

Abstract
Intrinsic resistance to agents targeting phosphatidylinositol-3-kinase (PI3K)/AKT pathway is one of the major challenges in cancer treatment with such agents. The objective of this study is to identify the genes or pathways that can be targeted to overcome the resistance of non-small cell lung cancer to the AKT inhibitor, MK2206, which is currently being evaluated in phase I and II clinical trials. Using a genome-wide small interfering RNA (siRNA) library screening and biological characterization we identified that inhibition of Thioredoxin Reductase-1 (TXNRD1), one of the key anti-oxidant enzymes, with siRNAs or its inhibitor, Auranofin, sensitized non-small cell lung cancer cells to MK2206 treatment in vitro and in vivo. We found that simultaneous inhibition of TXNRD1 and AKT pathways induced robust reactive oxygen species (ROS) production, which was involved in c-Jun N-terminal Kinase (JNK, MAPK8) activation and cell apoptosis. Furthermore we found that the synthetic lethality interaction between the TXNRD1 and AKT pathways occurred through the KEAP1/NRF2 cellular antioxidant pathway. Lastly, we found that synthetic lethality induced by TXNRD1 and AKT inhibitors relied on wild type KEAP1 function. Our study indicates that targeting the interaction between AKT and TXNRD1 antioxidant pathways with MK2206 and Auranofin, a FDA approved drug, is a rational strategy to treat lung cancer and that KEAP1 mutation status may offer a predicative biomarker for such combination approaches.
Keywords: Synthetic lethality, AKT, TXNRD1, MK2206, KEAP1
Introduction
Previous studies have established the critical role of the phosphatidylinositol-3-kinase (PI3K)/AKT pathway in cancer cell growth and survival; therefore, strategies that target this pathway have been developed and tested for cancer treatment, including lung cancer (1). MK2206 (Merck, Inc.), a small-molecule inhibitor that targets the AKT signaling pathway, is currently being tested in phase I and phase II clinical trials and has demonstrated promising results in terms of tumor shrinkage, disease stabilization and patient tolerability in patients with advanced solid tumor (2). We previously reported that a large subset of human none-small-cell-lung-cancer (NSCLC) cells do not respond to MK2206 (3). We and others have also reported that combination with other agents, such as AZD6244 (an inhibitor of mitogen-activated protein kinase kinase) and erlotinib (an epidermal growth factor receptor [EGFR] inhibitor), sensitized some lung cancer cells to MK2206 (3, 4). Those findings indicate that identification and inhibition of additional targets are needed to improve the efficacy of targeted therapeutic agents, such as MK2206.
Thioredoxin reductase and thioredoxin (TXNRD1/TXN) is one of the major thiol-dependent electron donor systems in cells; it plays a critical role in regulating the cellular redox environment and in a wide range of cellular activities, such as maintenance of viability and proliferation (5). Preclinical studies indicated that Auranofin, an orally active gold compound and a TXNRD1 inhibitor, induced apoptosis in cancer cells and enhanced the antitumor activities of chemotherapeutic agents (6-10). It is currently being tested in a phase II clinical trial for treatment of chronic lymphocytic leukemia (11), and was recently repurposed as an orphan drug for human amebiasis treatment (12). Other TXNRD1/TXN inhibitors, such as PG12, have also been tested clinically in solid tumors (13, 14). Kelch-like ECH-associated protein (KEAP1) and nuclear factor-erythroid-derived-related factor 2 (NRF2) pathways is another major cellular antioxidant pathways. Under normal conditions, KEAP1 binds to NRF2 and promotes degradation by cullin3 (Cul3)-based E3 ligase system (15). Activated NRF2 due to oxidative stress can stimulate multiple antioxidant molecules by binding to antioxidant response elements (AREs) (16, 17). Loss of function mutations of KEAP1 have been reported in many cancer types including NSCLC with frequency of about 19% (18-20). KEAP1 mutations cause constitutive activation of NRF2, which is associated with cancer progression, treatment resistance, and poor patient survival (21-23). Genome-wide small-interfering RNA (siRNA) library screening has been used to identify targets or networks that can be targeted for cancer therapy. siRNA library screening has also been used to identify synthetic lethality loci with specific drugs (24-27). Using genome-wide siRNA library screening and biochemical and biological methods, we found that inhibition TXNRD1 with siRNAs or inhibitor, auranofin sensitized lung cancer cells to MK2206 in vitro and in vivo. Our study indicate that Auranofin, a TXNRD1 inhibitor and clinically proven drug that has been used to treat rheumatoid arthritis for 25 years, could be repurposed for lung cancer therapy in combination with MK2206.
Materials and Methods
Cell lines
Human lung cancer cell lines HCC193, H1993, H460, H1299, H2126, H292, H1648, H838, H1395, H23, H358, H1703, H1666 and A549 were from the Hamon Center for Therapeutic Oncology Research at The University of Texas Southwestern Medical Center, Dallas, Texas. Cell lines were maintained in high-glucose RPMI 1640 supplemented with 5% fetal bovine serum and cultured at 37°C in a humidified atmosphere containing 5% CO2 and 95% air. Cell lines have been authenticated regularly with STR fingerprinting method.
siRNA library screening
We used the non-small-cell lung cancer cell line HCC193 for genome-wide siRNA screening, because it has a high level of p-AKT and the IC50 for MK2206 is 10 μM (3) The screening concentration of MK2206 is 1 μM, a dose can sufficiently inhibit phosphorylated AKT (p-AKT) in vitro and in vivo (2, 3). The screening was performed using a Dharmacon siRNA siGENOME (Dharmacon, Lafayette, CO) library containing 84,508 siRNAs with 4 unique siRNA duplexes that target each of 21,127 unique human genes. On day 1, 2000 cells were reverse transfected with 0.07 μl Dharmafect 4 in a 384-well plate, to a final concentration of 40 nM siRNA per well. MK2206 was added on day 3 at a 1μM final concentration. Cell growth was analyzed on day 5 using the CellTiter Blue method (Promega, WI). Raw data were log 2 transformed. Data were normalized using 16 negative controls in each plate. T-statistics were performed to compare the significant differences between the values of siRNA transfection and the negative control and siRNA transfection plus MK2206 treatment. P values were obtained using computed t-statistics. Beta-uniform mixture methods were used to account for multiple tests and estimate the false discovery rate (FDR).
Proliferation assay
The drug treatment protocol was reported in our previous publications (3, 28). In brief, 2000 cells was plated in 96-well plates and treated with the indicated doses of Auranofin (0-0.5 μM), MK2206 (0-2.5 μM), or both for 72 hours. A sulforhodamine B assay (SRB) was performed to determine cell viability. IC50 values of Auranofin and MK2206 in each cell line were calculated using Curve Expert 1.3 software and plotted in dose-response curves. Chou-Talalay method was used to calculate the two drug combination index (CI) using Calculsyn software (Biosoft, Cambridge, UK). (CI=0.2–0.9, synergism; CI<0.2, strong synergism) (29).
Western blotting analysis
Whole-cell lysates were collected using RIPA reagent. Western blotting analysis was performed with 40 μg of total cell proteins and antibodies against phosphorylated-c-Jun N-terminal kinase (p-JNK), JNK, poly (ADP-ribose) polymerase (PARP), p-AKT, AKT (Cell Signaling, Danvers, MA), KEAP1, NRF2 (Santa Cruz Biotechnology, Santa Cruz, CA), and TXNRD1 (Abcam, Cambridge, MA).
Gene knockdown and overexpression
Cells were transfected in 96-well or 6-well plate with siRNAs (40 nM) using Dharmafect 4, and 48 hours post transfection, cells were treated with dimethyl sulfoxide (DMSO) or MK2206 for additional 72 hours. For stable gene knockdown of KEAP1, shRNAs in lentivirus plasmids were purchased from Thermo Fisher Scientific (Waltham, MA), and lentiviruses were packaged using a kit from System Biosciences (Palo Alto, CA). Cells were infected with lentiviruses containing specific shRNAs, followed by selection with Puromycin (1μg/ml) for 4 weeks. For overexpression of dominant negative AKT (AKT-DN) or wild type KEAP1 (Dsred-KEAP1), plasmids of interest were transfected into HCC193, H1993 and H460 cells respectively with Lipofectamine 2000 followed by selection with G418 (500μg/ml) for 4 weeks.
Measuring ROS production
The ROS measurement method was described in our previous publication (30). In brief, cells (1×106/mL) were incubated in Hank’s balanced saline solution containing 50 mM 2’, 7’-dichlorfluorescein-diacetate (DCFHDA) for 30 minutes. The cell population fluorescence is proportional to the levels of intracellular ROS generated and was measured using fluorescence-activated sell sorting (FACS) (Becton Dickinson, Mountain View, CA) at 588 nm emission.
Real-time polymerase chain reaction (PCR)
The NRF2 downstream molecules glutamate-cysteine ligase catalytic (GCLC) subunit was analyzed using real-time PCR as described previously (28). The primers for GCLC forward, 5’-ctgttgcaggaaggcattgat-3’ and reverse, 5’-ttcaaacagtgtcagtgggtctct-3’. GAPDH, forward, 5′-atcccatcaccatcttccag-3′ and reverse, 5′-atgagtccttccacgatacc-3′.
Glutathione (GSH) assay
GSH levels were tested using the GSH assay kit (Sigma, St. Louis, MO). After the indicated treatments GSH levels were determined using the GSH-reductase recycling assay with 5, 5’-Dithiobis-(2-Nitrobenzoic Acid) (DNTB) as substrate in microtiter plates, following the manufacturer’s instructions.
TXNRD1 activity assay
Cells (0.5×108) were washed with PBS and extracted with CellLytic M. The lysates were centrifuged at 10,000 g for 10 minutes, and supernatants were used as the enzyme sample. Tumor tissues (from mice) were extracted with 4 volumes of 0.25X assay buffer containing protease inhibitor cocktail using a Potter-Elvehjem homogenizer. The samples were centrifuged for 15 minutes at 10,000 g. TXNRD1 activity was measured with a TXNRD1 activity assay kit (Sigma) using 5,5’-Dithiobis-(2-Nitrobenzoic Acid) (DNTB) as a substrate.
Animal experiments
Animal experiments were performed according to the approved protocol (10-11-11531) by The University of Texas MD Anderson Cancer Center (Houston, Texas) institutional review board and in accordance with the Guidelines for the Care and Use of Laboratory Animals published by the National Institutes of Health. A total of 2×106 H1993 cells were inoculated subcutaneously into the right dorsal flanks of nude mice. Treatment started when tumors reached a mean volume of approximately 40 mm3. Animals bearing xenograft tumors derived from the H1993 cell line were randomly assigned to 4 treatment groups, with 5 mice in each group: (1) MK2206 only (by oral, 25 mg/kg), (2) Auranofin only (5 mg/kg, by intraperitoneal), (3) MK2206 (25 mg/kg) and Auranofin (5 mg/kg), and (4) solvent only (control). Animals were treated daily, and tumor volumes were measured blindly every 3 days. The tumor volumes were calculated using the formula length × width2 × 0.52. Mice were euthanized when their tumor volume was larger than 2000 mm3. Mean tumor growth was analyzed using the Kruskal-Wallis test for overall significance among different treatment groups and Mann-Whitney U test for significance between two treatment groups (two tailed). Kaplan-Meier method was used to analyze the animal survival.
Examination of NRF2 subcellular localization
Immunofluorescence staining was performed to determine the subcellular localization of NRF2 in H1993 cells, before and after treatment as described previously (31). NRF2 subcellular localization was also examined with enhanced green fluorescent protein (EGFP) fusion protein (32). Images were obtained under a Carl Zeiss Deconvolution AxioVision 4 microscope (Carl Zeiss, Jena, Germany). Nuclear and cytoplasmic fractions were prepared with hypotonic lysis buffer (20mM Tris pH 7.5, 5mM. MgCl2, 5mM CaCl2, 1mM DTT, 1mM EDTA, and protease inhibitor cocktail. Cells were passed through 27-gauge needle on ice for 30 times followed by centrifuge at 250 × g for 5 minutes at 4°C. The supernatant was used as cytoplasmic fraction. The nuclear pellet was thoroughly washed in phosphate-buffered saline and nuclear protein was extracted with RIPA.
Luciferase reporter assay
Cells (2×104) were transfected with ARE-Luc plasmid (Dr. Donna Zhang, Arizona University, Tucson, Arizona) and a renilla-luciferase control plasmid in 12-well plate using Lipofectamine 2000. Forty-eight hours after transfection, cells were treated with DMSO or MK2206 (1 μM) for 1, 3, 6, or 24 hours. Cells were then collected for luciferase activity assay. Luciferase activity was normalized using a renilla-luciferase control.
Statistical analysis
T-statistics were used to determine the significant differences between the survival values of cells treated with negative control siRNA, targeted siRNA and targeted siRNA plus MK2206. P values were obtained using computed t-statistics. Beta-uniform mixture methods were used to account for multiple tests and estimate the FDR. The correlation between KEAP1 mutation and sensitivity to MK2206 and Auranofin was determined using the Fisher’s exact test with two-sided P values at 95% confidence interval. Online Ingenuity software was used to perform a signaling pathway analysis. The significance of the in vitro cell proliferation between and within different treatment groups were determined using Student’s t-test (two tailed). The significance of the in vivo animal study data was determined using the Kruskal-Wallis test for overall significance among different treatment groups and Mann-Whitney U test for significance between two treatment groups (two tailed). Kaplan-Meier method was used to analyze the animal survival.
Results
Synthetic lethality screening identified MK2206 synthetic lethal hits
To identify gene candidates that could be targeted to overcome the intrinsic resistance to MK2206 in NSCLC cells, genome-wide siRNA screening was performed with the Dharmacon siRNA siGENOME library in the NSCLC cell line HCC193. The false discovery rate (FDR) of 0.1 was used as the cut-off to define the significant hits. Using a P value of 0.007542048 and at least 50% percent of growth inhibition as the cut-off, we identified 156 hits. We analyzed the significant hits using Ingenuity Pathway Analysis software and found that targets in the NF-kB, autophagy, and cellular redox regulation pathways were synthetic lethal with MK2206 (Fig.1A and supplementary Table 1). Using cell proliferation assay and siRNA knockdown, we further validated the top 10 hits, including TXNRD1, CFLAR, ATP5J, SLC9A10, and CLN10. The results showed that knockdown of TXNRD1, CFLAR, SCLC9A10, AHCYL1, and CLN10 sensitized both HCC193 and H1993 cells to 1μM MK2206 (Figs. 1B and 1C).
Figure 1. RNAi library screening identified synthetic lethality hits.

(A) The network of synthetic lethality hits with AKT inhibition was analyzed using Ingenuity Pathway Analysis. (B and C) Validations of the synthetic lethality hits, cells were transfected with non-targeting control siRNAs and siRNAs from Dharmacon. Forty-eight hours after transfection, cells were treated with vehicle (DMSO) or MK2206 at a final concentration of 1 μM for another 48 hours. Cell proliferation was analyzed using an SRB colorimetric assay. Values are the mean ± SD of 3 assays, *, P < 0.05.
TXNRD1 knockdown sensitized cells to MK2206 treatment by inducing cell apoptosis
To determine whether TXNRD1 expression levels correlate with the sensitivity to MK2206, we examined the TXNRD1 and P-AKT expression in 4 MK2206 sensitive and 10 resistant NSCLC cell lines based on our previous published data (3). We found that the expression levels of TXNRD1 are low in all 4 MK2206 sensitive cell lines, whereas the expressions of TXNRD1 are significantly higher in 7 out 10 resistant cell lines (P<0.01), and there is a correlation of TXNRD1 levels with MK2206 resistance (r=0.66 (Fig. 2A). Knockdown of TXNRD1 with three different siRNAs significantly sensitized the HCC193 and H1993 cells (Figs. 2B and 2C). Cell apoptosis analysis by flow cytometry confirmed that TXNRD1 knockdown combined with MK2206 induced 10%-30% apoptosis in HCC193 and H1993 cells within 48 hours treatment, whereas no apoptosis was induced in the cells treated with MK2206 or siRNA only (Fig. 2D). PARP cleavage, an apoptosis marker was also observed in HCC193 and H1993 cells treated with TXNRD1 siRNA and MK2206 (Fig. 2E). Those results indicate that TXNRD1 knockdown sensitizes NSCLC cells to AKT inhibition by inducing cell apoptosis.
Figure 2. TXNRD1 knockdown sensitized cells to MK2206 and induced cell apoptosis.

(A) TXNRD1 expression level correlates with sensitivity to MK2206. Western blotting was performed to examine TXNRD1 and p-AKT levels in 4 MK2206 sensitive and 10 resistant cell lines (left); relative expression levels of TXNRD1 were calculated by normalization with loading control (right). (B) Knockdown of TXNRD1 with 3 individual siRNAs targeting TXNRD1, siRNA-T1, siRNA-T2, siRNA-T3 sensitized the cells to MK2206 treatment compared with 2 non-targeting siRNA controls, siRNA-C1 and siRNA-C2. (C) Western blotting analysis was performed to confirm the TXNRD1 knockdown. (D) Fluorescence-activated sell sorting was performed to analyze cell apoptosis in the cells treated with TXNRD1 siRNA and different doses of MK2206 as indicated. (E) Western blotting analysis was performed to examine PARP cleavage and p-AKT inhibition in the cells treated with TXNRD1 siRNA and different doses of MK2206.
TXNRD1 inhibition with Auranofin sensitized cells to MK2206 treatment in vitro and in vivo
To develop a treatment strategy that would be useful clinically, we tested the combination of MK2206 and Auranofin, a TXNRD1 inhibitor that is used for rheumatoid arthritis treatment. Inhibition of TXNRD1 by different doses of Auranofin was firstly confirmed in HCC193 cells. Approximately 80% percent of TXNRD1 activity was inhibited by 0.1 μM Auranofin (Fig. 3A), which is consistent with a previous report (33). At 0.1 μM, Auranofin did not affect the viability of HCC193 and H1993 cells as was shown in Fig. 3B. We used a dose of 0.1 μM which can inhibit TXNRD1 activity but not the cell proliferation for the combination with MK206. Combination of MK2206 (1 μM or 3 μM) and 0.1 μM Auranofin induced significant growth inhibition in HCC193 and H1993 cells, P<0.01 (Fig. 3C). Liquid colonogenic assay further confirmed that combination of 0.1 μM Auranofin and MK2206 significantly reduced the colony formation in HCC193 and H1993 cells more than 80% compared with vehicle or each single treatment alone, (P<0.01) (Figs. 3D and 3E).
Figure 3. MK2206 and Auranofin inhibited cancer cell growth in vitro and in vivo.
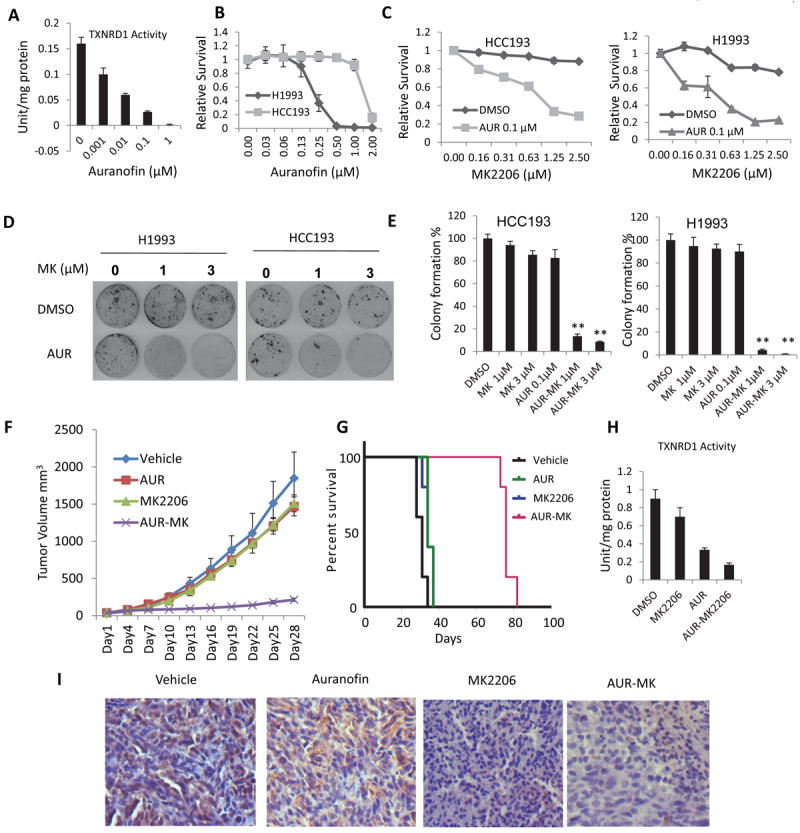
(A) Inhibition of TXNRD1 activity by Auranofin. (B) Dose responses of HCC193 and H1993 cells to Auranofin. (C) Auranofin sensitized the HCC193 (left) and H1993 (right) cells to MK2206. Values are the mean ± SD of 3 assays, **, P < 0.01. (D) Combination of Auranofin and MK2206 inhibited colony formation of HCC193 (left) and H1993 (right) cells. Cells (2000) were plated in 60 mm dishes and treated with DMSO or 0.1 μM Auranofin and different doses of MK2206 as indicated for 7 days. Cell colonies were stained with crystal violet and counted. (E) Relative colonies formation was calculated by normalization with control (DMSO) treatment as 100%. Values are the mean ± SD of 3 assays, **, P < 0.01. (F) Combination of Auranofin and MK2206 inhibited tumor growth in vivo. Values are the mean ± SD, n=5. AUR-MK vs vehicle: P=0.009; AUR-MK vs AUR: P=0.009; AUR-MK vs MK: P=0.009. (G) Combination of Auranofin and MK2206 prolonged animal survival compared with vehicle, Auranofin or MK2206 alone, P<0.001. (H) Auranofin inhibited TXNRD1 activity in vivo. Fresh tumor samples were obtained after 7 days of treatment, and tumor tissues were minced and assayed for TXNRD1 activity. (I) Inhibition of p-AKT in vivo, formalin-fixed, paraffin-embedded tissue sections of animal tumor tissue specimens were used for immunostaining with antibodies against p-AKT (Y473).
To determine the synthetic lethality induced by MK2206 and Auranofin in vivo, xenograft tumors were established with H1993 cells. Single drug treatment with MK2206 or Auranofin had no effect on tumor growth, but the combination significantly inhibited tumor growth by 85% in reduction of tumor volumes compared with each drug alone and 90% compared with vehicle treatment (P=0.009 in all three comparisons). Tumor growth inhibition by Auranofin or MK2206 alone was significant compared with vehicle only at a late time point (day 28), (AUR vs vehicle, P=0.023; MK vs vehicle, P=0.047), but the tumor volume reduction was very minimal (approximately 5%, Fig. 3F). This combination also significantly prolonged animal survival (median 76 days) compared with vehicle (median 31 days) or single drug treatment (median 34 days for both single treatments), P<0.001) (Fig. 3G). Significant Inhibition of TXNRD1 by Auranofin and Auranofin and MK2206 combination treatment was confirmed in animal tumor tissues (Fig. 3H). Immunohistochemical staining indicated that p-AKT was inhibited in the tumor tissue treated with MK2206 or the combination (Fig. 3I). These results indicate that the combination of MK2206 and Auranofin induces synthetic lethality in vivo.
Combination of Auranofin and MK2206 induced ROS production, which mediated cell apoptosis
Because TXNRD1/TNX is one of the major cellular redox regulatory systems that regulate cellular reactive oxygen species (ROS), we hypothesized that combination of Auranofin and MK2206 may disrupt the cellular ROS balance. Indeed, we found that MK2206 alone induced ROS production in HCC193 and H1993 cells by approximately 2 folds (Fig. 4A). The combination of Auranofin and MK2206 induced robust ROS production by 5 folds (P<0.05) (Fig. 4B). To determine whether ROS induction is essential for cell apoptosis induced by MK2206 and Auranofin, we treated cells with the ROS blocker nordihydroguaiaretic acid (NDGA) (20 μM). NDGA blocked the ROS production in HCC193 and H1993 cells induced by the combination (Fig. 4C), and reversed cell growth inhibition in both HCC193 and H1993 cells (Fig. 4D). High level of ROS can activate JNK pathway which mediated cell apoptosis (34). Indeed, we observed that combination of MK2206 and Auranofin induced activation and PARP cleavage in H1993 and HCC193 cells (Fig. 4E). In addition, JNK activation and PARP cleavage were blocked by ROS inhibitor NDGA (Fig. 4F). Those results indicated that combination of MK2206 and Auranofin induced ROS production, which mediated the cell apoptosis.
Figure 4. Auranofin and MK2206-induced cell apoptosis was mediated by ROS induction and JNK activation.

(A) MK2206 induced ROS production in H1993 and HCC193 cells. Cells were treated with 1 μM MK2206 for different time periods, and ROS levels were analyzed by staining with H2DCFDA. (B) Combination of Auranofin and MK2206 induced dramatic ROS production. Values are the mean ± SD of 3 assays. *, P < 0.05, **, P<0.01. (C) NDGA blocked ROS production mediated by Auranofin-MK2206 combination. Values are mean ± SD of 3 assays. *, P < 0.05, **, P<0.01. (D) Blocking ROS reduced proliferation inhibition induced by Auranofin and MK2206 combination in HCC193 and H1993 cells. **, P<0.01. (E) Combination of Auranofin and MK2206 induced JNK activation and PARP cleavage in H1993 and HCC193 cells. (F) Blocking ROS inhibited JNK activation and PARP cleavage induced by Auranofin and MK2206 combination. Cells were treated with DMSO or 0.1 μM Auranofin and different doses of MK2206, as indicated, for 48 hours, Cell lysates were collected for Western blotting analysis.
Synthetic lethality induced by Auranofin and MK2206 is through the KEAP1/NRF2 pathway
In addition to TXNRD1, KEAP1/NRF2 is another major cellular antioxidant system. Activated NRF2 translocates to the nucleus and stimulates gene expression, such as glutamate--cysteine ligase catalytic subunit (GCLC) and NAD(P)H dehydrogenase, quinone1 (NQO1) which mediate cellular antioxidant responses (35-37). To determine whether AKT inhibition induced ROS production was due to NRF2 inhibition, subcellular localization of NRF2 was examined using immunofluorescence staining. The results showed NRF2 was excluded from the nuclei in the H1993 cells treated with 1 μM MK2206 (Fig. 5A). Luciferase reporter assay confirmed that MK22006 inhibited GCLC promoter activity in HCC193 and H1993 cells (Fig. 5B) indicating that the transcriptional activity of NRF2 was inhibited by MK2206. Similarly, a dominant negative form of AKT (AKT-DN) inhibited the transcriptional activity of NRF2 in HCC193 and H1993 cells in a luciferase reporter assay (Figs. 5C and 5D), and reduced NRF2 in the nuclear fractions of H1993 cells (Fig. 5E). Down-regulation of GCLC mRNA was also observed in both HCC193 and H1993 cells with a time point dependent manner (Fig. 5F). Because GCLC is a rate-limiting molecule for GSH synthesis and GSH is the major ROS scavenger, we further determined GSH production in cells treated with MK2206. The GSH level was dramatically reduced in cells treated with MK2206 or combination in HCC193 and H1993 cells (Fig. 5G). These results indicate that MK2206 inhibited NRF2 transcriptional activity and its downstream molecules, such as GCLC, which subsequently reduced the GSH level and resulted in ROS induction.
Figure 5. MK2206 inhibited KEAP1/NRF2 pathway.
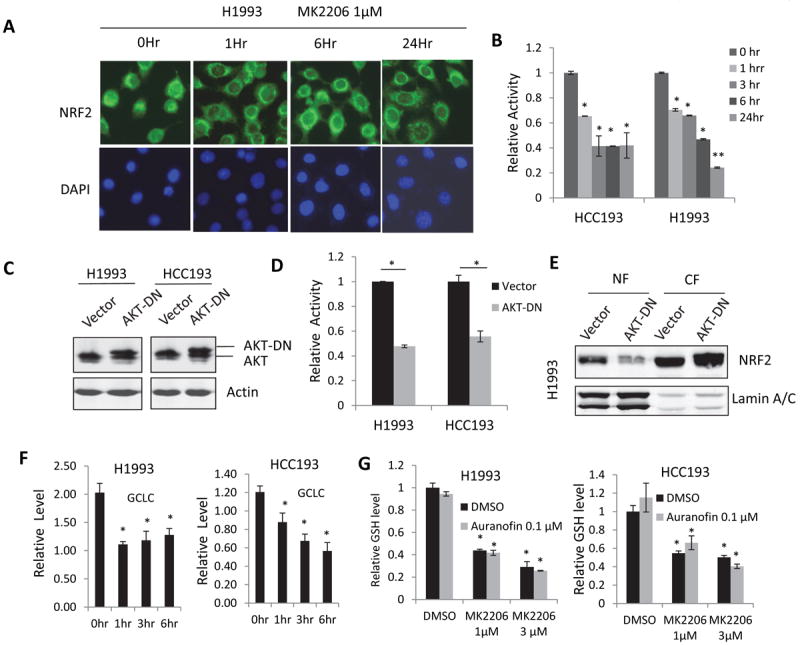
(A) MK2206 excluded NRF2 nuclear localization. H1993 cells were treated with 1 μM MK2206 for the indicated time periods and fixed and stained with NRF2 antibody and FITC-conjugated secondary antibody. (B) MK2206 inhibited NRF2 transcriptional activity. (C) Western blotting confirmed the overexpression of dominant negative AKT (AKT-DN) in H1993 and HCC193 cells. (D) Dominant negative AKT inhibited NRF2 transcriptional activity. (E) Overexpression of dominant negative AKT reduced nuclear NRF2 in H1993 cells. (F) MK2206 reduced GCLC mRNA in H1993 and HCC193 cells. Real-time PCR was performed to determine GCLC expression in cells treated with 1 μM MK2206 for the indicated time periods. (G) Total GSH level was reduced by MK2206 or Auranofin-MK2206 combination. HCC193 and H1993 cells were treated Auranofin, with or without different doses of MK2206, for 24 hours. GSH levels were analyzed using the GSH kit. All values are the mean ± SD of 3 assays, *, P < 0.05.
Synthetic lethality induced by MK2206 and Auranofin depends on the genetic status of KEAP1
The above results indicate that the synthetic lethality induced by MK2206 and Auranofin is mediated by ROS through concurrent inhibition of NRF2 and TXNRD1, two major cellular redox regulators. Because KEAP1 mutations cause constitutive activation of NRF2 (38, 39), which we expect will not be inhibited by MK2206 and thereby cause resistance to MK2206 and Auranofin combination. To test this hypothesis, we tested 5 additional KEAP1 wild type cell lines (H1299, H23, H358, H292, and H1703), 5 KEAP1 inactive mutant cell lines (H460, A549, H2126, H1648, and H838), and 2 heterozygous mutant cell lines (H1993 and H1395) for their sensitivity to treatment with MK2206, Auranofin, or both (18, 19). All KEAP1 wild-type cell lines and heterozygous mutant cell lines were sensitive to MK2206-Auranofin combination based on the IC50s of MK2206 which are bellow or close to 1 μM, a clinical achievable dose (Fig. 6A and supplementary Table 1). In contrast, all KEAP1 mutant cells were not responsive to the combination treatment (IC50s of MK2206 are more than the 3 μM) (Fig. 6B and Table 2). Statistical analysis revealed that the cell lines’ sensitivity to the combination of MK2206 and Auranofin was significantly correlated with the KEAP1 genetic status (P=0.0013); no correlation was found between cell lines’ sensitivity and other mutations, including KRAS and LKB1 mutations (Table 1). Combination of Auranofin and MK2206 induced PARP cleavage in KEAP1 wild type H1299 but not in KEAP1 mutant H460 cell lines (Fig. 6C). Our results indicate that synthetic lethality induced by MK2206 and Auranofin depends on the genetic status of KEAP1. Using an EGFP-NRF2 fusion protein, we confirmed that MK2206 induced NRF2 nuclear exclusion in KEAP1 wild-type H1299 cells but not in KEAP1 mutant H460 cells (Fig. 6D). Moreover, MK2206 could not inhibit NRF2 transcriptional activity by MK2206 in KEAP1 mutant H460 and A549 cells in a Luciferase reporter assay (Fig. 6E). These results indicate that constitutive activation of NRF2 due to inactive KEAP1 mutations cannot be overcome by MK2206 thus causing resistance to the Auranofin and MK2206 combination. To further confirm this, KEAP1was stably knocked down in KEAP1 wild-type H1299 cells (Fig. 6F, left). KEAP1 knockdown rendered cells resistance to the combination treatment (Fig. 6F, middle and right). Whereas overexpression of wild-type KEAP1 (32), DsRed-KEAP1 (D-KEAP1) fusion gene in KEAP1 mutant H460 cells sensitized the cells to this combination (Fig. 6G). The apoptosis maker, PARP cleavage was also induced by the combination treatment in H460 cells with overexpression of a wild type KEAP (S-Figure 1). These results confirmed that synthetic lethality induced by MK2206 and Auranofin depends on a functional KEAP1 gene. Collectively our results indicate that combination of Auranofin and MK2206 concurrently inhibits TXNRD1 and NRF2, two major cellular antioxidant systems in KEAP1 wild type NSCLC cells, and induces ROS production and cell apoptosis. A model of synthetic lethality induced by TXNRD1 and AKT inhibitor was shown in Figure 6H.
Figure 6. Synthetic lethality induced by Auranofin and MK2206 depends on KEAP1 genetic status.
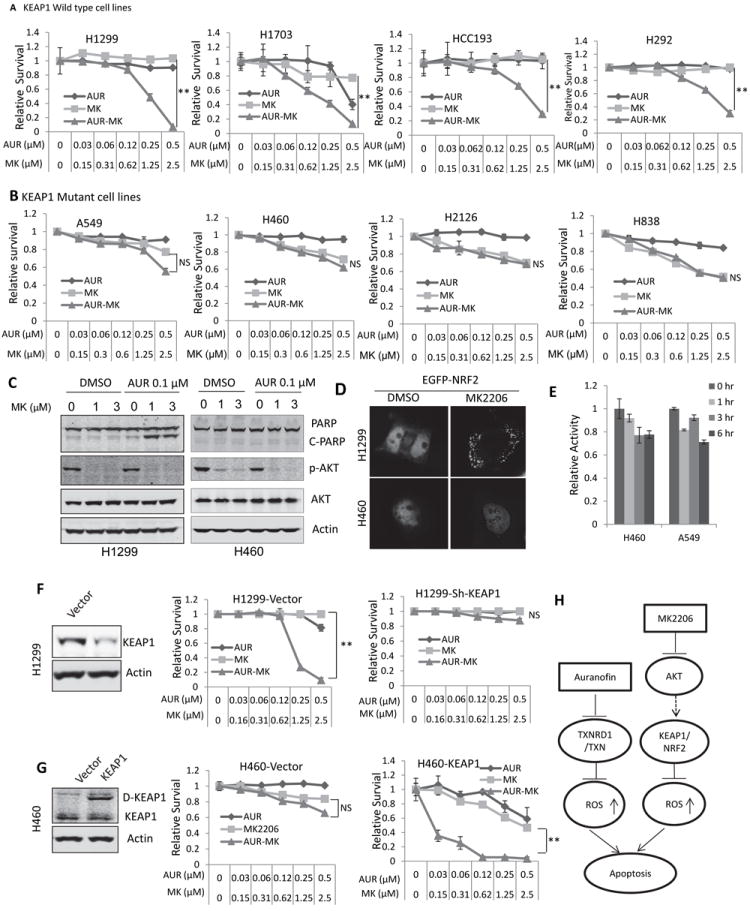
(A) Combination of Auranofin and MK2206 induced synthetic lethality in KEAP1 wild type NSCLC cell lines. (B) Combination of Auranofin and MK2206 did not induce synthetic lethality in KEAP1 mutant NSCLC cell lines. (C) Combination of Auranofin and MK2206 induced PARP cleavage, an apoptosis marker, in KEAP1 wild type H1299 but not KEAP1 mutant H460 cells. (D) MK2206 induced NRF2 nuclear exclusion in H1299 (KEAP1 wild type) but not H460 (KEAP1 mutant) cells. (E) MK2206 could not inhibit NRF2 transcriptional activity in KEAP1 mutant H460 and A549 cells.(F) KEAP1 knockdown in H1299 cells was validated using Western blotting (left); knockdown of KEAP1 rendered H1299 cells resistance to Auranofin and MK2206 combination (middle and right). (G) Overexpression of DsRed-KEAP1 (D-KEAP1) in H460 cells was confirmed by Western blotting (left); Overexpression of functional KEAP1 sensitized the cells to MK2206 and Auranofin combination (middle and right). All values are the mean ± SD of 3 assays, **, P < 0.01. (H) Proposed working model of synthetic lethality induced by Auranofin and MK2206.
Table 1.
Mutation statuses of KEAP1 correlate with the sensitivity to MK2206 and Auranofin combination.
| Cell line | KEAP1 | KRAS/NRAS | LKB1 | EGFR | P53 | IC50 of MK2206 | Sensitivity
|
|---|---|---|---|---|---|---|---|
| H460 | Mut | Mut | Mut | WT | WT | 4.04 | Resistant |
| A549 | Mut | Mut | Mut | WT | WT | 4.35 | Resistant |
| H838 | Mut | WT | DL | WT | Mut | 3.70 | Resistant |
| H2126 | Mut | WT | Mut | WT | Mut | 6.19 | Resistant |
| H1648 | Mut | WT | WT | WT | Mut | 4.96 | Resistant |
| H1395 | WT/mut | WT | DL | WT | WT | 0.59 | Sensitive |
| H1993 | WT/mut | WT | Mut | WT | Mut | 0.81 | Sensitive |
| H1299 | WT | Mut | WT | WT | WT | 1.38 | Sensitive |
| H1703 | WT | WT | WT | WT | Mut | 1.26 | Sensitive |
| H292 | WT | WT | WT | WT | WT | 0.76 | Sensitive |
| H23 | WT | Mut | Mut | WT | Mut | 0.29 | Sensitive |
| H358 | WT | Mut | WT | WT | WT | 1.33 | Sensitive |
| P | 0.0013 | 1.0 | 0.293 | 1.0 |
Note: Fisher’s exact test was used to determine the significant correlation between mutations status of KEAP1 gene and cell lines’ sensitivity to Auranofin and MK2206 combination. P<0.05 indicates significance.
DISCUSSION
In this study we performed a genome-wide siRNA library screen with NSCLC cell line HCC193 to identify the genes that can be targeted to sensitize the cells to AKT inhibitor MK2206. This screening revealed 156 MK2206 synthetic lethality hits (genes) with a false discover rate (FDR) of 0.1, and a minimum 50% inhibition with combination of siRNA and 1 μM MK2206. Those hits include previously reported autophagy-related genes, such as MAP1LC3C, cathepsin E, and cathepsin H, and components in the NF-κB pathway, such as CASP8 and CFLAR. Inhibition of autophagy could sensitize the cells to PI3K/AKT inhibitor treatment, which has been reported previously (40, 41). Other hits include ATPases and ATP synthase members, such as ATP5J and ATP6V1B1, both of which are H+ transporters. The synthetic lethality interaction between AKT and ATPases may occur through the autophagy pathway since previous studies reported that the vacuolar membrane H+-ATPase plays an important role in autophagy (42, 43). The synthetic lethality hits are related to different pathways, and many of those hits or pathways have crosstalk (44, 45). This screening result confirmed some previous finding such as synthetic lethality induced by autophagy and AKT inhibition but also revealed some novel targets, such as TXNRD1, the molecules in redox balance, suggesting potential synthetic lethal interactions between the redox regulation system and AKT pathways.
Previous studies indicate that other pathways, such as KEAP1/NRF2, may compensate some functions of the TXNRD1/TXN in cancer cells especially in vivo. In addition, oncogenic pathways such as KRAS and NF-KB also regulate the cellular redox balance through interaction with TXNRD1 or KEAP1/NRF2 (46-49), suggesting complicated interaction between oncogenic signaling pathways and cellular redox balance system. Here we showed that concurrently inhibited TXNRD1 and AKT pathways with Auranofin and MK2206 respectively induced robust ROS production and NSCLC cell apoptosis in vitro and tumor growth inhibition in vivo, which was not achieved by each drug alone. More importantly, NDGA, a ROS blocker, rescued the cell growth inhibition, JNK activation and PARP cleavage induced by the Auranofin and MK2206 combination. Our results indicate that interruption of the cellular redox balance plays an important role in the synthetic lethality induced by simultaneous inhibition of TXNRD1 and AKT. We found that MK2206 inhibited the NRF2 activity and reduced the GSH level through down-regulating GCLC expression, which is consistent with a recent study that inhibition of mammalian target of rapamycin (mTOR) reduced GSH level and induced ROS in KRAS mutant lung cancer (50). Since mTOR pathway is regulated by AKT, it is also possible that MK2206 induced ROS production may be mediated by both NRF2 and mTOR inhibition. Nevertheless, our results indicate that targeting the interactions between the PI3K/AKT oncogenic pathway and cellular antioxidant system, TXNRD1 and KEAP1/NRF2, will disrupt cancer redox homeostasis and might be a promising cancer therapeutic approach.
Loss of function mutations of KEAP1 occur in many cancer types and KEAP1 mutations cause constitutive activation of NRF2 (18-20). Because the combination of Auranofin and MK2206 induced synthetic lethality by simultaneous inhibition of the TXNRD1 and NRF2 pathways, we hypothesized that KEAP1 mutations might render cancer cells resistant to the combination of MK2006 and Auranofin. Our results showed that lung cancer cells with wild-type KEAP1 were particularly sensitive to MK2206 and Auranofin, whereas cells with mutant KEAP1 were not as sensitive. This finding was further supported by evidence that MK2206 induced NRF2 nuclear exclusion and transcriptional activity in KEAP1 wild-type cells but not in KEAP1 mutant cells. In addition, KEAP1 knockdown in KEAP1 wild-type cells or re-expression of functional KEAP1 in KEAP1 mutant cells reversed the sensitivity to Auranofin and MK2206 treatment. These findings indicate that the synthetically lethal activity induced by MK2206 and Auranofin depends on a functional KEAP1 gene. Further study of the genetic mutations in this pathway and their association with drug sensitivity may be useful for the identification of predictive biomarkers.
In summary, we identified a set of synthetic loci that could be targeted to improve the antitumor activity of AKT inhibitor MK2206. We discovered that the synthetic lethal interaction between the AKT pathway and TXNRD1 occurs through the KEAP1/NRF2 antioxidant system, and targeting such interaction with a combination of AKT inhibitor and Auranofin is a promising approach for lung cancer therapy, with KEAP1 mutation status as a genetic biomarker of drug sensitivity.
Supplementary Material
Acknowledgments
We thank Dr. Donna Zhang, Arizona University, Tucson, Arizona, for providing the ARE-LUC plasmid, Dr. Yue Xiong, University of North Carolina at Chapel Hill, North Carolina, for providing DesRed-KEAP1 and EGFP-NRF2 plasmids. We thank Drs. Michael Peyton, Wei Guo, Xiaoying Liu and Kai Xu for technique help. We thank Ann M Sutton for scientific editing.
Grant Support.
This work was supported in part by the National Institutes of Health through MD Anderson Cancer Center Core Grant CA-016672 - Lung Program, the Research Animal Support Facility, NIH/NCI Specialized Program of Research Excellence (SPORE) grant CA-070907 (to J. Minna and J. Roth), SPORE Development Award (to B. Dai) and NIH/NCI R01 grant CA-124951 (to B. Fang).
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Hennessy BT, Smith DL, Ram PT, Lu Y, Mills GB. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov. 2005;4:988–1004. doi: 10.1038/nrd1902. [DOI] [PubMed] [Google Scholar]
- 2.Yap TA, Yan L, Patnaik A, Fearen I, Olmos D, Papadopoulos K, et al. First-in-man clinical trial of the oral pan-AKT inhibitor MK-2206 in patients with advanced solid tumors. J Clin Oncol. 2011;29:4688–95. doi: 10.1200/JCO.2011.35.5263. [DOI] [PubMed] [Google Scholar]
- 3.Meng J, Dai B, Fang B, Bekele BN, Bornmann WG, Sun D, et al. Combination treatment with MEK and AKT inhibitors is more effective than each drug alone in human non-small cell lung cancer in vitro and in vivo. PLoS One. 2010;5:e14124. doi: 10.1371/journal.pone.0014124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jiang J, Mo ZC, Yin K, Zhao GJ, Lv YC, Ouyang XP, et al. Epigallocatechin-3-gallate prevents TNF-alpha-induced NF-kappaB activation thereby upregulating ABCA1 via the Nrf2/Keap1 pathway in macrophage foam cells. Int J Mol Med. 2012;29:946–56. doi: 10.3892/ijmm.2012.924. [DOI] [PubMed] [Google Scholar]
- 5.Powis G, Kirkpatrick DL. Thioredoxin signaling as a target for cancer therapy. Curr Opin Pharmacol. 2007;7:392–7. doi: 10.1016/j.coph.2007.04.003. [DOI] [PubMed] [Google Scholar]
- 6.Cox AG, Brown KK, Arner ES, Hampton MB. The thioredoxin reductase inhibitor auranofin triggers apoptosis through a Bax/Bak-dependent process that involves peroxiredoxin 3 oxidation. Biochem Pharmacol. 2008;76:1097–109. doi: 10.1016/j.bcp.2008.08.021. [DOI] [PubMed] [Google Scholar]
- 7.Hedstrom E, Eriksson S, Zawacka-Pankau J, Arner ES, Selivanova G. p53-dependent inhibition of TrxR1 contributes to the tumor-specific induction of apoptosis by RITA. Cell Cycle. 2009;8:3576–83. doi: 10.4161/cc.8.21.9977. [DOI] [PubMed] [Google Scholar]
- 8.Madeira JM, Gibson DL, Kean WF, Klegeris A. The biological activity of auranofin: implications for novel treatment of diseases. Inflammopharmacology. 2012;20:297–306. doi: 10.1007/s10787-012-0149-1. [DOI] [PubMed] [Google Scholar]
- 9.Marzano C, Gandin V, Folda A, Scutari G, Bindoli A, Rigobello MP. Inhibition of thioredoxin reductase by auranofin induces apoptosis in cisplatin-resistant human ovarian cancer cells. Free Radic Biol Med. 2007;42:872–81. doi: 10.1016/j.freeradbiomed.2006.12.021. [DOI] [PubMed] [Google Scholar]
- 10.Mirabelli CK, Johnson RK, Sung CM, Faucette L, Muirhead K, Crooke ST. Evaluation of the in vivo antitumor activity and in vitro cytotoxic properties of auranofin, a coordinated gold compound, in murine tumor models. Cancer Res. 1985;45:32–9. [PubMed] [Google Scholar]
- 11.Weir SJ, DeGennaro LJ, Austin CP. Repurposing approved and abandoned drugs for the treatment and prevention of cancer through public-private partnership. Cancer Res. 2012;72:1055–8. doi: 10.1158/0008-5472.CAN-11-3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Debnath A, Parsonage D, Andrade RM, He C, Cobo ER, Hirata K, et al. A high-throughput drug screen for Entamoeba histolytica identifies a new lead and target. Nat Med. 18:956–60. doi: 10.1038/nm.2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ramanathan RK, Stephenson JJ, Weiss GJ, Pestano LA, Lowe A, Hiscox A, et al. A phase I trial of PX-12, a small-molecule inhibitor of thioredoxin-1, administered as a 72-hour infusion every 21 days in patients with advanced cancers refractory to standard therapy. Invest New Drugs. 2012;30:1591–6. doi: 10.1007/s10637-011-9739-9. [DOI] [PubMed] [Google Scholar]
- 14.Jordan BF, Runquist M, Raghunand N, Gillies RJ, Tate WR, Powis G, et al. The thioredoxin-1 inhibitor 1-methylpropyl 2-imidazolyl disulfide (PX-12) decreases vascular permeability in tumor xenografts monitored by dynamic contrast enhanced magnetic resonance imaging. Clin Cancer Res. 2005;11:529–36. [PubMed] [Google Scholar]
- 15.Kobayashi A, Kang MI, Okawa H, Ohtsuji M, Zenke Y, Chiba T, et al. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol. 2004;24:7130–9. doi: 10.1128/MCB.24.16.7130-7139.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sykiotis GP, Bohmann D. Keap1/Nrf2 signaling regulates oxidative stress tolerance and lifespan in Drosophila. Dev Cell. 2008;14:76–85. doi: 10.1016/j.devcel.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sekhar KR, Crooks PA, Sonar VN, Friedman DB, Chan JY, Meredith MJ, et al. NADPH oxidase activity is essential for Keap1/Nrf2-mediated induction of GCLC in response to 2-indol-3-yl-methylenequinuclidin-3-ols. Cancer Res. 2003;63:5636–45. [PubMed] [Google Scholar]
- 18.Ohta T, Iijima K, Miyamoto M, Nakahara I, Tanaka H, Ohtsuji M, et al. Loss of Keap1 function activates Nrf2 and provides advantages for lung cancer cell growth. Cancer Res. 2008;68:1303–9. doi: 10.1158/0008-5472.CAN-07-5003. [DOI] [PubMed] [Google Scholar]
- 19.Singh A, Misra V, Thimmulappa RK, Lee H, Ames S, Hoque MO, et al. Dysfunctional KEAP1-NRF2 interaction in non-small-cell lung cancer. PLoS Med. 2006;3:e420. doi: 10.1371/journal.pmed.0030420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hammerman PS, Hayes DN, Wilkerson MD, Schultz N, Bose R, Chu A, et al. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489:519–25. doi: 10.1038/nature11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shibata T, Saito S, Kokubu A, Suzuki T, Yamamoto M, Hirohashi S. Global downstream pathway analysis reveals a dependence of oncogenic NF-E2-related factor 2 mutation on the mTOR growth signaling pathway. Cancer Res. 70:9095–105. doi: 10.1158/0008-5472.CAN-10-0384. [DOI] [PubMed] [Google Scholar]
- 22.Zhang P, Singh A, Yegnasubramanian S, Esopi D, Kombairaju P, Bodas M, et al. Loss of Kelch-like ECH-associated protein 1 function in prostate cancer cells causes chemoresistance and radioresistance and promotes tumor growth. Mol Cancer Ther. 9:336–46. doi: 10.1158/1535-7163.MCT-09-0589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shibata T, Ohta T, Tong KI, Kokubu A, Odogawa R, Tsuta K, et al. Cancer related mutations in NRF2 impair its recognition by Keap1-Cul3 E3 ligase and promote malignancy. Proc Natl Acad Sci U S A. 2008;105:13568–73. doi: 10.1073/pnas.0806268105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Astsaturov I, Ratushny V, Sukhanova A, Einarson MB, Bagnyukova T, Zhou Y, et al. Synthetic lethal screen of an EGFR-centered network to improve targeted therapies. Sci Signal. 2010;3:ra67. doi: 10.1126/scisignal.2001083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Luo J, Emanuele MJ, Li D, Creighton CJ, Schlabach MR, Westbrook TF, et al. A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell. 2009;137:835–48. doi: 10.1016/j.cell.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Turner NC, Lord CJ, Iorns E, Brough R, Swift S, Elliott R, et al. A synthetic lethal siRNA screen identifying genes mediating sensitivity to a PARP inhibitor. EMBO J. 2008;27:1368–77. doi: 10.1038/emboj.2008.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Whitehurst AW, Bodemann BO, Cardenas J, Ferguson D, Girard L, Peyton M, et al. Synthetic lethal screen identification of chemosensitizer loci in cancer cells. Nature. 2007;446:815–9. doi: 10.1038/nature05697. [DOI] [PubMed] [Google Scholar]
- 28.Dai B, Meng J, Peyton M, Girard L, Bornmann WG, Ji L, et al. STAT3 mediates resistance to MEK inhibitor through microRNA miR-17. Cancer Res. 2011;71:3658–68. doi: 10.1158/0008-5472.CAN-10-3647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 30.Wei X, Guo W, Wu S, Wang L, Huang P, Liu J, et al. Oxidative stress in NSC-741909-induced apoptosis of cancer cells. J Transl Med. 2010;8:37. doi: 10.1186/1479-5876-8-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Na HK, Kim EH, Jung JH, Lee HH, Hyun JW, Surh YJ. (-)-Epigallocatechin gallate induces Nrf2-mediated antioxidant enzyme expression via activation of PI3K and ERK in human mammary epithelial cells. Arch Biochem Biophys. 2008;476:171–7. doi: 10.1016/j.abb.2008.04.003. [DOI] [PubMed] [Google Scholar]
- 32.Furukawa M, Xiong Y. BTB protein Keap1 targets antioxidant transcription factor Nrf2 for ubiquitination by the Cullin 3-Roc1 ligase. Mol Cell Biol. 2005;25:162–71. doi: 10.1128/MCB.25.1.162-171.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Omata Y, Folan M, Shaw M, Messer RL, Lockwood PE, Hobbs D, et al. Sublethal concentrations of diverse gold compounds inhibit mammalian cytosolic thioredoxin reductase (TrxR1) Toxicol In Vitro. 2006;20:882–90. doi: 10.1016/j.tiv.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 34.Pham CG, Bubici C, Zazzeroni F, Papa S, Jones J, Alvarez K, et al. Ferritin heavy chain upregulation by NF-kappaB inhibits TNFalpha-induced apoptosis by suppressing reactive oxygen species. Cell. 2004;119:529–42. doi: 10.1016/j.cell.2004.10.017. [DOI] [PubMed] [Google Scholar]
- 35.Kobayashi A, Ohta T, Yamamoto M. Unique function of the Nrf2-Keap1 pathway in the inducible expression of antioxidant and detoxifying enzymes. Methods Enzymol. 2004;378:273–86. doi: 10.1016/S0076-6879(04)78021-0. [DOI] [PubMed] [Google Scholar]
- 36.Ishii T, Itoh K, Yamamoto M. Roles of Nrf2 in activation of antioxidant enzyme genes via antioxidant responsive elements. Methods Enzymol. 2002;348:182–90. doi: 10.1016/s0076-6879(02)48637-5. [DOI] [PubMed] [Google Scholar]
- 37.Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun. 1997;236:313–22. doi: 10.1006/bbrc.1997.6943. [DOI] [PubMed] [Google Scholar]
- 38.Nioi P, Nguyen T. A mutation of Keap1 found in breast cancer impairs its ability to repress Nrf2 activity. Biochem Biophys Res Commun. 2007;362:816–21. doi: 10.1016/j.bbrc.2007.08.051. [DOI] [PubMed] [Google Scholar]
- 39.Wakabayashi N, Itoh K, Wakabayashi J, Motohashi H, Noda S, Takahashi S, et al. Keap1-null mutation leads to postnatal lethality due to constitutive Nrf2 activation. Nat Genet. 2003;35:238–45. doi: 10.1038/ng1248. [DOI] [PubMed] [Google Scholar]
- 40.Fan QW, Cheng C, Hackett C, Feldman M, Houseman BT, Nicolaides T, et al. Akt and autophagy cooperate to promote survival of drug-resistant glioma. Sci Signal. 2010;3:ra81. doi: 10.1126/scisignal.2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Degtyarev M, De Maziere A, Orr C, Lin J, Lee BB, Tien JY, et al. Akt inhibition promotes autophagy and sensitizes PTEN-null tumors to lysosomotropic agents. J Cell Biol. 2008;183:101–16. doi: 10.1083/jcb.200801099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ramachandran N, Munteanu I, Wang P, Aubourg P, Rilstone JJ, Israelian N, et al. VMA21 deficiency causes an autophagic myopathy by compromising V-ATPase activity and lysosomal acidification. Cell. 2009;137:235–46. doi: 10.1016/j.cell.2009.01.054. [DOI] [PubMed] [Google Scholar]
- 43.Meyer H, Bug M, Bremer S. Emerging functions of the VCP/p97 AAA-ATPase in the ubiquitin system. Nat Cell Biol. 2012;14:117–23. doi: 10.1038/ncb2407. [DOI] [PubMed] [Google Scholar]
- 44.Gloire G, Piette J. Redox regulation of nuclear post-translational modifications during NF-kappaB activation. Antioxid Redox Signal. 2009;11:2209–22. doi: 10.1089/ars.2009.2463. [DOI] [PubMed] [Google Scholar]
- 45.Reynaert NL, van der Vliet A, Guala AS, McGovern T, Hristova M, Pantano C, et al. Dynamic redox control of NF-kappaB through glutaredoxin-regulated S-glutathionylation of inhibitory kappaB kinase beta. Proc Natl Acad Sci U S A. 2006;103:13086–91. doi: 10.1073/pnas.0603290103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang HC, Nguyen T, Pickett CB. Phosphorylation of Nrf2 at Ser-40 by protein kinase C regulates antioxidant response element-mediated transcription. J Biol Chem. 2002;277:42769–74. doi: 10.1074/jbc.M206911200. [DOI] [PubMed] [Google Scholar]
- 47.Huang HC, Nguyen T, Pickett CB. Regulation of the antioxidant response element by protein kinase C-mediated phosphorylation of NF-E2-related factor 2. Proc Natl Acad Sci U S A. 2000;97:12475–80. doi: 10.1073/pnas.220418997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thu KL, Pikor LA, Chari R, Wilson IM, Macaulay CE, English JC, et al. Genetic disruption of KEAP1/CUL3 E3 ubiquitin ligase complex components is a key mechanism of NF-kappaB pathway activation in lung cancer. J Thorac Oncol. 2011;6:1521–9. doi: 10.1097/JTO.0b013e3182289479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee DF, Kuo HP, Liu M, Chou CK, Xia W, Du Y, et al. KEAP1 E3 ligase-mediated downregulation of NF-kappaB signaling by targeting IKKbeta. Mol Cell. 2009;36:131–40. doi: 10.1016/j.molcel.2009.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.De Raedt T, Walton Z, Yecies JL, Li D, Chen Y, Malone CF, et al. Exploiting cancer cell vulnerabilities to develop a combination therapy for ras-driven tumors. Cancer Cell. 2011;20:400–13. doi: 10.1016/j.ccr.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.