

Abstract
Preterm birth (PTB) is a major global public health concern. However, little is known about the pathophysiology of spontaneous idiopathic PTB. We tested the hypothesis that rare variants in families would target specific genes and pathways that contribute to PTB risk in the general population. Whole-exome sequencing was performed on 10 PTB mothers from densely affected families including two mother-daughter pairs. We identified novel variants shared between the two mother-daughter pairs when compared to a 1000 Genomes Project background exome file and investigated these genes for pathway aggregation using the Kyoto Encyclopedia of Genes and Genomes (KEGG). Genes in enriched pathways were then surveyed in the other six PTB exomes and tested for association in a larger number of nuclear families. The KEGG complement and coagulation cascade was one of the most enriched pathways in our two mother-daughter pairs. When the six genes found in this pathway (CFH, CR1, F13B, F5, CR2, and C4BPA) were examined for novel missense variants, half of all the exomes harbored at least one. Association analysis of variants in these six gene regions in nuclear families from Finland (237 cases and 328 controls) found statistically significant associations after multiple test corrections in three CR1 SNPs; the strongest in an exonic missense SNP, rs6691117, p-value = 6.91e-5, OR = 1.71. Our results demonstrate the importance of the complement and coagulation cascades in the pathophysiology of PTB, and suggest potential screening and intervention approaches to prevent prematurity that target this pathway.
Keywords: CR1, Exome Sequencing, Association Study, Preterm Birth
INTRODUCTION
Preterm birth (PTB), defined as live birth before 37 weeks’ completed gestation, is the leading cause of infant mortality worldwide (Howson et al. 2012). Despite this major public health concern, little is known about the pathogenesis of PTB. The limited insight into PTB is contributed to by the fact that the mechanism for normal parturition in general is not known in humans. There have been a number of suggested pathways believed to play a role in PTB pathogenesis, but direct evidence for any of these is modest at best (Lockwood and Kuczynski 2001).
A number of lines of evidence suggest that PTB has a genetic component such as PTB aggregating in families, segregation analysis and genetic modeling (Plunkett et al. 2009; Chaudhari et al. 2008) Primarily through candidate gene studies, there have been a number of SNPs associated with PTB; however, contradictory evidence from replication studies exists, and none of these have large effect sizes or have implicated new mechanisms in parturition control (Plunkett and Muglia 2008).
With the advent of next-generation sequencing (NGS) and exon capture technology the ability to sequence a patient’s exome provides an important new approach to disease gene discovery. Whole-exome sequencing has been used to identify the casual variant/gene for a number of Mendelian diseases (Züchner et al. 2011; Ng et al. 2010). While the potential of whole-exome sequencing to identify the cause of complex diseases has been discussed, this approach has only been used sparingly such as in autism spectrum disorders (O’Roak et al. 2011).
In this study, we test the hypothesis that rare variants aggregate in specific genes and pathways that contribute to PTB risk. In order to test our hypothesis, we performed whole-exome sequencing in multiplex families with a history of spontaneous idiopathic PTB. We identified predicted deleterious variants aggregating in complement/coagulation pathway genes, and extended this observation to more common coding region variants to demonstrate a significant association of the complement receptor 1 (CR1) gene with spontaneous idiopathic preterm birth in a larger case-control dataset.
MATERIALS AND METHODS
Sample collection
Mothers of preterm or term infants were enrolled for genetic analysis by methods approved by Institutional Review Boards/Ethics Committees of University Central Hospital, Helsinki, University of Oulu, Cincinnati Children’s Hospital Medical Center, University of Iowa College of Medicine, and Vanderbilt University. DNA was extracted from whole blood or Oragene® saliva kits. Standard manufacturer protocols were followed.
Sample inclusion and exclusion criteria
Samples were selected for whole-exome sequencing based on a number of features, which we termed, “preterm birth load”. What we took into account was the shortest gestation in a pedigree, number of preterm children and whether the mother herself was born preterm. For this study we only sequenced case mothers.
For the Illumina and Affymetrix SNP genotyping arrays the inclusion and exclusion criteria are as described below. Case mothers were required to have a spontaneous preterm birth between 22–36 weeks gestation and additionally either the child or mother had to have a first degree relative with a history of PTB, or spontaneous idiopathic preterm birth less than 35 weeks of gestation. Families for whole exome sequencing were selected by more than one generation of the pedigree affected with an apparent maternal mode of transmission. Mothers were excluded if they had any medical indication for a preterm delivery, a multiple gestation pregnancy, or other identified risk from preterm birth such as recent trauma or clinical evidence of infection.
Exome capture
A total of 3 μg of genomic DNA was submitted to the Vanderbilt Genome Technology Core (GTC) for whole-exome capture. The Agilent Technologies 50 Mb SureSelect Human All Exome Kit was used to capture and amplify the submitted samples. Per the manufacturer’s website, the kit interrogates 1.22% of the human genomic regions corresponding to the NCBI Consensus CDS database (CCDS).
Whole-exome sequencing
The GTC performed the sequencing using an Illumina HiSeq and 100bp paired-end reads. This resulted in an average of 76× read-depth for all of our exome with a range between ~60–104×.
Sequencing quality control
The Genome Analysis Toolkit (GATK) using current best practices was used to trim, align and call variants in our exome sequence data against the Hg19 genome build.
VAAST analysis
Only variants which passed GATK QC were annotated against the Hg19 genome build using the Variant Annotation Tool (VAT). Once annotated the Variant Selection Tool (VST) was used to select variants that were shared between the mother-daughter pairs in family 1168 and 1281. Each set of these shared variants were then individually compared to a 1000 Genomes background VAAST background file that contained data from 1093 individuals using the Variant Analysis Tool (VAT). When VAT was run the codon-bias option and default additive genetic model were used. VAAST p-values were calculated using the fast genome-permutation option with 1e5 permutations.
KEGG pathway analysis of most significant VAAST p-value genes
All of the genes with the most significant p-value for each mother-daughter pair were searched against the KEGG Homo Sapiens pathways to test for pathway enrichment.
Genotyping of complement and coagulation factor pathway common variants
To efficiently and cost-effectively genotype common coding region variants in the genes identified in the exome sequencing studies, we analyzed 750 ng of genomic DNA at the Vanderbilt DNA core from women delivering preterm or at term in a case-control design on Illumina HumanExome beadchips per the manufacturer’s protocol. This analysis included 67 SNPs in the six gene emerging from the exome sequencing study. QC was performed in PLINK. We chose to not filter by Hardy-Weinberg equilibrium (HWE), but HWE checks were performed in order to see if our most significant SNPs were out of HWE in controls. In addition, samples previously genotyped on Affymetrix SNP Array 6.0 (Plunkett et al. 2011) were reanalyzed to interrogate noncoding SNPs in the CR1 region for association with PTB. For these samples, the PLINK ready BED files were processed for QC using PLINK. The QC steps were as follows and performed in the following order: removed SNPs with genotype frequency <95%, remove samples with <95% SNP calls, remove SNPs with MAF <5%, and remove SNPs with HWE p-values in controls <0.0001.
Statistical analysis
All SNP statistical analysis was performed using the software package PLINK v1.07. Demographic data was analyzed using SPSS Statistics V.20 and means were tested for a significant difference using a one-way analysis of variance (ANOVA). For association tests of complement/coagulation factor cascade SNPs, we used additive logistic regression adjusting for the factors shown to differ significantly between cases and controls (Table 1). Associations were considered statistically significant if they survived a Bonferroni correction for the number of SNPs tested.
Table 1.
Demographic information for Helsinki mothers genotyped on the Illumina exome beadchip. Numbers in the table are mean (standard deviation) except for dichotomous variables where percentages were used. P-values were determined using a one-way analysis of variance (ANOVA).
| Variable | Preterm (n=237) | Term (n=328) | P-value |
|---|---|---|---|
| Maternal age (yr) | 31.1 (5.0) | 31.5 (4.2) | 0.334 |
| Body Mass Index (kg/m2) | 23.5 (4.5) | 22.7 (3.1) | 0.012 |
| Parity (n) | 1.6 (0.89) | 1.5 (0.73) | 0.062 |
| Gravidity (n) | 2.1 (1.3) | 1.9 (1.0) | 0.050 |
| Birth weight (g) | 2337.0 (500.1) | 3578.7 (421.3) | <0.0001 |
| Birth length (cm) | 45.0 (2.8) | 50.3 (1.9) | <0.0001 |
| Alcohol use (%) | 5.2% | 1.5% | 0.012 |
| Tobacco use (%) | 8.7% | 2.7% | 0.002 |
Family based association test (FBAT)
Analyses were done using the Family Based Association Test (FBAT (Laird et al. 2000) focused on allele transmission from the maternal grandparents to the mother of preterm infants (born prior to 37 weeks of completed gestation). Subjects were recruited from the neonatal intensive care units at the University of Iowa and collaborating sites in United States and Denmark. All subjects were of European ancestry. Allelic variation was determined using the TaqMan genotyping system (Applied Biosystems, Foster City, California). Genotyping reactions were completed in 384-well plates containing dried sample DNA. Polymerase chain reactions (PCRs) were performed on an ABI GeneAmp9700 thermocycler using conditions set by the manufacturer. Allelic determination was carried out on an ABI 7900HT using the Sequence Detection Systems software (version 2.4, Applied Biosystems).
RESULTS
Analysis of shared variants in mother-daughter pairs
To identify potential rare variants that contribute to the risk for preterm birth, we analyzed two Finnish mother-daughter pairs (families 1168 and 1281) each of whom experienced preterm delivery (Figure 1). These families were selected from more than 100 pedigrees due to high penetrance of preterm birth to each mother, more than one affected generation with spontaneous idiopathic preterm birth phenotype, and exhibiting a maternal pattern of transmission. Variants were called using standard best-practice quality control (QC) thresholds in Genome Analysis Toolkit (GATK) (McKenna et al. 2010). Only variants that passed QC filters were then analyzed using the Variant Annotation, Analysis and Search Tool (VAAST) pipeline (Yandell et al. 2011). Each set of these variants were then individually compared to a 1000 Genomes background VAAST file that contained data from 1093 individuals.
Figure 1.

Pedigrees for the two mother-daughter pairs, family 1168(A) and family 1281 (B) subjected to whole-exome, sequencing. The initial case proband is indicated by the triangle and sequenced mothers by the black dot on the bottom right of the circle.
In family 1168 there were 202 genes/features, which had the most significant VAAST genome-wide permutated p-value (1.67e-06) (Supplemental Table 1). For family 1281 there were 275 genes with the most significant VAAST genome-wide permutated p-value (1.67e-06) (Supplemental Table 2). Examining the overlap of these gene lists reveals 163 genes that are common between the two families. The top genome-wide permutation p-value for both families are considered genome-wide significant due to the fact that it surpasses the Bonferroni corrected p-value for 20,000 genes, 2.5e-06.
Pathway analysis of the most significant genes in mother-daughter pairs
In order to glean insight into PTB pathophysiology we used a pathway analysis approach. The most significant genes for our two families were tested for pathway enrichment using the Kyoto Encyclopedia of Genes and Genomes (KEGG) (Kanehisa and Goto 2000).
Overall, for family 1168 the most significantly associated genes played a role in 102 pathways (Supplemental Table 3). The top three pathways were olfactory transduction, metabolic pathways and complement and coagulation cascades with 25, eight, and five genes respectively. In family 1281, there were 86 KEGG pathways with at least one gene, which had the most significant VAAST p-value (Supplemental Table 4). The top three pathways were olfactory transduction, focal adhesion and metabolic pathways with 20, six, and five genes respectively. The complement and coagulation cascades are in the next group of three pathways which each have four genes. Table 2 lists the genes in each family that were involved in the complement and coagulation cascades.
Table 2.
Complement and coagulation cascade genes from the KEGG analysis of the top p-value VAAST genes
| Gene | Gene Name | VAAST Score Family 1168 | VAAST Score Family 1281 | VAAST Rank Family 1168 | VAAST Rank Family 1281 | Total Missense SNPs for the other 6 exomes |
|---|---|---|---|---|---|---|
| CR1 | Complement component (3b/4b) receptor 1 (Knops blood group) | 174.31 | 71.81 | 12 | 74 | 60* φ |
| F5 | Coagulation factor V (proaccelerin, labile factor) | 91.08 | 53.38 | 45 | 130 | 44* |
| F13B | Coagulation factor XIII, B polypeptide | 57.60 | 57.60 | 64 | 86 | 6 |
| CR2 | Complement component (3d/Epstein Barr virus) receptor 2 | 56.76 | 39.38 | 70 | 162 | 30* |
| C4BPA | Complement component 4 binding protein, alpha | 23.64 | 38.25 | 182 | 170 | 7 |
| CFH | Complement factor H | n/a | 76.52 | n/a | 65 | 10* |
Contains variants predicted to be probably damaging
Contains variants predicted to be possibly damaging
Examination of complement and coagulation cascades in our other PTB exomes
We chose to focus our analysis on the KEGG complement and coagulation cascade instead of the two others that they shared, olfactory transduction and metabolic pathways, for three main reasons. First, whole-genome sequencing has shown that olfactory transduction genes harbor more predicted loss-of-function variants than expected when interrogating 1000 Genomes Project data (MacArthur et al. 2012); therefore we believed that these shared variants in the mother-daughter pairs were unlikely to be involved in PTB. Second, coagulation and immune activation (complement system) are two of the proposed pathways previously hypothesized to contribute to PTB (Lockwood and Kuczynski 2001; Wang et al. 2001). Third, prior modest associations exist between coagulation pathway genes, F5 (Hao et al. 2004; Velez et al. 2008), F7 (Härtel et al. 2005; Velez et al. 2008) F13A1 (Härtel et al. 2005) and PLAT (Velez et al. 2008) and PTB.
Using our two mother-daughter pairs as a “discovery” cohort we examined the six genes from the KEGG complement and coagulation cascades identified by VAAST in six other PTB exomes (5 Finnish, 1 European American). Because of the higher probability of being damaging, we initially focused on novel variants. Of the six exomes, three harbored novel variants. There were 19 total novel variants: 14 were unique, and half were missense SNPs. Using the in silico tool PolyPhen-2, we assessed the novel missense variants for potential to be deleterious using the HumDiv algorithm (Adzhubei et al. 2010). The only variant predicted to be “probably damaging” was a complement factor H (CFH) Thr956Met variant seen in a single family. All of the other novel missense variants were predicted to be “benign” (Supplemental Table 5).
We also analyzed all missense variants in these genes. All of the six genes harbored between six and 60 missense variants for a total of 157, and 36 were unique. We once again tested the potential for these variants to be deleterious using the PolyPhen-2 in silico tool’s HumDiv algorithm. Only half of the genes, CR1, CR2, and F5, contained missense variants that were predicted to be “probably damaging” and CR1 was the sole gene, which contained missense variants predicted to be “possibly damaging” by PolyPhen-2 (Table 2).
Interrogation of the complement and coagulation cascade in nuclear PTB mothers
Based upon the exome sequencing findings, we next tested the hypothesis that coding-region variants in the complement/coagulation cascade genes identified in the Finnish families contributed more broadly to the pathogenesis of preterm birth. We conducted an association study in 237 case and 328 control Finnish mothers. We performed additive logistic regression adjusting for the variables shown to differ significantly between the preterm and term mothers: body mass index (BMI), gravidity, ethanol use and smoking use. Our examination was focused on the six complement and coagulation cascade genes identified by VAAST in our two whole-exome families. In total, 67 coding region SNPs from the six gene regions were analyzed (Supplemental Table 6). The most significantly associated SNP was an exonic missense SNP, rs6691117, unadjusted p-value = 6.93e-5, OR = 1.74 (1.33, 2.29 95% CI); adjusted additive logistic regression p-value = 1.07e-4, OR = 1.73 (1.31, 2.29 95% CI) in CR1. This association withstands a conservative Bonferroni corrected p-value of 7.64e-4. Depending on the transcript, this SNP changes an isoleucine to a valine at amino acid position 1615 or 2065. Both of these substitutions are predicted to be “benign” by the HumDiv algorithm in PolyPhen-2.
Due to the robust association of the CR1 coding region SNP we applied a similar analysis to our Finnish mothers (252 cases/287 controls), which were previously genotyped on the Affymetrix 6.0 SNP arrays (Plunkett et al. 2011). We provide this additional analysis to explore whether our coding SNP, rs6691117, on the exome array may be tagging another variant elsewhere in the CR1 gene or its regulatory regions which would be detected with this more densely genotyped, largely noncoding variant array. We identified 103 SNPs in the region spanning 10 kb 5′ through 10 kb 3′ of the CR1 gene boundary (Supplemental Table 7). The most significantly associated SNP, rs10429953, was located in an intron of CR1 unadjusted additive logistic regression p-value = 1.31e-4, OR = 1.93. This p-value surpasses the Bonferroni corrected p-value of 4.85e-4 for 103 SNPs tested. In addition to the most significantly associated SNPs surpassing Bonferroni, the second highest associated SNP, rs10429943, also in a CR1 intron p-value = 3.74e-4 OR = 1.84 clears the threshold. However, these two SNPs are in strong linkage disequilibrium (r2 = 0.96) so this should be considered a single strong association in CR1 (Figure 2).
Figure 2.
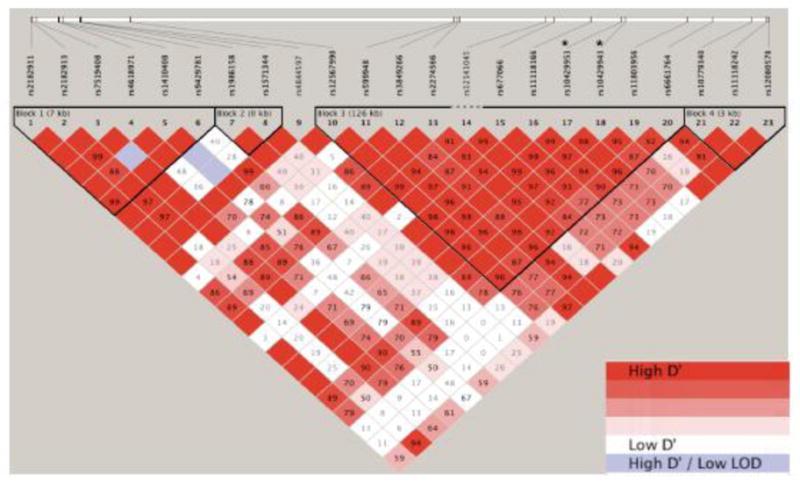
Haploview view linkage disequilibrium (LD) diagram showing D′ values for Affymetrix 6.0 SNP array samples in the CR1 gene region with the addition of 10 kb 5′ and 3′. The two intronic SNPs on the Affymetrix array are marked with an *. The intronic SNP on the Illumina exome beadchip resides in the same 126 kb LD block.
To gain additional evidence for the association of rs6691117 with preterm birth, we performed family-based association tests on a separate cohort of European ancestry mothers from the United States and Denmark. We genotyped the mothers of the preterm infants and their parents to measure over-transmission of the risk promoting minor allele. We found marginal significance for over-transmission in this cohort consisting of 52 informative trios from 852 individuals in 278 nuclear families (p = 0.06, one-tailed). Limiting our analysis to a group of mothers at relatively low risk for environmental exposures to enhance the potential contribution of genetic factors (Iowa and Denmark), we found a more significant association of rs6691117 with preterm birth (p = 0.01, one-tailed) in a cohort of 153 families with 17 informative trios.
DISCUSSION
To our knowledge, this is the first report using whole-exome sequencing to interrogate for rare variants that aggregate in specific genes and pathways in the complex disease of PTB. We used our results from the exome sequencing of mother-daughter pairs in highly affected families to expand to candidate gene/pathway association studies using genotyping arrays. This more focused analysis enhances the likelihood to detect significant variants in moderately sized nuclear datasets, and should accelerate gene discovery. We believe that this approach can be effectively applied to other complex disorders.
CR1, and the complement/coagulation factor pathway, as revealed by our exome sequencing and follow-up analysis, provides a biologically plausible pathway related to adverse pregnancy outcomes. There is a growing body of literature describing activation of the complement system and adverse pregnancy outcomes, including PTB (Vaisbuch et al. 2010; Soto et al. 2005). There have been a number of reports illustrating that an increased level of fragment Bb (FBb) early in pregnancy is associated with an increased risk of PTB < 34 weeks (Lynch et al. 2008). FBb is a marker of alternative pathway complement activation. In addition to increased FBb, increased maternal plasma levels of complement C3a during the first trimester have been associated with an increased risk of a number of adverse pregnancy outcomes including PTB (Lynch et al. 2011). It has also been shown that erythrocyte membrane complement receptor 1 (CR1) levels are reduced during pregnancy and reach their nadir during the third trimester (Imrie et al. 1996). In this study, we found a significant association between three SNPs in CR1 and an increased risk of PTB. However, when we examine linkage disequilibrium all three of the SNPs are in a large 126 kb linkage block (Figure 2). The strongest association residing in the coding region variant suggests that the other non-coding SNPs are detected based on tagging this variant.
While predicted to be “benign” the isoleucine to valine exonic SNP in our exome arrays may be functional; such a subtle substitution would be misclassified by in silico tools such as PolyPhen-2 (Adzhubei et al. 2010) and SIFT (Ng and Henikoff 2003). Evidence for functional consequences of the SNP leading to the isoleucine to valine change in CR1 we identified in our exome arrays is its association with alteration in erythrocyte sedimentation rate (ESR), as detected in a large genome-wide association study (Kullo et al. 2011). The most significantly associated SNP (rs6691117) identified in our study is associated with a decreased ESR. As CR1 on erythrocytes leads to increased clearance of immune complexes to limit their deposition in vessel walls, we would predict greater systemic inflammation or coagulability resulting from our risk-promoting allele. ESR normally increases in pregnancy due to increased fibrinogen levels and the need for clearance of immune complexes (van den Broe and Letsky 2001); attenuating this process by less functional CR1 variants would be predicted to increase risk for adverse pregnancy outcomes such as preterm birth. Alternatively, this SNP may be tagging a different causative variant in the gene. It should be noted that the predicted damaging SNP rs2274567, resulting in a histidine to arginine change, is in linkage disequilibrium with rs6691117, and is associated with similar changes in ESR (Kullo et al. 2011).
In conclusion, this is the first report of using whole-exome sequencing to interrogate for rare variants that aggregate in specific genes and pathways in the complex disease of PTB. We believe that our results strengthen the argument that the complement and coagulation cascade are involved in the pathophysiology of PTB, and suggest potential screening and intervention approaches to prevent prematurity, which target this pathway (Ricklin and Lambris 2007). Possible future interventions include the use of soluble CR1 (Weisman et al. 1990) or monoclonal antibodies to C5 (Wang et al. 1995) to limit complement activation and inflammation or enhance clearance, which our risk allele may compromise. While there are FDA approved anti-C5 antibodies, eculizumab (Zareba 2007), soluble CR1 is still in the clinical trial stage. Future functional studies will be essential to determine the specific mechanisms by which these pathways increase prematurity risk.
Supplementary Material
Supplemental Table 1. VAAST genes with most significant p-values in family 1168 sorted by rank
Supplemental Table 2. VAAST genes with most significant p-values in family 1281 sorted by rank
Supplemental Table 3. KEGG pathways with more than three genes from the list of the most significant p-value genes for family 1168
Supplemental Table 4. KEGG pathways that contained more than three genes from the most significant p-value genes for family 1281
Supplemental Table 5. Novel missense variants in the six other exomes for the six complement and coagulation cascade genes identified by VAAST (for the PolyPhen-2 prediction the HumDiv algorithm was used).
Supplemental Table 6. Complete unadjusted and adjusted additive logistic regression association results for Finnish mothers’ complement and coagulation factor exome SNPs on Illumina arrays sorted by adjusted p-value. The gene listed is that in which the SNP is located and in parentheses is the VAAST gene the SNP was selected to interrogate within the 10 kb 5′ and 3′ buffer.
Supplemental Table 7. Complete unadjusted additive logistic association results for Finnish mothers’ Affymetrix 6.0 SNP arrays sorted by p-value. The gene listed is that in which the SNP is located and in parentheses is the VAAST gene the SNP was selected to interrogate within the 10kb 5′ and 3′ buffer.
Acknowledgments
Funding: National Institutes of Health and March of Dimes.
This work was supported by grants from the March of Dimes (LJM and JCM) and NIH (T32 GM07347 to JJM; R01 HD52953 to JCM; and MOD grant FY10-180 T32 HD068256 to CEG).
Footnotes
The authors declare that they have no conflict of interest.
References
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nature Methods. 2010;7(4):248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhari BP, Plunkett J, Ratajczak CK, Shen TT, DeFranco EA, Muglia LJ. The genetics of birth timing: insights into a fundamental component of human development. Clin Genet. 2008;74(6):493–501. doi: 10.1111/j.1399-0004.2008.01124.x. [DOI] [PubMed] [Google Scholar]
- Hao K, Wang X, Niu T, Xu X, Li A, Chang W, Wang L, Li G, Laird N, Xu X. A candidate gene association study on preterm delivery: application of high-throughput genotyping technology and advanced statistical methods. Hum Mol Genet. 2004;13(7):683–691. doi: 10.1093/hmg/ddh091. [DOI] [PubMed] [Google Scholar]
- Härtel C, von Otte S, Koch J, Ahrens P, Kattner E, Segerer H, Möller J, Diedrich K, Göpel W. Polymorphisms of haemostasis genes as risk factors for preterm delivery. Thrombosis and haemostasis. 2005;94(1):88–92. doi: 10.1267/THRO05010088. [DOI] [PubMed] [Google Scholar]
- Howson CP, Kinney MV, Lawn J. Born Too Soon: The Global Action Report on Preterm Birth. World Health Organization; Geneva: 2012. March of Dimes, PMNCH, Save the Children, WHO. [Google Scholar]
- Imrie H, McGonigle T, Liu D, Jones D. Reduction in erythrocyte complement receptor 1 (CR1, CD35) and decay accelerating factor (DAF, CD55) during normal pregnancy. Journal of reproductive immunology. 1996;31 (3):221–227. doi: 10.1016/0165-0378(96)00977-1. [DOI] [PubMed] [Google Scholar]
- Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Research. 2000;28 (1):27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kullo IJ, Ding K, Shameer K, McCarty CA, Jarvik GP, Denny JC, Ritchie MD, Ye Z, Crosslin DR, Chisholm RL, Manolio TA, Chute CG. Complement receptor 1 gene variants are associated with erythrocyte sedimentation rate. American journal of human genetics. 2011;89(1):131–138. doi: 10.1016/j.ajhg.2011.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird NM, Horvath S, Xu X. Implementing a unified approach to family-based tests of association. Genet Epidemiol. 2000;19(Suppl 1):S36–42. doi: 10.1002/1098-2272(2000)19:1+<::AID-GEPI6>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Lockwood CJ, Kuczynski E. Risk stratification and pathological mechanisms in preterm delivery. Paediatr Perinat Epidemiol. 2001;15(Suppl 2):78–89. doi: 10.1046/j.1365-3016.2001.00010.x. [DOI] [PubMed] [Google Scholar]
- Lynch AM, Gibbs RS, Murphy JR, Byers T, Neville MC, Giclas PC, Salmon JE, Van Hecke TM, Holers VM. Complement activation fragment Bb in early pregnancy and spontaneous preterm birth. American journal of obstetrics and gynecology. 2008;199(4):354 e351–358. doi: 10.1016/j.ajog.2008.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch AM, Gibbs RS, Murphy JR, Giclas PC, Salmon JE, Holers VM. Early elevations of the complement activation fragment C3a and adverse pregnancy outcomes. Obstetrics and gynecology. 2011;117(1):75–83. doi: 10.1097/AOG.0b013e3181fc3afa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacArthur DG, Balasubramanian S, Frankish A, Huang N, Morris J, Walter K, Jostins L, Habegger L, Pickrell JK, Montgomery SB, Albers CA, Zhang ZD, Conrad DF, Lunter G, Zheng H, Ayub Q, DePristo MA, Banks E, Hu M, Handsaker RE, Rosenfeld JA, Fromer M, Jin M, Mu XJ, Khurana E, Ye K, Kay M, Saunders GI, Suner M-M, Hunt T, Barnes IHA, Amid C, Carvalho-Silva DR, Bignell AH, Snow C, Yngvadottir B, Bumpstead S, Cooper DN, Xue Y, Romero IG, Consortium GP, Wang J, Li Y, Gibbs RA, Mccarroll SA, Dermitzakis ET, Pritchard JK, Barrett JC, Harrow J, Hurles ME, Gerstein MB, Tyler-Smith C. A systematic survey of loss-of-function variants in human protein-coding genes. Science (New York, NY) 2012;335(6070):823–828. doi: 10.1126/science.1215040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Research. 2010;20(9):1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng PC, Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Research. 2003;31 (13):3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng SB, Buckingham KJ, Lee C, Bigham AW, Tabor HK, Dent KM, Huff CD, Shannon PT, Jabs EW, Nickerson DA, Shendure J, Bamshad MJ. Exome sequencing identifies the cause of a mendelian disorder. Nat Genet. 2010;42(1):30–35. doi: 10.1038/ng.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Roak BJ, Deriziotis P, Lee C, Vives L, Schwartz JJ, Girirajan S, Karakoc E, Mackenzie AP, Ng SB, Baker C, Rieder MJ, Nickerson DA, Bernier R, Fisher SE, Shendure J, Eichler EE. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nature Genetics. 2011;43(6):585–589. doi: 10.1038/ng.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plunkett J, Doniger S, Orabona G, Morgan T, Haataja R, Hallman M, Puttonen H, Menon R, Kuczynski E, Norwitz E, Snegovskikh V, Palotie A, Peltonen L, Fellman V, DeFranco EA, Chaudhari BP, McGregor TL, McElroy JJ, Oetjens MT, Teramo K, Borecki I, Fay J, Muglia L. An Evolutionary Genomic Approach to Identify Genes Involved in Human Birth Timing. PLoS Genetics. 2011;7(4):e1001365. doi: 10.1371/journal.pgen.1001365.t002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plunkett J, Feitosa MF, Trusgnich M, Wangler MF, Palomar L, Kistka ZA-F, DeFranco EA, Shen TT, Stormo AED, Puttonen H, Hallman M, Haataja R, Luukkonen A, Fellman V, Peltonen L, Palotie A, Daw EW, An P, Teramo K, Borecki I, Muglia LJ. Mother’s genome or maternally-inherited genes acting in the fetus influence gestational age in familial preterm birth. Hum Hered. 2009;68(3):209–219. doi: 10.1159/000224641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plunkett J, Muglia LJ. Genetic contributions to preterm birth: implications from epidemiological and genetic association studies. Ann Med. 2008;40(3):167–195. doi: 10.1080/07853890701806181. [DOI] [PubMed] [Google Scholar]
- Ricklin D, Lambris JD. Complement-targeted therapeutics. Nature Biotechnology. 2007;25(11):1265–1275. doi: 10.1038/nbt1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soto E, Romero R, Richani K, Espinoza J, Nien JK, Chaiworapongsa T, Santolaya-Forgas J, Edwin SS, Mazor M. Anaphylatoxins in preterm and term labor. Journal of perinatal medicine. 2005;33(4):306–313. doi: 10.1515/JPM.2005.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaisbuch E, Romero R, Erez O, Mazaki-Tovi S, Kusanovic JP, Soto E, Dong Z, Chaiworapongsa T, Kim SK, Ogge G, Pacora P, Yeo L, Hassan SS. Activation of the alternative pathway of complement is a feature of pre-term parturition but not of spontaneous labor at term. American journal of reproductive immunology (New York, NY : 1989) 2010;63(4):318–330. doi: 10.1111/j.1600-0897.2009.00800.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Broe NR, Letsky EA. Pregnancy and the erythrocyte sedimentation rate. BJOG : an international journal of obstetrics and gynaecology. 2001;108 (11):1164–1167. [PubMed] [Google Scholar]
- Velez DR, Fortunato SJ, Thorsen P, Lombardi SJ, Williams SM, Menon R. Preterm birth in Caucasians is associated with coagulation and inflammation pathway gene variants. PloS one. 2008;3(9):e3283. doi: 10.1371/journal.pone.0003283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zuckerman B, Kaufman G, Wise P, Hill M, Niu T, Ryan L, Wu D, Xu X. Molecular epidemiology of preterm delivery: methodology and challenges. Paediatr Perinat Epidemiol. 2001;15(Suppl 2):63–77. doi: 10.1046/j.1365-3016.2001.00009.x. [DOI] [PubMed] [Google Scholar]
- Wang Y, Rollins SA, Madri JA, Matis LA. Anti-C5 monoclonal antibody therapy prevents collagen-induced arthritis and ameliorates established disease. Proceedings of the National Academy of Sciences of the United States of America. 1995;92 (19):8955–8959. doi: 10.1073/pnas.92.19.8955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisman HF, Bartow T, Leppo MK, Marsh HC, Carson GR, Concino MF, Boyle MP, Roux KH, Weisfeldt ML, Fearon DT. Soluble human complement receptor type 1: in vivo inhibitor of complement suppressing post-ischemic myocardial inflammation and necrosis. Science (New York, NY) 1990;249 (4965):146–151. doi: 10.1126/science.2371562. [DOI] [PubMed] [Google Scholar]
- Yandell M, Huff C, Hu H, Singleton M, Moore B, Xing J, Jorde LB, Reese MG. A probabilistic disease-gene finder for personal genomes. Genome Research. 2011;21(9):1529–1542. doi: 10.1101/gr.123158.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zareba K. Eculizumab: A novel therapy for paroxysmal nocturnal hemoglobinuria. Drugs Today. 2007 doi: 10.1358/dot.2007.43.8.1130446. [DOI] [PubMed] [Google Scholar]
- Züchner S, Dallman J, Wen R, Beecham G, Naj A, Farooq A, Kohli MA, Whitehead PL, Hulme W, Konidari I, Edwards YJK, Cai G, Peter I, Seo D, Buxbaum JD, Haines JL, Blanton S, Young J, Alfonso E, Vance JM, Lam BL, Periak-Vance MA. Whole-Exome Sequencing Links a Variant in DHDDS to Retinitis Pigmentosa. American journal of human genetics. 2011;88(2):201–206. doi: 10.1016/j.ajhg.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table 1. VAAST genes with most significant p-values in family 1168 sorted by rank
Supplemental Table 2. VAAST genes with most significant p-values in family 1281 sorted by rank
Supplemental Table 3. KEGG pathways with more than three genes from the list of the most significant p-value genes for family 1168
Supplemental Table 4. KEGG pathways that contained more than three genes from the most significant p-value genes for family 1281
Supplemental Table 5. Novel missense variants in the six other exomes for the six complement and coagulation cascade genes identified by VAAST (for the PolyPhen-2 prediction the HumDiv algorithm was used).
Supplemental Table 6. Complete unadjusted and adjusted additive logistic regression association results for Finnish mothers’ complement and coagulation factor exome SNPs on Illumina arrays sorted by adjusted p-value. The gene listed is that in which the SNP is located and in parentheses is the VAAST gene the SNP was selected to interrogate within the 10 kb 5′ and 3′ buffer.
Supplemental Table 7. Complete unadjusted additive logistic association results for Finnish mothers’ Affymetrix 6.0 SNP arrays sorted by p-value. The gene listed is that in which the SNP is located and in parentheses is the VAAST gene the SNP was selected to interrogate within the 10kb 5′ and 3′ buffer.