

Hepatocellular carcinoma (HCC) is the third leading cause of cancer death and the fifth most common solid tumor worldwide [1], [2]. Liver tumorigenesis is a multistep process in which external stimuli such as chronic inflammation or cirrhosis lead to the development of clonal populations of dysplastic hepatocytes that accumulate genetic changes and evolve into malignant foci [2]. Among the most common risk factors for HCC pathogenesis include viral hepatitis, alcoholism, and obesity [1], [3].
The same insults that predispose to HCC are known to induce endoplasmic reticulum (ER) stress pathways. One such pathway, known as the unfolded protein response (UPR), is triggered by the accumulation of incompletely folded proteins in the ER lumen [4]–[6]. Stimulation of the UPR results in the activation of three transmembrane proteins that induce downstream effectors to alter gene expression and ultimately modulate ER function. One of these UPR transmembrane proteins is protein kinase RNA (PKR)-like ER kinase (PERK), which phosphorylates eIF2α, leading to a transient translational blockade. A related pathway that shares transcriptional targets with the UPR is the integrated stress response (ISR) pathway. When triggered by viral infection or amino acid starvation the ISR also initiates eIF2α-dependent signaling events [7]. Although the UPR and ISR pathways are active in distinct human tumor types and the UPR is implicated in HCC [8]–[10], their relative contribution to the pathogenesis of HCC has remained uncharacterized.
In this issue of PLOS Genetics, Rutkowski and colleagues (DeZwaan-McCabe et al., [11]) sought to determine whether the UPR pathway was induced in murine liver tumors that developed in a Sleeping Beauty (SB) transposon-induced insertional mutagenesis screen [12], [13]. The application of transposon-based approaches to cancer gene identification provides a powerful opportunity to examine the consequences of specific mutations in the context of in vivo tumor development [14]. Whole transcriptome sequencing of liver tumors generated in an SB-mediated liver tumorigenesis screen identified an induction of C/EBP Homologous Protein (CHOP), a stress-regulated transcription factor, in multiple SB-induced tumors. Upon further analysis, components of the two PERK-independent arms of the UPR pathway were not altered at the transcript level, leading the authors to further investigate the role of the ISR and CHOP in HCC.
CHOP, which has a diverse repertoire of transcriptional targets and modes of transcriptional modulation, was previously known to mediate apoptosis in response to ER stress [15]–[17]. Accordingly, several studies implicate CHOP as a putative tumor suppressor. In contrast to this, chromosomal translocations fusing CHOP to FUS/TLS and EWS have been identified in several cancers, hinting that CHOP may also play an oncogenic role in tumorigenesis in certain contexts [18], [19].
The Integrated Stress Response in HCC: Not Just CHOPped Liver
Consistent with a pro-oncogenic role for CHOP, McCabe et al. [11] hypothesized that CHOP contributes to the pathogenesis of HCC in vivo by promoting apoptosis, inflammation, fibrosis, compensatory proliferation, and development of liver tumors ( Figure 1 ). Consistent with this hypothesis, global deletion of Chop in mice attenuated these sequelae following treatment with the chemical carcinogen diethylnitrosamine (DEN). Following administration of the hepatotoxin carbon tetrachloride in wild-type mice, the authors observed an association of CHOP-positive foci with increased fibrosis. Staining of human HCC samples with a CHOP antibody revealed CHOP-positive foci in tumors and significantly less staining in normal liver. These results suggest that activation of CHOP promotes HCC progression. Moreover, these findings provide the first link between CHOP and liver oncogenesis.
Figure 1. The role of CHOP in HCC pathogenesis.
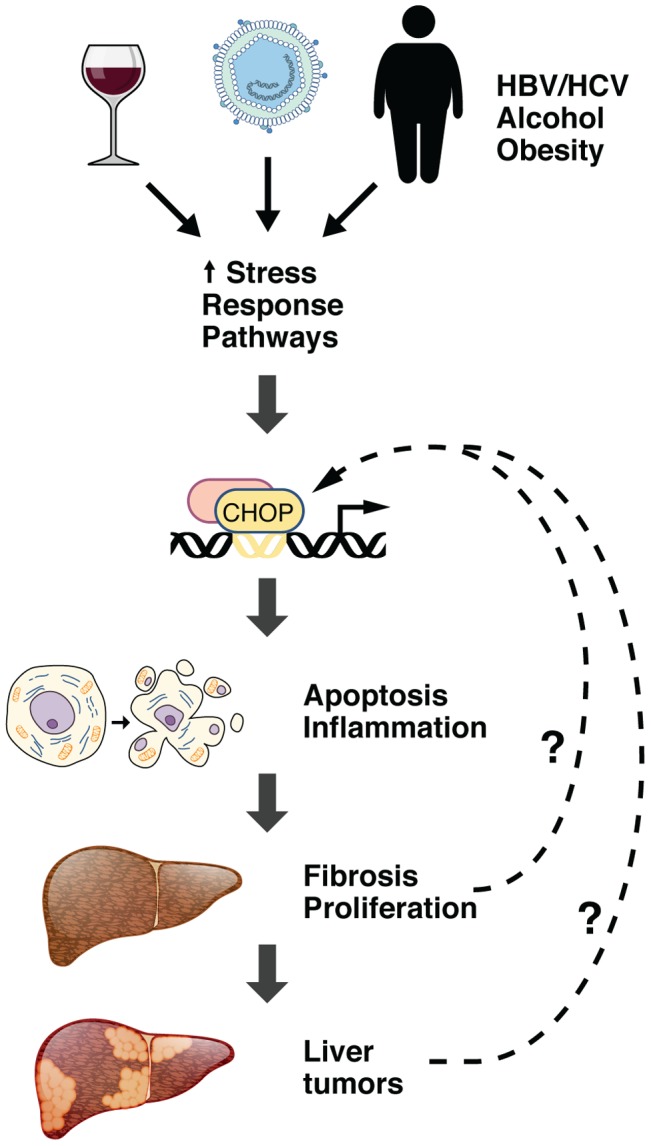
Hepatocyte injury, including toxicity from viral hepatitis, obesity, or alcoholism, promotes the induction of canonical ER stress pathways, including the UPR and ISR. This results in induction of the CHOP transcription factor, which stimulates cell death and provokes an inflammatory response. Inflammation may further stimulate fibrosis and compensatory proliferation, leading to the development of liver tumor nodules. It is also possible that stress caused by fibrosis and proliferation of dysplastic hepatocytes in HCC may induce expression of CHOP, further amplifying its effect on tumorigenesis.
Gene expression profiling of liver mRNA from Chop-null and wild-type mice in the absence of hepatotoxic challenge revealed that deletion of Chop reduced the levels of basal inflammatory signaling genes. This is consistent with an important role for CHOP in promoting inflammation after liver injury. Interestingly, genes encoding ribosomal proteins were significantly increased in liver tumors derived from DEN-treated Chop-null animals relative to tumors that developed in wild-type animals. None of these genes harbored canonical CHOP binding sites, leaving the question of how this occurs unresolved. This represents the first evidence that CHOP can reduce translation by suppressing expression of ribosomal proteins. However, this is consistent with the general role of the ISR as an inhibitor of translation. Further studies are needed to fully elucidate how CHOP affects the translational machinery and the resulting effects on translational output.
The authors of this study present several lines of evidence consistent with an oncogenic role for CHOP in promoting HCC. Their findings suggest that induction of CHOP is a common feature of liver cancer caused by viral infection, alcoholism, and obesity. Recently, a novel framework has been proposed suggesting that cancer cells exhibit hallmarks of chronic stress induced by DNA damage, proteotoxic stress created by accumulation of unfolded protein aggregates, metabolic stress, and oxidative stress [20]. Additional experiments are therefore warranted to determine whether CHOP induction is a causative event that promotes liver tumorigenesis and/or a consequence of the immense cellular stress that cells are subjected to as hepatocytes acquire mutations and undergo the multistep progression to HCC. This will require the generation of inducible and tissue-specific transgenic mouse models, which are currently lacking. Temporal manipulation of CHOP expression in the liver could also tease out whether CHOP promotes the initiation of HCC, or if it enhances tumorigenesis after dysplastic liver nodules form.
Given the resistance to HCC-associated phenotypes observed in Chop-null animals and the discovery of human HCC-associated CHOP expression, this stress-responsive transcription factor may serve as a useful biomarker for liver cancer. However, several important questions remain. For example, is CHOP-mediated apoptosis of hepatocytes the major initiating event that triggers the cycle of subsequent inflammation, fibrosis, and ultimately HCC initiation? Or does hepatocyte-specific expression of CHOP indirectly stimulate inflammation, perhaps through cytokine release, initiating the inflammation-tumorigenesis sequence? The analysis of CHOP target genes that mediate these effects in HCC will shed light on these issues. Perhaps most intriguingly, the identity of the eIF2α kinase that leads to CHOP induction in liver cancer remains unknown. PERK is one candidate, and it would be useful to determine whether PERK inhibitors will blunt CHOP expression and ameliorate HCC in mouse models. Thus, further investigation of the pro- and anti-oncogenic functions of CHOP is likely to reveal important new insights into the pathogenesis of liver cancer and other tumor types.
Funding Statement
The authors received no specific funding for this article.
References
- 1. Altekruse SF, McGlynn KA, Reichman ME (2009) Hepatocellular carcinoma incidence, mortality, and survival trends in the United States from 1975 to 2005. J Clin Oncol 27: 1485–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Thorgeirsson SS, Grisham JW (2002) Molecular pathogenesis of human hepatocellular carcinoma. Nat Genet 31: 339–346. [DOI] [PubMed] [Google Scholar]
- 3. Starley BQ, Calcagno CJ, Harrison SA (2010) Nonalcoholic fatty liver disease and hepatocellular carcinoma: a weighty connection. Hepatology 51: 1820–1832. [DOI] [PubMed] [Google Scholar]
- 4. Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, et al. (2000) Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell 6: 1099–1108. [DOI] [PubMed] [Google Scholar]
- 5. Rutkowski DT, Hegde RS (2010) Regulation of basal cellular physiology by the homeostatic unfolded protein response. J Cell Biol 189: 783–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Walter P, Ron D (2011) The unfolded protein response: from stress pathway to homeostatic regulation. Science 334: 1081–1086. [DOI] [PubMed] [Google Scholar]
- 7. Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, et al. (2003) An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell 11: 619–633. [DOI] [PubMed] [Google Scholar]
- 8. Bobrovnikova-Marjon E, Grigoriadou C, Pytel D, Zhang F, Ye J, et al. (2010) PERK promotes cancer cell proliferation and tumor growth by limiting oxidative DNA damage. Oncogene 29: 3881–3895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dong D, Ni M, Li J, Xiong S, Ye W, et al. (2008) Critical role of the stress chaperone GRP78/BiP in tumor proliferation, survival, and tumor angiogenesis in transgene-induced mammary tumor development. Cancer Res 68: 498–505. [DOI] [PubMed] [Google Scholar]
- 10. Shuda M, Kondoh N, Imazeki N, Tanaka K, Okada T, et al. (2003) Activation of the ATF6, XBP1 and grp78 genes in human hepatocellular carcinoma: a possible involvement of the ER stress pathway in hepatocarcinogenesis. J Hepatol 38: 605–614. [DOI] [PubMed] [Google Scholar]
- 11. DeZwaan-McCabe D, Riordan JD, Arensdorf AM, Icardi MS, Dupuy AJ, et al. (2013) The stress-regulated transcription factor CHOP promotes hepatic inflammatory gene expression, fibrosis, and oncogenesis. PLoS Genet 9: e1003937 doi:10.1371/journal.pgen.1003937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dupuy AJ, Rogers LM, Kim J, Nannapaneni K, Starr TK, et al. (2009) A modified sleeping beauty transposon system that can be used to model a wide variety of human cancers in mice. Cancer Res 69: 8150–8156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Riordan JD, Keng VW, Tschida BR, Scheetz TE, Bell JB, et al. (2013) Identification of Rtl1, a retrotransposon-derived imprinted gene, as a novel driver of hepatocarcinogenesis. PLoS Genet 9: e1003441 doi:10.1371/journal.pgen.1003441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. O'Donnell KA, Keng VW, York B, Reineke EL, Seo D, et al. (2012) A Sleeping Beauty mutagenesis screen reveals a tumor suppressor role for Ncoa2/Src-2 in liver cancer. Proc Natl Acad Sci U S A 109: E1377–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Marciniak SJ, Yun CY, Oyadomari S, Novoa I, Zhang Y, et al. (2004) CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev 18: 3066–3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yamaguchi H, Wang HG (2004) CHOP is involved in endoplasmic reticulum stress-induced apoptosis by enhancing DR5 expression in human carcinoma cells. J Biol Chem 279: 45495–45502. [DOI] [PubMed] [Google Scholar]
- 17. Zinszner H, Kuroda M, Wang X, Batchvarova N, Lightfoot RT, et al. (1998) CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev 12: 982–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Crozat A, Aman P, Mandahl N, Ron D (1993) Fusion of CHOP to a novel RNA-binding protein in human myxoid liposarcoma. Nature 363: 640–644. [DOI] [PubMed] [Google Scholar]
- 19. Rabbitts TH, Forster A, Larson R, Nathan P (1993) Fusion of the dominant negative transcription regulator CHOP with a novel gene FUS by translocation t(12;16) in malignant liposarcoma. Nat Genet 4: 175–180. [DOI] [PubMed] [Google Scholar]
- 20. Luo J, Solimini NL, Elledge SJ (2009) Principles of cancer therapy: oncogene and non-oncogene addiction. Cell 136: 823–837. [DOI] [PMC free article] [PubMed] [Google Scholar]