

Abstract
Aims
Congenital coronary artery anomalies produce serious events that include syncope, arrhythmias, myocardial infarction, or sudden death. Studying the mechanism of coronary development will contribute to the understanding of the disease and help design new diagnostic or therapeutic strategies. Here, we characterized a new calcineurin–NFAT signalling which specifically functions in the epicardium to regulate the development of smooth muscle wall of the coronary arteries.
Methods and results
Using tissue-specific gene deletion, we found that calcineurin–NFAT signals in the embryonic epicardium to direct coronary smooth muscle cell development. The smooth muscle wall of coronary arteries fails to mature in mice with epicardial deletion of calcineurin B1 (Cnb1), and accordingly these mutant mice develop cardiac dysfunction with reduced exercise capacity. Inhibition of calcineurin at various developmental windows shows that calcineurin–NFAT signals within a narrow time window at embryonic Day 12.5–13.5 to regulate coronary smooth muscle cell development. Within the epicardium, NFAT transcriptionally activates the expression of Smad2, whose gene product is critical for transducing transforming growth factor β (TGFβ)–Alk5 signalling to control coronary development.
Conclusion
Our findings demonstrate new spatiotemporal and molecular actions of calcineurin–NFAT that dictate coronary arterial wall development and a new mechanism by which calcineurin–NFAT integrates with TGFβ signalling during embryonic development.
Keywords: Epicardial, Calcineurin–NFAT, Coronary artery, Smooth muscle cell Differentiation, Smad2
1. Introduction
Embryonic development requires intense signalling within and between tissues to organize the formation of organs, including the coronary arteries. Formation of the coronary vasculature is essential to support cardiac growth and function.1,2 Anomalies of coronary arteries can cause cardiac ischaemia, particularly during exercise, which can then precipitate infarction, arrhythmias, syncope, or sudden cardiac death.3 Congenital coronary artery anomalies are found in 0.64–5.6% of patients undergoing coronary angiography,4 and ∼20% of coronary anomalies produce life-threatening events.3 This creates a serious medical burden to our society, given that coronary artery anomalies are the second most common cause of sudden cardiac death in young athletes.5 Therefore, studying how cells signal to direct coronary development is crucial to further our understanding of congenital coronary anomalies and the pathobiology of coronary arteries.
Coronary arteries are composed of three tissue layers: endothelium, smooth muscle cell (SMC) layer, and the adventitial layer.2 Development of coronary arteries is a complex developmental process: the endothelium is the first layer formed, and the endothelial plexuses then remodel and recruit SMCs and fibroblasts to form the smooth muscle wall and adventitia of mature coronary arteries.6 SMCs of coronary arteries have a fundamental role in cardiac function. These cells maintain coronary vascular tone to regulate cardiac perfusion. During exercise, coronary smooth muscle relaxes to dilate the arteries, thereby increasing the blood supply to the heart muscle and enabling the heart to pump more vigorously to meet the circulatory demand. Abnormalities of SMCs are crucial in many pathological processes, which include hypertension, atherosclerosis, and vascular restenosis after coronary artery grafting or stenting surgery.7 Coronary SMCs originate developmentally from the embryonic epicardium. During development, the epicardial cells undergo epithelial-to-mesenchymal transformation (EMT) and through this process give rise to mesenchymal cells that migrate into the myocardium. Within the myocardium, the epicardially derived mesenchymal cells associate with coronary endothelial cells and then differentiate into smooth muscle and adventitial cells of coronary arteries. Such complex developmental process requires signalling regulation from the endothelium, myocardium, and epicardium.6,8
Many signalling pathways, including the transforming growth factor (TGFβ1-3), bone morphemic protein (BMP), fibroblast growth factor, sonic hedgehog (Shh), and Wnt/β-catenin, are essential for the development of coronary vasculature.9–15 These pathways are involved in cell growth, migration, or differentiation during embryonic heart development. For example, the TGFβ family of signalling molecules plays an important role in coronary development controlled by the epicardial progenitor cells. Mice lacking epicardial Tgfbr3 (TGFβ receptor III) or Alk5 (a type I TGFβ receptor) show defects in the migration of epicardially derived mesenchymal cells or abnormalities in coronary SMC differentiation.9,16 Although the signalling events may occur in different cardiac tissues during coronary development, the temporal actions and interactions among these signalling pathways remain largely unknown.
Here, we show a new epicardial pathway calcineurin–NFAT that cooperates with TGFβ-Alk5 signalling within the epicardium to regulate coronary development. Calcineurin is a phosphatase that is activated by signals from cell surface receptors or ion channels.17 Once activated, calcineurin dephosphorylates the NFAT transcription factors, triggering NFAT proteins to translocate into the nucleus to control target gene expression. Our studies show that calcineurin–NFAT signals within a distinct developmental window to organize coronary arterial wall development. Within the epicardium, calcineurin–NFAT activates the expression of Smad2 to control the transduction of TGFβ-Alk5 signalling essential for coronary artery development.
2. Methods
More detailed methods were described in Supplementary material online.
2.1. Mice
All mouse strains were maintained in outbred backgrounds. The Cnb1f/f,18 Nfatc1f/f,19 Sm22aCre,20 and Gata5Cre21 strains are previously described. The date of observing a vaginal plug was set as E0.5, and embryonic development was confirmed by ultrasonography before sacrificing pregnant mice.22 All animal studies were approved by Stanford University School of Medicine.
2.2. Exercise tolerance test
Maximum exercise capacity was measured by using a rodent treadmill equipped with an electrical stimulus (Columbus Instruments, Columbus, OH, USA). Animals were familiarized with running on the treadmill before exercise testing. The exercise protocol consisted of progressive increase of the belt speed from 7.5 to 20 m/min by 2.5 m/min and incline from 4 to 20° by 2° at 3 min intervals. Total exercise time was recorded as the elapsed time to exhaustion. Exhaustion, determined by an observer blinded to control and mutant group, was defined as the point at which the animals could not keep pace with the treadmill and had no response to the electrical stimulus. Data presented are the average of the two runs for each mouse.
2.3. Echocardiography
The echocardiographer was blinded to the genotypes of the mice tested. Transthoracic ultrasonography with a GE Vivid 7 ultrasound platform (GE Health Care, Milwaukee, WI, USA) and a 13 MHz transducer was used to measure left ventricular function. The left ventricular function was assessed by the M-mode scanning of the left ventricular chamber, standardized by two-dimensional, short-axis views of the left ventricle at the mid-papillary muscle level. P-values were calculated by Student's t-test. Error bars indicate standard error of mean.
2.4. Histology, immunostaining, trichrome staining, and Weigert's Resorcin-Fuchsin staining
Histology, immunostaining, trichrome staining, and Weigert's Resorcin-Fuchsin (Cat#: 26370, Electron Microscopy Sciences) staining were performed as described.20,23,24 The primary antibodies used for immunostaining included: rabbit anti-Calcineurin (07-068 Upstate), rabbit anti-Myh11 (BT-562, Biomedical), rabbit anti-Calponin (ab46794, Abcam), mice anti-alpha smooth muscle actin (α-SMA) (A2547, Sigma), rabbit anti-Mmp9 (ab38898, Abcam), rabbit anti-p-Smad2/3 (sc-11769-R, Santa Cruz Biotechnology), anti-β-catenin (ab6302, Abcam), rabbit anti-p-Smad1/5/8 (#9511, cell signaling), rabbit anti-Smad2 (ab33875, Abcam), and rabbit anti-Smad3 (ab28379, Abcam).
2.5. Cyclosporin treatment
Pregnant females were injected with cyclosporin A (CsA) intraperitoneally during the time windows indicated in Figure 4J. CsA was administered as described previously.25 CsA was administered at a dose of 50 mg/kg through i.p. injection twice a day at 9:00–10:00 a.m. and at 19:00–20:00 p.m. Newborn mice were harvested for staining. Control females were injected with phosphate-buffered saline at the same time points.
Figure 4.

Epicardial calcineurin regulates coronary smooth muscle development. (A and B) Haematoxylin and eosin sections of P1 heart coronary artery. Scale bar: 100 μm. (C–H) Immunostaining of α-SMA, Myh11 and Calponin in P1 hearts of littermate control and Gata5Cre;Cnb1f/f mice. Red: α-SMA, Myh11, or Calponin. Blue: DAPI nuclear staining. Scale bar: 100 μm. (I) Quantitation of smooth muscle cells per coronary vessel by the measurement of sections stained as in (C–H). Data shown are means ± SEM. P-value: Student's t-test. Error bar: SEM, standard error of the mean. (J) Summary of the CsA treatment protocol. (K–N) Immunostaining of Myh11 in P1 heart of control and CsA-treated mice. Red: Myh11 immunostaining. Blue: DAPI nuclear staining. Scale bar: 100 μm.
2.6. Reporter assay
The Smad2 promoter (7.5 kb, −7028 to +490) was cloned into pREP4-Luc vector as described previously.24 The construct was then transfected into 293 T cells with lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) along with pREP7-RL as a transfection efficiency control, constitutive nuclear Nfatc4 (Nfatc4 SS17,1680AA)26 expression vector with the appropriate empty vector control. Luciferase activity was measured and normalized to a cotransfected Renilla luciferase construct using the Dual-Luciferase Reporter System (Promega, Madison, WI, USA).
2.7. Reverse transcription-quantitative PCR (RT–qPCR)
RNA extraction was performed using TRIzol (Invitrogen) and 150 ng of purified RNA were used as a template to synthesize cDNA using the Reverse Transcription Mix (Bio-rad). RT–qPCR reactions were performed using SYBR green master mix (BioRad, Hercules, CA, USA) with an Eppendorf realplex, and the primer sets were tested to be quantitative. Primer Threshold cycles and melting curve measurements were performed with software. The primers used in the assay are described in Supplementary material online, Table S1. P-values were calculated by Student's t-test. Error bars indicate standard error of mean.
2.8. Chromatin immunoprecipitation-quantitative PCR
The chromatin immunoprecipitation (ChIP) assay was described previously.23,24 Chromatin was sonicated to generate average fragment sizes of 200–600 bp, and immunoprecipitated using anti-Nfatc4 antibody (ab62613, Abcam), and normal rabbit IgG. Isolation and purification of immunoprecipitated and input DNA were done according to the manufacturer's protocol (Magna ChIP Protein G Magnetic Beads, Cat# 16-662, Millipore), and RT–qPCR analysis of immunoprecipitated DNA was performed. PCR signals of individual ChIP reaction was standardized to its own input PCR signals and normalized to IgG ChIP PCR signals. PCR primers (listed in Supplementary material online, Table S2) were designed to amplify the conserved promoter regions containing NFAT binding site of rat Smad2.
3. Results
3.1. Epicardial Cnb1-null mice exhibit cardiac dysfunction with reduced exercise capacity
We used Gata5Cre21 and loxP-flanked alleles of Cnb118 to disrupt calcineurin (Cnb1) in the developing epicardium. Immunostaining showed that Cnb1 was present in the developing heart, including the endocardium, myocardium, and epicardium (Figure 1A). In Gata5Cre;Cnb1f/f, Cnb1 was absent in the epicardium but present in the myocardium and endocardium (Figure 1B), and quantitation showed an 99.3% efficiency of Cnb1 disruption in the epicardium at E11.5 (data not shown). Although previous studies suggested that Gata5Cre may direct gene deletion in the myocardium,21 we did not observe any ectopic deletion of Cnb1 in the myocardium by Gata5Cre in our inbred strains of mice; neither was there Gata5Cre activity detected in the myocardium (Supplementary material online, Figure S1). The Gata5Cre;Cnb1f/f mice were born at expected Mendelian ratios, and on average the mutant mice had reduced body weight at 2 months of age, some of the mutant mice were easily identified because of their smaller body size (Figure 1C). The heart ventricle/body-weight ratio of all Gata5Cre;Cnb1f/f mice, however, was comparable with that of control littermates (Figure 1D). Histological analysis showed that the thickness of trabeculae and myocardial wall was normal in the Gata5Cre;Cnb1f/f hearts at E16.5, P1, and 2 months of age (Supplementary material online, Figure S2A–J). No apoptotic cells were detected in either control or mutant hearts by TUNEL staining (Supplementary material online, Figure 3A–C). Treadmill exercise analysis27 showed that the Gata5Cre;Cnb1f/f mice had severely reduced exercise capacity by the age of 2 months (Figure 1E), suggesting cardiac dysfunction. Echocardiography showed that Gata5Cre;Cnb1f/f mice had 33% reduction of left ventricular fractional shortening, accompanied by 25% increase in end-systolic left ventricular diameter (LVIDs) with preservation of end-diastolic left ventricular diameter (LVIDd) (Figure 2A–E). Cardiac catheterization of the left ventricle Gata5Cre;Cnb1f/f mice showed that the maximal rates of generating systolic pressure (+dP/dt) and of diastolic relaxation (−dP/dt) were reduced by 23 and 21%, respectively (Figure 2F–H). RT–qPCR of cardiac stress markers—Nppa (ANF) and Nppb (BNP)—showed that both Nppa and Nppb mRNA were highly elevated in the adult Gata5Cre;Cnb1f/f hearts (Figure 2I). Collectively, these findings indicate that mice lacking epicardial Cnb1 develop heart failure in adulthood.
Figure 1.

Gross phenotype of epicardium-restricted Cnb1 mutant mice. (A and B) Immunostaining of Cnb1 in E11.5 hearts of littermate control (A) and Gata5Cre;Cnb1f/f (B) mice. White arrow indicates the epicardial (Epi) and endocardial (Endo) border. Red: Cnb1. Blue: DAPI nuclear staining. Scale bar: 100 μm. (C) Body weight distribution of littermate control and Gata5Cre;Cnb1f/f mice. Data represented are means ± SEM. P-value: Student's t-test. Error bar: SEM, standard error of the mean. Line represents the mean of body weight. (D) Heart ventricle to body-weight ratio of 2-month-old littermate control was comparable with that of the Gata5Cre;Cnb1f/f mice. Data shown are means ± SEM. P-value: Student's t-test. Error bar: SEM, standard error of the mean. (E) Running time on the treadmill of littermates at 2 month of age. Data represented are means ± SEM. P-value: Student's t-test. Error bar: SEM, standard error of the mean.
Figure 2.

Epicardial deletion of Cnb1 causes heart dysfunction. (A and B) Representative echocardiographic image of the left ventricle of littermate control and Gata5Cre;Cnb1f/f mice at 2 month of age. (C) Echocardiographic measurement of fractional shortening of the left ventricle of 2-month-old littermate control and Gata5Cre;Cnb1f/f mice. Data represented are means ± SEM. P-value: Student's t-test. Error bar: SEM, standard error of the mean. (D and E) Echocardiographic measurement of LVIDd and LVIDs of the left ventricle of 2-month-old littermate control and Gata5Cre;Cnb1f/f mice. Data shown are means ± SEM. P-value: Student's t-test. Error bar: SEM, standard error of the mean. (F and G) Representative dP/dt tracing of the left ventricle of 2-month-old littermate control and Gata5Cre;Cnb1f/f mice. Y-axis: mmHg/s. X-axis: time (s). (H) Maximal dP/dt of the left ventricle of 2-month-old littermate control and Gata5Cre;Cnb1f/f mice. Data represented are means ± SEM. P-value: Student's t-test. Error bar: SEM, standard error of the mean. (I) mRNA expression of Nppa and Nppb in 2-month-old hearts of littermate control and Gata5Cre;Cnb1f/f mice. Data shown are means ± SEM. P-value: Student's t-test. Error bar: SEM, standard error of the mean.
3.2. Epicardial Cnb1-null hearts show increased elastin and collagen deposition in coronary arteries
Histological analysis of Gata5Cre;Cnb1f/f hearts showed extensive interstitial fibrosis (Figure 3A, B, and G), as well as intense collagen and elastin deposition (Figure 3C–F) in the coronary arteries. These results suggest impaired regulation of extracellular matrix (ECM) in the mutant hearts. To identify the developmental causes of ECM defects, we examined embryonic day 15.5 (E15.5) hearts for the expression of ECM-related genes. These genes included Col1a1, Col3a1, Timp1, Mmp-9, and Osf2.28 Among these genes, only Mmp9, a key metalloprotease that degrades elastin and collagen,29,30 had 62% reduction (P = 0.009) in E15.5 Gata5Cre;Cnb1f/fhearts; the other genes had no significant changes (Figure 3H). Immunostaining confirmed the reduction of Mmp9 in the mutant E15.5 hearts (Figure 3I and J). The down-regulation of Mmp9 provides an explanation for the increased ECM deposition around coronary vessels.
Figure 3.
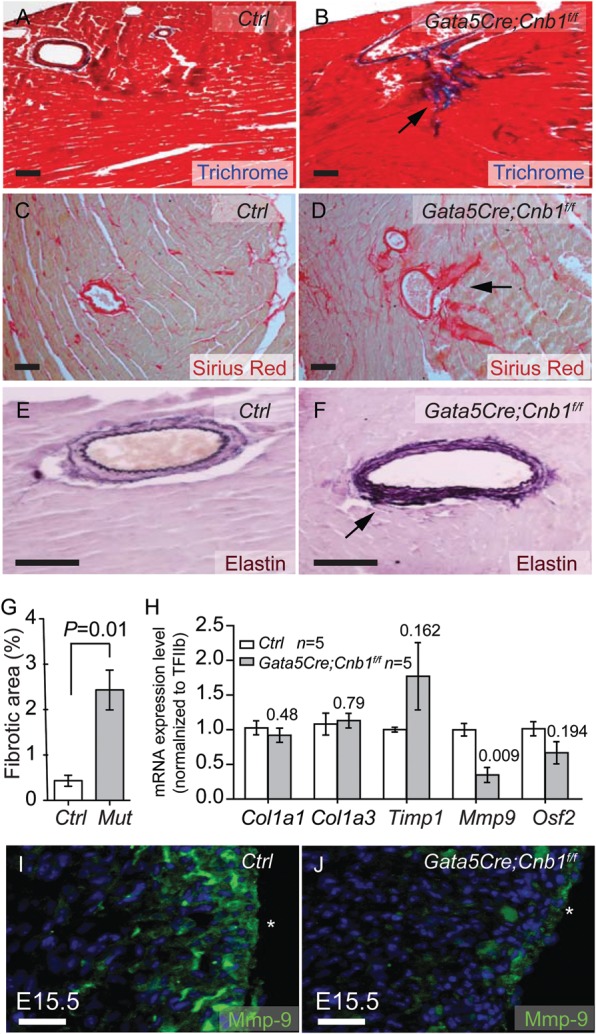
Epicardial deletion of Cnb1 induces heart ECM deposition and fibrosis. (A–D) Trichrome staining (A and B, red: cardiomyocytes. Blue: fibrosis) and Sirus red staining (C and D, red: Collagen) of fibrosis in 2-month-old littermate control and Gata5Cre;Cnb1f/f hearts. Scale bar: 100 μm. (E and F) Weigert's Resorcin Fuchsin staining of elastic fibers in 2-month-old littermate control and Gata5Cre;Cnb1f/f hearts. Dark blue: elastic tissues. Pink: tissue elements. Scale bar: 100 μm. (G) The quantitation of the fibrotic area by the measurement of sections stained as in (A) and (B). Data shown are means ± SEM. P-value: Student's t-test. Error bar: SEM, standard error of the mean. (H) Expression of Col1a1, Col1a3, Timp1, Mmp9, Osf2 in E15.5 hearts of littermate control and Gata5Cre;Cnb1f/f mice. Data represented are means ± SEM. P-value: Student's t-test. Error bar: SEM, standard error of the mean. (I and J) Immunostaining of Mmp9 in E15.5 heart of littermate control and mutant mice. Green: Mmp9. Blue: DAPI nuclear staining. *Epicardial border. Scale bar: 100 μm.
3.3. Epicardial Cnb1 is required for the differentiation of coronary SMCs
Given that embryonic epicardium gives rise to fibroblasts and coronary SMCs, deletion of Cnb1 in the epicardium may cause abnormalities of these two cell populations. We first investigated the function of cardiac fibroblasts by testing their ability to synthesize and secrete ECM proteins. Immunostaining of periostin, a component of the ECM specifically secreted by cardiac fibroblasts,31 showed the normal distribution and abundance of periostin in the mutant hearts relative to that of littermate controls (Supplementary material online, Figure S4A and B). Furthermore, Col1a1 and Col1a3, which are the most highly secreted collagens by cardiac fibroblasts, had normal expression in the mutant hearts (Figure 3H). These findings suggest normal fibroblast function in Gata5Cre;Cnb1f/f hearts. To test coronary SMC development, we first analysed the patterning of coronary arteries by vascular casting.32 The patterning of right and left coronary arteries in mutant mice was comparable with that of littermate control mice at 2 months of age (Supplementary material online, Figure S5A–E). Histology of coronary arteries showed that the smooth muscle layer had comparable thickness in the control and mutant hearts (Figure 4A–B). We then examined SMC differentiation by testing their expression of differentiation markers, including alpha smooth muscle actin (α-SMA), smooth muscle myosin heavy chain (Myh11), and Calponin (Figure 4C–H). Immunostaining showed that the expression of those markers was greatly reduced in coronary arteries of Gata5Cre;Cnb1f/f mice at postnatal Day 1 (P1) (Figure 4C–H; Supplementary material online, Figure S6A and B). Despite the abnormal differentiation, the number of coronary SMCs that faintly expressed the markers in the mutant hearts was comparable with the number of well-differentiated SMCs in the control hearts (Figure 4I). The normal amount but poor differentiation of mutant coronary SMCs suggests that there is no defect of epicardially derived mesenchymal cells to migrate to the arteries but a defect of those cells to differentiate into mature coronary SMCs in the epicardial Cnb1-null hearts.
3.4. Epicardial calcineurin–NFAT signals at E12.5–13.5 for coronary smooth muscle development
To define the developmental window of Cnb1 action for coronary SMC development, we used CsA to inhibit calcineurin activity in embryos at different gestational ages.23,25 Pregnant mice were treated with CsA at various developmental windows (E11.5–12.0, E12.5–13.0, and E13.5–14.0), and embryos exposed to CsA or control vehicles were harvested at P1 for coronary SMC immunostaining (Figure 4J). Pups exposed to CsA at E11.5–12.0 and E13.5–14.0 exhibited normal Myh11 in coronary SMCs (Figure 4K, L, and N). Conversely, pups with CsA exposure at E12.5–13.0 showed drastic reduction of Myh11 in coronary SMCs (Figure 4M). These observations indicate a specific temporal requirement of calcineurin–NFAT signalling beginning at E12.5 for coronary smooth muscle development.
3.5. Nfatc1 is not essential in the epicardium for heart or coronary development
Previous work suggested that disruption of Nfatc1 in the epicardium causes embryonic lethal by E18.5, resulting in the reduction of COL1A1 protein and cardiac fibrous matrix formation.33 However, this report is inconsistent with the lack of Nfac1 expression in the epicardium as shown by many other studies.23,25,34,35 To address the contradiction, we further tested the role of epicardial Nfatc1 in heart development and used the Nfatc1f/f mice19 to generate epicardial-specific Nfatc1 knockout mice (Gata5Cre;Nfatc1f/f). We found that these mice lived to adulthood and were indistinguishable from their littermate controls. They exercised normally on the treadmill (Figure 5A), and the Gata5Cre;Nfatc1f/f hearts had normal left ventricular fractional shortening (Figure 5B), indicating the absence of cardiac dysfunction. Histological analysis showed normal ECM in the heart and coronary arteries (Figure 5C and D). Myh11 immunostaining showed normal coronary SMC development in P1 Gata5Cre;Nfatc1f/f hearts (Figure 5E and F). These observations indicate that Nfatc1 is not essential in the epicardium for heart or coronary development or embryonic survival, which is in accord with the absence of Nfatc1 expression in the epicardium.23,25,34,35 Given that Nfatc3/c4 are expressed in the epicardium,25 epicardial calcineurin likely signals through Nfatc3/4 to regulate coronary smooth muscle development.
Figure 5.

Calcineurin controls TGFβ/Smad2 signalling to regulate coronary SMC development. (A) The exercise treadmill test of 2-month-old littermate control and Gata5Cre;Nfatc1f/f mice. Data shown are means ± SEM. P-value: Student's t-test. Error bar: SEM, standard error of the mean. (B) Echocardiographic measurement of fractional shortening of the left ventricle of 2-month-old littermate control and Gata5Cre;Nfatc1f/f mice. P-value: Student's t-test. Error bar: SEM, standard error of the mean. (C and D) Trichrome staining of fibrosis in 2-month-old littermate control and Gata5Cre;Nfatc1f/f hearts. Red: cardiomyocytes. Blue: fibrosis. Scale bar: 100 μm. (E and F) Immunostaining of Myh11 in P1 heart of littermate control and Gata5Cre;Nfatc1f/f mice. Brown: Myh11. Blue: haematoxylin nuclear staining. Scale bar: 100 μm. (G and H) Immunostaining of Myh11 in P1 heart of littermate control and Sm22aCre;Cnb1f/f mice. Brown: Myh11. Blue: haematoxylin nuclear staining. Scale bar: 100 μm. (I and J) Immunostaining of β-catenin in E12.5 hearts of littermate control and Gata5Cre;Cnb1f/f mice. Green: β-catenin immunostaining. Blue: DAPI nuclear staining. Scale bar: 100 μm. (K and L) Immunostaining of p-Smad1/5/8 in E12.5 hearts of littermate control and Gata5Cre;Cnb1f/f mice. Brown: p-Smad1/5/8 staining. Blue: haematoxylin nuclear staining. Scale bar: 100 μm. (M and N) Co-immunostaining of α-SMA and p-Smad2/3 in P1 hearts of littermate control and Gata5Cre;Cnb1f/f mice. Green: α-SMA staining, Red: p-Smad2/3 staining. Blue: DAPI nuclear staining. Scale bar: 100 μm. Arrow indicates the coronary artery smooth muscle layer. (O–R) Immunostaining of Smad2 and Smad3 in E12.5 hearts of littermate control and Gata5Cre;Cnb1f/f mice. Green: Smad2, Smad3 immunostaining. Scale bar: 100 μm. Blue: DAPI nuclear staining. (S and T) Quantitation of Smad2, Smad3 mRNA in control or CsA-treated (500 ng/mL) rat EMC cells. n = 5. Data shown are means ± SEM. P-value: Student's t-test. Error bar: SEM, standard error of the mean. (U) Western blot analysis of Smad2, Smad3 and pSmad2/3 in control or CsA-treated (500 ng/mL) rat EMC cells.
3.6. Calcineurin does not function in smooth muscle or myocardial cells for coronary development
To exclude that calcineurin might function in smooth muscle or myocardial cells for coronary smooth muscle development, we used a Sm22aCre mouse line to delete Cnb1 in smooth muscle and myocardial cells.20,24,36 We found that Sm22aCre;Cnb1f/f mice lived to adulthood without defects in coronary SMC differentiation (Figure 5G and H). These observations further support a specific role of calcineurin in the epicardial or epicardially derived mesenchymal cells for coronary wall development.
3.7. Calcineurin–NFAT activates Smad2 in the epicardium
Epicardial Wnt/β-catenin, TGFβ, and BMP signalling are essential for epicardial progenitor cells to differentiate into coronary SMCs.9–12 To test if these signalling events were disrupted in mice lacking epicardial Cnb1, we examined epicardial Wnt/β-catenin, TGFβ, and BMP signalling by testing its downstream effectors—β-catenin, activated Smad2/3 (p-Smad2/3), and activated Smad1/5/8 (p-Smad1/5/8). By immunostaining, we observed no difference of β-catenin and p-Smad1/5/8 between Gata5Cre;Cnb1f/f and control littermates at E12.5 (Figure 5I–L). Conversely, p-Smad2/3 was nearly absent in the nuclei of coronary SMCs of Gata5Cre;Cnb1f/f mice (Figure 5M and N), indicating severely compromised TGFβ–Smad2/3 signalling in the absence of calcineurin. We next tested the expression of Smad2 and Smad3 in the developing epicardium. Smad2 proteins were dramatically diminished in the epicardium of E12.5 Gata5Cre;Cnb1f/f mice (Figure 5O and P), whereas Smad3 proteins were normal (Figure 5Q and R). Furthermore, in a rat epicardial cell line,37 inhibition of calcineurin by CsA caused down-regulation of Smad2 and phosphoSmad2/3 but without changes of Smad3, as shown by RT–qPCR and/or western blot analysis (Figure 5S–U). Both the in vivo and in vitro data, therefore, indicate that calcineurin is essential for Smad2 expression in the epicardium.
3.8. Epicardial NFAT directly regulates Smad2 expression for coronary arterial development
To determine whether Nfat transcriptionally activated Smad2 expression, we examined the binding of Nfatc4, which is predominantly expressed in the epicardium,25 to the Smad2 promoter in rat epicardial/mesothelial cells (EMCs).37 With sequence alignment, we identified five regions (s1–s5), which contains putative NFAT binding sites (GGAAA), in the ∼5.5 kb of rat Smad2 promoter that are evolutionarily conserved among rhesus macaque, rat, and human (Figure 6A). ChIP assay using anti-Nfatc4 antibody showed that Nfatc4 was associated with s1–s5 regions, but not the non-conserved region in EMCs (Figure 6B). To test if such Nfatc4 binding to Smad2 was controlled by calcineurin, we treated EMCs with CsA to inactivate calcineurin and then performed the ChIP assay. CsA treatment, indeed, abolished Nfatc4's binding to these Smad2 promoter sites, indicating that calcineurin signals Nfatc4 to target Smad2 promoter. To further test the transcriptional activity of Nfatc4 on the Smad2 promoter, we cloned 7.5 kb of Smad2 upstream promoter (−7028 to +490) into episomal luciferase reporter pREP424 and then transfected the constructs into 293 T cells. We used 293 T cells as a surrogate for reporter assays because EMCs were resistant to transfection by various methods attempted. By measuring the luciferase activity, we found that the expression of constitutively nuclear Nfatc4 caused 2.3-fold increase of the Smad2 promoter activity (Figure 6C). These ChIP studies and reporter assays, together with the in vivo observations, suggest that calcineurin activates NFATc in the epicardial cells to transcriptionally activate Smad2.
Figure 6.

Calcineurin–NFAT directly regulates Smad2 expression in epicardial cells. (A) Sequence alignment of the Smad2 promoter from rat, human and rhesus macaque. Peak heights indicate degree of sequence homology. Black boxes (s1–s5) are regions of high sequence homology containing Nfat binding site (GGAAA) and were further analysed by ChIP. Red, promoter elements. Green, transposons/simple repeats. (B) Quantitation of Nfatc4 ChIP on the Smad2 promoter in control or 500 ng/mL CsA-treated rat EMC cells. Data shown are means ± SEM. P-value: Student's t-test. Error bar: SEM, standard error of the mean. (C) Luciferase reporter assay of the Smad2 promoter (7.5 kb, −7028 to +490) in 293 T cells. Data shown are means ± SEM. P-value: Student's t-test. Error bar: SEM, standard error of the mean. (D) Calcineurin–NFAT signalling functions during distinct time windows to regulate cardiovascular development. (E) Working model of cross-talk between calcineurin–NFAT and TGFβ signalling and molecular signals between epicardial and myocardial cells during coronary vessel development. EMT, epithelial to mesenchymal transition; TF, transcription factor; CaN, calcineurin; P, phosphate.
4. Discussion
Our studies show that anomalous coronary arterial wall development results in cardiac dysfunction and exercise intolerance in adult. Here, we demonstrate a previously unknown temporospatial, molecular function of calcineurin–NFAT signalling in the epicardial progenitor cells to direct coronary arterial wall formation (Figure 6D). Combined with previous studies that show how the endothelial calcineurin–NFAT signal controls coronary endothelial angiogenesis,25 the current studies indicate that NFAT signalling functions in distinct tissues and at different times to organize the sequence of coronary angiogenesis and arterial wall development. NFAT signalling first functions in endothelial cells at E10.5–E11.5 to pattern coronary endothelial tubes, and it then activates the epicardial/epicardially derived mesenchymal cells at E12.5–E13.5 to differentiate into SMCs that assemble with the endothelial tubes to form mature coronary arteries (Figure 6D).
Such sequential NFAT signalling from the endothelium to the epicardium to orchestrate coronary development is reminiscent of the sequential NFAT signalling between the endocardium and myocardium to direct distinct morphogenetic steps of heart valve development.23 Myocardial NFAT signals at E9.5 to initiate EMT of atrioventricular cushions, and endocardial NFAT signals at E11.5 to direct the elongation of heart valve leaflets (Figure 6D). These four waves of NFAT signalling at different spatiotemporal domains of the developing heart thus provide a model in which calcineurin–NFAT determines the sequence of morphogenetic events of cardiovascular tissues.
Calcineurin in the epicardium or epicardium-derived cells controls the expression of Mmp-9 in the heart. Because Mmp-9 is a key metalloprotease that degrades elastin and collagen,29,30 the reduction of Mmp-9 in epicardial Cnb1-null hearts results in increased elastin and collagen deposition around the coronary arteries. Such excessive collagen deposition and the lack of maturation of coronary SMCs, in combination, impair the dilatation of coronary arteries in response to increased myocardial demand. During exercise, coronary arteries would normally dilate to increase cardiac perfusion to meet the increased metabolic demand of cardiomyocytes. Such dilation of coronary arteries requires the relaxation of coronary SMCs. In the mutant heart, the abnormal coronary SMCs with fibrosis may not only impair coronary muscle relaxation, but also increase the stiffness of coronary arteries, leading to failure of coronary artery dilatation and thus exercise-induced cardiac dysfunction observed in the mutant mice.
Our studies show a previously unknown molecular interaction by which calcineurin–NFAT controls the expression of Smad2, a critical intermediate that transduces TGFβ signalling. In mice TGF-β1, TGF-β receptors, and Smad proteins are all essential for embryonic vascular development. Loss of any of these components leads to defects in SMC recruitment and/or differentiation during vascular development.38 The TGFβ family of signalling molecules also plays an important role in coronary development controlled by the epicardial progenitor cells. Epicardial Tgfbr3-null mice display diminished invasion of the epicardially derived mesenchymal cells into the myocardium, resulting in abnormal coronary vessel formation.16 In mice lacking epicardial Alk5, a type I TGFβ receptor, the coronary SMCs are reduced in amount and fail to mature,9 a phenotype also observed the epicardial Cnb1-null hearts (current studies). The finding that calcineurin–NFAT controls Smad2 expression in the epicardium, coupled with the similar phenotype of epicardial Alk5-null heart, suggest that calcineurin–NFAT cooperates with TGFβ–Alk5 signalling through Smad2 in the epicardium to regulate coronary smooth muscle development (Figure 6E).
Besides enabling TGFβ–Alk5 to signal through Smad2, the calcineurin–NFAT pathway may respond to Shh and vascular endothelial growth factor (VEGF) to regulate coronary arterial wall development. Shh released from the epicardium can activate smoothen in the myocardial cells to trigger the expression and release VEGF,13 which is capable of activating the calcineurin–NFAT pathway.25 VEGF from the myocardium may then activate the endothelial calcineurin–NFAT25 and epicardial calcineurin–NFAT, respectively, to promote coronary endothelial patterning and smooth muscle wall formation. Further studies will be needed to elucidate how the calcineurin–NFAT may orchestrate Shh/smoothen–VEGF and TGFβ signalling to organize the interactions among epicardial, myocardial, and endothelial cells during coronary artery development.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Funding
C-P.C. was supported by March of Dimes Foundation (MOD), the American Heart Association (AHA) Established Investigator Award, the Oak foundation, California Institute of Regenerative Medicine,National Institutes of Health (NIH, R01HL118087), Heart Center Research Program of the Stanford Child Health Research Institute, the Indiana University Health–Indiana University School of Medicine Strategic Research Initiative, and the Indiana University Physician-Scientist Initiative, endowed by Lilly Endowment, Inc. M.Z. was supported by Marie Curie Fellowship of the European Commission; C-J.L. by Stanford graduate fellowship; Y.X. by fellowships from the Oak Foundation, AHA, and Lucile Packard Children's Foundation; C.S. by MOD and NIH postdoctoral Fellowships; B.Z. by NIH (HL078881 and HL111770).
Supplementary Material
Acknowledgements
The Gata5-Cre mouse line was a generous gift from Dr Pilar Ruiz-Lozano (Stanford University, CA, USA).
Conflict of interest: none declared.
References
- 1.Lavine KJ, Ornitz DM. Shared circuitry: developmental signaling cascades regulate both embryonic and adult coronary vasculature. Circ Res. 2009;104:159–169. doi: 10.1161/CIRCRESAHA.108.191239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reese DE, Mikawa T, Bader DM. Development of the coronary vessel system. Circ Res. 2002;91:761–768. doi: 10.1161/01.res.0000038961.53759.3c. [DOI] [PubMed] [Google Scholar]
- 3.Yildiz A, Okcun B, Peker T, Arslan C, Olcay A, Bulent Vatan M. Prevalence of coronary artery anomalies in 12,457 adult patients who underwent coronary angiography. Clin Cardiol. 2010;33:E60–E64. doi: 10.1002/clc.20588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rigatelli G, Rigatelli A, Cominato S, Panin S, Nghia NT, Faggian G. A clinical-angiographic risk scoring system for coronary artery anomalies. Asian Cardiovasc Thorac Ann. 2012;20:299–303. doi: 10.1177/0218492312437880. [DOI] [PubMed] [Google Scholar]
- 5.Basso C, Maron BJ, Corrado D, Thiene G. Clinical profile of congenital coronary artery anomalies with origin from the wrong aortic sinus leading to sudden death in young competitive athletes. J Am Coll Cardiol. 2000;35:1493–1501. doi: 10.1016/s0735-1097(00)00566-0. [DOI] [PubMed] [Google Scholar]
- 6.Olivey HE, Svensson EC. Epicardial-myocardial signaling directing coronary vasculogenesis. Circ Res. 2010;106:818–832. doi: 10.1161/CIRCRESAHA.109.209197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gittenberger-de Groot AC, DeRuiter MC, Bergwerff M, Poelmann RE. Smooth muscle cell origin and its relation to heterogeneity in development and disease. Arterioscler Thromb Vasc Biol. 1999;19:1589–1594. doi: 10.1161/01.atv.19.7.1589. [DOI] [PubMed] [Google Scholar]
- 8.Wada AM, Willet SG, Bader D. Coronary vessel development: a unique form of vasculogenesis. Arterioscler Thromb Vasc Biol. 2003;23:2138–2145. doi: 10.1161/01.ATV.0000098645.38676.CC. [DOI] [PubMed] [Google Scholar]
- 9.Sridurongrit S, Larsson J, Schwartz R, Ruiz-Lozano P, Kaartinen V. Signaling via the tgf-beta type i receptor alk5 in heart development. Dev Biol. 2008;322:208–218. doi: 10.1016/j.ydbio.2008.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zamora M, Manner J, Ruiz-Lozano P. Epicardium-derived progenitor cells require beta-catenin for coronary artery formation. Proc Natl Acad Sci USA. 2007;104:18109–18114. doi: 10.1073/pnas.0702415104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sanchez NS, Barnett JV. Tgfbeta and bmp-2 regulate epicardial cell invasion via tgfbetar3 activation of the par6/smurf1/rhoa pathway. Cell Signal. 2012;24:539–548. doi: 10.1016/j.cellsig.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Galvin KM, Donovan MJ, Lynch CA, Meyer RI, Paul RJ, Lorenz JN, et al. A role for smad6 in development and homeostasis of the cardiovascular system. Nat Genet. 2000;24:171–174. doi: 10.1038/72835. [DOI] [PubMed] [Google Scholar]
- 13.Lavine KJ, Long F, Choi K, Smith C, Ornitz DM. Hedgehog signaling to distinct cell types differentially regulates coronary artery and vein development. Development. 2008;135:3161–3171. doi: 10.1242/dev.019919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lavine KJ, White AC, Park C, Smith CS, Choi K, Long F, et al. Fibroblast growth factor signals regulate a wave of hedgehog activation that is essential for coronary vascular development. Genes Dev. 2006;20:1651–1666. doi: 10.1101/gad.1411406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lavine KJ, Yu K, White AC, Zhang X, Smith C, Partanen J, et al. Endocardial and epicardial derived fgf signals regulate myocardial proliferation and differentiation in vivo. Dev Cell. 2005;8:85–95. doi: 10.1016/j.devcel.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 16.Compton LA, Potash DA, Brown CB, Barnett JV. Coronary vessel development is dependent on the type iii transforming growth factor beta receptor. Circ Res. 2007;101:784–791. doi: 10.1161/CIRCRESAHA.107.152082. [DOI] [PubMed] [Google Scholar]
- 17.Crabtree GR, Olson EN. Nfat signaling: choreographing the social lives of cells. Cell. 2002;109(Suppl):S67–S79. doi: 10.1016/s0092-8674(02)00699-2. [DOI] [PubMed] [Google Scholar]
- 18.Neilson JR, Winslow MM, Hur EM, Crabtree GR. Calcineurin b1 is essential for positive but not negative selection during thymocyte development. Immunity. 2004;20:255–266. doi: 10.1016/s1074-7613(04)00052-4. [DOI] [PubMed] [Google Scholar]
- 19.Aliprantis AO, Ueki Y, Sulyanto R, Park A, Sigrist KS, Sharma SM, et al. Nfatc1 in mice represses osteoprotegerin during osteoclastogenesis and dissociates systemic osteopenia from inflammation in cherubism. J Clin Invest. 2008;118:3775–3789. doi: 10.1172/JCI35711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stankunas K, Hang CT, Tsun ZY, Chen H, Lee NV, Wu JI, et al. Endocardial brg1 represses adamts1 to maintain the microenvironment for myocardial morphogenesis. Dev Cell. 2008;14:298–311. doi: 10.1016/j.devcel.2007.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Merki E, Zamora M, Raya A, Kawakami Y, Wang J, Zhang X, et al. Epicardial retinoid x receptor alpha is required for myocardial growth and coronary artery formation. Proc Natl Acad Sci USA. 2005;102:18455–18460. doi: 10.1073/pnas.0504343102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chang CP, Chen L, Crabtree GR. Sonographic staging of the developmental status of mouse embryos in utero. Genesis. 2003;36:7–11. doi: 10.1002/gene.10186. [DOI] [PubMed] [Google Scholar]
- 23.Chang CP, Neilson JR, Bayle JH, Gestwicki JE, Kuo A, Stankunas K, et al. A field of myocardial-endocardial nfat signaling underlies heart valve morphogenesis. Cell. 2004;118:649–663. doi: 10.1016/j.cell.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 24.Hang CT, Yang J, Han P, Cheng HL, Shang C, Ashley E, et al. Chromatin regulation by brg1 underlies heart muscle development and disease. Nature. 2010;466:62–67. doi: 10.1038/nature09130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zeini M, Hang CT, Lehrer-Graiwer J, Dao T, Zhou B, Chang CP. Spatial and temporal regulation of coronary vessel formation by calcineurin-nfat signaling. Development. 2009;136:3335–3345. doi: 10.1242/dev.037903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang TT, Xiong Q, Graef IA, Crabtree GR, Chow CW. Recruitment of the extracellular signal-regulated kinase/ribosomal s6 kinase signaling pathway to the nfatc4 transcription activation complex. Mol Cell Biol. 2005;25:907–920. doi: 10.1128/MCB.25.3.907-920.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Biolo A, Greferath R, Siwik DA, Qin F, Valsky E, Fylaktakidou KC, et al. Enhanced exercise capacity in mice with severe heart failure treated with an allosteric effector of hemoglobin, myo-inositol trispyrophosphate. Proc Natl Acad Sci USA. 2009;106:1926–1929. doi: 10.1073/pnas.0812381106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li YY, McTiernan CF, Feldman AM. Interplay of matrix metalloproteinases, tissue inhibitors of metalloproteinases and their regulators in cardiac matrix remodeling. Cardiovasc Res. 2000;46:214–224. doi: 10.1016/s0008-6363(00)00003-1. [DOI] [PubMed] [Google Scholar]
- 29.Johnson C, Galis ZS. Matrix metalloproteinase-2 and -9 differentially regulate smooth muscle cell migration and cell-mediated collagen organization. Arterioscler Thromb Vasc Biol. 2004;24:54–60. doi: 10.1161/01.ATV.0000100402.69997.C3. [DOI] [PubMed] [Google Scholar]
- 30.Whiteside EJ, Jackson MM, Herington AC, Edwards DR, Harvey MB. Matrix metalloproteinase-9 and tissue inhibitor of metalloproteinase-3 are key regulators of extracellular matrix degradation by mouse embryos. Biol Reprod. 2001;64:1331–1337. doi: 10.1095/biolreprod64.5.1331. [DOI] [PubMed] [Google Scholar]
- 31.Snider P, Standley KN, Wang J, Azhar M, Doetschman T, Conway SJ. Origin of cardiac fibroblasts and the role of periostin. Circ Res. 2009;105:934–947. doi: 10.1161/CIRCRESAHA.109.201400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chang CP. Analysis of the patterning of cardiac outflow tract and great arteries with angiography and vascular casting. Methods Mol Biol. 2012;843:21–28. doi: 10.1007/978-1-61779-523-7_3. [DOI] [PubMed] [Google Scholar]
- 33.Combs MD, Braitsch CM, Lange AW, James JF, Yutzey KE. Nfatc1 promotes epicardium-derived cell invasion into myocardium. Development. 2011;138:1747–1757. doi: 10.1242/dev.060996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.de la Pompa JL, Timmerman LA, Takimoto H, Yoshida H, Elia AJ, Samper E, et al. Role of the nf-atc transcription factor in morphogenesis of cardiac valves and septum. Nature. 1998;392:182–186. doi: 10.1038/32419. [DOI] [PubMed] [Google Scholar]
- 35.Ranger AM, Grusby MJ, Hodge MR, Gravallese EM, de la Brousse FC, Hoey T, et al. The transcription factor nf-atc is essential for cardiac valve formation. Nature. 1998;392:186–190. doi: 10.1038/32426. [DOI] [PubMed] [Google Scholar]
- 36.Umans L, Cox L, Tjwa M, Bito V, Vermeire L, Laperre K, et al. Inactivation of smad5 in endothelial cells and smooth muscle cells demonstrates that smad5 is required for cardiac homeostasis. Am J Pathol. 2007;170:1460–1472. doi: 10.2353/ajpath.2007.060839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wada AM, Smith TK, Osler ME, Reese DE, Bader DM. Epicardial/mesothelial cell line retains vasculogenic potential of embryonic epicardium. Circ Res. 2003;92:525–531. doi: 10.1161/01.RES.0000060484.11032.0B. [DOI] [PubMed] [Google Scholar]
- 38.Goumans MJ, Mummery C. Functional analysis of the tgfbeta receptor/smad pathway through gene ablation in mice. Int J Dev Biol. 2000;44:253–265. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.