated frequently in a variety of human tumors by epigenetic silencing and displays tumor suppressor activity by positively regulating cell death [23]. Indeed, it was shown that loss of DAPK1 expression increases the resistance of CLL cells to apoptosis, and low levels of DAPK1 are associated with a CLL phenotype [24, 25]. Similar to DAPK1, DAPK2 participates in different cell death pathways, such as in TNF-α/FASR-, E2F1-, or epigallocatechin-3-gallate-mediated cell death [26–29]. Moreover, we reported previously a specific function for DAPK2 as an enhancer of neutrophil and erythroid differentiation [30, 31]. These data suggest that loss of DAPK2 expression may contribute to myeloid leukemogenesis.
To unravel for the first time possible DAPK2 inhibitory mechanisms in AML, we analyzed DAPK2 expression in large cohorts of AML patients with clearly defined chromosomal aberrations and mutations. We found particular low levels of DAPK2 in APL, as well as C/EBPα-mutated AML patients, and describe for the first time PML-RARα-mediated repression of DAPK2. Moreover, we demonstrate that induction of DAPK2 during neutrophil differentiation of APL cells is dependent on PU.1 and C/EBPα levels.
MATERIALS AND METHODS
Primary cells, cell lines, and cell culture conditions
Fresh leukemic blast cells from untreated AML patients at diagnosis obtained at the Inselspital Bern (Switzerland) were classified according to the French-American-British classification and cytogenetic analysis. All leukemia samples had blast counts >90% after separation of mononuclear cells using a Ficoll gradient (Lymphoprep; Axon Lab AG, Switzerland), as described previously [32].
Protocols and use of 67 human samples acquired in Bern were approved by the Cantonal Ethical Committee at the Inselspital. A cohort of 101 samples from patients with a diagnosis of primary AML was enrolled on HOVON/SAKK Protocols −04, −04A, −29, and −42 (available at www.hovon.nl) between 1987 and 2006 [32–36]. All patients provided written, informed consent in accordance with the Declaration of Helsinki. Our findings were validated in a second AML cohort of 175 patients from the Munich Leukemia Laboratory. Patient data from the different cohorts are summarized in Supplemental Tables 1–3.
The isolation of primary neutrophils (purity >95%) was performed by separating blood cells from healthy donors using polymorphprep (Axon Lab AG). CD34+ cells from cord blood or bone marrow were isolated as described [30].
The human APL cell lines NB4, NB4-R2, and HT93 and the CML cell lines K562-CEBPα-p42-ER, K562-CEBPα-p30-ER, K562-CEBPα-BRM2-ER, and K562-empty-ER, as well as the human non-small cell lung cancer cell line H1299 and the human promonocytic cell line U937, were maintained in RPMI 1640 with 10% FCS, 50 U/mL penicillin, and 50 μg/mL streptomycin in a 5% CO2-95% air-humified atmosphere at 37°C. 293T cells were maintained in DMEM (Sigma-Aldrich, St. Louis, MO, USA), supplemented with 5% FBS, 1% penicillin/streptomycin, and 1% Hepes (Sigma-Aldrich), and kept in 7.5% CO2 at 37°C.
For differentiation experiments, APL cells were seeded at a density of 0.2–0.4 × 106/ml and treated with 1μM ATRA (dissolved in DMSO) as indicated. Successful neutrophil differentiation of cells was assessed by light microscopy using May-Grünwald-Giemsa (Merck, Darmstadt, Germany)-stained cells and by increased expression of CD11b, CEBPE, and G-CSFR mRNA expression. K562 CEBPα-ER cells were differentiated as described [37] by adding 5μM 4-OHT to the media.
condHoxb8-immortalized murine neutrophil progenitor cells (SCF-condHoxb8) were generated to bone marrow of C57BL/6 WT mice based on the protocol by Wang et al. [38]. In contrast to Wang et al. [38], who used an estrogen-responsive Hoxb8-ER fusion protein, we used untagged WT Hoxb8 under the control of the 5× upstream activating sequence yeast promoter. Hoxb8 expression is induced by the permanently expressed GAL4-DBD_ER-LBD T2 mutant_VP16 TD (GEV16) transcription factor in the presence of 4-OHT [39, 40]. The cells were maintained in RPMI 1640 with 10% FCS, 5% of CHO/SCF conditioned medium as a source for murine SCF, 0.1 μM 4-OHT, 50 U/mL penicillin, and 50 μg/mL streptomycin in a 5% CO2-95% air-humified atmosphere at 37°C. SCF-condHoxb8 cells were differentiated, as described [38], by removing 4-OHT from the medium in the presence of SCF. Neutrophil differentiation was assessed by Gr-1 surface marker and G-Csfr mRNA expression.
ChIP
ChIP assays were done according to the protocol provided by Activ Motif (Carlsbad, CA, USA). Following DNA purification, PCR was performed using a JumpStart Taq (Sigma-Aldrich) and the following primers: DAPK2 promoter 700 bp forward 5′-GAGAAGGCGTGATGGTGAGAG-3′, reverse 5′-AGGAAGCCCCACTGAGGAATAGG-3′; DAPK2 promoter 900 bp forward 5′-CATGGGTGACTTAGGGATGG-3′, reverse 5′-ACTTGGGAATGGGTTCCTCT-3′; DAPK2-negative control forward 5′-GGTGGCTATCAACAGAAGAA-3′, reverse 5′-ACTATATGTTGGCGTTCTGG-3′. Anti-PU.1, anti-RARA, and anti-PML (Santa Cruz Biotechnology, Santa Cruz, CA, USA) were used for ChIP assays.
Reporter assays and transient transfections
The two DAPK2 promoter constructs used in this study have been described earlier [28]. H1299 cells were transfected with lipofectamine (Invitrogen, Carlsbad, CA, USA), according to the manufacturer's protocol. Briefly, cells were transfected with 40 ng reporter, 80 ng or 120 ng effectors, and 5 ng pRL-TK expression plasmid for Renilla luciferase (Promega, Madison, WI, USA). Reporter expression was analyzed using the Dual-Glo luciferase assay system (Promega). Firefly luciferase activity of each sample was normalized to its Renilla luciferase activity, and the fold activation was obtained by setting the value of empty vector control to 1.0.
TaqMan LDA and qPCR
RNA extraction, RT-PCR and LDA measurements, and data analysis were done as described [32]. Total RNA was extracted using the RNeasy Mini Kit and the RNase-Free DNase Set, according to the manufacturer's protocol (Qiagen, Hombrechtikon, Switzerland). Total RNA was reverse-transcribed using random primers (Roche Diagnostics, Indianapolis, IN, USA) and MMLV RT (Promega). PCR and fluorescence detection were performed using the ABI PRISM 7700 Sequence Detection System (Applied Biosystems, Rotkreuz, Switzerland). For quantification of DAPK2, CEBPE, G-CSFR (CSF3R), and CEBPα mRNA in human cells, we used TaqMan Gene Expression Assays Hs00204888_m1, Hs00357657_m1, Hs00167918_m1, and Hs00269972_s1 (Applied Biosystems), respectively. HMBS and PU.1 primers and probes have been described [32]. For quantification of Gapdh, Dapk2, Pu.1, and G-Csfr (Csf3r) mRNA in mouse SCF-condHoxb8 neutrophil progenitor cells, we used TaqMan Gene Expression Assays Mm01143545_m1, Mm00802402_m1, Mm03048233_m1, and Mm00432735_m1 (Applied Biosystems), respectively.
Cell lysate preparation and Western blotting
Whole cell extracts were prepared using RIPA lysis buffer, supplemented with 8 M urea, according to the protocol found at www.abcam.com (Abcam, Cambridge, MA, USA). Total protein (40–60 μg) was loaded on a 10% or 12% denaturing polyacrylamide gel. Blots were incubated with the primary antibodies in TBS 0.05% Tween-20/5% milk overnight at 4°C, incubated with secondary antibodies goat anti-rabbit IRDye 800CW and goat anti-mouse IRDye 680LT (LI-COR Biosciences, Lincoln, NE, USA) at 1:5000–10,000 for 1 h at room temperature, and analyzed using the Odyssey infrared imaging system detection (LI-COR Biosciences). Primary antibodies used were anti-DAPK2 1:500 (ProSci, Poway, CA, USA), anti-PU.1 1:1000 (Cell Signaling Technology, Danvers, MA, USA), anti-RARA (Santa Cruz Biotechnology), anti-C/EBPα 1:500 (Abcam), and anti-GAPDH 1:5000 (Millipore, Darmstadt, Germany). All antibodies used were compatible for mouse and human tissue.
Lenti-/retrovirus preparation and transduction of target cells
Lentiviral vectors expressing shRNAs targeting PU.1 (SHCLNG-NM_003120), PML (SHCLNG-NM_002675), DAPK2 (SHCLNG-NM_014326), or C/EBPα (SHCLNG-NM_004364) were purchased from Sigma-Aldrich. All vectors contain a puromycin antibiotic-resistance gene for selection of transduced mammalian cells. Lentivirus production and transduction were done as described [30, 41]. The following shRNA target sequence pLKO.1-puro lentiviral vectors were used to target PU.1 (shPU1_1: NM_003120.1-256s1c1 or shPU1_2: NM_003120.1-928s1c1), PML (shPML_1: combination of NM_002675.3-680s21c1 and NM_002675.3-1982s21c1 or shPML_2: combination of NM_003120.1-256s1c1 and NM_003120.1-928s1c1), DAPK2 (shDAPK2_1: NM_014326.x-759s1c1 or shDAPK2_2: NM_014326.x-1735s1c1), or C/EBPα (shC/EBPα_1: NM_004364.2-942s1c1 or shC/EBPα_2: NM_004364.2-1335s1c1) in NB4 and HT93 APL cells. Transduced HT93 and NB4 cell populations were selected with 1.5 μg/ml puromycin for 4 days, and knockdown efficiency was assessed by qPCR and Western blot analysis. Ectopic DAPK2 expression using lentiviral vectors was done as described [30].
The pCl10A1 retroviral packaging plasmid [42] was cotransfected with pBabe-puro, pBabe-PUER-puro, or pLPCX/HA-PML-RARΑ (kindly provided by Dr. H. Yoshida) in 293T cells. After 24 h, the cell-culture medium was removed, and cells were incubated for 2 min with PBS/15% glycerol was removed, and fresh media were added. Viral supernatants were harvested 48 h later. NB4 and U937 cells were transduced for 24 h in the presence of 8 μg/mL polybrene. Transduced NB4 and U937 cell populations were selected with 1.5 μg/ml puromycin for 4 days.
Statistical analysis
Nonparametric MWU tests were applied to compare the difference between two groups using Prism software. P < 0.05 was considered statistically significant.
RESULTS
Significant repression of DAPK2 in primary AML patient samples with particularly low levels in APL
The mechanism leading to low DAPK2 expression in AML patients remained unidentified. In a first approach to test if recurrent chromosomal aberrations in AML are associated significantly with low DAPK2 expression, we determined DAPK2 expression in 168 AML patient samples from the Bern and HOVON/SAKK studies with well-defined molecular subtypes. DAPK2 mRNA levels in AML samples were compared with that in granulocytes from healthy donors (n=24) and in CD34+ progenitor cells (n=5). We could detect DAPK2 in 151/168 primary AML patients (Supplemental Table 1), 24/24 granulocytes from healthy donors, and five of five CD34+ progenitor cells. DAPK2 mRNA is downregulated significantly in AML patient samples and CD34+ progenitor cells compared with granulocytes from healthy donors (Fig. 1A, left panel; P<0.001). Interestingly, no such down-regulation in AML was found for the DAPK2 relative DAPK1 (Supplemental Fig. 1), pointing to a specific role of DAPK2 in granulocytic differentiation. A more detailed analysis of DAPK2 mRNA expression based on recurrent genetic abnormalities revealed significantly lower DAPK2 mRNA levels in t(15;17) APL compared with t(8;21), inv(16), normal or complex karyotype AML patients, as well as nonleukemic CD34+ progenitor cells (Fig. 1A, right panel). With the use of data from a microarray profiling experiment, performed in samples from a second cohort of 175 AML patients (Munich cohort; ref. [43]), we were able to validate our findings from the first patient cohort (Fig. 1B). Furthermore, PU.1 expression correlates positively with DAPK2 expression in AML patient samples (Fig. 1C).
Figure 1. DAPK2 is down-regulated in AML.

(A) DAPK2 mRNA levels in AML blasts from the HOVON/SAKK/Bern cohort in granulocytes from healthy donors and in CD34+ progenitor cells were quantified using qPCR. The relative mRNA expression levels were given as differences in comparative threshold (dCt) values compared with mRNA levels for the housekeeping genes GAPDH and ABL1. Values are the differences in Ct values between DAPK2 and the housekeeping genes and ABL1 and GAPDH. ABL1=Abelson tyrosine-protein kinase 1, CK=complex karyotype, G=granulocytes, NK=normal karyotype. (B) DAPK2 mRNA expression in AML patient samples from the Munich cohort was measured by microarray hybridization. (C) Positive correlation between DAPK2 and PU.1 mRNA levels in the HOVON/SAKK/Bern AML patient cohorts (n=133), as measured by qPCR. (D) DAPK2 mRNA levels during a short-term follow-up of two newly diagnosed APL patients treated with ATRA were evaluated. Values are normalized to the housekeeping genes ABL1 and HMBS and are given as n-fold increase relative to Day (d) 0 of treatment. (E) DAPK2 mRNA levels in a long-term follow-up of three APL patients receiving ATRA (45 mg/m2) daily for up to 60 days. Values are the differences in Ct values between DAPK2 and the housekeeping genes ABL1 and HMBS. (F and G) DAPK2 is up-regulated during neutrophil differentiation of APL cells. DAPK2 mRNA and protein levels in NB4, HT93, and ATRA-resistant NB4-R2 APL cells were measured by qPCR and Western blotting, respectively. mRNA levels were normalized to the HMBS housekeeping gene and the nontreated control cells. Results of at least three independent experiments are shown as n-fold regulation to control cells. Total protein was extracted and submitted to immunoblotting using anti-DAPK2. GAPDH is shown as a loading control. MWU: *P < 0.05; **P < 0.01; ***P < 0.001.
With the support of our findings of DAPK2 expression associated with PML-RARα expression and block in granulocytic differentiation, we found increased DAPK2 mRNA levels in five APL patients undergoing ATRA therapy. In two APL patients under ATRA therapy, DAPK2 message was induced ten- to 52-fold on Days 5 and 7, respectively (Fig. 1D). A similar induction of DAPK2 mRNA was seen in three APL patients after finishing ATRA therapy (mean 1.2 months; Fig. 1E).
To investigate further DAPK2 regulation in granulocytic differentiation of APL cells, we used the NB4 and HT93 APL t(15:17) cell line models to measure DAPK2 induction upon ATRA-induced neutrophil differentiation. We used qPCR and Western blot analysis to measure the expression of DAPK2 during ATRA-induced differentiation. In NB4 cells, we observed a 76- and 210-fold induction of DAPK2 at Days 4 and 6, respectively, paralleled by DAPK2 protein expression (Fig. 1F, left panel). Importantly, DAPK2 expression was not changed in ATRA-resistant NB4-R2 cells, excluding that the DAPK2 induction seen in NB4 parental cells represents a nonspecific stress response to ATRA rather than being an effector of neutrophil differentiation (Fig. 1F, right panel). In addition, we found a similar DAPK2 mRNA and protein induction in HT93 cells, a second APL cell line model (Fig. 1G).
Together, our data clearly argue for an association of PML-RARα expression with low DAPK2 levels in APL.
PML-RARα represses DAPK2 expression
Given the significantly lower DAPK2 transcript levels in APL compared with other AML subtypes, we were asking whether PML-RARα is a transcriptional repressor of DAPK2. With the use of Matinspector 8.0 software, we identified two putative RAREs at positions −867 bp and −733 bp upstream of the transcriptional start site (+1) in the DAPK2 promoter (Fig. 2A). To verify direct binding of PML-RARα to these sites in the DAPK2 promoter, we performed ChIP assays using NB4 cell lysates and anti-PML and anti-RARα antibodies. Anti-histone 3 and IgG pull-downs were used as positive and negative controls, respectively. Precipitated chromatin was used to amplify a 240- or 270-bp fragment of the DAPK2 promoter containing one of the putative RAREs. ChIP analysis revealed in vivo binding of PML-RARα to one of the two putative RAREs in the DAPK2 promoter (Fig. 2B). As an additional negative control for the ChIP assay, we used primers that amplify a region 1.6 kb downstream of the GC box in the DAPK2 promoter (Fig. 2B, bottom panel).
Figure 2. PML-RARα inhibits the transcription of DAPK2 in APL cells.

(A) Schematic representation of putative RAREs in the proximal DAPK2 promoter. (B) ChIP assays using NB4 cells. Histone 3 (His) and IgG served as positive and negative controls, respectively. PCR was performed using primers encompassing the −867 or the −733 PML-RARα-binding site in the DAPK2 promoter and an unrelated sequence 1.6 kb downstream of the GC box as a negative control. (C) Confirmation of ectopic PML-RARα expression. PML-RARα RNA levels were evaluated using qPCR. mRNA levels were normalized to the HMBS housekeeping gene. Results are shown as n-fold regulation compared with NB4 APL cells. Und., Undetectable. (D) Western blot analysis of DAPK2 and PML-RARα expression in U937 PML-RARα cells. Total protein was extracted and submitted to immunoblotting using anti-DAPK2 and anti-RARα. GAPDH is shown as a loading control. (E) Inhibiting PML-RARα increases DAPK2 expression. NB4 cells were transduced with shRNA targeting PML, and PML-RARα knockdown efficiency was confirmed at the mRNA level. DAPK2 mRNA levels were measured by qPCR. DAPK2 expression was normalized to the HMBS housekeeping gene. Results are shown as n-fold regulation compared with control-transduced NB4 SHC002 cells. (F) Western blot analysis of DAPK2 and PML-RARα expression in NB4 PML knockdown cells. MWU: *P < 0.05.
To prove further the inhibitory effect of PML-RARα on DAPK2 transcription, we expressed PML-RARα ectopically in PML-RARα-negative U937 AML cells using a retroviral vector. The transduction efficiency was determined by qPCR and Western blotting (Fig. 2C and D, top panel). As expected, PML-RARα was not detectable in control-transduced U937 cells but showed a threefold higher expression of PML-RARα in PML-RARα-transduced U937 compared with NB4 APL cells (Fig. 2C). Expression of PML-RARα in U937 cells repressed endogenous DAPK2 expression significantly (Fig. 2D). Next, we knocked down PML-RARα in NB4 cells using two different sets of lentiviral vectors expressing shRNAs targeting the PML moiety of PML-RARα. Successful PML-RARα knockdown was shown at the mRNA and protein levels (Fig. 2E and F, top panel). In line with our findings of reduced DAPK2 levels upon ectopic expression of PML-RARα, specific inhibition of endogenous PML-RARα by RNA interference in NB4 cells resulted in increased DAPK2 protein expression (Fig. 2F). Taken together, our results indicate that DAPK2 is repressed by the leukemic fusion protein PML-RARα in APL.
PU.1 is a direct transcriptional regulator of DAPK2
PU.1 coactivates genes crucial for myeloid differentiation [1, 2]. In addition, RARE sites often coexist with nearby PU.1-binding motifs in a large number of genes, and PML-RARα represses PU.1-mediated transactivation of these genes [11]. In addition, PU.1 transcription is repressed by PML-RARA, which is reflected by particularly low PU.1 levels in APL compared with other AML subtypes, and we found a positive association of PU.1 and DAPK2 mRNA in AML [10, 44, 45]. Thus, we asked whether DAPK2 is a direct PU.1 target and screened the DAPK2 promoter for potential PU.1-binding motifs. We identified three putative PU.1-binding sites in the DAPK2 promoter at positions −551 bp, −565 bp, and −807 bp relative to the transcriptional start site (Fig. 3A). We next performed ChIP analysis to address whether PU.1 binds to the DAPK2 promoter in vivo. Indeed, we found that PU.1 occupies the −551/−565-bp sites but not the −807-bp PU.1 site of the promoter (Fig. 3B). To test whether PU.1 can activate the DAPK2 promoter, we used a 2.1-kb DAPK2 promoter reporter construct containing all PU.1-binding sites as well as a deletion construct without any of the PU.1-binding sites. Cotransfection of different concentrations of a PU.1 expression plasmid, together with the 2.1-kb DAPK2 promoter reporter into H1299 cells, revealed a significant, dose-dependent activation of the DAPK2 promoter. A short DAPK2 promoter reporter lacking all putative PU.1-binding sites was not activated by PU.1 (Fig. 3C), indicating that PU.1 not only binds to but also activates the DAPK2 promoter.
Figure 3. PU.1 is a transcriptional regulator of DAPK2 during neutrophil differentiation.

(A) Schematic representation of putative PU.1-binding sites in the proximal DAPK2 promoter. (B) PU.1 ChIP assays using NB4 cells. Histone 3 and IgG served as positive and negative controls, respectively. PCR was performed using primers encompassing the −807 or the −565 and the −551 PU.1-binding site in the DAPK2 promoter and an unrelated sequence 1.6 kb downstream of the GC box which served as a negative control. (C) PU.1 activates the DAPK2 promoter (Prom 1/2). Transactivation assays of H1299 cells transiently transfected with PU.1 expression or pcDNA3.1 empty vector together with a long or short DAPK2 promoter reporter construct as indicated. The promoter activity is shown as relative light units and was normalized to pcDNA3.1-transfected cells. (D) Induction of ectopic PU.1 increases DAPK2 mRNA levels. NB4 cells were transduced with an inducible PU1-ER-expressing vector. Cells were treated with 4-OHT to induce PU.1 translocation to the nucleus. DAPK2 mRNA levels were measured by qPCR, and values were normalized to the HMBS housekeeping gene. Results are given as n-fold regulation compared with untreated, control-transduced NB4 pBabe cells. (E) Induction of G-CSFR mRNA is shown as a control for PU.1 transcriptional activity. Analysis as in D. MWU: *P < 0.05; **P < 0.01; ***P < 0.001.
To determine whether PU.1 alone is sufficient to up-regulate DAPK2, we transduced NB4 with a tamoxifen-inducible PU.1-ER fusion protein. Upon 4-OHT treatment for 24 h, we detected a significant, 1.9-fold induction of DAPK2 in NB4-PU.1-ER cells compared with control-transduced cells (Fig. 3D). As a control for PU.1 transcriptional activity, we measured induction of the known PU.1 target G-CSFR [2] (Fig. 3E).
Next, we generated NB4 and HT93 PU.1 knockdown cells using two different lentiviral constructs expressing shRNAs targeting PU.1 to analyze PU.1-dependent DAPK2 induction. PU.1 knockdown efficiency was evaluated by qPCR and Western blotting (Fig. 4A–C). Impaired neutrophil differentiation of NB4 PU.1 knockdown cells was shown by measuring the neutrophil markers CEBPE and G-CSFR (Supplemental Fig. 2A). Inhibiting PU.1 in NB4 and HT93 APL cells attenuated DAPK2 induction significantly at the mRNA (P<0.001) and protein levels upon ATRA-induced neutrophil differentiation (Fig. 4B–E).
Figure 4. Knocking down PU.1 attenuates DAPK2 induction significantly during neutrophil differentiation.

(A and B) NB4 cells were transduced with two independent shRNAs targeting PU.1 (shPU.1_1/_2) or with a nontargeting shRNA control (SHC002). NB4 control and PU.1 knockdown cells were differentiated toward neutrophils for 4 days using ATRA. (A) PU.1 mRNA levels were measured by qPCR. Values were normalized to the HMBS housekeeping gene. Results are shown as n-fold regulation compared with untreated NB4 SHC002 control cells. (B) DAPK2 mRNA levels analyzed as described for PU.1. (C) Western blot analysis of DAPK2 and PU.1 expression in NB4 PU.1 knockdown cells during neutrophil differentiation. Total protein was extracted and subjected to immunoblotting using anti-DAPK2 and anti-PU.1 antibodies. GAPDH is shown as a loading control. (D) HT93 APL cells were transduced with lentviral vectors expressing shRNAs targeting PU.1 as in A. DAPK2 mRNA levels in HT93 PU.1 knockdown or control cells upon neutrophil differentiation were determined as in A. (E) Western blot analysis of DAPK2 and PU.1 expression in PU.1 knockdown and control HT93 APL cells upon ATRA-induced neutrophil differentiation. Analysis as in C. (F and G) Knocking down Pu.1 in conditionally immortalized murine neutrophil progenitor cells (SCF-condHoxb8) impairs Dapk2 induction. SCF-condHoxb8 cells were transduced with a shRNA targeting Pu.1. Neutrophil differentiation was induced by removing 4-OHT from the cell culture medium. Six days later, cells were analyzed for DAPK2 and PU.1 expression. Pu.1 (F) and Dapk2 (G) mRNA levels were determined by qPCR. Values were normalized to the housekeeping gene Gapdh. Results are shown as n-fold regulation compared with undifferentiated SCF-condHoxb8 cells growing in the presence of 4-OHT. (H) Dapk2 and Pu.1 Western blot analysis of SCF-condHoxb8 cells treated as in G. (I and J) Attenuated neutrophil differentiation of SCF-condHoxb8 cells upon 4-OHT removal, as seen by reduced G-Csfr mRNA and Gr-1 cell-surface expression. ctrl, Control; SHC, SHC002. MWU: *P < 0.05; ***P < 0.001.
To test PU.1-dependent expression of DAPK2 in non-APL neutrophil differentiation, we analyzed DAPK2 levels in condHoxb8-immortalized murine neutrophil progenitors, a noncancerous model for neutrophil differentiation [39, 40]. In the presence of 4-OHT, the differentiation of these cells is blocked as a result of the expression of exogenous Hoxb8, an oncogene that promotes self-renewal of myeloid progenitors. To induce neutrophil differentiation, 4-OHT is removed from the medium, resulting in a rapid shutdown of Hoxb8 expression, and terminal differentiation of mature neutrophils accompanied with an increase of PU.1 and DAPK2 levels within 6 days (Supplemental Fig. 2B–F). We generated SCF-condHoxb8 Pu.1 knockdown cells using one of our lentiviral vectors expressing a shRNA targeting an identical nucleotide sequence in human and mouse PU.1. Knockdown efficiency was assessed by qPCR and Western blot (Fig. 4F and H). Dapk2 mRNA and protein induction were reduced significantly in SCF-condHoxb8 Pu.1 knockdown compared with control cells during neutrophil differentiation (Fig. 4G and H). Moreover, knocking down Pu.1 in SCF-condHoxb8 cells attenuated neutrophil differentiation as seen by significantly lower mRNA induction of the differentiation maker G-Csfr (Csf3r) and Gr-1 expression (Fig. 4I and J). Altogether, our results show that PU.1 is a direct transcriptional regulator of DAPK2 in granulocytic differentiation.
DAPK2 is a relevant downstream effector of PU.1 during neutrophil differentiation
To evaluate whether DAPK2 is a relevant downstream effector of PU.1 during neutrophil differentiation, we performed a rescue experiment by ectopically expressing DAPK2 in NB4 PU.1 knockdown cells. DAPK2 exogenous expression as well as PU.1 knockdown efficiency during neutrophil differentiation were confirmed by qPCR and Western blotting (Fig. 5A and B). Neutrophil differentiation of NB4 cells was assessed by CEBPE and CD11b expression. Expression of the neutrophil markers CEBPE and CD11b were increased significantly in NB4 PU.1 knockdown cells expressing ectopic DAPK2 (Fig. 5C and D). Importantly, expressing DAPK2 in PU.1 knockdown NB4 cells partially rescued neutrophil differentiation, as seen in a marked increase in metamyelocytes and mature neutrophils compared with the control cells upon ATRA-induced neutrophil differentiation (Fig. 5E).
Figure 5. DAPK2 is a relevant PU.1 target during neutrophil differentiation of APL cells.
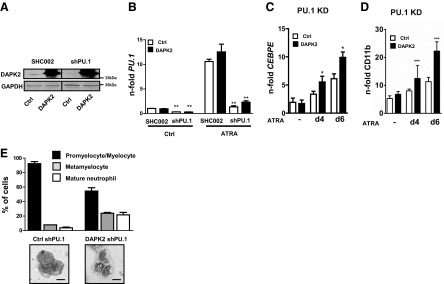
(A–E) Ectopic expression of DAPK2 in NB4 APL cells lacking PU.1 partially rescues neutrophil differentiation. NB4 PU.1 knockdown cells were transduced with a control or a DAPK2-expressing lentiviral vector. Expression and knockdown efficiency were measured by Western blot (A) or qPCR (B). Cells were treated for 6 days with 1 μM ATRA. (C and D) Increased expression of the CEBPE (C) or neutrophil marker CD11b (D) in DAPK2-rescued NB4 PU.1 knockdown (KD) cells, treated with ATRA for 4 and 6 days. Analysis as in A. MWU: *P < 0.05; **P < 0.01; ***P < 0.001. (E) May-Grünwald Giemsa staining of cytospin cells was performed. A minimum of 100 cells was counted in three optical fields of at least two independent experiments.
Together, our results revealed that DAPK2 induction during neutrophil differentiation of APL and murine neutrophil progenitor cells is PU.1-dependent. Furthermore, our rescue experiments suggest that DAPK2 is a crucial downstream effector of PU.1 during neutrophil differentiation.
C/EBPα-dependent induction of DAPK2 during neutrophil differentiation
Further analysis of AML samples from the Bern and HOVON/SAKK studies that were molecularly characterized for C/EBPα mutations (n=6) revealed significantly lower DAPK2 levels in AML patients with mutated C/EBPα compared with those negative for any relevant mutations (Fms-like tyrosine kinase 3, nucleophosmin 1, or C/EBPα; n=91) and thus, carrying a WT C/EBPα gene (Fig. 6A). To assess the regulation of DAPK2 by C/EBPα, we used K562 leukemic cell lines expressing inducible, full-length C/EBPα (K562-C/EBPα-p42-ER), a truncated isoform (K562-C/EBPα-p30-ER), or a p42 isoform with a mutation preventing interaction with E2F and neutrophil differentiation (K562-C/EBPα-BRM2-ER). Upon tamoxifen-induced translocation of these C/EBPα-ER fusion proteins to the nucleus, a significant, 2.9-fold transcriptional activation of DAPK2 was seen in C/EBPα-p42 WT cells but not in any of the control cell lines (Fig. 6B). Induction of the C/EBPα-regulated gene G-CSFR (CSF3R) served as a positive control (Fig. 6C). Secondly, we knocked down C/EBPα using two independent shRNAs in NB4 and HT93 APL cells. Knockdown efficiency was measured by qPCR and Western blot (Fig. 6D, E, G, and H). Lack of C/EBPα reduced induction of DAPK2 significantly during ATRA-induced neutrophil differentiation (Fig. 6F and I). In summary, we provide evidence that induction of DAPK2 in myeloid cells is dependent, directly or indirectly, on C/EBPα—yet another key transcription factor in granulopoiesis.
Figure 6. DAPK2 levels correlate with C/EBPα inactivation in AML.

(A) Down-regulation of DAPK2 mRNA levels in C/EBPα-mutated (CEBPA mut) AML patients. DAPK2 mRNA levels in AML blast cells were quantified using qPCR. Values are the differences in Ct values between DAPK2 and the housekeeping genes ABL1 and GAPDH. (B) K562 leukemic cells expressing inducible WT C/EBPα (p42), the N-terminus truncated form (p30), or the C-terminus mutated form (BRM2) were treated with 5μM 4-OHT to induce the translocation of the C/EBPα-ER proteins to the nucleus. Activation of WT but not the truncated C/EBPα proteins resulted in a significant increase of DAPK2 mRNA levels. (C) Induction of the C/EBPα target gene G-CSFR is shown as a control for C/EBPα transcriptional activity. (D and E) NB4 APL cells were transduced with two independent shRNAs (shC/EBPα_1/_2) targeting C/EBPα and differentiated for 4 days using ATRA. C/EBPα knockdown efficiency was analyzed by qPCR and Western blotting. Transcript values were normalized to the housekeeping gene HMBS and are given as n-fold regulation compared with ATRA-treated control cells. Total protein was extracted and subjected to immunoblotting using anti-C/EBPα antibody. GAPDH is shown as a loading control. (F) Knocking down C/EBPα results in significantly decreased induction of DAPK2 message upon neutrophil differentiation of NB4 cells. Analysis as in C. (G and H) HT93 APL cells were transduced with two independent shRNAs (shC/EBPα_1/_2) targeting C/EBPα. Treatment and C/EBPΑ mRNA measurements were done as in D and E. (I) Knocking down C/EBPα results in significantly decreased induction of DAPK2 message upon neutrophil differentiation of HT93 APL cells. Treatment and analysis as in C. MWU: *P < 0.05; **P < 0.01; ***P < 0.001.
DISCUSSION
APL is caused by PML-RARα driven, abnormal accumulation of immature promyelocytic blast cells with increased cell survival. Interestingly, DAPK2 is not only a proapoptotic protein, but it also enhances neutrophil differentiation [30]. Inactivation of such a gene at the nexus of myeloid cell survival and differentiation is likely to have a major impact on leukemogenesis. To get a first glimpse at molecular mechanisms inhibiting DAPK2 transcription, we determined DAPK2 expression in 343 primary AML patients with clearly defined genetic lesions. Our data demonstrating significantly lower DAPK2 levels in clinical APL samples compared with normal granulocytes, as well as compared with other AML subtypes, let to the hypothesis that DAPK2 is a PML-RARα-repressed gene. This is clearly supported by our findings in HT93 and NB4 APL cell lines that show the induction of DAPK2 upon ATRA-induced neutrophil differentiation. As these results in APL cell lines may not entirely reflect the in vivo situation of this disease, we confirmed our data in samples from APL patients during ATRA therapy. We found increased DAPK2 message in five of five APL patients receiving oral ATRA. These clinical findings support our experimental data, showing a strong association of DAPK2 expression with neutrophil differentiation and direct PML-RARα-mediated repression of DAPK2 in APL. Moreover, we found a functional RARE in the DAPK2 promoter identifying DAPK2 as novel PML-RARα-repressed gene in APL. This puts DAPK2 in line with the antiapoptotic effect of PML-RARα in APL blasts via attenuating p53 activity or by increasing the ratio of the antiapoptotic dNp73 to the proapoptotic TAp73 [46, 47].
Previously, it was found that most PU.1-binding motifs coexist with RAREs, resulting in PML-RARα-mediated inhibition of PU.1-regulated promoters [11]. We found two functional PU.1-binding sites in close proximity to the RARE found in the DAPK2 promoter. Therefore, DAPK2 might represent yet another gene that is repressed by PML-RARα via PU.1 inactivation. On the other hand, PML-RARα is a direct transcriptional repressor of PU.1 [10], and low DAPK2 levels in APL may be attributed to low expression of PU.1. Our DAPK2 expression-profiling experiments not only revealed significantly lower DAPK2 levels in APL but also in AML in general. Therefore, additional DAPK2 repression mechanisms must be operative in AML. Another leukemic fusion protein, AML1-ETO, directly inhibits PU.1 in t(8;21)-positive AML patients, may explain the low DAPK2 expression levels in this AML subtype [48]. In addition, we found significantly lower DAPK2 levels in C/EBPα-mutated AML patient samples and showed CEBPα-dependent regulation of DAPK2 in AML cells. C/EBPα-mediated regulation of DAPK2 is likely to be direct, based on a genome-wide C/EBPα, DNA-binding analysis by ChIP sequencing [49]. Moreover, C/EBPα expression is suppressed in AML patients bearing the translocations t(8;21), t(3;21), or inv(16), and the reduced C/EBPα levels in these AML subtypes may contribute to the reduction in DAPK2 expression [50]. Additionally, C/EBPα is a positive regulator of PU.1, and deregulated C/EBPα expression might affect DAPK2 transcription indirectly via PU.1 [51]. Lastly, we identified earlier the KLF6 transcription factor as a positive regulator of DAPK2, and its low expression in AML may contribute to the low DAPK2 levels in this hematological disorder [28, 52].
The restoration of DAPK2 expression in PU.1 knockdown APL cells revealed that DAPK2 is a relevant PU.1 target during neutrophil differentiation. Although, re-expressing DAPK2 did not fully rescue neutrophil differentiation in PU.1 impaired APL cells, it clearly shows the differentiation potential of DAPK2. How might DAPK2 support neutrophil differentiation? A prerequisite of ATRA-induced APL differentiation is a G1 cell-cycle arrest often meditated by cyclin-dependent kinase inhibitors, such as p15INK4b or p21WAF1/CIP1 [53, 54]. Interestingly, other DAPK family members, namely DAPK1 and zipper-interacting protein kinase, phosphorylate p21WAF1/CIP1 [55, 56] and thereby, contribute to cell-cycle arrest. Our preliminary data show binding of DAPK2 to p15INK4b and p21WAF1/CIP1 (data not shown), and we suggest that DAPK2, among others, is needed for the cell-cycle arrest in terminal differentiation of neutrophils.
In contrast to DAPK2, the tumor suppressor DAPK1 is not down-regulated significantly in AML patient samples compared with granulocytes from healthy donors. DAPK1 is often inactivated by promoter hypermethylation in cancer [23], but in line with our results, no methylation of the DAPK1 promoter was found in AML and MDS [57]. Earlier, we found a particularly high expression of Dapk2 in murine hematopoietic tissue compared with Dapk1, pointing at a specific function of DAPK2 in hematopoiesis [31]. Altogether, DAPK1, as opposed to its relative DAPK2, may not play an essential role in neutrophil differentiation and AML pathology.
DAPK family members not only participate in classical cell death pathways but also contribute to autophagy induction. DAPK1, for example, binds and activates the key autophagy gene beclin-1 [58], and overexpression of DAPK2 did induce autophagy in 293T human embryonic kidney cells [59]. Linking autophagy to APL, two recent studies show that autophagy contributes to ATRA-induced neutrophil differentiation of APL cells by degrading aggregated PML-RARα proteins [60, 61]. Therefore, it is tempting to speculate that the induction of DAPK2 during neutrophil differentiation might contribute to autophagy activation. Consistently, we found significantly reduced LC3B dot formation, a marker for activation of autophagy, in NB4 DAPK2 knockdown cells compared with control cells during neutrophil differentiation (data not shown).
In conclusion, by showing that DAPK2 is a PML-RARα-repressed gene in APL, we provide the first mechanism explaining low DAPK2 levels in AML. Most likely, PML-RARα represses DAPK2 transcription by direct inhibition or indirectly, by repressing its transcriptional activator PU.1. Furthermore, the regulation of DAPK2 by two myeloid master genes, such as PU.1 and C/EBPα, strongly supports an important function for this kinase in myeloid differentiation.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants from the Swiss National Science Foundation 31003A_129702 (to M.F.F. and M.P.T.); Swiss National Science Foundation 31003A_129955 (to M.P.T.); Marlies-Schwegler Foundation, Ursula-Hecht-Foundation for Leukemia Research, and Bernese Foundation of Cancer Research (to M.F.F.); Werner and Hedy Berger-Janser Foundation of Cancer Research (to M.F.F. and M.P.T.); Bern University Research Foundation (to M.P.T.); and Joyce Klein Stock Gift and U.S. National Institutes of Health grant R01HL091219 (to B.E.T.).
Deborah Shan is gratefully acknowledged for excellent technical support. We are grateful to Dr. Hitoshi Yoshida for providing us with pLPCX/HA-PML-RARα retroviral vectors.
The online version of this paper, found at www.jleukbio.org, includes supplemental information.
- 4-OHT
- 4-hydroxytamoxifen
- AML1
- acute myeloid leukemia 1 protein
- APL
- acute promyelocytic leukemia
- ChIP
- chromatin immunoprecipitation
- CLL
- chronic lymphocytic leukemia
- condHoxb8
- conditional homeobox b8
- DAPK2
- death-associated protein kinase 2
- ETO
- eight-twenty-one
- Gr-1
- granulocyte differentiation antigen 1
- HMBS
- hydroxymethylbilane synthase
- HOVON
- Hemato Oncology Foundation for Adults in The Netherlands
- Hoxb8
- homeobox b8
- LDA
- low-density array
- MDS
- myelodysplastic syndrome
- MWU
- Mann-Whitney U
- PML
- promyelocytic leukemia
- qPCR
- real-time quantitative PCR
- RARA
- retinoic acid receptor α
- RARE
- retinoic acid responsive element
- SAKK
- Swiss Group for Clinical Cancer Research Cooperative
- SCF
- stem cell factor
- shRNA
- short hairpin RNA
AUTHORSHIP
M.H. performed the experimental research, interpreted the data, and drafted the article. E.A.F., A.M.S., and A.B. performed ChIP and luciferase assays. P.J.M.V. and T.H. provided patient samples, analyzed patient data, and revised the article. B.E.T. provided essential PU.1 reagents and revised the article. T.K. provided murine SCF-condHoxb8 neutrophil progenitor cells and protocols and revised the article. G.B. provided K562 C/EBPα-ER cells and revised the article. H-U.S. and M.F.F. instigated the experimental design and revised the drafted article. M.P.T. designed the project, prepared lentiviral vectors, analyzed data, and gave final approval of the submitted manuscript.
DISCLOSURES
The authors declare no conflicts of interest.
REFERENCES
- 1. Hohaus S., Petrovick M. S., Voso M. T., Sun Z., Zhang D. E., Tenen D. G. (1995) PU. 1 (Spi-1) and C/EBP α regulate expression of the granulocyte-macrophage colony-stimulating factor receptor α gene. Mol. Cell. Biol. 15, 5830–5845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Smith L. T., Hohaus S., Gonzalez D. A., Dziennis S. E., Tenen D. G. (1996) PU. 1 (Spi-1) and C/EBP α regulate the granulocyte colony-stimulating factor receptor promoter in myeloid cells. Blood 88, 1234–1247 [PubMed] [Google Scholar]
- 3. Zhang D. E., Zhang P., Wang N. D., Hetherington C. J., Darlington G. J., Tenen D. G. (1997) Absence of granulocyte colony-stimulating factor signaling and neutrophil development in CCAAT enhancer binding protein α-deficient mice. Proc. Natl. Acad. Sci. USA 94, 569–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zhang D. E., Hetherington C. J., Meyers S., Rhoades K. L., Larson C. J., Chen H. M., Hiebert S. W., Tenen D. G. (1996) CCAAT enhancer-binding protein (C/EBP) and AML1 (CBF α2) synergistically activate the macrophage colony-stimulating factor receptor promoter. Mol. Cell. Biol. 16, 1231–1240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pahl H. L., Scheibe R. J., Zhang D. E., Chen H. M., Galson D. L., Maki R. A., Tenen D. G. (1993) The proto-oncogene PU. 1 regulates expression of the myeloid-specific CD11b promoter. J. Biol. Chem. 268, 5014–5020 [PubMed] [Google Scholar]
- 6. Brugnoli F., Lambertini E., Varin-Blank N., Piva R., Marchisio M., Grassilli S., Miscia S., Capitani S., Bertagnolo V. (2010) Vav1 and PU. 1 are recruited to the CD11b promoter in APL-derived promyelocytes: role of Vav1 in modulating PU. 1-containing complexes during ATRA-induced differentiation. Exp. Cell Res 316, 38–47 [DOI] [PubMed] [Google Scholar]
- 7. De Thé H., Chen Z. (2010) Acute promyelocytic leukaemia: novel insights into the mechanisms of cure. Nat. Rev. Cancer 10, 775–783 [DOI] [PubMed] [Google Scholar]
- 8. Mi J-Q., Li J-M., Shen Z-X., Chen S-J., Chen Z. (2012) How to manage acute promyelocytic leukemia. Leukemia 26, 1743–1751 [DOI] [PubMed] [Google Scholar]
- 9. Ablain J., de The H. (2011) Revisiting the differentiation paradigm in acute promyelocytic leukemia. Blood 117, 5795–5802 [DOI] [PubMed] [Google Scholar]
- 10. Mueller B. U., Pabst T., Fos J., Petkovic V., Fey M. F., Asou N., Buergi U., Tenen D. G. (2006) ATRA resolves the differentiation block in t(15;17) acute myeloid leukemia by restoring PU. 1 expression. Blood 107, 3330–3338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang K., Wang P., Shi J., Zhu X., He M., Jia X., Yang X., Qiu F., Jin W., Qian M., et al. (2010) PML/RARα targets promoter regions containing PU. 1 consensus and RARE half sites in acute promyelocytic leukemia. Cancer Cell 17, 186–197 [DOI] [PubMed] [Google Scholar]
- 12. Pabst T., Mueller B. U., Zhang P., Radomska H. S., Narravula S., Schnittger S., Behre G., Hiddemann W., Tenen D. G. (2001) Dominant-negative mutations of CEBPA, encoding CCAAT/enhancer binding protein-α (C/EBPα), in acute myeloid leukemia. Nat. Genet. 27, 263–270 [DOI] [PubMed] [Google Scholar]
- 13. Chim C. S., Wong A. S. Y., Kwong Y. L. (2002) Infrequent hypermethylation of CEBPA promotor in acute myeloid leukaemia. Br. J. Haematol. 119, 988–990 [DOI] [PubMed] [Google Scholar]
- 14. Figueroa M. E., Wouters B. J., Skrabanek L., Glass J., Li Y., Erpelinck-Verschueren C. A. J., Langerak A. W., Löwenberg B., Fazzari M., Greally J. M., et al. (2009) Genome-wide epigenetic analysis delineates a biologically distinct immature acute leukemia with myeloid/T-lymphoid features. Blood 113, 2795–2804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jost E., do O. N., Wilop S., Herman J. G., Osieka R., Galm O. (2009) Aberrant DNA methylation of the transcription factor C/EBPα in acute myelogenous leukemia. Leuk. Res. 33, 443–449 [DOI] [PubMed] [Google Scholar]
- 16. Lu Y., Chen W., Chen W., Stein A., Weiss L. M., Huang Q. (2010) C/EBPA gene mutation and C/EBPA promoter hypermethylation in acute myeloid leukemia with normal cytogenetics. Am. J. Hematol. 85, 426–430 [DOI] [PubMed] [Google Scholar]
- 17. Pabst T., Mueller B. U., Harakawa N., Schoch C., Haferlach T., Behre G., Hiddemann W., Zhang D. E., Tenen D. G. (2001) AML1-ETO downregulates the granulocytic differentiation factor C/EBPα in t(8;21) myeloid leukemia. Nat. Med. 7, 444–451 [DOI] [PubMed] [Google Scholar]
- 18. Helbling D., Mueller B. U., Timchenko N. A., Hagemeijer A., Jotterand M., Meyer-Monard S., Lister A., Rowley J. D., Huegli B., Fey M. F., et al. (2004) The leukemic fusion gene AML1-MDS1-EVI1 suppresses CEBPA in acute myeloid leukemia by activation of calreticulin. Proc. Natl. Acad. Sci. USA 101, 13312–13317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Helbling D., Mueller B. U., Timchenko N. A., Schardt J., Eyer M., Betts D. R., Jotterand M., Meyer-Monard S., Fey M. F., Pabst T. (2005) CBFB-SMMHC is correlated with increased calreticulin expression and suppresses the granulocytic differentiation factor CEBPA in AML with inv(16). Blood 106, 1369–1375 [DOI] [PubMed] [Google Scholar]
- 20. Durual S., Rideau A., Ruault-Jungblut S., Cossali D., Beris P., Piguet V., Matthes T. (2007) Lentiviral PU. 1 overexpression restores differentiation in myeloid leukemic blasts. Leukemia 21, 1050–1059 [DOI] [PubMed] [Google Scholar]
- 21. Schepers H., Wierenga A. T. J., van Gosliga D., Eggen B. J. L., Vellenga E., Schuringa J. J. (2007) Reintroduction of C/EBPα in leukemic CD34+ stem/progenitor cells impairs self-renewal and partially restores myelopoiesis. Blood 110, 1317–1325 [DOI] [PubMed] [Google Scholar]
- 22. Bialik S., Kimchi A. (2006) The death-associated protein kinases: structure, function, and beyond. Annu. Rev. Biochem. 75, 189–210 [DOI] [PubMed] [Google Scholar]
- 23. Gozuacik D., Kimchi A. (2004) Autophagy as a cell death and tumor suppressor mechanism. Oncogene 23, 2891–2906 [DOI] [PubMed] [Google Scholar]
- 24. Raval A., Tanner S. M., Byrd J. C., Angerman E. B., Perko J. D., Chen S-S., Hackanson B., Grever M. R., Lucas D. M., Matkovic J. J., et al. (2007) Downregulation of death-associated protein kinase 1 (DAPK1) in chronic lymphocytic leukemia. Cell 129, 879–890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lin Y., Hupp T. R., Stevens C. (2010) Death-associated protein kinase (DAPK) and signal transduction: additional roles beyond cell death. FEBS J. 277, 48–57 [DOI] [PubMed] [Google Scholar]
- 26. Inbal B., Shani G., Cohen O., Kissil J. L., Kimchi A. (2000) Death-associated protein kinase-related protein 1, a novel serine/threonine kinase involved in apoptosis. Mol. Cell. Biol. 20, 1044–1054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shohat G., Shani G., Eisenstein M., Kimchi A. (2002) The DAP-kinase family of proteins: study of a novel group of calcium-regulated death-promoting kinases. Biochim. Biophys. Acta 1600, 45–50 [DOI] [PubMed] [Google Scholar]
- 28. Britschgi A., Trinh E., Rizzi M., Jenal M., Ress A., Tobler A., Fey M. F., Helin K., Tschan M. P. (2008) DAPK2 is a novel E2F1/KLF6 target gene involved in their proapoptotic function. Oncogene 27, 5706–5716 [DOI] [PubMed] [Google Scholar]
- 29. Britschgi A., Simon H-U., Tobler A., Fey M. F., Tschan M. P. (2010) Epigallocatechin-3-gallate induces cell death in acute myeloid leukaemia cells and supports all-trans retinoic acid-induced neutrophil differentiation via death-associated protein kinase 2. Br. J. Haematol. 149, 55–64 [DOI] [PubMed] [Google Scholar]
- 30. Rizzi M., Tschan M. P., Britschgi C., Britschgi A., Hügli B., Grob T. J., Leupin N., Mueller B. U., Simon H-U., Ziemiecki A., et al. (2007) The death-associated protein kinase 2 is up-regulated during normal myeloid differentiation and enhances neutrophil maturation in myeloid leukemic cells. J. Leukoc. Biol. 81, 1599–1608 [DOI] [PubMed] [Google Scholar]
- 31. Fang J., Menon M., Zhang D., Torbett B., Oxburgh L., Tschan M., Houde E., Wojchowski D. M. (2008) Attenuation of EPO-dependent erythroblast formation by death-associated protein kinase-2. Blood 112, 886–890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Federzoni E. A., Valk P. J. M., Torbett B. E., Haferlach T., Löwenberg B., Fey M. F., Tschan M. P. (2012) PU. 1 is linking the glycolytic enzyme HK3 in neutrophil differentiation and survival of APL cells. Blood 119, 4963–4970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Breems D. A., Boogaerts M. A., Dekker A. W., Van Putten W. L. J., Sonneveld P., Huijgens P. C., Van der Lelie J., Vellenga E., Gratwohl A., Verhoef G. E. G., et al. (2005) Autologous bone marrow transplantation as consolidation therapy in the treatment of adult patients under 60 years with acute myeloid leukaemia in first complete remission: a prospective randomized Dutch-Belgian Haemato-Oncology Co-operative Group (HOVON) and Swiss Group for Clinical Cancer Res. (SAKK) trial. Br. J. Haematol. 128, 59–65 [DOI] [PubMed] [Google Scholar]
- 34. Löwenberg B., Boogaerts M. A., Daenen S. M., Verhoef G. E., Hagenbeek A., Vellenga E., Ossenkoppele G. J., Huijgens P. C., Verdonck L. F., van der Lelie J., et al. (1997) Value of different modalities of granulocyte-macrophage colony-stimulating factor applied during or after induction therapy of acute myeloid leukemia. J. Clin. Oncol. 15, 3496–3506 [DOI] [PubMed] [Google Scholar]
- 35. Löwenberg B., van Putten W., Theobald M., Gmür J., Verdonck L., Sonneveld P., Fey M., Schouten H., de Greef G., Ferrant A., et al. (2003) Effect of priming with granulocyte colony-stimulating factor on the outcome of chemotherapy for acute myeloid leukemia. N. Engl. J. Med. 349, 743–752 [DOI] [PubMed] [Google Scholar]
- 36. Ossenkoppele G. J., Graveland W. J., Sonneveld P., Daenen S. M. G. J., Biesma D. H., Verdonck L. F., Schaafsma M. R., Westveer P. H. M., Peters G. J., Noordhuis P., et al. (2004) The value of fludarabine in addition to ARA-C and G-CSF in the treatment of patients with high-risk myelodysplastic syndromes and AML in elderly patients. Blood 103, 2908–2913 [DOI] [PubMed] [Google Scholar]
- 37. Pulikkan J. A., Peramangalam P. S., Dengler V., Ho P. A., Preudhomme C., Meshinchi S., Christopeit M., Nibourel O., Müller-Tidow C., Bohlander S. K., et al. (2010) C/EBPα regulated microRNA-34a targets E2F3 during granulopoiesis and is down-regulated in AML with CEBPA mutations. Blood 116, 5638–5649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wang G. G., Calvo K. R., Pasillas M. P., Sykes D. B., Häcker H., Kamps M. P. (2006) Quantitative production of macrophages or neutrophils ex vivo using conditional Hoxb8. Nat. Methods 3, 287–293 [DOI] [PubMed] [Google Scholar]
- 39. Vince J. E., Wong W. W-L., Khan N., Feltham R., Chau D., Ahmed A. U., Benetatos C. A., Chunduru S. K., Condon S. M., McKinlay M., et al. (2007) IAP antagonists target cIAP1 to induce TNFα-dependent apoptosis. Cell 131, 682–693 [DOI] [PubMed] [Google Scholar]
- 40. Gurzeler U., Rabachini T., Dahinden C. A., Salmanidis M., Brumatti G., Ekert P. G., Echeverry N., Bachmann D., Simon H. U., Kaufmann T. (2013) In vitro differentiation of near-unlimited numbers of functional mouse basophils using conditional Hoxb8. Allergy 68, 604–613 [DOI] [PubMed] [Google Scholar]
- 41. Tschan M. P., Fischer K. M., Fung V. S., Pirnia F., Borner M. M., Fey M. F., Tobler A., Torbett B. E. (2003) Alternative splicing of the human cyclin D-binding Myb-like protein (hDMP1) yields a truncated protein isoform that alters macrophage differentiation patterns. J. Biol. Chem. 278, 42750–42760 [DOI] [PubMed] [Google Scholar]
- 42. Naviaux R. K., Costanzi E., Haas M., Verma I. M. (1996) The pCL vector system: rapid production of helper-free, high-titer, recombinant retroviruses. J. Virol. 70, 5701–5705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Haferlach T., Kohlmann A., Wieczorek L., Basso G., Kronnie G. T., Béné M-C., De Vos J., Hernández J. M., Hofmann W-K., Mills K. I., et al. (2010) Clinical utility of microarray-based gene expression profiling in the diagnosis and subclassification of leukemia: report from the International Microarray Innovations in Leukemia Study Group. J. Clin. Oncol. 28, 2529–2537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Martens J. H. A., Brinkman A. B., Simmer F., Francoijs K-J., Nebbioso A., Ferrara F., Altucci L., Stunnenberg H. G. (2010) PML-RARα/RXR alters the epigenetic landscape in acute promyelocytic leukemia. Cancer Cell 17, 173–185 [DOI] [PubMed] [Google Scholar]
- 45. Jenal M., Batliner J., Reddy V. A., Haferlach T., Tobler A., Fey M. F., Torbett B. E., Tschan M. P. (2010) The anti-apoptotic gene BCL2A1 is a novel transcriptional target of PU. 1. Leukemia 24, 1073–1076 [DOI] [PubMed] [Google Scholar]
- 46. Insinga A., Monestiroli S., Ronzoni S., Carbone R., Pearson M., Pruneri G., Viale G., Appella E., Pelicci P., Minucci S. (2004) Impairment of p53 acetylation, stability and function by an oncogenic transcription factor. EMBO J. 23, 1144–1154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lucena-Araujo A. R., Panepucci R. A., dos Santos G. A., Jácomo R. H., Santana-Lemos B. A., Lima A. S., Garcia A. B., Araújo A. G., Falcão R. P., Rego E. M. (2008) The expression of ΔNTP73, TATP73 and TP53 genes in acute myeloid leukaemia is associated with recurrent cytogenetic abnormalities and in vitro susceptibility to cytarabine cytotoxicity. Br. J. Haematol. 142, 74–78 [DOI] [PubMed] [Google Scholar]
- 48. Vangala R. K., Heiss-Neumann M. S., Rangatia J. S., Singh S. M., Schoch C., Tenen D. G., Hiddemann W., Behre G. (2003) The myeloid master regulator transcription factor PU. 1 is inactivated by AML1-ETO in t(8;21) myeloid leukemia. Blood 101, 270–277 [DOI] [PubMed] [Google Scholar]
- 49. Trompouki E., Bowman T. V., Lawton L. N., Fan Z. P., Wu D-C., DiBiase A., Martin C. S., Cech J. N., Sessa A. K., Leblanc J. L., et al. (2011) Lineage regulators direct BMP and Wnt pathways to cell-specific programs during differentiation and regeneration. Cell 147, 577–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Paz-Priel I., Friedman A. (2011) C/EBPα dysregulation in AML and ALL. Crit. Rev. Oncog. 16, 93–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kummalue T., Friedman A. D. (2003) Cross-talk between regulators of myeloid development: C/EBPα binds and activates the promoter of the PU. 1 gene. J. Leukoc. Biol. 74, 464–470 [DOI] [PubMed] [Google Scholar]
- 52. Humbert M., Halter V., Shan D., Laedrach J., Leibundgut E. O., Baerlocher G. M., Tobler A., Fey M. F., Tschan M. P. (2011) Deregulated expression of Kruppel-like factors in acute myeloid leukemia. Leuk. Res. 35, 909–913 [DOI] [PubMed] [Google Scholar]
- 53. Liu J., Bi G., Wen P., Yang W., Ren X., Tang T., Xie C., Dong W., Jiang G. (2007) Down-regulation of CD44 contributes to the differentiation of HL-60 cells induced by ATRA or HMBA. Cell. Mol. Immunol. 4, 59–63 [PubMed] [Google Scholar]
- 54. Dimberg A., Bahram F., Karlberg I., Larsson L-G., Nilsson K., Oberg F. (2002) Retinoic acid-induced cell cycle arrest of human myeloid cell lines is associated with sequential down-regulation of c-Myc and cyclin E and posttranscriptional up-regulation of p27(Kip1). Blood 99, 2199–2206 [DOI] [PubMed] [Google Scholar]
- 55. Fraser J. A., Hupp T. R. (2007) Chemical genetics approach to identify peptide ligands that selectively stimulate DAPK-1 kinase activity. Biochemistry 46, 2655–2673 [DOI] [PubMed] [Google Scholar]
- 56. Burch L. R., Scott M., Pohler E., Meek D., Hupp T. (2004) Phage-peptide display identifies the interferon-responsive, death-activated protein kinase family as a novel modifier of MDM2 and p21WAF1. J. Mol. Biol. 337, 115–128 [DOI] [PubMed] [Google Scholar]
- 57. Claus R., Hackanson B., Poetsch A. R., Zucknick M., Sonnet M., Blagitko-Dorfs N., Hiller J., Wilop S., Brümmendorf T. H., Galm O., et al. (2012) Quantitative analyses of DAPK1 methylation in AML and MDS. Int. J. Cancer 131, E138–E142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zalckvar E., Berissi H., Mizrachy L., Idelchuk Y., Koren I., Eisenstein M., Sabanay H., Pinkas-Kramarski R., Kimchi A. (2009) DAP-kinase-mediated phosphorylation on the BH3 domain of beclin 1 promotes dissociation of beclin 1 from Bcl-XL and induction of autophagy. EMBO Rep. 10, 285–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Inbal B., Bialik S., Sabanay I., Shani G., Kimchi A. (2002) DAP kinase and DRP-1 mediate membrane blebbing and the formation of autophagic vesicles during programmed cell death. J. Cell Biol. 157, 455–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Isakson P., Bjørås M., Bøe S. O., Simonsen A. (2010) Autophagy contributes to therapy-induced degradation of the PML/RARA oncoprotein. Blood 116, 2324–2331 [DOI] [PubMed] [Google Scholar]
- 61. Wang Z., Cao L., Kang R., Yang M., Liu L., Zhao Y., Yu Y., Xie M., Yin X., Livesey K. M., et al. (2011) Autophagy regulates myeloid cell differentiation by p62/SQSTM1-mediated degradation of PML-RARα oncoprotein. Autophagy 7, 401–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.