

The common toxicities observed during clinical trials of targeted therapies for advanced renal cell carcinoma as well as specific reported side effects are discussed, and recommended strategies to anticipate the occurrence of key adverse events, minimize the risk for increasing adverse event severity, and manage toxicities to help optimize treatment outcomes in patients with advanced renal cell carcinoma are highlighted.
Keywords: Adverse event, Toxicity, Renal cell carcinoma, Kidney cancer, Vascular endothelial growth factor, Mammalian target of rapamycin, mTOR
Abstract
Targeted therapy for advanced renal cell carcinoma (RCC) has recently expanded the available treatment options for patients with these malignancies. The rapid introduction of novel treatment options into clinical practice within a relatively short time frame has created some new challenges pertaining to adverse event (AE) management in patients with advanced RCC. Accumulating safety data from the pivotal phase III clinical trials of the anti–vascular endothelial growth factor (VEGF) antibody bevacizumab plus interferon, VEGF receptor tyrosine kinase inhibitors (sunitinib, sorafenib, and pazopanib), and mammalian target of rapamycin inhibitors (temsirolimus and everolimus) have served to characterize the toxicity profiles of these novel agents. Overall, it is evident that RCC-directed targeted therapy differs from immunotherapy and cytotoxic chemotherapy in terms of a number of unique nonhematologic AEs (some of which have not been traditionally encountered in oncology practice) and that there are distinctions within and across the various classes of agents with respect to the most prominent AEs and the risk for less common but serious complications. Although treatment-associated AEs are common, the majority of AEs reported during clinical trial experiences were grade 1 or 2 in severity and manageable with intervention in the form of supportive measures and/or dosage modification. Therefore, despite the relatively complex AE profiles of RCC-directed targeted therapy, patient education, consistent monitoring with a focus on early detection by health care providers (oncologists, general physicians, nurses), and the application of emerging AE management strategies may allow for prolonged treatment in most patients with advanced RCC.
Introduction
The benefits of immunotherapy for advanced renal cell carcinoma (RCC), the mainstay of systemic therapy for many years, have been limited by the modest antitumor activity and characteristically poor tolerability of cytokine-based regimens [1]. RCC-directed targeted therapies are now offering clinical benefits over immunotherapy, in terms of both better efficacy and safety profiles that have the potential to allow for a prolonged duration of treatment when adverse events (AEs) are managed appropriately. Despite the gains in efficacy that have been collectively documented across the various newly approved regimens, including the combination of bevacizumab plus interferon [2, 3], the vascular endothelial growth factor receptor tyrosine kinase inhibitors (VEGFR TKIs) sunitinib [4, 5], sorafenib [6, 7], and pazopanib [8], and the mammalian target of rapamycin (mTOR) inhibitors temsirolimus [9] and everolimus [10, 11], therapeutic benefits are of a primarily palliative nature. Thus, prompt detection and effective management of AEs are a fundamental component of targeted therapy for advanced RCC in clinical practice.
This article discusses the common toxicities observed during these clinical trial experiences as well as specific reported side effects, while also highlighting recommended strategies to anticipate the occurrence of key AEs, minimize the risk for increasing AE severity, and manage toxicities to help optimize treatment outcomes in patients with advanced RCC.
Phase III Safety and Tolerability Data for Targeted Agents Approved for Treatment of RCC
Table 1 [4–14] provides an overview of the patient populations, treatment regimens, and safety assessments of the pivotal phase III clinical trials of RCC targeted therapy, which randomized a total of approximately 4,500 patients with advanced RCC with varying degrees of prior treatment exposure. Study treatment was allowed to continue until disease progression or unacceptable toxicity, except in the Avastin and Roferon in Renal Cell Carcinoma (AVOREN) trial (BO17705) of bevacizumab plus interferon [2, 3]. Accumulating safety data from these targeted therapies demonstrate distinctions within and across the various classes of agents with respect to the most commonly observed AEs and the risk for less common but serious complications. Determining individual patient risk and managing these toxicities are essential to optimize patient benefit.
Table 1.
Pivotal trials of targeted therapy for renal cell carcinoma: Treatment and safety assessment details
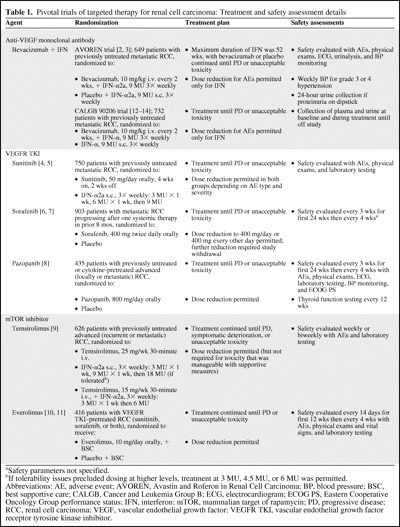
aSafety parameters not specified.
bIf tolerability issues precluded dosing at higher levels, treatment at 3 MU, 4.5 MU, or 6 MU was permitted.
Abbreviations: AE, adverse event; AVOREN, Avastin and Roferon in Renal Cell Carcinoma; BP, blood pressure; BSC, best supportive care; CALGB, Cancer and Leukemia Group B; ECG, electrocardiogram; ECOG PS, Eastern Cooperative Oncology Group performance status; IFN, interferon; mTOR, mammalian target of rapamycin; PD, progressive disease; RCC, renal cell carcinoma; VEGF, vascular endothelial growth factor; VEGFR TKI, vascular endothelial growth factor receptor tyrosine kinase inhibitor.
Bevacizumab Plus Interferon
Phase III RCC Clinical Trial AE Summary
All-grade AEs occurring in ≥30% and grade 3 or 4 AEs occurring in ≥5% of bevacizumab plus interferon–treated patients in the AVOREN trial are shown in Figure 1 [2, 3]. In the AVOREN trial, the interferon component was limited to a 1-year duration, with continued use of bevacizumab or placebo permitted beyond the 1-year period [2, 3]. Consistent with the known toxicity profile of interferon, fatigue and asthenia were among the most commonly reported all-grade and grade 3 or 4 AEs regardless of whether patients received bevacizumab plus interferon or placebo with or without interferon [3, 13]. The addition of bevacizumab to interferon led to higher overall incidences of hypertension, proteinuria, and bleeding: 26%, 18%, and 33%, respectively, versus 9%, 3%, and 9%, respectively with placebo plus interferon [3]. Based on a recently updated report, grade 3 or 4 hypertension and proteinuria occurred in 6% and 8% of bevacizumab plus interferon recipients, respectively [2]. Arterial thromboembolic events (ATEs), gastrointestinal perforation, wound-healing complications, and congestive heart failure (CHF), all previously associated with bevacizumab in clinical trials in other tumor populations, occurred at overall and grade 3 or 4 incidences ≤1% in the advanced RCC AVOREN trial [3]. In the Cancer and Leukemia Group B (CALGB) 90206 trial, bevacizumab plus interferon was most commonly associated with fatigue, proteinuria, nausea, neutropenia, and hypertension [13]. The incidences of grade ≥3 hypertension (11% versus 0%), anorexia (17% versus 8%), fatigue (37% versus 30%), and proteinuria (15% versus <1%) were significantly greater with bevacizumab plus interferon than with interferon alone [13]. The overall and grade 3 or 4 incidences of cardiac ischemia/infarction, left ventricular dysfunction, and gastrointestinal perforation were ≤1%. Thromboses/embolisms were observed in 4% of patients receiving bevacizumab plus interferon, and 2% experienced grade ≥3 [13]. In both studies, the incidence of drug-related deaths was 1%–2% regardless of treatment arm [2, 3, 13].
Figure 1.
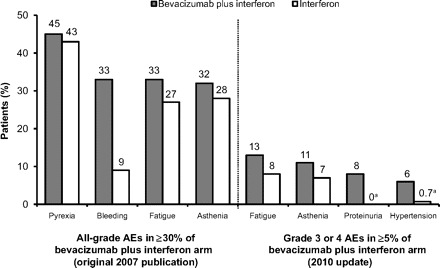
Most prominent adverse events (AEs) with bevacizumab plus interferon in the Avastin and Roferon in Renal Cell Carcinoma pivotal trial [2, 3].
aIncidences based on original 2007 publication (not reported in 2010 update).
Recommended AE Monitoring and Management
The product labeling for bevacizumab contains no dose reduction recommendations (with AEs to be managed instead by treatment interruption or discontinuation), and the risks for gastrointestinal perforation, surgery and wound-healing complications, and hemorrhage are highlighted as boxed warnings [15]. It is recommended that the use of bevacizumab be avoided in patients with serious hemorrhage or recent hemoptysis and discontinued in patients developing gastrointestinal perforation or wound dehiscence [15]. To reduce the risk for surgical and wound-healing complications, bevacizumab should be discontinued at least 28 days prior to elective surgery and restarted ≥28 days postsurgery, and only after complete wound healing has occurred [15]. Blood pressure (BP) monitoring on an every-2-week or every-3-week basis and urinalysis to quantify urine protein are to be included in the routine monitoring of patients undergoing bevacizumab therapy, with temporary suspension of treatment warranted in patients developing uncontrolled severe hypertension or moderate proteinuria [15]. In the event of hypertensive crisis/encephalopathy or nephrotic syndrome, discontinuing bevacizumab is the appropriate course of action [15]. Additional complications that occur infrequently but for which discontinuation would be warranted include nongastrointestinal fistula formation, a severe ATE (e.g., myocardial or cerebral infarction), and reversible posterior leukoencephalopathy syndrome, for which the incidence has been <0.1% across various clinical trials [15]. Severe infusion reactions, which have been rarely reported, necessitate temporary suspension of therapy, with the recommendation to stop the infusion and administer medical intervention as appropriate [15].
VEGFR TKIs
The AE profiles of the three RCC-approved VEGFR TKIs based on the published pivotal trials are summarized in Table 2, providing insight into the safety and tolerability of sunitinib as first-line therapy, sorafenib in patients with progressive disease after one prior systemic therapy (mainly cytokine-based regimens), and pazopanib in a mixed population of previously untreated and cytokine-pretreated patients.
Table 2.
Most prominent adverse events with VEGFR TKIs in pivotal trials in renal cell carcinoma
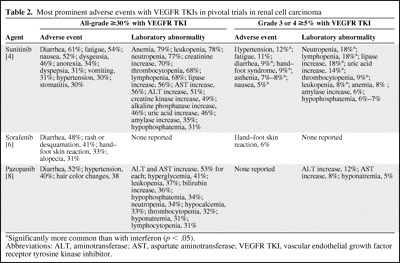
aSignificantly more common than with interferon (p < .05).
Abbreviations: ALT, aminotransferase; AST, aspartate aminotransferase; VEGFR TKI, vascular endothelial growth factor receptor tyrosine kinase inhibitor.
Sunitinib
Phase III RCC Clinical Trial AE Summary
Key notable clinical AEs observed with sunitinib treatment included diarrhea (61%), fatigue (54%), hypertension (30%), stomatitis (30%), hand–foot syndrome (29%), and asthenia (20%) [4]. Sunitinib was associated with higher incidences of treatment-related AEs than with interferon, with the most pronounced differences in the overall incidences of diarrhea (61% versus 15%) and taste disturbance (46% versus 15%) [4]. The most common grade 3 or 4 treatment-related AEs were hypertension (12%), followed by fatigue (11%), diarrhea (9%), and hand–foot syndrome (9%) [4]. Grade 3 or 4 laboratory abnormalities during sunitinib therapy included neutropenia, lymphopenia, and lipase elevation (each in ∼18% of patients) and hyperuricemia (grade 4 incidence, 14%) [4]. Most grade 3 or 4 AE and laboratory abnormality incidences were significantly higher (p < .05) with sunitinib than with interferon, except for fatigue (similar between groups) and lymphopenia (significantly higher with interferon) [4]. Hypothyroidism and left ventricular ejection fraction (LVEF) decline were reported in 14% and 13% of sunitinib recipients, respectively (2% and 3%, respectively, with interferon) [4]; most cases of these AEs were grade 1 or 2, with grade 3 incidences of 2% and 3%, respectively. Regarding LVEF reduction, three patients were described as having left ventricular dysfunction and one developed CHF [16]. Three deaths (<1%) were treatment related: one in the sunitinib group and two in the interferon group (cardiovascular) [4].
In an open-label trial in which 4,564 patients with metastatic RCC received sunitinib therapy on a compassionate-use basis, the most common any-grade AEs were diarrhea (44%) and fatigue (37%); grade 3 or 4 AEs included fatigue (8%) and thrombocytopenia (8%) [17].
A meta-analysis of the risk for ATE posed by sunitinib and sorafenib was recently published, capturing a total of 10 reports of phase II and III oncology clinical trials and expanded-access programs for which ATE data were available [18]. Regarding sunitinib, the ATE incidence was 1.3% (95% confidence interval [CI], 1.0%–1.6%) across four clinical trials in patients with RCC (two trials—one phase II trial and an expanded-access program), neuroendocrine tumors (phase II trial), and gastrointestinal stromal tumors (GISTs) (phase III trial), with a relative risk of 2.4 (95% CI, 0.12–49.4) when compared with the placebo control arm of the GIST trial [18].
Recommended AE Monitoring and Management
In addition to hypertension, requiring BP monitoring (either daily or multiple times per week) and antihypertensive management as needed (including temporary suspension to allow for control of severe hypertension) [16], further sunitinib-associated cardiovascular risks in the form of LVEF reduction and dose-dependent QT interval prolongation with potential torsade de pointes (<0.1%) necessitate close patient monitoring [16]. In this regard, considerations for follow-up include periodic monitoring of LVEF and electrocardiogram and electrolyte monitoring (for early detection of QT interval prolongation) [16]. Obtaining a baseline LVEF should be considered even in the absence of cardiac risk factors [16]. Because significant cardiac events within the prior year precluded clinical trial participation, the extent to which patients with pre-existing cardiac conditions have a higher risk for sunitinib-induced LVEF reduction or CHF is unknown, requiring clinicians to evaluate the risk–benefit ratio for each individual patient [16]. Patients developing signs and/or symptoms of thyroid dysfunction should undergo laboratory testing to evaluate potential sunitinib-associated thyroid dysfunction, the management of which would follow standard practice guidelines [16]. Routine CBCs together with physical examinations may allow for early detection of hemorrhagic events; the potential for adrenal hemorrhage, albeit observed only in animal studies, has led to the recommendation of adrenal function monitoring in situations of stress (e.g., surgery, trauma, or severe infection) [16].
Additional practical AE management strategies for use in sunitinib-treated patients have been described [19]. For example, medical management of hypertension must take into account the potential impact of cytochrome P450 (CYP)3A4 inhibitors or inducers on sunitinib metabolism [19]. Managing fatigue likewise may be challenging because of the inherent difficulty in identifying the primary underlying cause(s), and close monitoring involving a combination of clinical assessment/counseling (focused on characterizing the severity and quality-of-life [QOL] impact of fatigue) and laboratory evaluation (hemoglobin, thyroid function) may prove useful in this regard [19]. Sunitinib dose reduction may play a role in the management of hypertension and fatigue depending on the issues surrounding the individual patient (e.g., QOL-impairing fatigue, inability to avoid concurrent antihypertensive CYP3A4 inhibitors or inducers) [19]. Palliative treatment for hand–foot syndrome includes moisturizers, foot and hand care products, and medication for pain management [13]. For patients who experience grade ≥3 skin toxicity, treatment interruption and dose reduction may be necessary. Generalized skin rashes can be treated with moisturizing lotions [20].
Interferon-Related AE Summary
Interferon-related AEs in these studies included fatigue, chills/fever/asthenia, anemia, and nausea in many patients; most were grade 1 or 2, but grade ≥3 AEs were not uncommon [2–4, 6, 13]. In general, these toxicities are considered manageable, with fatigue often being the most challenging [1].
Sorafenib
Phase III RCC Clinical Trial AE Summary
In the recently published final Treatment Approaches in Renal Cancer Global Evaluation Trial (TARGET) analysis, sorafenib was associated with higher AE incidences than with placebo, including diarrhea (48% versus 11%), fatigue (29% versus 16%), hypertension (17% versus 1%), and various dermatologic-type reactions; that is, rash/desquamation (41% versus 13%), hand–foot skin reactions (33% versus 8%), alopecia (31% versus 4%), pruritus (17% versus 4%), and dry skin (13% versus 3%) (Fig. 2) [6]. Grade 3 or 4 AEs were infrequent, and most commonly hand–foot syndrome with sorafenib, 6%, versus 0.4% with placebo [6]. Also with sorafenib, there were increased incidences of grade 3/4 hypertension (4% vs. 0% with placebo) and cardiac ischemia/infarction (4.9% vs. 0.4% with placebo) [6]. In patients initially randomized to placebo who crossed over to sorafenib, the AE incidences were notably similar to those in the sorafenib arm; in this subset, the incidences of grade 3 or 4 hypertension and cardiac ischemia/infarction were 4% and 1.4%, respectively [6]. No treatment-related deaths were reported [6]. A retrospective analysis of 169 patients in the TARGET study who received sorafenib treatment for >1 year confirmed that diarrhea, rash, hand–foot syndrome, and fatigue were the most common mild to moderate AEs, suggesting that long-term use of sorafenib does not exacerbate any drug-related toxicities [21].
Figure 2.
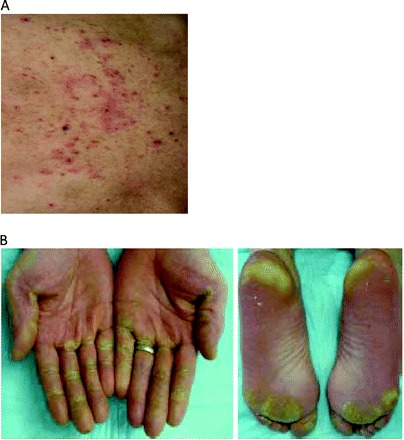
Skin rashes include (A) erythema, maculopapular dermatitis, or seborrheic dermatitis and (B) hand–foot syndrome.
(A) Reprinted with permission from Sarkodie T, Ross P. Late presentation of sorafenib-associated rash: a case report. J Med Case Reports 2010;4:338–343. (B) Reprinted from Lacouture ME, Reilly LM, Gerami P et al. Hand foot skin reaction in cancer patients treated with the multikinase inhibitors sorafenib and sunitinib. Ann Oncol 2008;19:1955–1961, by permission of Oxford University Press.
In the open-label, expanded-access Advanced Renal Cell Carcinoma Sorafenib program, 2,504 patients in the U.S. and Canada were permitted access to sorafenib prior to regulatory approval [22]. The AE profile in this large patient cohort was similar to the results from the phase III TARGET trial, with the most common AEs being hand–foot syndrome (18%), rash (14%), hypertension (12%), fatigue (11%), diarrhea (8%), and nausea (5%). Grade 3 hypertension was reported in 5% of patients receiving expanded-access sorafenib.
In the aforementioned meta-analysis of sunitinib and sorafenib, the ATE incidence was 1.7% (95% CI, 1.1%–2.4%) across six reports in patients with RCC (four trials), hepatocellular carcinoma (one trial), and non-small cell lung cancer (one trial) [16]. When assessing data from the TARGET study and a similar placebo-controlled trial of sorafenib in patients with advanced hepatocellular carcinoma, the relative risk for an ATE was 3.1 (95% CI, 1.2–7.9) with sorafenib versus placebo, which was not significantly higher than the relative risk of 2.4 observed with sunitinib [18]. Considering both sunitinib and sorafenib data together, the relative risk for an ATE was significant at 3.0 (95% CI, 1.3–7.4; p = .015)—relative risks of 6.0 and 2.1 in the RCC and non-RCC populations, respectively (despite the numerical difference between the cancer types, this was not statistically significant) [18].
Recommended AE Monitoring and Management
Similar to the guidelines for sunitinib, potential hypertension should be watched closely with regular BP monitoring. Given that sorafenib-associated hypertension has typically occurred early in the course of treatment, specific guidelines are to monitor BP weekly for the first 6 weeks and then periodically thereafter, instituting antihypertensive therapy if needed [23]. The product labeling for sorafenib also provides detailed management recommendations for skin toxicity, which is also typically seen during the first 6 weeks of treatment [23] and may require palliative treatment or dose interruption/reduction as described above. Sorafenib discontinuation is warranted in patients who develop the rare occurrence of gastrointestinal perforation (<1%) and should be a consideration in patients who experience cardiac ischemia and/or infarction (either temporary or permanent discontinuation) or bleeding necessitating medical intervention [23]. Although treatment interruption is recommended around the time of major surgery, more specific guidance is precluded by the limited experience with sorafenib in this setting [23].
Pazopanib
Phase III RCC Clinical Trial AE Summary
Diarrhea and hypertension were the most prominent AEs with pazopanib, both overall (52% and 40%, respectively, versus 9% and 10%, respectively, with placebo) and when considering grade 3 or 4 events only (4% for both, versus <1% for both with placebo) [8]. Other gastrointestinal AEs and fatigue/asthenia were also more frequent with pazopanib, but the grade 3 or 4 incidences were ≤3% [8]. Hemorrhage (any site) occurred in 13% of pazopanib recipients versus 5% of placebo recipients [8]. With pazopanib, increases in serum transaminase levels were the most common type of all-grade (53% for alanine aminotransferase [ALT] and 53% for aspartate aminotransferase) and grade 3 or 4 (12% and 8%, respectively) laboratory abnormalities [8]. Leukopenia, neutropenia, and thrombocytopenia each occurred in approximately one third of pazopanib recipients, compared with 5%–6% of placebo recipients; however, the grade 3 or 4 incidences of these hematologic abnormalities were low (0%–2%) in both arms [8]. Elevations in ALT ≥3× the upper limit of normal were observed in 18% of pazopanib-treated patients, and most cases occurred within the first 4 months of therapy [8].
Recommended AE Monitoring and Management
The AE warnings in the product labeling for pazopanib share similarities with those for other VEGFR TKIs, including the need for monitoring and treatment of hypertension and thyroid function testing (for hypothyroidism) [24]. Routine liver function tests and urinalyses are specifically recommended in light of reports of severe and fatal hepatotoxicity (boxed warning) and grade 3 or 4 proteinuria during pazopanib treatment [24]. In addition, a cautious approach is suggested in patients at higher risk for QT prolongation (which may include an electrocardiogram and electrolyte monitoring) [24]. Use of pazopanib should be avoided in patients with a history of hemoptysis, cerebral or clinically significant gastrointestinal hemorrhage, or an ATE in the past 6 months [24], and caution is advised in those with an earlier ATE history (or with an elevated risk) or who are considered at heightened risk for gastrointestinal perforation or fistula (incidence of 0.9% during pazopanib treatment, including a 0.3% incidence of fatal events) [24]. Although not specifically recommended when considering sunitinib and sorafenib as patient treatment options, ATE and gastrointestinal perforation are class effects and these recommendations should be considered with any VEGFR TKI therapy.
mTOR Inhibitors
AE data from phase III clinical trials of the mTOR inhibitors temsirolimus and everolimus are summarized in Table 3, representing different settings in terms of prior treatment exposure; that is, temsirolimus in a poor-prognosis population with no prior systemic therapy and everolimus in patients previously treated with the VEGFR TKIs sunitinib, sorafenib, or both.
Table 3.
Most prominent adverse events with mTOR inhibitors in pivotal trials in renal cell carcinoma
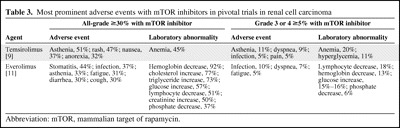
Abbreviation: mTOR, mammalian target of rapamycin.
Temsirolimus
Phase III RCC Clinical Trial AE Summary
In the Global Advanced Renal Cell Carcinoma (ARCC) trial in previously untreated patients with poor-prognosis disease, asthenia was the most prominent AE across all three arms, including a 51% incidence with temsirolimus monotherapy, versus 64% with interferon monotherapy [9]. Rash was the second most common AE with temsirolimus monotherapy, 47%, versus 6% with interferon monotherapy [9]. All-grade AE incidences with temsirolimus monotherapy were >30% for anemia (45%), nausea (37%), and anorexia (32%) and >25% for pain (28%), dyspnea (28%), diarrhea (27%), infection (27%), peripheral edema (27%), hyperlipidemia (27%), hyperglycemia (26%), and cough (26%) [9]. Most of these AEs were seen less frequently with interferon, except for anemia and gastrointestinal AEs. In addition, stomatitis was more common with temsirolimus (20%, versus 4% with interferon) [9], as were drug-related renal AEs (25%, versus 12% with interferon) and creatinine increase in particular (11%, versus 4% with interferon) [20]. Anemia was the most common grade 3 or 4 AE with temsirolimus monotherapy (20%), followed by asthenia (11%) and hyperglycemia (11%) [9]. Other grade 3 or 4 AEs occurring in ≥5% of temsirolimus recipients were dyspnea (9%), pain (5%), and infection (5%) [9].
Four temsirolimus recipients developed drug-related pneumonitis of grade 1 at week 18 (n = 1), grade 2 at week 9 (n = 1), grade 2 at week 9 that progressed to grade 3 (n = 1), and grade 3 at week 41 that progressed to grades 4 and 5 (n = 1) [25]. Although the one death was classified as being from progressive disease, the relative contribution of the underlying progressive pneumonitis was not altogether clear [25]. Of note, in these patients, there were associations between cough and grade ≥2 pneumonitis as well as between dyspnea and pneumonitis of progressive severity [25]. One additional case of drug-related pneumonitis was reported, of grade 2 severity at week 28 in the interferon group [25].
Recommended AE Monitoring and Management
Temsirolimus-associated infusion reactions can be managed by stopping the infusion and administering an antihistamine, with the potential for restarting the infusion after 30–60 minutes with additional antihistamine support and at a slower rate based on physician discretion [26]. The risks for bowel perforation and abnormal wound healing are elevated during temsirolimus therapy [26]. Patients presenting with symptoms of fever, abdominal pain, bloody stools, and/or acute abdomen should be promptly evaluated for potential bowel perforation, and a cautious approach should be applied during the perioperative period [26]. CBCs should be performed to monitor for anemia (among the most prominent AEs of temsirolimus), with use of transfusions and erythropoietin as needed, and for thrombocytopenia (albeit less frequently occurring than anemia) [25]. Renal function monitoring during temsirolimus therapy is essential because of the heightened propensity for renal toxicity with temsirolimus than with interferon in the ARCC trial [25], with some rapidly progressive, dialysis-unresponsive fatalities [26]. Other components of routine monitoring include glucose and lipid profiles (instituting dietary and/or medical intervention for hyperglycemia or hyperlipidemia as needed) [25, 26] and close evaluation for immunosuppression-related infection and clinical or radiographic evidence of pneumonitis [26]. Based on the ARCC clinical trial experience, continuing treatment without modification in asymptomatic patients with radiographic changes and temporarily interrupting therapy in patients with limited symptoms may be feasible [25]. Conversely, patients with worsening symptoms plus deteriorating pulmonary function or with underlying pulmonary conditions (in whom ruling out infectious etiology may be of particular importance) may be appropriate candidates for temsirolimus discontinuation [25].
Everolimus
Phase III RCC Clinical Trial AE Summary
In the final safety analysis of the Renal Cell Cancer Treatment with Oral RAD001 Given Daily 1 (RECORD-1) trial in VEGFR TKI–pretreated metastatic RCC patients, stomatitis and infections were the most prominent all-grade AEs with everolimus (44% and 37%, respectively), more common than with placebo (8% and 18%, respectively), but mostly of grade 1 or 2 severity [11]. Grade 3 or 4 AEs in ≥5% of everolimus recipients were infections (10%), dyspnea (7%), and fatigue (5%) [11]. Laboratory abnormalities were also more common with everolimus, including hemoglobin (92%) and lymphocyte (51%) reductions as well as increases in cholesterol (77%), triglycerides (73%), and glucose (57%) [11]. Most laboratory abnormalities were grade 1 or 2, although grade 3 or 4 incidences were >10% for hemoglobin (13%), lymphocyte (18%), and glucose (15%–16%) abnormalities [11].
Noninfectious pneumonitis was reported in 37 everolimus recipients (grade 1, n = 9; grade 2, n = 18; grade 3, n = 10), for a 14% incidence, versus 0% with placebo [11, 27]. The median time to onset of this toxicity was 108 days, with a wide range of 24–257 days, frequent association with cough, dyspnea, or both (51%, 43%, or 32% of patients, respectively), and substantial variability with respect to radiographic patterns (ground-glass to diffuse infiltrates) [27]. In five of the 10 patients who developed grade 3 pneumonitis, there was radiological evidence of pneumonitis before the initiation of everolimus therapy [27].
Recommended AE Monitoring and Management
Patient education surrounding the heightened risk and early signs/symptoms of oral ulceration, infection, and noninfectious pneumonitis has the potential to lead to presentation at an earlier grade. Topical remedies are appropriate for stomatitis/mucositis, and it is further recommended that alcohol- or peroxide-containing mouthwashes and antifungal agents (in the absence of confirmed fungal infection) be avoided [28]. Infections should be promptly managed [28]. To minimize the risks associated with invasive fungal infections, they should be fully treated before initiating everolimus treatment and warrant the discontinuation of everolimus (and use of antifungal therapy) if emerging during treatment. The recommended management of noninfectious pneumonitis is dependent on the severity of the associated symptoms, with limited symptoms allowing for continuation of therapy, patients with moderate symptoms potentially benefiting from interruption, and severe symptoms warranting a combination of everolimus discontinuation and corticosteroid therapy [28]. Even in cases of severe noninfectious pneumonitis, it may be feasible to restart therapy at a reduced dose depending on patient-specific considerations [28]. Given the various laboratory abnormalities that have been reported during everolimus therapy, periodic CBCs, renal function testing, and fasting glucose and lipid profiles are recommended [28]. In the event that glucose or lipid levels are elevated at baseline, treatment to normal levels prior to initiating everolimus is preferable [28].
More specific guidelines for managing key everolimus-associated AEs (based on the RECORD-1 experience) were recently developed by a multidisciplinary panel of experts, with management of stomatitis, noninfectious pneumonitis, and infection by grade summarized in Table 4 [29].
Table 4.
Recommended adverse event management in everolimus-treated renal cell carcinoma patients based on RECORD-1 experience [29]
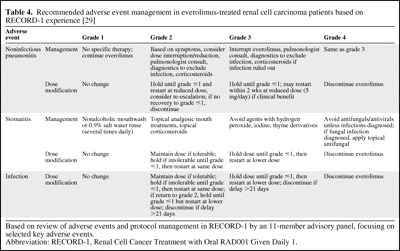
Based on review of adverse events and protocol management in RECORD-1 by an 11-member advisory panel, focusing on selected key adverse events.
Abbreviation: RECORD-1, Renal Cell Cancer Treatment with Oral RAD001 Given Daily 1.
Relationship Between RCC Targeted Therapy–Associated AEs and Efficacy
The relationship between AEs and the efficacy of targeted agents in patients with RCC has been investigated. In a retrospective analysis of sunitinib in 40 patients with cytokine-refractory RCC, only hypertension, particularly grade 3, was associated with a higher treatment response rate [30]. A similar finding was demonstrated in a prospective study of 43 patients with metastatic RCC treated with bevacizumab. In that study, a significantly longer median time to progression was observed for patients with hypertension than for patients with BP <150/100 mmHg (8.1 versus 4.2; p = .036) [31]. Ravaud and Sire [32] evaluated hypertension and efficacy in 93 patients receiving either sunitinib, sorafenib, or bevacizumab as first-, second-, or third-line therapy. Of the evaluable patients with grade ≥2 hypertension, 88% had a clinical benefit (defined as an objective response or stable disease) and 53% benefited for ≥6 months, versus 55% and 35%, respectively, for patients with no significant change in BP. More recently, the predictive power of hypertension was evaluated in a retrospective analysis of the phase III CALGB 90206 study, which demonstrated that patients on bevacizumab plus interferon who developed grade ≥2 hypertension had significantly greater progression-free survival (PFS) and overall survival times than patients who did not develop hypertension [13].
Relationship Between RCC Targeted Therapy–Associated Pharmacokinetics/Pharmacodynamics and AEs
As part of the clinical development of the molecularly targeted anticancer agents, research efforts have been seeking to elucidate the relationship between the pharmacokinetics/pharmacodynamics (PK/PD) and clinical activity and AE profiles. Recently, a PK/PD meta-analysis of the relationships between clinical endpoints and sunitinib exposure in patients with advanced solid tumors was published, with data derived from six clinical trials in patients with metastatic RCC, GIST, or various solid tumors [33]. Sunitinib doses were in the range of 25–150 mg, administered either daily or every other day as 4 weeks on–2 weeks off or 2 weeks on–2 weeks off schedules or daily as a 2 weeks on–1 week off schedule [33]. Relationships for tolerability endpoints were identified between exposure (total drug steady-state area under the curve [AUCss]) and incidence, but not severity, of fatigue [33]. The results of a simulation model of 1,000 patients, as illustrated in Figure 3, demonstrated that patients who developed fatigue reached the maximum grade of severity after the first cycle of sunitinib [33]. In the RCC subset, this model determined that the probability of experiencing grade ≥1 fatigue was 57% for 25 mg/day sunitinib and 74% for 50 mg/day sunitinib [33]. Regarding the absolute neutrophil count (ANC), there was a direct relationship with the cumulative AUC of the total drug and the ANC [33]. In the ANC modeling, RCC patients with a baseline ANC of 5 per nl who are subsequently treated with 25 mg/day, 50 mg/day, and 75 mg/day sunitinib are expected to experience reduction to 4.2, 3.5, and 2.7 per nl, respectively [33]. Data for the relationship between total drug concentration and diastolic BP was evaluable for all patients irrespective of tumor type, with estimated maximum elevations in diastolic BP of 5 mmHg during treatment with 25 mg/day sunitinib and 8 mmHg during treatment with 50 mg/day sunitinib [33].
Figure 3.
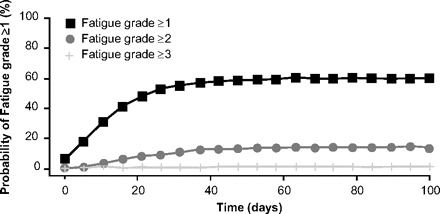
The probability of experiencing fatigue (from sunitinib) over time using 1,000 simulated patients [33].
From Houk BE, Bello CL, Poland B et al. Relationship between exposure to sunitinib and efficacy and tolerability endpoints in patients with cancer: Results of a pharmacokinetic/pharmacodynamic meta-analysis. Cancer Chemother Pharmacol 2010;66:357–371. Figure 10, reprinted with kind permission from Springer Science and Business Media © Springer-Verlag, 2009.
A recent study also evaluated the association between thyroid dysfunction and PFS in patients receiving sunitinib for metastatic RCC. Bladou et al. [34] evaluated thyroid function in 111 patients with metastatic RCC who received 50 mg/day sunitinib for 4 weeks with a 2-week rest. Approximately half (52%) of the patients in that study developed thyroid dysfunction; of those with hypothyroidism, 93% received thyroid replacement. No differences in the PFS times were noted between patients with thyroid dysfunction who were treated and those without thyroid dysfunction.
Boni et al. [35] conducted an evaluation of the population PK of temsirolimus, estimating PK parameters and PD associations of exposure parameters with safety (and clinical activity) from a randomized, double-blind, multicenter trial. In the dose-escalation phase II trial, 50 patients were treated with temsirolimus at doses of 25 mg, 75 mg, or 250 mg i.v. once weekly [35]. The temsirolimus AUC was significantly correlated with AE severity, particularly for thrombocytopenia (p = .007), but also for pruritus and hyperlipemia (both p < .05), and with AE duration for thrombocytopenia and dry mouth (both p < .05) [35]. In analyzing cumulative exposures for temsirolimus, there were significant associations between the cumulative AUC and AE severity for acne, infection, mucositis, and nail discoloration (all p < .01), as well as for pruritus, maculopapular rash, and cough (all p ≤ .05) [35]. Greater exposure was likewise associated with a longer duration of rash (p < .001), anorexia (p = .001), and hyperglycemia, diarrhea, and maculopapular rash (all p < .05) [35].
PK data for various doses of sorafenib are forthcoming from a phase II trial of intrapatient dose escalation of sorafenib monotherapy in previously untreated patients with metastatic RCC [36]. In that European multicenter trial, approximately 80 patients received the recommended starting dose of 400 mg twice daily for 28 days, 600 mg twice daily for the next 28 days, and finally 800 mg twice daily until disease progression or unacceptable toxicity. The design of the study was supported by a smaller phase II trial in 44 cytokine-pretreated patients, in which dose escalation from 400 mg twice daily to 600 or 800 mg twice daily was feasible in 93% of patients [37]. Substantial antitumor activity was suggested by an objective response rate of 55% (including complete and partial response rates of 16% and 39%, respectively) and a median PFS interval of 8.4 months, with no apparent deleterious effect of dose escalation on the therapeutic balance in terms of the frequency and severity of AEs [37].
Summary
Based on the phase III clinical trials that formed the basis of their regulatory approval, AEs with targeted therapies for advanced RCC are common but generally acceptable and manageable, typically occurring at a severity of grade 1 or 2. Importantly, however, there are notable AE-related similarities and differences within and across the various classes of agents currently available for use in community practice. Fatigue has been commonly reported across the various agents, as have gastrointestinal complaints (particularly, diarrhea with the VEGFR TKIs and stomatitis with everolimus). Overall, the use of bevacizumab or a VEGFR TKI predisposes patients to cardiovascular complications (most commonly hypertension, but also in the form of ATEs, with LVEF reductions and QT prolongation also described for sunitinib), bleeding/hemorrhage, and gastrointestinal perforation (most commonly described with bevacizumab but with some reports during VEGFR TKI therapy). Dermatologic reactions of varying types have ranked among the most common AEs with VEGFR TKIs, including hand–foot syndrome with sunitinib and sorafenib and a particularly high frequency of rash/desquamation with sorafenib. Overall, whereas sunitinib has been associated with the highest incidences of hematologic and other laboratory abnormalities, the newest VEGFR TKI, pazopanib, appears to have a heightened propensity for producing grade 3 or 4 transaminase elevations. Key class effects for the mTOR inhibitors include noninfectious pneumonitis, infections related to the immunosuppressive properties of these agents, and glucose and lipid abnormalities. Within the mTOR inhibitor class, key drug-specific toxicities are stomatitis/mucositis for everolimus and hypersensitivity reactions for temsirolimus.
Given the frequency and potential severity of the AEs characterized to date, the importance of understanding the toxicity profiles of these agents and close vigilance to allow for prompt detection and efficient management cannot be overstated. In this regard, patient-focused education (with key principles for the most common AEs outlined in Table 5) and sufficient institutional resources to maintain a good patient relationship with the health care provider (i.e., nurses and/or other staff to answer patient concerns and triage AEs) are of the utmost importance for optimizing dosing, the duration of therapy, and, ultimately, duration of life and QOL in the advanced RCC population.
Table 5.
Examples of patient education points for common targeted therapy–associated toxicity [19]
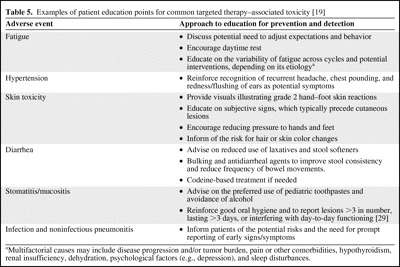
aMultifactorial causes may include disease progression and/or tumor burden, pain or other comorbidities, hypothyroidism, renal insufficiency, dehydration, psychological factors (e.g., depression), and sleep disturbances.
Acknowledgments
The author takes full responsibility for the content of the paper but thanks Laurie Orloski, Pharm.D., and Amy Zannikos, Pharm.D., supported by Novartis Pharmaceuticals Corporation, for their assistance in preparing an initial draft of the manuscript.
References
- 1.Jonasch E, Haluska FG. Interferon in oncological practice: Review of interferon biology, clinical applications, and toxicities. The Oncologist. 2001;6:34–55. doi: 10.1634/theoncologist.6-1-34. [DOI] [PubMed] [Google Scholar]
- 2.Escudier B, Bellmunt J, Négrier S, et al. Phase III trial of bevacizumab plus interferon alfa-2a in patients with metastatic renal cell carcinoma (AVOREN): Final analysis of overall survival. J Clin Oncol. 2010;28:2144–2150. doi: 10.1200/JCO.2009.26.7849. [DOI] [PubMed] [Google Scholar]
- 3.Escudier B, Pluzanska A, Koralewski P, et al. AVOREN Trial investigators. Bevacizumab plus interferon alfa-2a for treatment of metastatic renal cell carcinoma: A randomised, double-blind phase III trial. Lancet. 2007;370:2103–2111. doi: 10.1016/S0140-6736(07)61904-7. [DOI] [PubMed] [Google Scholar]
- 4.Motzer RJ, Hutson TE, Tomczak P, et al. Overall survival and updated results for sunitinib compared with interferon alfa in patients with metastatic renal cell carcinoma. J Clin Oncol. 2009;27:3584–3590. doi: 10.1200/JCO.2008.20.1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Motzer RJ, Hutson TE, Tomczak P, et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N Engl J Med. 2007;356:115–124. doi: 10.1056/NEJMoa065044. [DOI] [PubMed] [Google Scholar]
- 6.Escudier B, Eisen T, Stadler WM, et al. Sorafenib for treatment of renal cell carcinoma: Final efficacy and safety results of the phase III Treatment Approaches in Renal Cancer Global Evaluation Trial. J Clin Oncol. 2009;27:3312–3318. doi: 10.1200/JCO.2008.19.5511. [DOI] [PubMed] [Google Scholar]
- 7.Escudier B, Eisen T, Stadler WM, et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med. 2007;356:125–134. doi: 10.1056/NEJMoa060655. [DOI] [PubMed] [Google Scholar]
- 8.Sternberg CN, Davis ID, Mardiak J, et al. Pazopanib in locally advanced or metastatic renal cell carcinoma: Results of a randomized phase III trial. J Clin Oncol. 2010;28:1061–1068. doi: 10.1200/JCO.2009.23.9764. [DOI] [PubMed] [Google Scholar]
- 9.Hudes G, Carducci M, Tomczak P, et al. Global ARCC Trial. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N Engl J Med. 2007;356:2271–2281. doi: 10.1056/NEJMoa066838. [DOI] [PubMed] [Google Scholar]
- 10.Motzer RJ, Escudier B, Oudard S, et al. RECORD-1 Study Group. Efficacy of everolimus in advanced renal cell carcinoma: A double-blind, randomised, placebo-controlled phase III trial. Lancet. 2008;372:449–456. doi: 10.1016/S0140-6736(08)61039-9. [DOI] [PubMed] [Google Scholar]
- 11.Motzer RJ, Escudier B, Oudard S, et al. Phase 3 trial of everolimus for metastatic renal cell carcinoma: Final results and analysis of prognostic factors. Cancer. 2010;116:4256–4265. doi: 10.1002/cncr.25219. [DOI] [PubMed] [Google Scholar]
- 12.Rini BI, Halabi S, Rosenberg JE, et al. Bevacizumab plus interferon alfa compared with interferon alfa monotherapy in patients with metastatic renal cell carcinoma: CALGB 90206. J Clin Oncol. 2008;26:5422–5428. doi: 10.1200/JCO.2008.16.9847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rini BI, Halabi S, Rosenberg JE, et al. Phase III trial of bevacizumab plus interferon alfa versus interferon alfa monotherapy in patients with metastatic renal cell carcinoma: Final results of CALGB 90206. J Clin Oncol. 2010;28:2137–2143. doi: 10.1200/JCO.2009.26.5561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rini BI, Halabi S, Taylor J, et al. Cancer and Leukemia Group B 90206: A randomized phase III trial of interferon-alpha or interferon-alpha plus anti-vascular endothelial growth factor antibody (bevacizumab) in metastatic renal cell carcinoma. Clin Cancer Res. 2004;10:2584–2586. doi: 10.1158/1078-0432.ccr-03-0605. [DOI] [PubMed] [Google Scholar]
- 15.Avastin® [prescribing information] South San Francisco, CA: Genentech, Inc.; 2009. [Google Scholar]
- 16.Sutent® [prescribing information] New York: Pfizer, Inc.; 2010. [Google Scholar]
- 17.Gore ME, Szczylik C, Porta C, et al. Safety and efficacy of sunitinib for metastatic renal-cell carcinoma: An expanded-access trial. Lancet Oncol. 2009;10:757–763. doi: 10.1016/S1470-2045(09)70162-7. [DOI] [PubMed] [Google Scholar]
- 18.Choueiri TK, Schutz FAB, Je Y, et al. Risk of arterial thromboembolic events with sunitinib and sorafenib: A systematic review and meta-analysis of clinical trials. J Clin Oncol. 2010;28:2280–2285. doi: 10.1200/JCO.2009.27.2757. [DOI] [PubMed] [Google Scholar]
- 19.Négrier S, Ravaud A. Optimisation of sunitinib therapy in metastatic renal cell carcinoma: Adverse-event management. Eur J Cancer Suppl. 2007;5:12–19. [Google Scholar]
- 20.Hutson TE, Figlin RA, Kuhn JG, et al. Targeted therapies for metastatic renal cell carcinoma: An overview of toxicity and dosing strategies. The Oncologist. 2008;13:1084–1096. doi: 10.1634/theoncologist.2008-0120. [DOI] [PubMed] [Google Scholar]
- 21.Hutson TE, Bellmunt J, Porta C, et al. Long-term safety of sorafenib in advanced renal cell carcinoma: Follow-up of patients from phase III TARGET. Eur J Cancer. 2010;46:2432–2440. doi: 10.1016/j.ejca.2010.06.121. [DOI] [PubMed] [Google Scholar]
- 22.Stadler WM, Figlin RA, McDermott DF, et al. ARCSS Study Investigators. Safety and efficacy results of the Advanced Renal Cell Carcinoma Sorafenib expanded access program in North America. Cancer. 2010;116:1272–1280. doi: 10.1002/cncr.24864. [DOI] [PubMed] [Google Scholar]
- 23.Nexavar® [prescribing information] Wayne, NJ: Bayer HealthCare Pharmaceuticals Inc.; 2009. [Google Scholar]
- 24.Votrient® [prescribing information] Research Triangle Park, NC: GlaxoSmithKline; 2009. [Google Scholar]
- 25.Bellmunt J, Szczylik C, Feingold J, et al. Temsirolimus safety profile and management of toxic effects in patients with advanced renal cell carcinoma and poor prognostic features. Ann Oncol. 2008;19:1387–1392. doi: 10.1093/annonc/mdn066. [DOI] [PubMed] [Google Scholar]
- 26.Torisel® [prescribing information] Philadelphia: Wyeth Pharmaceuticals Inc.; 2008. [Google Scholar]
- 27.White DA, Camus P, Endo M, et al. Noninfectious pneumonitis after everolimus therapy for advanced renal cell carcinoma. Am J Respir Crit Care Med. 2010;182:396–403. doi: 10.1164/rccm.200911-1720OC. [DOI] [PubMed] [Google Scholar]
- 28.Afinitor® [prescribing information] East Hanover, NJ: Novartis Pharmaceuticals Corp.; 2009. [Google Scholar]
- 29.Porta C, Ravaud A, Osanto S, et al. Recommendations for adverse event management in patients with renal cell carcinoma treated with everolimus: Safety data from the RECORD-1 trial. Poster presented at the 8th International Kidney Cancer Symposium; September 25–26, 2009; Chicago, IL. [Google Scholar]
- 30.Rixe O, Billemont B, Izzedine H. Hypertension as a predictive factor of sunitinib activity. Ann Oncol. 2007;18:1117. doi: 10.1093/annonc/mdm184. [DOI] [PubMed] [Google Scholar]
- 31.Bono P, Elfving H, Utriainen T, et al. Hypertension and clinical benefit of bevacizumab in the treatment of advanced renal cell carcinoma. Ann Oncol. 2009;20:393–394. doi: 10.1093/annonc/mdn729. [DOI] [PubMed] [Google Scholar]
- 32.Ravaud A, Sire M. Arterial hypertension and clinical benefit of sunitinib, sorafenib and bevacizumab in first and second-line treatment of metastatic renal cell cancer. Ann Oncol. 2009;20:966–967. doi: 10.1093/annonc/mdp201. [DOI] [PubMed] [Google Scholar]
- 33.Houk BE, Bello CL, Poland B, et al. Relationship between exposure to sunitinib and efficacy and tolerability endpoints in patients with cancer: Results of a pharmacokinetic/pharmacodynamic meta-analysis. Cancer Chemother Pharmacol. 2010;66:357–371. doi: 10.1007/s00280-009-1170-y. [DOI] [PubMed] [Google Scholar]
- 34.Bladou F, Gravis G, Sabatier R, et al. Hypothyroidism and survival during sunitinib therapy in metastatic renal cell carcinoma (mRCC): A prospective observational analysis [abstract e15013] J Clin Oncol. 2010;28(15 suppl):856s. [Google Scholar]
- 35.Boni JP, Leister C, Bender G, et al. Population pharmacokinetics of CCI-779: correlations to safety and pharmacogenomic responses in patients with advanced renal cancer. Clin Pharmacol Ther. 2005;77:76–89. doi: 10.1016/j.clpt.2004.08.025. [DOI] [PubMed] [Google Scholar]
- 36.ClinicalTrials.gov. A Phase II, Multi-Centre, Open-Label Study to Assess the Efficacy, Safety, Tolerability and Pharmacokinetics of Intrapatient Dose Escalation of Sorafenib as First Line Treatment for Metastatic Renal Cell Carcinoma [Identifier, NCT00618982] [accessed August 26, 2010]. Available at http://www.clinicaltrials.gov/ct2/show/NCT00618982?term=00618982&rank=1.
- 37.Amato RJ, Harris P, Dalton M, et al. A phase II trial of intra-patient dose-escalated sorafenib in patients (pts) with metastatic renal cell cancer [abstract 5026] J Clin Oncol. 2007;25(18 suppl):241s. [Google Scholar]