

Abstract
Interferon beta (IFN-β) is widely used to ameliorate disease progression in patients with Multiple Sclerosis. IFN-β has a short half-life in humans, necessitating frequent administration for optimum effectiveness. Covalent modification of IFN-β with polyethylene glycol (PEG) improves the pharmacokinetic properties of the protein, but can adversely affect the protein's in vitro bioactivity. Random modification of lysine residues in IFN-β with amine-reactive PEGs decreased the in vitro bioactivity of the protein 50-fold, presumably due to modification of lysine residues near critical receptor binding sites. PEGylated IFN-β proteins that retained high in vitro bioactivity could be obtained by selective modification of the N-terminus of the protein with PEG. Here we use site-specific PEGylation technology (targeted attachment of a cysteine-reactive-PEG to an engineered cysteine residue in IFN-β) to identify several additional amino acid positions where PEG can be attached to IFN-β without appreciable loss of in vitro bioactivity. Unexpectedly, we found that most of the PEG-IFN-β analogs showed 11- to 78-fold improved in vitro bioactivities relative to their unPEGylated parent proteins and to IFN-β-1b. In vivo studies showed that a lead PEG-IFN-β protein had improved pharmacokinetic properties compared to IFN-β and was significantly more effective than IFN-β at inhibiting growth of a human tumor xenograft in athymic mice.
Introduction
Interferon beta (IFN-β) is a 20-kDa cytokine that exhibits immunomodulatory, antiviral, and antiproliferative effects on many cell types. IFN-β is used clinically to ameliorate disease progression in patients with multiple sclerosis (Comi 2000; Li and others 2001; Hughes 2003), and has shown promise as a treatment for hepatitis B, hepatitis C, and various cancers (Johns and others 1992; Lindner and Borden 1997; Qin and others 1998; Munoz and others 2002; Chan and others 2007). Recombinant IFN-β-1a (glycosylated) and IFN-β-1b (nonglycosylated) have circulating half-lives of <5 h in humans and serum levels of the proteins are barely detectable following subcutaneous (sc) or intramuscular administration (Goldstein and others 1989; Liberati and others 1992; Salmon and others 1996). Biological responses to IFN-β (intracellular 2′,5′–oligoadenylate synthetase and secreted neopterin) peak 24 h following sc or intramuscular administration and return to baseline by 4 days (Goldstein and others 1989; Salmon and others 1996). Multiple frequent injections (every other day) of IFN-β are required to maintain elevated pharmacodynamic parameters (Witt and others 1993), and seem to improve efficacy in multiple sclerosis patients (Blumhardt 2002). These studies suggest that longer acting forms of IFN-β that provide patients with more constant exposure to the protein may prove more efficacious than existing IFN-β therapies.
The pharmacological properties of many protein therapeutics, including IFN-β, can be improved by modification of the proteins with polymers, such as polyethylene glycol (PEG) (Abuchowski and others 1984; Katre and others 1987; Kinstler and others 1996; Bailon and others 2001; Harris and others 2001; Pepinsky and others 2001; Baker and others 2006; Basu and others 2006; Bell and others 2008). Commonly, proteins are modified with amine-reactive PEGs, which typically attach to lysine residues and the N-terminal amino acid. IFN-β contains 11 lysine residues, in addition to the N-terminal amine. Over half of these lysine residues are located in the alpha helical regions of IFN-β thought to be involved in receptor binding (Senda and others 1995; Karpusas and others 1997). Random modification of IFN-β-1b with amine-reactive 40 kDa-PEGs yields a heterogeneous product mixture with >50-fold reduced in vitro biological activity (Basu and others 2006). Loss of in vitro bioactivity could be largely avoided by selective modification of the N-terminal amino acid of IFN-β-1b with a 40 kDa-PEG (Basu and others 2006). No loss of in vitro bioactivity was observed when the N-terminus of human IFN-β-1a was modified with a 20 kDa-PEG, but a 6-fold loss of bioactivity was observed when the N-terminus was modified with a 30 kDa-PEG and complete loss of in vitro bioactivity was observed when the N-terminus was modified with a 40 kDa-PEG (Pepinsky and others 2001).
In this report, we use a different PEGylation technology, site-specific PEGylation (Goodson and Katre 1990), to modify cysteine analogs of IFN-β-1b with cysteine-reactive maleimide-PEGs. This method permits selective modification of internal amino acids, as well as the N-terminus and C-terminus of IFN-β-1b. We identify several amino acid positions in IFN-β where cysteine residues can be introduced and modified with PEG (PEGylated) without appreciable loss of in vitro bioactivity. Unexpectedly, we find that most of the PEGylated IFN-β-1b cysteine analogs have 11- to 78-fold improved bioactivities compared to their unPEGylated parent proteins and IFN-β-1b in certain in vitro biological assays. In vivo studies showed that a lead PEG IFN-β-1b cysteine analog had improved pharmacokinetic properties compared to IFN-β-1b in rats, and was signficantly more effective than IFN-β-1b at inhibiting growth of a human tumor xenograft in athymic mice.
Materials and Methods
Cloning of IFN-β-1b and construction of IFN-β-1b cysteine analogs
DNA encoding human IFN-β was amplified using the polymerase chain reaction method from human genomic DNA (Clontech Laboratories, Inc., Mountain View, CA). Genes encoding IFN-β-1b, which contains a substitution of serine for cysteine-17 (referred to as C17S), and IFN-β-1b cysteine analogs were created by site-directed polymerase chain reaction-based mutagenesis methods (Higuchi 1990; Horton 1993). Correct DNA sequences for the genes were confirmed. IFN-β-1b and IFN-β-1b cysteine analogs were expressed in Escherichia coli strain W3110 using an E. coli tryptophan operon promoter expression vector that carries an ampicillin resistance gene. The IFN-β-1b (F111C) analog also was expressed in E. coli strain W3110 using phage T5 promoter expression vectors containing ampicillin or tetracycline resistance genes. Growth and expression of the tryptophan promoter strains was performed essentially as described (Porter and others 1986), using media containing 100 μg/mL ampicillin. The T5 promoter strains were grown in Luria Broth media supplemented with 100 μg/mL ampicillin or 10 μg/mL tetracycline and expression of the IFN-β-1b (F111C) protein induced with isopropyl-β-D-thiogalactopyranoside. The induced cells were harvested by centrifugation and the cell pellets stored at −80°C until further processed.
Purification of IFN-β-1b and IFN-β-1b cysteine analogs
The IFN-β-1b proteins were partially purified using a previously described sodium dodecyl sulfate (SDS), butanol extraction, and methanol precipitation method (Hanisch and Fernandes 1984). The precipitated IFN-β-1b proteins from a 1.2 L culture were dissolved in 10 mL of 6 M guanidine, 5 mM cysteine, 20 mM Tris at pH 8.0. Each protein was refolded by dilution into 60 mL of 50% sucrose, 2 mM cystine, 20 mM Tris at pH 8.0. The refold mixture was incubated at 4°C overnight, then diluted into 600 mL of 1 M NaCl, 50 mM sodium phosphate, pH 7.2 (Buffer A) and applied to a 5 mL Blue-Sepharose HiTrap column (GE Healthcare, Piscataway, NJ). Bound proteins were eluted with a step gradient of 100% Buffer B (50% ethylene glycol, 1 M sodium chloride, 50 mM sodium phosphate, pH 7.2). Column fractions were analyzed by nonreducing SDS-polyacrylamide gel electrophoresis (PAGE) and fractions enriched for the refolded IFN-β-1b proteins pooled and stored at −20°C or −80°C. Protein concentrations were determined using a Bradford dye binding assay (Bio-Rad Laboratories, Richmond, CA) or a MicroBCA assay kit (Pierce Chemical Company, Rockford, IL), using bovine serum albumin as a standard.
PEGylation of IFN-β-1b cysteine analogs
Linear 20 kDa- and 30 kDa-maleimide PEGs, and a branched 40 kDa-maleimide-PEG were purchased from NOF Corporation (Irvine, CA). A linear 5 kDa-maleimide PEG was purchased from Nektar, Inc. (Huntsville, AL). The purified proteins were mixed with a 10-fold molar excess of maleimide-PEG, a 10-fold excess of Tris (2-carboxyethylphosphine) hydrochloride, and 2 mM EDTA at pH 8. After 1 h, an additional 10-fold excess of maleimide-PEG was added and the reaction allowed to proceed for an additional hour. The reaction was terminated by diluting the mixture with Buffer A (20 mM Tris, pH 8.0) before loading on a 1 or 5 mL S-Sepharose HiTrap column (GE Healthcare). The column was eluted with a linear gradient of 0%–50% Buffer B (1 M NaCl, 20 mM Tris, pH 8.0). Zwittergent 3–14 (0.05% v/v final concentration) was added to the column buffers in most experiments. Column fractions were analyzed by nonreducing SDS-PAGE and fractions containing monoPEGylated IFN-β proteins were pooled and stored frozen. Concentrations of the PEGylated proteins were determined using the Micro BCA protein assay kit or by UV absorbance. The mass of the PEGylated proteins is stated as the mass of the protein portion only and does not include the mass contributed by the attached PEG moiety.
In vitro bioactivity assays
The human Daudi (catalogue number CCL-213) and NIH:OVCAR-3 (catalogue number HTB-161) cell lines were obtained from the American Type Culture Collection (Manassas, VA). In vitro cell growth inhibition assays using human Daudi cells were performed as described previously (Rosendahl and others 2005a). NIH:OVCAR-3 cells were maintained in cell maintenance media [(alpha minimal essential medium (MEM) supplemented with 10% (v/v) fetal bovine serum, 50 units/mL penicillin, 50 μg/mL streptomycin, and 2 mM glutamine (or 2 mM Glutamax; Invitrogen Corporation, Carlsbad, CA)]. For bioassays, NIH:OVCAR-3 cells were trypsinized, washed, and resuspended at a concentration of 8×104/mL in cell maintenance media. Fifty μL (4×103 cells) of the cell suspension was aliquotted per test well of a flat bottom 96 well tissue culture plate. Serial 3-fold dilutions of the protein samples were prepared in cell maintenance media. Serial dilutions of recombinant human IFN-β-1b (Betaseron®; Bayer Healthcare Pharmaceuticals, Emeryville, CA) were analyzed in parallel. A 40 kDa-PEG was diluted in cell maintenance media and tested like the protein samples to control for potential PEG interference in the assay. Fifty μL of the diluted protein samples were added to the test wells and the plates incubated at 37°C in a humidified 5% CO2 tissue culture incubator. Protein samples were assayed in triplicate wells. After 4 days, 20 μL of CellTiter 96® AQueous One Solution (Promega Corporation, Madison, WI) was added to each well and the plates incubated at 37°C in the tissue culture incubator for 1–4 h. Absorbance of the wells, which is proportional to cell number, was read at 490 nm using a microplate reader. Control wells contained media but no cells. Mean absorbance values for the triplicate control wells were subtracted from mean values obtained for the test wells.
Pharmacokinetic evaluation of IFN-β-1b and 40 kDa-PEG-F111C in rats
Animal experiments were performed with the approval of Premier Laboratory's Institutional Animal Care and Use Committee. Male Sprague Dawley rats (N=3/protein) were obtained from Harlan Sprague Dawley, Inc. (Indianapolis, IN) and weighed ∼350g each. Rats received a single intravenous (iv) or sc injection of the test proteins and Day 0. Blood samples were obtained before drug administration and at various times postinjection for plasma preparation. Plasma levels of IFN-β-1b were quantitated using VeriKine™ Human IFN-β Serum ELISA Kits (PBL InterferonSource, Piscatawy, NJ; catalog number 41415-1). The linear range of this ELISA kit is 2.3 to 150 pg/mL. F111C and PEG-F111C were not detected by this ELISA kit, apparently due to failure of the coating monoclonal antibody on the plate to react with the proteins. Plasma levels of PEG-F111C were measured by incubating plasma samples with an anti-PEG ELISA kit (Epitomics, Inc., Burlingame, CA, catalog number 6100-1) to capture the protein from the plasma samples, followed by detection of the protein using the secondary, wash, and detection reagents from the VeriKine Human IFN-β Serum ELISA kit. Serial dilutions of PEG-F111C (from 0.31 to 15 ng/mL) were used to prepare a standard curve for use in this ELISA. Pharmacokinetic parameters of the test proteins were calculated using the WinNonlin Professional version 5.3 software program (Pharsight, Inc., Mountain View, CA), using noncompartmental methods.
Mouse tumor xenograft study
Female athymic nude (nu/nu) mice, 5 to 6 weeks of age and weighing ∼23 g at study initiation, were obtained from Charles River Laboratories (Wilmington, MA). NIH:OVCAR-3 cells were harvested by trypsinization, washed, and resuspended at a concentration of 2.5×107 cells/mL in a 50:50 mixture of MEM media and Matrigel (BD Biosciences, Bedford, MA). Mice were randomized to different test groups according to body weight on day 0. Each mouse received a single sc injection of 0.2 mL of the MEM media/Matrigel mixture containing 5×106 cells in the axillary area on day 0. Mice (10–11/group) received sc injections (0.2 mL/animal) in the abdomen of vehicle solution (phosphate-buffered saline) or 15 μg/injection of Betaseron or 40 kDa-PEG-F111C beginning on day 1 and continuing through day 69 using a 3 times per week dosing schedule (Monday, Wednesday, Friday). The proteins were formulated in phosphate-buffered saline. Tumors were measured using calipers at 3–4 days intervals for 10 weeks post injection. Tumor volume was determined using a formula of [(width×width)×length)/2]. Differences between groups were compared using a student's t-test, with significance set at P≤0.05.
Results
IFN-β is a globular protein comprised of 5 alpha helical regions (termed A-E from the N-terminus of the protein) joined by nonhelical loops (Karpusas and others 1997). IFN-β contains 3 native cysteine residues at positions 17, 31, and 141. Cysteine-31 and cysteine-141 form a critical disulfide bond required for biological activity (Shepard and others 1981). The unpaired cysteine residue, cysteine-17 (C17), is partially buried in the hydrophobic core of the protein and is located in Helix A, which has been implicated in receptor binding; therefore, C17 is not an optimum position for attachment of a large PEG molecule. We replaced C17 with a serine residue (C17S) using site-directed mutagenesis, creating IFN-β (C17S), which is identical in sequence to IFN-β-1b (Betaseron), one of the forms of IFN-β approved for use in humans (Plosker 2011).
We constructed 17 IFN-β-1b cysteine muteins, each of which contains a single added cysteine residue (Table 1). The cysteine analogs were constructed in an IFN-β-1b background so that the proteins contained only a single unpaired cysteine residue. We constructed 1 cysteine substitution analog in the region preceding helix A (M1C), 1 cysteine substitution analog in the first amino acid position of Helix A (S2C), 1 cysteine substitution at the last amino acid in helix A (W22C), 4 substitutions in the A-B loop (Q23C, L24C, N25C, and G26C), 1 substitution at the N-linked glycosylation site (N80C), 7 substitutions in the C-D loop (E109C, D110C, F111C, T112C, R113C, G114C, and K115C), and 1 substitution at the C-terminus of the protein (N166C). We also created an analog in which we added a cysteine residue following the C-terminal amino acid N166 (referred to as *167C). M1 of the mature IFN-β-1b protein was used as the initiator methionine for expression of the proteins in E. coli. For the M1C analog we added an initiator methionine preceding the C1 amino acid.
Table 1.
In Vitro Bioactivities of Interferon Beta (IFN-β)-1b, IFN-β-1b Cysteine Muteins, and PEGylated IFN-β-1b Cysteine Muteins
| Protein | Cysteine location | IC50 (ng/mL) | 5 kDa-PEG protein IC50 (ng/mL) | 20 kDa-PEG protein IC50 (ng/mL) |
|---|---|---|---|---|
| IFN-β-1ba |
— |
2.0±1.0 |
|
|
| IFN-β-1bb |
— |
2.9±0.1 |
|
|
| M1C |
N-terminus |
1.0±0 |
|
|
| S2C |
A Helix |
0.7±0.2 |
|
0.6±0.1 |
| W22C |
A Helix |
0.9±0.1 |
|
0.8±0.3 |
| Q23C |
A-B loop |
7.8±2.0 |
|
|
| L24C |
A-B loop |
1.5±0.6 |
|
0.1±0.0 |
| N25C |
A-B loop |
7.8±2.0 |
0.2±0.1 |
0.1±0.0 |
| G26C |
A-B loop |
10.3±0.5 |
|
|
| N80C |
C Helix |
10.0±0.0 |
|
|
| E109C |
C-D loop |
7.3±0.5 |
|
|
| D110C |
C-D loop |
4.0±1.6 |
0.1±0 |
|
| F111C |
C-D loop |
1.2±0.5 |
|
0.1±0.0 |
| T112C |
C-D loop |
2.2±1.0 |
0.1±0.1 |
0.2±0.1 |
| R113C |
C-D loop |
2.5±1.9 |
|
0.1±0.1 |
| G114C |
C-D loop |
3.7±0.8 |
0.6±0.2 |
0.1±0.0 |
| K115C |
C-D loop |
2.3±1.5 |
|
0.1±0.0 |
| N166C |
C-terminus |
4.8±2.1 |
|
|
| *167C | C-terminus | 2.9±1.0 | 0.2±0.1 |
Serial dilutions of each protein were incubated with human Daudi B cells in 96 well tissue culture plates at 37°C in a humidified tissue culture incubator. After 4 days, CellTiter 96® AQueous One Solution was added to each well and the absorbance, which is proportional to cell number, was measured. Absorbance values were used to calculate an IC50 value for each protein. Data are means±SD for at least 3 assays for each protein.
Betaseron.
IFN-β-1b prepared by us.
IFN-β-1b and IFN-β-1b cysteine analogs were expressed as cytoplasmic proteins in E. coli strain W3110 using tryptophan or T5 promoter expression vectors. All of the IFN-β-1b proteins were recovered predominantly in the insoluble fraction of cell lysates (data not shown). The proteins were partially purified by solubilizing the proteins in SDS, extracting the proteins with butanol, and precipitating them with methanol. The partially purified IFN-β-1b proteins were then refolded and purified by Blue Sepharose column chromatography. Recoveries of most of the cysteine analogs were similar to that of IFN-β-1b, indicating that the added cysteine residue did not significantly interfere with proper folding or disulfide bonding. Most of the IFN-β-1b cysteine analogs were recovered as monomers that comigrated with IFN-β-1b under reducing and nonreducing SDS-PAGE conditions. Figure 1 shows nonreducing SDS-PAGE analysis of representative purified proteins. In addition to the major 19.3 kDa species, a few of the purified cysteine analogs (and often IFN-β-1b prepared by us) contained minor species in the 19–21 kDa molecular weight range that may be protein species in which the native disulfide bond between cysteine-31 and cysteine-141 did not form properly, protein species in which cysteine-31 or cysteine-141 formed an incorrect disulfide bond with the added cysteine residue in the proteins, or proteolytic fragments.
FIG. 1.

Nonreducing sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) analysis of purified Interferon beta (IFN-β)-1b and 12 IFN-β-1b cysteine analogs. Molecular weight markers (MW) are indicated to the left.
In vitro bioactivities of the purified proteins were measured in a cell growth inhibition assay using the human Daudi B cell line, the proliferation of which is inhibited by IFN-β. Dose response curves for representative cysteine analogs are shown in Fig. 2. IFN-β-1b (Betaseron) and IFN-β-1b made by us had mean IC50 values of 2.0 and 2.9 ng/mL, respectively (Table 1). Twelve of the cysteine muteins (M1C, S2C, W22C, L24C, D110C, F111C, T112C, R113C, G114C, K115C, N166C, and *167C) had IC50 values that were comparable (within 2-fold) to that of IFN-β-1b, whereas 5 muteins (Q23C, N25C, G26C, N80C, and E109C) had IC50 values that were increased 3- to 5- fold relative to IFN-β-1b, indicating a modest reduction in bioactivity (Table 1).
FIG. 2.

Dose response curves for IFN-β-1b (Betaseron), IFN-β-1b prepared by us (BBT IFN-β-1b), and representative IFN-β-1b cysteine analogs and PEGylated cysteine analogs for inhibiting in vitro proliferation of human Daudi B cells. Daudi cells were incubated with serial dilutions of the indicated test proteins and incubated for 4 days at 37°C in a tissue culture incubator. On day 4, cell number was quantified using Cell-Titer AqueousOne solution. Absorbance on the Y-axis is proportional to cell number. (A) Compares in vitro bioactivities of IFN-β-1b (Betaseron), BBT IFN-β-1b, and the T112C protein. (B) Compares IFN-β-1b (Betaseron), N25C, and 5 kDa-PEG-N25C. (C) Compares F111C and F111C modified with 20 kDa-, 30 kDa-, and 40 kDa-PEGs. Data are means±SD of triplicate wells for each protein dilution. SD generally were <10% of the means.
Representative cysteine analogs from different regions of the protein were modified with linear 5 kDa- and 20 kDa-PEGs as described in Materials and Methods. The F111C protein was also modified with a linear 30 kDa-PEG and a branched 40 kDa-PEG. The PEGylation reactions yielded predominantly monoPEGylated proteins, which were purified from unPEGylated protein by S-Sepharose column chromatography. Nonreducing SDS-PAGE analysis of the purified PEG-F111C proteins is shown in Fig. 3. All but 3 of the PEGylated proteins had IC50 values of 0.1 to 0.2 ng/mL in the Daudi cell assay, indicating that most of the PEGylated proteins were 11- to 78-fold more potent than their unPEGylated parent proteins and IFN-β-1b (Table 1 and Fig. 2). Similar results were obtained when several of the proteins were modified with a larger 20 kDa-PEG (Table 1) and when the F111C protein was modified with 30 kDa- and 40 kDa-PEGs [IC50 values of 0.1±0.0 and 0.2±0.1 ng/mL, respectively (Fig. 2)]. Two exceptions were PEG-S2C and PEG-W22C, which displayed comparable in vitro bioactivities as their unPEGylated parent proteins and IFN-β-1b in the Daudi cell assay. The third exception was the 5 kDa-PEG-G114C protein, which displayed a 6-fold lower IC50 value (0.6 ng/mL) than the unPEGylated G114C protein (IC50 value of 3.7 ng/mL). The G114C protein modified with a 20 kDa-PEG had an even lower IC50 value of 0.1 ng/mL. Visual inspection of the assay wells confirmed that Daudi cell numbers in the wells mirrored the absorbance values for the wells, indicating that the increased potencies of most of the PEG-proteins was not an artifact due to PEG interfering with the dye reaction. We also confirmed that PEG (a 40 kDa-PEG) did not inhibit Daudi cell proliferation when tested in the assay at concentrations ranging from 0.012 to 1,000 ng/mL (data not shown).
FIG. 3.
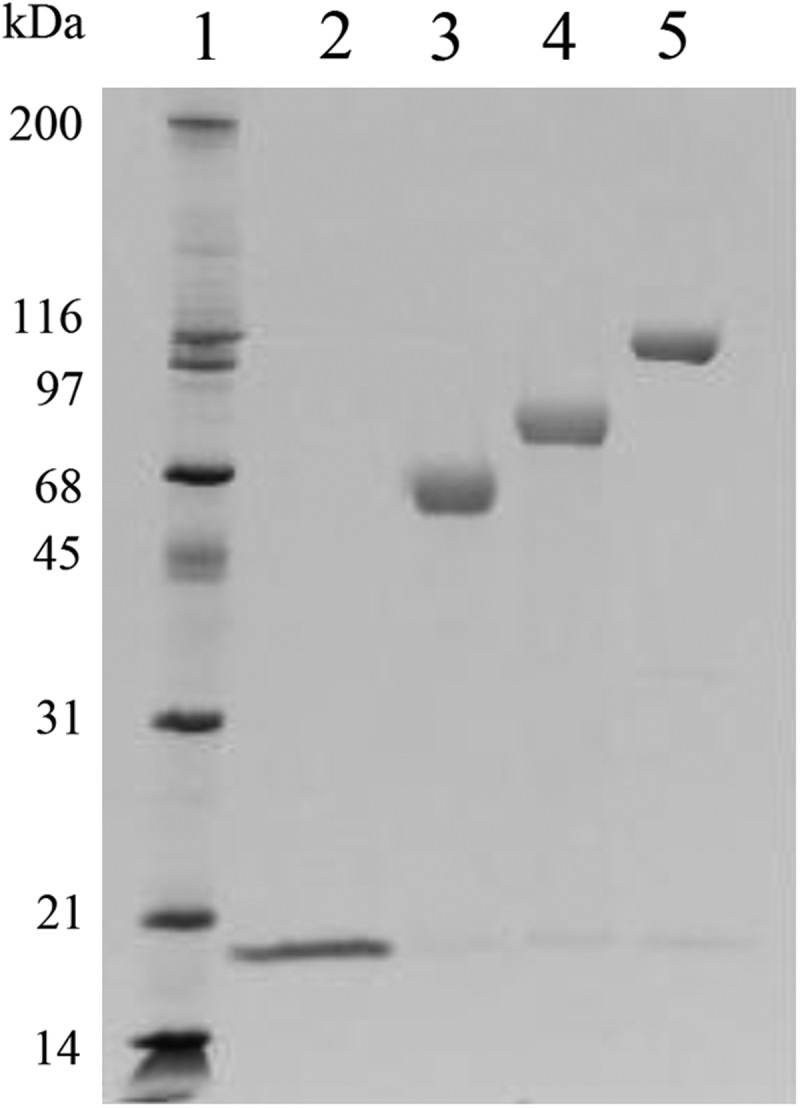
Nonreducing SDS-PAGE analysis of purified F111C and PEGylated F111C proteins. Lane 1, molecular weight markers; Lane 2, F111C, Lane 3, 20 kDa-PEG-F111C; Lane 4, 30 kDa-PEG-F111C; Lane 5, 40 kDa-PEG-F111C.
Pharmacokinetic studies of 40 kDa-PEG-F111C and IFN-β-1b in rats
The F111C analog was chosen for further study because it refolded efficiently and retained high in vitro bioactivity when modified with a 40 kDa-PEG. Pharmacokinetic properties of IFN-β-1b (Betaseron) and 40 kDa-PEG-F111C were compared following a single iv or sc administration to rats. Plasma levels of the proteins were measured by ELISA at various points postadministration and are presented in Fig. 4A (iv administration) and 4B (sc administration). Selected pharmacokinetic parameters for the proteins are summarized in Table 2 (iv) and Table 3 (sc). Following iv administration, IFN-β-1b was cleared rapidly from the rats and was undetectable 48 h postadministration. In contrast, 40 kDa-PEG-F111C was cleared more slowly and was easily detected up to 96 h postadministration. Terminal half-lives for 40 kDa-PEG-F111C was about 3-fold longer than that of IFN-β-1b (17.7 h versus 6.4 h). Systemic exposure, as measured by the area under the curve from time 0 to infinity (AUC 0→∞), was 41-fold greater for 40 kDa-PEG-F111C compared to IFN-β-1b. Similar results were obtained following sc administration. Peak plasma levels of IFN-β-1b occurred 1 h postadministration and the protein was cleared rapidly, with a terminal half-life of 4 h. 40 kDa-PEG-F111C was absorbed more slowly than IFN-β-1b and did not reach peak plasma levels until 24 h post-administration. The terminal half-life of 40 kDa-PEG-F111C was 17.5 h following sc administration. Peak plasma levels of IFN-β-1b following sc administration were only about 1% of the levels obtained following iv administration, whereas 40 kDa-PEG-F111C peak plasma levels following sc administration were about 5% of the levels reached following iv administration. Systemic exposure, as measured by AUC 0→∞, was 150-fold greater for 40 kDa-PEG-F111C compared to IFN-β-1b following sc injection. Bioavailability of 40 kDa-PEG-F111C was 12.5% following sc administration, compared to 3.4% for IFN-β-1b.
FIG. 4.

Clearance of IFN-β-1b (Betaseron) and 40 kDa-PEG F111C following intravenous (A) and subcutaneous (B) administration to rats. Proteins were dosed at 100 μg/kg and were quantitated in plasma samples by ELISA. Data are means±SD for 3 rats/group.
Table 2.
Selected Pharmacokinetic Parameters for IFN-β-1b and 40 kDa-PEG-F111C Following Intravenous Administration of 100 μg/kg of the Proteins to Rats
| Parameter | IFN-β-1b (Betaseron) | 40 kDa-PEG F111C |
|---|---|---|
| Cmax (ng/mL) |
367 |
1,331 |
| AUC 0→∞ (hr x ng/mL) |
557 |
22,827 |
| CL (mL/h/kg) |
179 |
4.4 |
| Vss (mL/kg) |
440 |
86 |
| T1/2 (h) | 6.4 | 17.7 |
Rats (N=3 males/group) received a single intravenous injection of 100 μg protein/kg. Blood samples were obtained predose and from 0.25–96 h postinjection and plasma levels of the proteins were measured by ELISA. Plasma levels were averaged for the 3 rats in each group and used to calculate pharmacokinetic parameters using WinNonlin software.
Cmax, maximal blood concentration; AUC 0→∞, area under the curve from time 0 to infinity; CL, clearance; Vss, volume at steady state; T1/2, terminal half-life.
Table 3.
Selected Pharmacokinetic Parameters for IFN-β-1b and 40 kDa-PEG-F111C Following Subcutaneous Administration of 100 μg/kg of the Proteins to Rats
| Parameter | IFN-β-1b (Betaseron) | 40 kDa-PEG F111C |
|---|---|---|
| Tmax (h) |
1 |
24 |
| Cmax (ng/mL) |
4.3 |
67 |
| AUC 0→∞ (hr x ng/mL) |
19.2 |
2,857 |
| CL/F (mL/h/kg) |
5,210 |
35 |
| T1/2 (h) | 4 | 17.5 |
Rats (N=3 males/group) received a single subcutaneous (sc) injection of 100 μg protein/kg. Blood samples were obtained predose and from 0.25–96 h postinjection and plasma levels of the proteins were measured by ELISA. Plasma levels were averaged for the 3 rats in each group and used to calculate pharmacokinetic parameters using WinNonlin software.
Tmax, time to maximal blood concentration; Cmax, maximal blood concentration; AUC 0→∞, area under the curve from time 0 to infinity; CL/F, apparent clearance after sc administration; T1/2, terminal half-life.
Inhibition of tumor growth in mice by IFN-β-1b and 40 kDa-PEG-F111C
The relative in vivo potencies of IFN-β-1b (Betaseron) and 40 kDa-PEG-F111C were assessed by their ability to inhibit growth of human NIH: OVCAR-3 ovarian cancer cells in athymic mice. Human IFN-β is not active in mice and the growth inhibitory effects of the proteins are due to their direct antiproliferative action on the transplanted human NIH: OVCAR-3 cells. IFN-β-1b and 40 kDa-PEG-F111C inhibited in vitro growth of NIH: OVCAR-3 cells to similar extents (Fig. 5). In contrast to the Daudi cell assay, IFN-β-1b and the PEG-F111C proteins had comparable IC50 values in the NIH: OVCAR-3 assay (0.8±0.1 ng/mL for IFN-β-1b, 0.3±0.1 ng/mL for 30 kDa-PEG-F111C, and 0.6±0.3 ng/mL for 40 kDa-PEG-F111C).
FIG. 5.

Dose response curves for IFN-β-1b (Betaseron) and PEG-F111C proteins for inhibiting in vitro proliferation of human NIH: OVCAR-3 cells. NIH: OVCAR-3 cells were incubated with serial dilutions of the indicated test proteins for 4 days at 37°C in a tissue culture incubator. On day 4, cell number was quantified as described in the legend to Fig. 2. Data are means±SD of triplicate wells for each protein dilution. SD generally were <10% of the means.
The tumor study was performed by implanting mice (10–11/group) with 5×106 NIH:OVCAR-3 cells in Matrigel on day 0. Treatment with the test proteins began the following day and continued until day 69 using a 3X/week dosing regimen. Different groups of mice received sc injections of vehicle solution, or 15 μg/injection of 40 kDa-PEG-F111C or IFN-β-1b (Betaseron). Tumor volumes increased in all the mice for the first few weeks of the study (Fig. 6). Tumor volumes continued to increase until study termination in mice treated with vehicle solution or IFN-β-1b, although tumors grew more slowly on average in mice treated with IFN-β-1b. At study termination on day 70, tumor volumes and tumor weights in mice treated with IFN-β-1b were 60% and 63% smaller, respectively, than tumor volumes and tumor weights in mice treated with vehicle solution (Table 4). All of the mice treated with IFN-β-1b or vehicle had detectable/viable tumors at study termination. In contrast, tumor volumes decreased after about day 40 in mice treated with 40 kDa-PEG-F111C and there were no detectable tumors in any of the mice treated with 40 kDa-PEG-F111C protein by study termination on day 70 (Table 4). Final tumor volumes and tumor weights at necropsy were significantly smaller (P<0.01) in animals treated with 40 kDa-PEG-F111C than in animals treated with the IFN-β-1b or vehicle solution (Table 4).
FIG. 6.

Inhibition of human tumor xenograft growth in athymic mice by IFN-β-1b (Betaseron) and 40 kDa-PEG-F111C. Athymic mice received an injection of 5×106 NIH: OVCAR-3 cells on day 0. Mice received 3X/week subcutaneous injections of vehicle solution or 15 μg protein/injection of IFN-β-1b (Betaseron) or 40 kDa-PEG-F111C and tumor volumes were measured over time. Data are means±SE for 10 to 11 mice per group.
Table 4.
Effects of 3x/Week sc Administration of Vehicle Solution, IFN-β-1b (Betaseron) and 40 kDa-PEG-F111C on Growth of Human NIH: OVCAR-3 Tumors in Athymic Mice
| Treatment group | Day 70 tumor volume (cm3) | Day 70 tumor weight (g) | Animals with visible tumors on day 70 |
|---|---|---|---|
| Vehicle |
1.24±0.56 |
1.40±0.56 |
10/10 |
| IFN-β-1b (Betaseron) |
0.49±0.12a |
0.52±0.15a |
10/10 |
| 40 kDa-PEG-F111C | 0±0.0a,b | 0±0.0a,b | 0/11 |
Female athymic mice (N=10–11/group) were implanted with NIH: OVCAR-3 cells on day 0. Starting on day 3, mice received 3x/week sc injections of the test proteins or vehicle solution. Tumor volume was measured at weekly intervals. At sacrifice on day 70, final tumor volumes and weights were measured.
P<0.01 versus vehicle.
P<0.01 versus IFN-β.
Discussion
The studies presented here demonstrate the feasibility of creating IFN-β-1b cysteine analogs that have in vitro bioactivities comparable to wild type IFN-β-1b. We identified several regions (the N-terminal region preceding helix A, the ends of Helix A, the A-B and C-D loops, and the C-terminus) where cysteine residues can be introduced without appreciably reducing biological activity. We showed that it is possible to PEGylate representative cysteine muteins in these regions and that the PEGylated proteins retain high specific activities in vitro. The high in vitro potencies of the IFN-β-1b cysteine analogs and their PEGylated derivatives make it unlikely that the proteins are grossly misfolded or have the PEG attached to C31 or C141 rather than the added cysteine, since the C31-C141 disulfide bond is absolutely required for biological activity of IFN−β (Shepard and others 1981). An unexpected result from this study was the finding that most of the PEGylated cysteine analogs possessed 11- to 78-fold higher potencies (lower IC50 values) in the Daudi cell growth inhibition assay than their unPEGylated parent proteins or IFN-β-1b. The reason for this will require further study. N-terminally PEGylated IFN-β-1b is more stable than IFN-β-1b at 37°C (Basu and others 2006), which theoretically could improve in vitro bioactivities of the PEG-proteins. However, IFN-β-1b and N-terminally PEGylated IFN-β-1b possessed similar potencies in an in vitro antiviral assay (Basu and others 2006). We observed no significant improvement in bioactivity for 2 of the PEG proteins, PEG-S2C and PEG-W22C, in the Daudi cell assay. The PEG attachment site in N-terminally PEGylated IFN-β-1b (Basu and others 2006) is at the S2 amino acid due to removal of the N-terminal methionine during synthesis of the protein in E. coli. The 2 PEG proteins we studied that showed no significant improvement in bioactivity in the Daudi cell assay (PEG-S2C and PEG-W22C) have the PEG attached to amino acids at the N-terminus of the protein or near a rigid alpha helix. The improved in vitro bioactivities we observed were primarily when PEG was attached to internal amino acid positions in IFN-β-1b, most of which were located in flexible, nonhelical regions of the protein (Karpusas and others 1997). Therefore, location of the PEG attachment site seems to strongly influence bioactivities of the PEG-IFN-β-1b analogs in the Daudi cell bioassay. In contrast to results obtained in the Daudi cell assay, IFN-β-1b and the PEG-F111C proteins had similar in vitro potencies (IC50 values) in the NIH: OVCAR-3 cell bioassay. This finding suggests a difference in the structure or signaling pathways of the IFN-β receptor in Daudi and NIH: OVCAR-3 cells. Differences in IFN-β receptor numbers or densities between Daudi and NIH:OVCAR-3 cells also could potentially explain differences in in vitro potencies of the PEG- IFN-β-1b analogs on these 2 cell lines.
IFN-β is a member of the GH supergene family, which comprises over 20 structurally related cytokines and growth factors (Mott and Campbell 1995). Our mutagenesis and PEGylation results with IFN-β-1b are similar to results obtained with other members of the growth hormone supergene family, such as alpha IFN, growth hormone, erythropoietin, granulocyte colony-stimulating factor, and granulocyte-macrophage colony-stimulating factor (Doherty and others 2005; Rosendahl and others 2005a, 2005b; Long and others 2006; Cox and others 2007; Bell and others 2008). For each of these proteins we find that cysteine residues can be introduced into the nonhelical regions of the proteins and PEGylated without appreciable loss of in vitro bioactivity. The nonhelical regions of the proteins possess largely unordered structures that appear to be conducive to cysteine substitutions and PEGylation without apparently causing significant alterations to the receptor binding regions, which tend to be located within the helical regions (Mott and Campbell 1995).
The 40 kDa-PEG-F111C protein had a longer circulating half-life, slower clearance, improved systemic exposure, and increased bioavailability compared to IFN-β-1b in rats. Similar results were reported for N-terminally PEGylated IFN-β-1a and PEGylated IFN-β-1b in animals (Pepinsky and others 2001; Basu and others 2006; Hu and others 2011) and more recently in humans (Hu and others 2012). N-terminally PEGylated IFN-β-1a was found to be more effective than IFN-β-1a at inhibiting angiogenesis in mice (Baker and others 2006). Our data extends this finding by demonstrating that PEGylation significantly improves the ability of IFN-β-1b to inhibit growth of human tumor cells in athymic mice. IFN-β is known to inhibit growth of human tumor cells in vitro and in athymice mice (Johns and others 1992; Lindner and Borden 1997). However, none of the previous in vivo studies showed complete elimination of tumor burden by IFN-β. 40 kDa-PEG-F111C administration resulted in complete tumor elimination in 11/11 treated mice compared to 0/10 mice treated with vehicle or IFN-β-1b. This finding suggests that the failure of previous studies to demonstrate complete tumor elimination with IFN-β proteins may be due to the poor pharmacokinetic properties and inadequate systemic exposure of IFN-β rather than due to inability to IFN-β to completely kill the tumor cells. PEGylation has been shown to dramatically improve the antitumor bioactivity of IFN alpha proteins (Bailon and others 2001; Bell and others 2008).
Other researchers were not successful at obtaining monoPEGylation of IFN-β-1b cysteine analogs using maleimide-PEGs (Basu and others 2006). The 3 cysteine analogs they examined were a substitution of cysteine for N80, the addition of a cysteine preceding the first amino acid (presumably corresponding to our M1C), and addition of a cysteine following the last amino acid (corresponding to our *167C). They reported that the PEGylation reactions yielded multiple PEGylated species, indicating modification of the native C31 and/or C141 residues, in addition to the introduced cysteine residue (Basu and others 2006). The purification buffers used in this study included the detergent Zwittergent. In pilot studies, we found that inclusion of Zwittergent in the PEGylation reactions led to a lack of specificity in the PEGylation reactions and to mutiPEGylated proteins. For this reason, we purified the IFN-β-1b cysteine analogs using buffers and column conditions that did not require Zwittergent. Once the cysteine analogs were PEGylated and the PEG reaction components diluted, the proteins appeared to be stable in buffers containing Zwittergent. We hypothesize that Zwittergent may be denaturing the proteins during the PEGylation reaction, which includes a partial reduction step, making the native cysteine residues available for reaction with the maleimide-PEG reagent.
IFN-β is widely used to treat Multiple Sclerosis. Patients treated with IFN-β-1b and many patients treated with IFN-β-1a must administer the drug every other day by sc injection. More frequent administration of IFN-β improves patient outcomes (Blumhardt 2002). Development of longer-acting forms of IFN-β that require less frequent dosing will provide Multiple Sclerosis patients with a more convenient treatment for their disease. The improved in vivo efficacy seen with PEGylated forms of IFN-α in humans (Zeuzem and others 2000) raises the possibility that longer-acting forms of IFN-β also may prove more efficacious than current IFN-β products in Multiple Sclerosis patients.
Acknowledgments
This work was supported by grants 1R43 NS040205 and 2R44 NS040205 to G. C. from the National Institute of Neurological Disorders and Stroke of the National Institutes of Health. The publication's contents are solely the responsibility of the authors and do not necessarily represent the official views of the National Institute of Neurological Disorders and Stroke.
Author Disclosure Statement
J.L., S.E., M.R., J.B., D.D., and G.C. are employees or former employees of Bolder BioTechnology, Inc. EC has a financial interest in Premier Laboratory, LLC, which received financial compensation from Bolder BioTechnology, Inc. to perform the animal studies.
References
- Abuchowski A, Kazo GM, Verhoest CR, van Es T, Kafkewitz D, Nucci ML, Viau AT, Davis FF. 1984. Cancer therapy with chemically modified enzymes. I. Antitumor properties of polyethylene glycol-asparaginase conjugates. Cancer Biochem Biophys 7:175–186 [PubMed] [Google Scholar]
- Bailon P, Palleroni A, Schaffer CA, Spence CL, Fung W-J, Porter JE, Ehrlich GK, Pan W, Xu Z-X, Modi MW, Farid A, Berthold W, Graves M. 2001. Rational design of a potent, long-lasting form of interferon: a 40 kDa branched polyethylene glycol-conjugated interferon-α2a for the treatment of hepatitis C. Bioconjug Chem 12:195–202 [DOI] [PubMed] [Google Scholar]
- Baker DP, Lin EY, Lin K, Pellegrini M, Petter RC, Chen LL, Arduini RM, Brickelmaier M, Wen D, Hess DM, Chen L, Grant D, Whitty A, Gill A, Lindner DJ, Pepinsky RB. 2006. N-terminally PEGylated human interferon-beta-1a with improved pharmacokinetic properties and in vivo efficacy in a melanoma angiogenesis model. Bioconjug Chem 17:179–188 [DOI] [PubMed] [Google Scholar]
- Basu A, Yang K, Wang M, Liu S, Chintala R, Palm T, Zhao H, Peng P, Wu D, Zhang Z, Hua J, Hsieh MC, Zhou J, Petti G, Li X, Janjua A, Mendez M, Liu J, Longley C, Zhang Z, Mehlig M, Borowski V, Viswanathan M, Filpula D. 2006. Structure-function engineering of interferon-beta-1b for improved stability, solubility, potency, immunogenicity, and pharmacokinetic properties by site-selective monoPEGylation. Bioconjug Chem 17:618–630 [DOI] [PubMed] [Google Scholar]
- Bell SJ, Fam CM, Chlipala EA, Carlson SJ, Lee JI, Rosendahl MS, Doherty DH, Cox GN. 2008. Enhanced circulating half-life and anti-tumor activity of a site-specific pegylated interferon - α protein therapeutic. Bioconjug Chem 19:299–305 [DOI] [PubMed] [Google Scholar]
- Blumhardt LD. 2002. Once weekly interferon beta for multiple sclerosis is superseded by higher and more frequent dosing. Int J Clin Pract Suppl 131:9–16 [PubMed] [Google Scholar]
- Chan HL, Ren H, Chow WC, Wee T. 2007. Randomized trial of interferon beta with or without ribavirin in asian patients with chronic hepatitis C. Hepatology 46:315–323 [DOI] [PubMed] [Google Scholar]
- Comi G. 2000. Why treat early multiple sclerosis patients? Curr Opin Neurol 13:235–240 [DOI] [PubMed] [Google Scholar]
- Cox GN, Rosendahl MS, Chlipala EA, Smith DJ, Carlson SJ, Doherty DH. 2007. A long-acting, monoPEGylated human growth hormone analog is a potent stimulator of weight gain and bone growth in hypophysectomized rats. Endocrinology 148:1590–1597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doherty DH, Rosendahl MS, Smith DJ, Hughes JM, Chlipala EA, Cox GN. 2005. Site-specific PEGylation of engineered cysteine analogues of recombinant human granulocyte-macrophage colony-stimulating factor. Bioconjug Chem 16:1291–1298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein D, Sielaff KM, Storer BE, Brown RR, Datta SP, Witt PL, Teitelbaum AP, Smalley RV, Borden EC. 1989. Human biologic response modification by interferon in the absence of measurable serum concentrations: a comparative trial of subcutaneous and intravenous interferon-beta serine. J Natl Cancer Inst 81:1061–1068 [DOI] [PubMed] [Google Scholar]
- Goodson RJ, Katre NV. 1990. Site-directed pegylation of recombinant interleukin-2 at its glycosylation site. Biotechnology 8:343–346 [DOI] [PubMed] [Google Scholar]
- Hanisch WH, Fernandes PM. 1984. Process for the recovery of human beta interferon-like polypeptides. United States Patent Number 4,462,940
- Harris JM, Martin NE, Modi M. 2001. Pegylation: a novel process for modifying pharmacokinetics. Clin Pharmacokinet 40:539–551 [DOI] [PubMed] [Google Scholar]
- Higuchi R. 1990. Recombinant PCR. In: Innis MA, Gelfand DH, Sninsky JJ, White TJ, eds. PCR protocols: a guide to methods and applications, Chapter 22. San Diego, CA: Academic Press; p 177–183 [Google Scholar]
- Horton RM. 1993. In vitro recombination and mutagnesis of DNA. SOIng together tailor-made genes. In: White BA, ed. Methods in molecular biology, Vol. 15, PCR protocols: current methods and applications, Chapter 25. Totawa, NJ: Humana Press; p 251–266 [DOI] [PubMed] [Google Scholar]
- Hu X, Miller L, Richman S, Hitchman S, Glick G, Liu S, Zhu Y, Crossman M, Nestorov I, Gronke RS, Baker DP, Rogge M, Subramanyan M, Davar G. 2012. A novel PEGylated interferon Beta-1a for multiple sclerosis: safety, pharmacology and biology. J Clin Pharmacol 338:984–996 [DOI] [PubMed] [Google Scholar]
- Hu X, Olivier K, Polack E, Crossman M, Zokowski K, Gronke RS, Parker S, Li Z, Nestorov I, Baker DP, Clarke J, Subramanyan M. 2011. In vivo pharmacology and toxicology evaluation of polyethylene glycol-conjugated interferon β1a. JPET 338:984–996 [DOI] [PubMed] [Google Scholar]
- Hughes RA. 2003. Interferon beta 1a for secondary progressive multiple sclerosis. J Neurol Sci 206:199–202 [DOI] [PubMed] [Google Scholar]
- Johns TG, Mackay IR, Callister KA, Hertzog PJ, Devenish RL, Linnane AW. 1992. Antiproliferative potencies of interferons on melanoma cell lines and xenografts: higher efficacy of interferon beta. J Natl Cancer Inst 84:1185–1190 [DOI] [PubMed] [Google Scholar]
- Karpusas M, Nolte M, Benton CB, Meier W, Lipscomb WN, Goelz S. 1997. The crystal structure of human interferon beta at 2.2-A resolution. Proc Natl Acad Sci U S A 94:11813–11818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katre NV, Knauf MJ, Laird WJ. 1987. Chemical modification of recombinant interleukin 2 by polyethylene glycol increases its potency in the murine Meth A sarcoma model. Proc Natl Acad Sci U S A 84:1487–1491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinstler OB, Brems DN, Lauren SL, Paige AG, Hamburger JB, Treuheit MJ. 1996. Characterization and stability of N-terminally PEGylated rhG-CSF. Pharm Res 13:996–1002 [DOI] [PubMed] [Google Scholar]
- Li DK, Zhao GJ, Paty DW. 2001. The University of British Columbia MS/MRI Analysis Research Group and the SPECTRIMS Study Group. Randomized controlled trial of interferon beta-1a in secondary progressive MS: MRI results. Neurology 56:1501–1513 [DOI] [PubMed] [Google Scholar]
- Liberati AM, Horisberger MA, Palmisano L, Astolfi S, Nastari A, Mechati S, Villa A, Mancini S, Arzano S, Grignani F. 1992. Double-blind randomized phase I study on the clinical tolerance and biological effects of natural and recombinant interferon-beta. J Interfer Res 12:329–336 [DOI] [PubMed] [Google Scholar]
- Lindner DJ, Borden EC. 1997. Synergistic antitumor effects of a combination of interferon and tamoxifen on estrogen receptor-positive and receptor-negative human tumor cell lines in vivo and in vitro. J Interfer Cytokine Res 17:681–693 [DOI] [PubMed] [Google Scholar]
- Long DL, Doherty DH, Eisenberg SP, Smith DJ, Rosendahl MS, Christensen KR, Edwards DP, Chlipala EA, Cox GN. 2006. Design of homogeneous, monoPEGylated erythropoietin analogs with preserved in vitro bioactivity. Exp Hematol 34:697–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mott HR, Campbell ID. 1995. Four-helix bundle growth factors and their receptors: protein-protein interactions. Curr Opin Struct Biol 5:114–121 [DOI] [PubMed] [Google Scholar]
- Munoz R, Castellano G, Fernandez I, Alvarez MV, Marcos MS, Cuenca B, Solis-Herruzo JA. 2002. A pilot study of beta interferon for treatment of patients with chronic hepatitis B who fail to respond to alpha interferon. J Hepatol 37:655–659 [DOI] [PubMed] [Google Scholar]
- Pepinsky RB, Lepage DJ, Gill A, Chakraborty A, Vaidyanathan S, Grren M, Baker D, Whalley E, Hochman PS, Martin P. 2001. Improved pharmacokinetic properties of a polyethylene glycol-modified form of interferon-β-1a with preserved in vitro bioactivity. JPET 297:1059–1066 [PubMed] [Google Scholar]
- Plosker GL. 2011. Interferon -β-1b: a review of its use in multiple sclerosis. CNS Drugs 25:67–88 [DOI] [PubMed] [Google Scholar]
- Porter AG, Bell LD, Adair J, Catlin GH, Clarke J, Davies JA, Dawson K, Derbyshire R, Doel SM, Dunthorne L, Finlay M, Hall J, Houghton M, Hynes C, Lindley I, Nugent M, O'Neill GJ, Smith JC, Stewart A, Tacon W, Viney J, Warburton N, Boseley PG, McCullagh KG. 1986. Novel modified beta-interferons: gene cloning, expression, and biological activity in bacterial extracts. DNA 5:137–148 [DOI] [PubMed] [Google Scholar]
- Qin XQ, Tao N, Dergay A, Moy P, Fawell S, Davis A, Wilson JM, Barsoum J. 1998. Interferon-beta gene therapy inhibits tumor formation and causes regression of established tumors in immune-deficient mice. Proc Natl Acad Sci U S A 95:14411–14416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosendahl MS, Doherty DH, Smith DJ, Carlson SJ, Chlipala EA, Cox GN. 2005a. A long-acting, highly potent interferon α-2 conjugate created using site-specific PEGylation. Bioconjug Chem 16:200–207 [DOI] [PubMed] [Google Scholar]
- Rosendahl MS, Doherty DH, Smith DJ, Bendele AM, Cox GN. 2005b. Site-specific protein PEGylation: application to cysteine analogs of recombinant human granulocyte colony-stimulating factor. BioProcess Int 3:52–62 [PMC free article] [PubMed] [Google Scholar]
- Salmon P, Le Cotonnec JY, Galazka A, Abdul-Ahad A, Darragh A. 1996. Pharmacokinetics and pharmacodynamics of recombinant human interferon-beta in healthy male volunteers. J Interfer Cytokine Res 16:759–764 [DOI] [PubMed] [Google Scholar]
- Senda T, Shimazu T, Matsuda S, Kawano G, Shimizu H, Nakamura KT, Mitsui Y. 1995. Three-dimensional crystal structure of recombinant murine interferon-beta. EMBO J 11:3193–3201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepard HM, Leung D, Stebbing N, Goeddel DV. 1981. A single amino acid change in IFN-beta abolishes its antiviral activity. Nature 294:563–565 [DOI] [PubMed] [Google Scholar]
- Witt PL, Storer BE, Bryan GT, Brown RR, Flashner M, Larocca AT, Colby CB, Borden EC. 1993. Pharmacodynamics of biological response in vivo after single and multiple doses of interferon-beta. J Immunother 13:191–200 [DOI] [PubMed] [Google Scholar]
- Zeuzem S, Feinman SV, Rasenack J, Heathcote EJ, Lai MY, Gane E, O'Grady J, Reichen J, Diago M, Lin A, Hoffman J, Brunda MJ. 2000. Peginterferon alfa-2a in patients with chronic hepatitis C. N Engl J Med 343:1666–1672 [DOI] [PubMed] [Google Scholar]