

Background: UCP1 transports protons across the inner mitochondrial membrane and generates heat.
Results: Oligomeric forms of UCP1 transported protons across phospholipid bilayers and the conformation and proton transport activity were affected by cardiolipin.
Conclusion: UCP1 is functional in its monomeric and associated forms in membranes.
Significance: Existence of functional associated forms of UCP1 provides insights into the mechanism of action of UCPs in mitochondria.
Keywords: Cardiolipin, Circular Dichroism (CD), Protein Folding, Protein Self-assembly, Proton Transport, Uncoupling Proteins, Mitochondrial Membrane Proteins, Protein Reconstitution, Proteoliposomes, Structure-Function Relationship
Abstract
Uncoupling protein-1 (UCP1) is abundantly expressed in the mitochondrial inner membrane of brown adipose tissues and has an important role in heat generation, mediated by its proton transport function. The structure and function of UCP1 are not fully understood, partially due to the difficulty in obtaining native-like folded proteins in vitro. In this study, using the auto-induction method, we have successfully expressed UCP1 in Escherichia coli membranes in high yield. Overexpressed UCP1 in bacterial membranes was extracted using mild detergents and reconstituted into phospholipid bilayers for biochemical studies. UCP1 was folded in octyl glucoside, as indicated by its high helical content and binding to ATP, a known UCP1 proton transport inhibitor. Reconstituted UCP1 in phospholipid vesicles also exhibited highly helical structures and proton transport that is activated by fatty acids and inhibited by purine nucleotides. Self-associated functional forms of UCP1 in lipid membranes were observed for the first time. The self-assembly of UCP1 into tetramers was unambiguously characterized by circular dichroism and fluorescence spectroscopy, analytical ultracentrifugation, and semi-native gel electrophoresis. In addition, the mitochondrial lipid cardiolipin stabilized the structure of associated UCP1 and enhanced the proton transport activity of the protein. The existence of the functional oligomeric states of UCP1 in the lipid membranes has important implications for understanding the structure and proton transport mechanism of this protein in brown adipose tissues as well as structure-function relationships of other mammalian UCPs in other tissues.
Introduction
Located in the inner mitochondrial membrane (IMM)3 of brown adipose tissue, UCP1 mediates proton leak that uncouples respiratory electron transport from ATP synthesis and produces heat (1, 2). UCP1-mediated proton leak is activated by fatty acids (FAs) and inhibited by purine nucleotides (2). The heat generation from UCP1-mediated proton transport is associated with non-shivering thermogenesis in infants and cold-acclimatized mammals (1, 2). Thermogenic activity of UCP1 has also been shown to reduce obesity and improve insulin sensitivity (3, 4). Thus, understanding the structure and function of this protein is essential for drug development and therapeutic applications. Consisting of 306 amino acids (molecular mass ∼ 33 kDa), UCP1 shares common structural features with members of the SLC25 (solute carrier protein) family (2, 5). The structure of UCP1 contains three repeated domains (tripartite structure), each of which is composed of two hydrophobic transmembrane α-helical regions spanning the inner mitochondrial membrane (2, 5). Currently, there is no high resolution structure available for UCP1. It has been suggested that the structures of UCP1 and other human UCPs (UCP2, -3, -4, and -5) are comparable to each other and to the crystal structure of the ADP/ATP carrier (AAC) (5, 6). Recently, using molecular fragment searching solution NMR, the structure of UCP2 was reported to be similar to that of AAC (7).
The structure and function of UCP1 can be strongly influenced by the surrounding phospholipid molecules in the IMM (8, 9). Cardiolipin (CL, [(1′,3′-bis(1,2-dioleoyl-snglycero-3-phospho)-sn-glycerol (sodium salt))] is a major lipid component of the IMM that accounts for ∼20 mol% of the total lipid content (8, 9). Each CL molecule is composed of two phosphatidylglycerols connected through a central glycerol backbone to form a dimeric structure. The conical shape and negative charge of CL at physiological pH play a key role in mitochondrial membrane morphology and complex protein organization in the IMM (9, 10). It has been observed that mutations leading to low expression and structural defects of CL can be pathogenic (11, 12). In addition, certain mitochondrial carrier proteins require CL for their transport activity in liposome systems (13–16). The monomeric form of AAC was bound to three CL molecules in its crystal structure (16). Therefore, CL could have multiple roles in the IMM, including its participation in mitochondrial carrier protein assembly, mitochondrial shape, and signal transduction (8, 9). Given its localization in the IMM, interaction of CL with UCP1 and its influence on this protein structure and function would not be surprising. In a previous study we reported that the proton transport rates of UCP2 and UCP5 in lipid vesicles were increased in the presence of CL (17).
A major hurdle in understanding the structure and mechanism of ion transport of UCP1 is the challenge of obtaining appropriately folded pure protein (18, 19). Recombinant protein expression in bacterial systems is an efficient and inexpensive method for protein production. However, refolding of UCP1 from inclusion bodies could only yield partially folded proteins in mild detergents and liposomes (6, 17, 19). The recent new development of bacterial strains, fusion proteins, and alternative expression methods provide other possibilities for recombinant UCP1 to be targeted toward the inner membrane of Escherichia coli (20–22). Cell-free expression systems have also been used for the production of UCP1 (23). In this study we report the use of the small periplasmic leader sequence PelB, provided by the pET26b(+) vector, as a means to target UCP1-His6 toward the inner membrane of E. coli. Recombinant UCP1 was obtained in high yield from the bacterial inner membrane using the auto-induction method to achieve expression. The purified recombinant UCP1 was appropriately folded in the detergent octyl glucoside (OG) and phospholipid vesicles. More importantly, self-assembly of UCP1 in lipid membranes was observed using a combination of spectroscopic and electrophoretic analyses. In addition, the role of CL in structural organization and proton transport activity of UCP1 was investigated in detail. Overall, this study provides essential information on the folding, structural aspects, and proton transport mechanism of UCP1 in the IMM.
EXPERIMENTAL PROCEDURES
UCP Constructs and Chemicals
The human UCP1 cDNA clone was purchased from ATTC (Manassas, VA) and received in a pCR-BluntII-TOPO vector. The construct was subcloned into the pET21d vector (Novagen) and modified to contain an N-terminal His tag followed by the tobacco etch virus protease recognition site (ENLYFQG) as previously described (6). The His6-ENLYFQG-hUCP1 coding sequence was subcloned into the pET26b(+) vector (a gift from Dr. Joel Weadge, Wilfrid Laurier University). The construct was then transformed into E. coli BL21 CodonPlus (DE3)-RIPL for production of the recombinant fusion protein containing an N-terminal PelB leader sequence fused to the His6-ENLYFQG-hUCP1 protein (recombinant UCP1). To verify the identity of the recombinant protein, the recombinant cDNA in the pET26 expression vector was sequenced. Egg yolk l-α-lecithin (Sigma) contained at least 60% (by weight) phosphatidylcholine. The remaining 40% was composed mostly of phosphatidylethanolamine and other lipids. Phospholipids POPC and CL were obtained from Avanti Polar (Alabaster, AL). C8E4 was obtained from Bachem (Torrance, CA). DDM was from Calbiochem-EMD Biosciences (Gibbstown, NJ). CHAPS and OG were from Affymetrix (Santa Clara, CA). The fluorescent probe SPQ (99% purity) was from Biotium Inc. (Burlington, ON). All other chemicals were from Sigma.
Expression, Membrane Extraction, and Purification of UCP1
Recombinant UCP1 was overexpressed in E. coli either by the conventional use of isopropyl β-d-thiogalactoside (IPTG) (0.1 and 1 mm) at various temperatures (16, 25, and 37 °C) or using the auto-induction method (24). When protein was expressed by the auto-induction method, the bacterial culture was grown in the auto-induction media (1% Tryptone, 0.5% yeast extract, 1 mm MgSO4, 0.5% glycerol, 0.05% glucose, 0.2% lactose, 25 mm (NH4)2SO4, 50 mm KH2PO4, 50 mm Na2HPO4) at 22 °C for 20 h before collection. Bacterial cells were collected by centrifugation at 5000 × g for 10 min (4 °C). Cell pellets were resuspended in extraction buffer (EB) (20 mm Tris-HCl, 500 mm NaCl, 5 mm MgCl2, pH 8.0). Bacterial cell lysis was achieved using a French press operating at 16,000 p.s.i. in the presence of cOmplete protease inhibitor mixture (EDTA-free) (Roche Applied Science), DNase (0.5 mg/ml), and lysozyme (0.2 mg/ml). The cell lysate was cleared of cell debris and inclusion bodies by centrifugation (20,000 × g, 20 min, 4 °C). Subsequently, the supernatant was ultracentrifuged at 256,631 × g for 30 min to collect the bacterial membranes in the pellet fraction. The enrichment of membranes in the pellet fraction was confirmed using the NADH oxidase activity assay (described below).
Bacterial membranes were solubilized in EB supplement with 1% (w/v) LDAO for 3 h at 4 °C before purifying recombinant UCP1. The mixture was spun at 10,000 × g for 10 min to remove insoluble particles. The supernatant containing solubilized recombinant UCP1 was then subjected to nickel-nitrilotriacetic acid chromatography for purification under non-denaturing conditions (1% LDAO, 20 mm Tris-HCl, 500 mm NaCl, 1 mm [Tris (hydroxypropyl) phosphine], pH 8.0). The concentrations of imidazole for binding, washing, and eluting steps were 20, 40, and 400 mm, respectively. Detergent exchange was performed on the column at the washing step. Purity and quantity of eluted proteins were determined by SDS-PAGE and protein concentration assays (Bradford or Lowry assays), respectively. The pooled eluted fractions containing purified UCP1 were desalted using Econo-Pac 10DG Columns (Bio-Rad). The final purified protein (∼0.2 mg/ml) was stored in desalting buffer (50 mm NaCl, 20 mm Tris-HCl, 1% glycerol, and 1% detergent, pH 8.0) and kept at −80 °C.
Western Blot Analysis
Approximately 10–50 μg of total proteins were run on a 12% SDS-PAGE gel and transferred (110 min, 15 V) to nitrocellulose membranes using the semi-dry technique. The membrane was stained with Amido Black to confirm the efficiency of transfer, and the membrane was blocked overnight at 4 °C in TBS-buffer containing 5% skim milk and 0.05% Tween 20. Two primary antibodies were used to confirm the presence of hUCP1-His6 protein, including mouse IgG2b anti-histidine (Millipore) and rabbit IgG anti-UCP1/2/3 (Santa Cruz Biotechnology, Inc.). Peroxidase-conjugated antibodies raised either against mouse or rabbit (Rockland) were used as secondary antibodies. Immunoreaction was visualized by luminescence using ECL Western blot reagent (GE Healthcare).
NADH Oxidase Activity Assay
After the fractionation of E. coli to isolate bacterial membranes, an NADH oxidase activity assay was used to determine the relative enrichment of membranes in each fraction (25). The membrane-embedded enzyme activity was analyzed based on its catalytic activity of converting NADH from its reduced to oxidized forms. A decrease in A340 represents NADH oxidase activity. Briefly, 0.15 mg of total protein from either the supernatant or membrane fraction was added to 12 mm β-NADH in 50 mm Tris-HCl, pH 7.5, and the absorbance at 340 nm was monitored for a period of 30 min. The rate of decrease in A340 is proportional to NADH oxidase activity. Specific enzyme activity was calculated per mg of protein (ϵNADH = 6220 m−1cm−1).
Semi-native Polyacrylamide Gel Electrophoresis Analysis
The amount of SDS in PAGE was significantly reduced to provide “semi-native” conditions (26). Thus, SDS was only included in the gel and running buffer at a concentration of 2 mm (as compared with 35 mm in denaturing SDS-PAGE). SDS was omitted from the sample loading buffer. In addition, protein samples were not heated before SDS-PAGE. All samples were incubated at room temperature for 5–10 min before loading the gel. SDS-PAGE gels were stained with Coomassie Brilliant Blue R-250.
Proteolytic Digestion and Mass Spectrometry Analysis
To confirm the identity of the recombinant protein in bands of different molecular weights, purified protein bands resolved on semi-native SDS-PAGE were excised, trypsin-digested, and analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS) in a data-dependent acquisition. Briefly, trypsin-digested fractions were injected and separated using a NanoAcquity (Waters) LC-MS/MS equipped with a 25 cm × 75-μm inner diameter C18 column. MS analysis was done on a QExactive (Thermo Scientific) using higher energy collision dissociation. De novo sequencing of fractionated proteins was performed with Peak Studio 6.0 Software using the Uniprot Swiss database for Homo sapiens, with a false discovery rate of <1%. In addition, post-translational modifications were incorporated in the search algorithm.
Analytical Ultracentrifugation
Analytical ultracentrifugation of UCP1 in 1% OG solution (20 mm Tris, 50 mm NaCl, 1% glycerol, pH 8.0) was carried out using a Beckman Optima XL-A analytical ultracentrifuge, with an An-60Ti four-hole rotor. A sedimentation velocity experiment was performed at 50,000 rpm at 20 °C. Absorbance of UCP1 samples and blank buffer were taken at 3-min interval at 280 nm for 5 h. In the sedimentation equilibrium experiment, two samples of UCP1 (4.7 and 3.1 μm) were spun at 10,000, 13,000, and 16,000 rpm at 4 °C. Absorbance at 280 nm was recorded. The SEDNTERP software was used to evaluate the solvent density, viscosity, and the partial specific volume of UCP1 based on its amino acid sequence (27). At 20 °C the solvent density and partial specific volume of UCP1 were calculated to be 1.00373 and 0.7456 ml/g, respectively. At 4 °C, the solvent density and partial specific volume of UCP1 were 1.00551 g/ml and 0.7388 ml/g, respectively. Partial specific volume of 0.83 ml/mg was used for the OG detergent. Analysis of the sedimentation velocity experiment was done using SEDFIT fitted to the continuous distribution model c(s) (28). Analysis of the sedimentation equilibrium experiment was performed using the Origin MicroCal XL-A/CL-I Data Analysis Software Package Version 4.0.
Reconstitution of UCP1 into Liposomes
The reconstitution of purified UCP1 into liposomes for spectroscopic and transport studies was described previously (17). Briefly, lipids were dissolved in chloroform, dried overnight under a vacuum, and rehydrated in the desired buffer. Phospholipids were solubilized in C8E4 to a final detergent/phospholipid ratio of 2.5 by weight. Purified proteins were then added to the mixed lipid/detergent micelles in each particular experiment. For ion transport experiments, the fluorescent probe SPQ was included in the internal buffer entrapped inside the liposomes at a concentration of 3 mm. Protein-free liposome controls were prepared in parallel for all experiments. Liposomes or proteoliposomes were formed spontaneously after detergent removal by SM-2 Biobeads (Bio-Rad). In ion transport assays, the external SPQ probe was removed using a coarse Sephadex G25–300 (GE Healthcare) spin column.
CD Spectroscopic Measurements
Far-UV CD spectra were measured on an Aviv 215 spectropolarimeter (Aviv Biomedical, NJ). Ellipticity was reported as mean residue ellipticity, [θ]. Far-UV CD and near-UV CD measurements were carried out in 0.1- and 0.5-cm path-length quartz cells, respectively, at 1 nm resolution (25 °C). The reported spectra are an average of at least eight scans. Secondary structure content of proteins was estimated from backbone CD spectra using the deconvolution program CDSSTR, and the analysis was based on a set of 48 reference proteins and performed on the Dichroweb web site (29, 30). Thermal denaturation of UCP1 in detergents and liposomes was monitored by the protein ellipticity at 222 nm, from 15 to 90 °C.
Fluorescence Measurements
Steady-state fluorescence measurements were performed using a Cary Eclipse spectrophotometer (Varian, CA). Excitation bandwidth slit for all measurements was 5 nm, and a scan speed of 600 nm/min was used throughout the experiments (25 °C).
Proton Transport Measurements
Proton transport measurements were performed as described previously (17). Liposomes or proteoliposomes were formed after reconstitution, trapping the fluorescent probe SPQ inside. Fluorescence of SPQ (λex = 347 nm; λem = 442 nm) was quenched by TES anion (TES−). In each transport assay, 40 μl of proteoliposomes (0.5 mg lipid) (20 mm TES, 54 mm tetraethylammonium sulfate, and 0.7 mm EDTA, pH 7.2) were incubated in 1.96 ml of external media (20 mm TES, 54 mm K2SO4 and 0.7 mm EDTA, pH 7.2). The osmotic pressure was kept equal across the lipid membrane at the beginning of the measurements. Ion transport in each experiment was driven by the influx of K+ from the external buffer mediated by the K+-ionophore valinomycin. Upon the inward diffusion of K+ across the membrane, osmotic balance was disrupted, and UCP1 transported protons out of the lipid vesicles to offset this membrane potential difference. In a low pH environment, TES (pKa of TES ∼ 7.4) is fully protonated and does not affect the fluorescence of SPQ. Upon the loss of a proton (at high pH), the TES anion quenches SPQ fluorescence. Lauric acid (LA) and palmitic acid (PA) were added at 100 μm to activate proton transport. All ion transport data were corrected by subtraction from the nonspecific proton leak and calibrated for the SPQ fluorescence response and internal volume of proteoliposomes. The final protein content in proteoliposomes was calculated using a modified Lowry concentration assay, as described previously (17).
Amino Acid Sequence Analysis and Structural Modeling of Neuronal UCPs
Protein sequence analysis and primary sequence alignment of UCPs and AAC were performed using T-Coffee (31). The three-dimensional structural model of UCP1 was obtained using MODELLER 9.9 (32) after the sequence alignment based on the crystal structure of AAC (PBD ID 2C3E) (16). Structural models were viewed by PyMOL (33).
RESULTS
Optimization of UCP1 Expression in Bacterial Membranes
Overexpressed recombinant UCP1 was targeted to bacterial membranes using the periplasmic leader sequence PelB provided by the pET26b(+) vector (Fig. 1). Production of UCP1 in E. coli BL21 CodonPlus (DE3)-RIPL was monitored under various expression temperatures (37, 25, and 16 °C) and IPTG concentrations (1 and 0.1 m). No additional growth was observed after the addition of IPTG (absorbance ∼ 0.5 over a 24-h growth period). This implies that strong expression of UCP1 could be toxic to the cells. To overcome this problem, auto-induction was used as a milder expression method. UCP1, overexpressed with this method, was detected in its monomeric and associated (dimeric and tetrameric) forms when extracted from the bacterial membranes using semi-native SDS-PAGE and Western blot analysis, probed with both anti-histidine (Fig. 2A) and anti-UCP1/2/3 (data not shown) antibodies. In addition, mass spectrometric analysis of the expressed recombinant protein verified the identification of human UCP1 without any additional post-translational modification. The mass spectrometric analysis also confirmed the identity of UCP1 in all monomeric, dimeric, and tetrameric bands (data not shown). IPTG-induced expression of UCPs in previous studies has resulted in targeting to inclusion bodies (6, 17, 18, 34). The more gradual protein expression offered by auto-induction may prevent overloading of the biosynthetic machinery and stimulation of stress responses due to the gradual reduction of glucose-mediated catabolite repression (24). The identity of the fraction in which UCP1 was enriched was verified to be the bacterial membrane fraction using an NADH oxidase activity assay (Fig. 2, B and C). Consequently, the use of PelB leader sequence together with the auto-induction method allows overexpression of UCP1 in the bacterial membranes.
FIGURE 1.
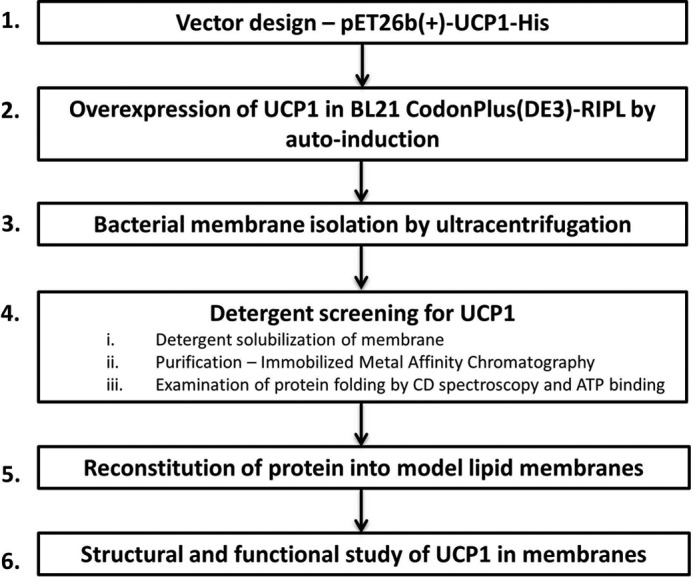
Schematics of UCP1 expression, purification, and reconstitution. Detailed protocols for each step are provided under “Experimental Procedures.”
FIGURE 2.

Expression of UCP1 in bacterial membranes and refolding of the protein in the detergent OG. A, Western blot analysis of UCP1 in bacterial membranes (Mem.) and supernatant (Sup.) of E. coli using mouse IgG2b anti-histidine antibody, detected by chemiluminescence using a horseradish peroxidase-linked secondary antibody. Molecular mass markers (kDa) are indicated to the left. B, NADH oxidase activity assay of the E. coli supernatant and membrane fractions obtained after expression of recombinant UCP1 using the auto-induction method. The assay measures the conversion of NADH from its reduced to oxidized forms, indicative in the decrease of A340 over time. C, average specific NADH oxidase activity (units/mg protein) of the supernatant and bacterial membrane fractions shown in B. D, far-UV CD spectra of 3–5 μm UCP1 folded in 1% OG in buffer solution in the absence and presence of 100 μm ATP at 25 °C. The inset of D shows the corresponding near-UV CD spectra. E, comparative thermal denaturation of UCP1 in 1% OG in the absence and presence of 50 μm ATP. The normalized molar ellipticity was detected at 222 nm, and Tm was calculated using the Hill equation to fit the data. The arrow indicates the increase in Tm (or increase in the protein thermal stability).
Folding of UCP1 in Detergents
The folding of UCP1 was analyzed in four different detergents: LDAO, DDM, CHAPS, and OG (Fig. 1). The secondary structure of UCP1 in each detergent was examined by far-UV CD spectroscopy, and the helical content for each was estimated (Table 1). Among the four detergents, DDM induced the least helical conformation in UCP1, and the protein exhibited a “β-sheet-like” conformation in this medium. In addition, the protein stability in DDM was poor, resulting in protein aggregation/precipitation after only a few hours at 4 °C. UCP1 in LDAO and CHAPS detergents was only partially helical (Table 1). In contrast, UCP1 in 1% OG exhibited the highest α-helical content, with a negative maximum ellipticity at 222 nm, a shoulder at 208 nm, and a positive maximum ellipticity around 195 nm (Fig. 2D). Deconvolution of the CD spectra of UCP1 in OG revealed a 67% α-helical content (Table 1). To further characterize the folding of UCP1 in detergents, thermal unfolding experiments were performed in which the ellipticity of UCP1 spectra at 222 nm was monitored in the 15–90 °C temperature range (Fig. 2E; data are only shown for OG). Among the four protein-detergent systems, only UCP1 in OG exhibited a characteristically cooperative unfolding curve. The melting point (Tm) of UCP1 in OG was 59.2 ± 1.1 °C, indicating that the protein structure was relatively stable in this detergent. Other evidence for the proper folding of UCP1 in OG was that of binding to its proton transport inhibitor, ATP. This interaction was better detected in the near-UV region, which is sensitive to changes in the local environment of the aromatic amino acids influenced by binding (Fig. 2D, inset). Furthermore, binding of ATP to UCP1 in OG resulted in a more stable conformation, indicated by the increase in the Tm to 61.5 ± 0.8 °C (Fig. 2E). Collectively, the data suggest that UCP1 remained folded in a native-like functional form when extracted from bacterial membranes using OG.
TABLE 1.
Secondary structure composition of UCP1 in detergents and lipid vesicles
Deconvolution of CD spectra was performed using the CDSSTR program on the Dichroweb website (see “Experimental Procedures”). The values represent the percentage of secondary structure composition. NRMSD, normalized root mean square deviation, denotes the best fit between the calculated and experimental CD spectra. PC, l-α-phosphatidylcholine.
| Protein environment | α-Helix | β-Strand | Turn | Random | NRMSD |
|---|---|---|---|---|---|
| Detergents | |||||
| DDM micelles | 16 | 26 | 20 | 38 | 0.026 |
| CHAPS micelles | 22 | 27 | 23 | 28 | 0.022 |
| LDAO micelles | 25 | 23 | 20 | 32 | 0.018 |
| OG micelles | 67 | 5 | 13 | 15 | 0.012 |
| OG micelles + ATP | 66 | 6 | 16 | 17 | 0.011 |
| Lipid bilayers | |||||
| l-α-Lecithin | 77 | 5 | 8 | 8 | 0.001 |
| l-α-Lecithin + ATP | 76 | 10 | 8 | 7 | 0.001 |
| Soy PC vesicles | 75 | 8 | 8 | 8 | 0.001 |
| POPC vesicles | 82 | 5 | 7 | 6 | 0.004 |
| POPC + 5% CL | 83 | 5 | 7 | 5 | 0.004 |
| POPC + 5% CL + ATP | 84 | 5 | 8 | 4 | 0.003 |
| POPC + 10% CL | 83 | 6 | 4 | 5 | 0.001 |
| POPC + 20% CL | 85 | 5 | 7 | 3 | 0.005 |
| POPC + 50% CL | 82 | 8 | 15 | 6 | 0.002 |
Reconstituted UCP1 Is Highly Helical in Phospholipid Bilayers and Transports Protons
The structure and ion transport function of reconstituted UCP1 were studied in different lipid systems, including egg yolk l-α-lecithin, soybean l-α-phosphatidylcholine extract, and synthetic POPC bilayers (Fig. 1). Although the far-UV CD spectra of UCP1 in lipid membranes were comparable to the spectrum in OG, protein ellipticity was enhanced in the phospholipid dispersions (Fig. 3A). This enhancement in ellipticity translates to a higher helical content of UCP1 in lipid bilayers (Table 1). Reconstituted UCP1 in liposomes was also able to bind ATP, as indicated in the change in the protein binding microenvironment observed in the near-UV CD spectra (Fig. 3A, inset). Moreover, upon the addition of ATP to the reconstituted protein, a decrease in the intensity of the fluorescence spectra of UCP1 in the lipid bilayers (mainly contributed by the two Trp residues located close to the nucleotide binding site) was observed, implying a conformational change (Fig. 3B).
FIGURE 3.

Reconstitution of UCP1 into egg yolk l-α-lecithin vesicles; conformation and ion transport analysis. A, comparative far-UV CD spectra of reconstituted UCP1 in l-α-lecithin vesicles in the absence and presence of 100 μm ATP at 25 °C. The inset of A shows the corresponding near-UV CD spectra. B, fluorescence spectra (excited at 280 nm) of reconstituted UCP1 in l-α-lecithin vesicles in the absence and presence of 100 μm ATP. When UCP1 was excited at 295 nm (specific to Trp), similar spectral characteristics were observed (data not shown). C, schematic of proton transport assays. The fluorescent probe SPQ was incubated with TES buffer inside the liposomes/proteoliposomes. Upon the efflux of protons mediated by UCP1, TES buffer is deprotonated and TES anion quenches SPQ. D, recorded H+ efflux through UCP1 reconstituted in phospholipid vesicles. Proton flux shown was activated by the addition of 100 μm PA. Valinomycin was added at 0.4 μm to initiate the membrane potential difference needed for triggering the proton transport. The nonspecific proton leak was subtracted from the proton transport rate. In the presence of 500 μm ATP, the UCP1-mediated proton transport was completely inhibited, and the proton transport rate was comparable to that of the nonspecific leakage rate. The inset shows comparative proton transport activity of UCP1 in the absence and presence of 500 μm ATP. An average protein-free liposome blank displays nonspecific proton leakage (dashed line).
To examine the transmembrane proton transport function of UCP1, purified recombinant UCP1 was reconstituted into liposomes containing the fluorescence probe SPQ trapped in their interior lumen (17) (Fig. 3C). FA-dependent UCP1-mediated proton flux was driven by diffusion of K+ from the external buffer into the proteoliposomes in the presence of the ionophore valinomycin (Fig. 3C). Two FAs were used as UCP1 proton transport activators: LA (C12 chain) and PA (C16 chain). Both FAs activated the proton transport mediated by UCP1, resulting in an efflux of protons across the bilayer membrane (Fig. 3, C and D). The UCP1 proton transport rates were higher when activated by PA. In egg yolk l-α lecithin vesicles, PA (100 μm) activated UCP1 proton transport at a rate of 3.8 ± 0.6 μmol·min−1·mg protein−1, which is more than 6 times higher than the rate of proton transport activated by LA at the same concentration (0.6 ± 0.3 μmol·min−1·mg protein−1). This observation is consistent with previous studies on the effect of FA chain length on the proton transport rate of UCPs (35). Furthermore, in the presence of 500 μm ATP, complete inhibition of LA-activated and or PA-activated proton transport was observed (Fig. 3D). Thus, ATP could interact with the purine nucleotide binding site and induce conformational change in UCP1, leading to the inhibition of the proton transport pathway. Overall, the membrane-extracted UCP1 folded with a high helical content in lipid bilayers and was functional in proton transport and inhibited by ATP.
Associated State of UCP1 Monitored by CD and Fluorescence Spectroscopy, Semi-native SDS-PAGE, and Analytical Ultracentrifugation
UCP1 in detergents and liposomes displayed high helical contents. However, the far-UV CD spectra of UCP1 did not resemble a typical α-helical protein. The typical CD spectrum of an α-helical protein has two negative maxima, at 208 and 222 nm (with equal or more intense negative ellipticity at 208 nm) and a positive maximum around 192 nm. In both detergent and lipid vesicle milieus, UCP1 possessed the characteristics of an α-helix, with the π → π* exciton split bands at ∼190 and 208 nm and the n → π* transition at ∼222 nm (Figs. 2D, 3A, and 4A). However, compared with a typical α-helical CD spectrum, the parallel band at ∼208–210 nm exhibited a shoulder-like negative maximum with decreased ellipticity (Figs. 2D, 3A, and 4A). The θ208/θ222 ratio of UCP1 spectra in these environments was less than 1 (Fig. 4B). It has been proposed that the θ208/θ222 ratio could be used to distinguish coiled coil motifs from independent helices (36–38). With multi-transmembrane domain proteins such as UCP1, the low ellipticity ratio could be due to oligomerization of monomers and/or packing of helical domains within a monomer. Under our experimental conditions on semi-native SDS-PAGE, the pure reconstituted UCP1 appeared as the only electrophoretic band at ∼140 kDa (∼4 × Mr UCP1) (Fig. 4C). Western blot analysis further confirmed that this tetrameric state of UCP1 originated from the expressed protein in bacterial membrane and not as an artifact from detergent solubilization (Fig. 2A). Tetrameric forms of UCP1 were also detected in bacterial membrane protein extracts in OG (data not shown). Therefore, expressed UCP1 was already in a mixture of monomeric and associated (dimeric and tetrameric) forms in the bacterial membranes, and these molecular forms were preserved during protein purification and reconstitution into liposomes.
FIGURE 4.

Titration of UCP1 in POPC liposomes with SDS. A, far-UV CD spectra of reconstituted UCP1 in POPC liposomes titrated with SDS (0–84 mm). Ai shows the low SDS region of the spectra in greater detail (0–2 mm) where increased SDS enhanced negative ellipticity at both 208 and 222 nm. Aii shows the high SDS region (2–84 mm) in greater detail, where increased SDS resulted in decreased negative ellipticity at 222 nm but did not affect the ellipticity at 208 nm. B, plot of θ208/θ222 versus SDS concentration reveals a cooperative dissociation of UCP1 tetramers in the presence of SDS. The θ208/θ222 ratio was normalized and K1/2 (SDS at 50% protein dissociation) was calculated using the Hill fitting (K1/2 = 1.42 ± 0.14 mm). C, corresponding semi-native SDS-PAGE (12%) of reconstituted UCP1 in POPC liposomes during SDS titration experiment, stained with Coomassie Brilliant Blue. UCP1 tetramers (T) were the only species detected in POPC liposomes when treated with loading buffer lacking SDS. As the SDS concentration increased beyond the CMC, transition toward dominantly UCP1 dimers (D) was observed. At 84 mm SDS (∼40× CMC of SDS), UCP1 tetramers were completely absent, UCP1 dimers became prominent, and a small concentration of monomers (M) was present.
To further examine and confirm the tetrameric state of UCP1 in detergents and liposomes, analytical ultracentrifugation was performed for UCP1 in 1% OG detergent. Sedimentation velocity analysis revealed a predominant (∼90%) homogenous species at 6.5 S corresponding to ∼180 kDa (Fig. 5). This molecular mass corresponds with the expected size of a tetramer of UCP1 (with monomeric Mr UCP1 of 37.2) in OG micelles (8 kDa/micelle). In addition, sedimentation equilibrium analysis of 4.7 and 3.1 μm UCP1 in 1% OG resulted in a monodisperse species with an average molecular mass of 177 kDa, which is also consistent with the size of tetrameric UCP1 (data not shown).
FIGURE 5.

Sedimentation velocity experiment of UCP1 in 1% OG solution. A, the plot shows a superposition of a selection of experimental (dots) and fitted (lines) absorbance profile at 280 nm during ultracentrifugation at 50,000 rpm of UCP1 in OG. Scans were taken every 3 min as described under “Experimental Procedures.” B, superimposition of differences between the experimental and fitted curves. C, sedimentation velocity analysis made in terms of a continuous distribution of species.
To analyze the dissociation/unfolding pathway and stability of UCP1 tetramers, we performed an SDS titration on the reconstituted UCP1 in lipid bilayers and monitored the changes in the protein structure using CD and fluorescence spectroscopy. Fig. 4A shows the far-UV CD spectra of the UCP1 tetramer being dissociated with increasing SDS concentration. In the lower SDS concentration range (0–2 mm), the far-UV CD spectra of UCP1 exhibited characteristics of an oligomeric-associated conformation, as shown by a negative maximum at 222 nm and a negative shoulder at 208–210 nm. As SDS concentration increased from 0 to 2 mm, negative ellipticity at 208 and 222 nm was enhanced (Fig. 4Ai). Deconvolution of CD spectra of UCP1 in this region revealed an increase in helical content, indicating a less constrained helical conformation. The negative shoulder at 210 nm blue-shifted toward 208 nm and gradually became a visible negative maximum (Fig. 4Ai). These results suggest the formation of a more relaxed helical conformation (fewer intermolecular interactions between helices and more independence for monomers) (Fig. 4). Increase in the SDS concentration above 2 mm did not affect the θ208 ellipticity of the protein, whereas the θ222 negative ellipticity further decreased (Fig. 4Aii). Plotting θ208/θ222 ratio versus SDS revealed a two-state cooperative dissociation curve for UCP1 in lipid bilayers, with a dissociation constant of K1/2 (50% dissociated) of 1.42 ± 0.14 mm SDS (Fig. 4B). This concentration is close to the CMC of SDS in buffer (50 mm NaCl, 20 mm Tris, pH 8.0). The CMC for SDS in this buffer was 2 ± 0.5 mm (at 25 °C) and was determined by isothermal titration calorimetry as described previously (39) (data not shown). Results from semi-native SDS-PAGE revealed a tetrameric form of reconstituted UCP1 being dissociated and unfolded into dimers and monomers with increasing concentrations of SDS (Fig. 4C). At 4 mm SDS, a mix of tetramers and dimers was observed that corresponds to the CD spectral ratio of UCP1, θ208/θ222, reaching the value of 1 (Fig. 4, B and C). At the end point of the SDS titration curve (84 mm), no tetramer was detected by SDS-PAGE and Western blotting. UCP1 dimers were the predominant molecular form at this point along with some UCP1 monomers (Fig. 4C). The value of the θ208/θ222 in the CD spectra exceeds 1 at this SDS concentration, which is consistent with a full dissociation of the tetrameric state of UCP1 to dimers and monomers. In this way the change in the CD spectra of UCP1 during the SDS titration correlated with the changes in its associated state, observed using semi-native SDS-PAGE.
Additional evidence for dissociation of UCP1 oligomers was also provided by intrinsic fluorescence spectra of the protein at increasing SDS concentrations (Fig. 6). The Trp fluorescence (λex = 295 nm) spectra exhibited an increase in intensity and a conspicuous red shift in the maximum emission in the presence of SDS (Fig. 6). Plotting of the fluorescence intensity versus SDS concentration revealed a three-state transition curve of UCP1 during SDS titration (Fig. 6A). The first transition, with K1/2 = 1.3 ± 0.1 mm SDS, corresponded to the dissociation of UCP1 tetramers into dimers (compare Figs. 6Ai and 4B). The second transition (not clearly detected in the CD measurements, Fig. 4B), with K1/2 = 23 ± 1 mm SDS, was interpreted as the dissociation of UCP1 dimers (Fig. 6Aii). Plotting the λmax of Trp versus SDS provided information about the microenvironment of Trp residues (Fig. 6B). The maximum emission of Trp residues experienced a red shift from ∼331 to ∼338 nm at SDS concentrations close to 20 mm, indicating that the Trp residues became more exposed to a more polar environment (Fig. 6B). This shift of emission λmax of Trp occurred at an SDS concentration close to the second transition point observed for the UCP1 fluorescence intensity (∼15–18 mm) (Fig. 6B). The fluorescence results together with the previous data from semi-native SDS-PAGE (Figs. 6 and 4C, respectively) suggest two distinct transitions for UCP1. The first transition occurred at the low SDS (close to CMC), where UCP1 tetramers dissociated into their corresponding dimeric units upon formation of SDS micelles. During this transition, the Trp residues remained in a weakly polar environment, as indicated by the emission λmax value at ∼331 nm. The second transition occurred at a much higher SDS (∼10× CMC), where UCP1 dimers were further transformed to a combination of loose dimeric units and monomers. In comparison with the first transition, at the second transition, Trp residues were exposed to a more polar environment (λmax ∼ 338 nm).
FIGURE 6.

Dissociation of UCP1 tetramers in POPC liposomes by SDS, monitored by Trp fluorescence. A, plot of maximum fluorescence of UCP1 (excited at 295 nm) versus SDS revealed a three-state dissociation process for UCP1 tetramers in lipid vesicles. The inset (Ai) shows the maximum fluorescence intensity of UCP1 in the low SDS region (0–10 mm), interpreted as transition of UCP1 tetramers toward tight dimeric units. The inset (Aii) shows the fluorescence intensity of UCP1 in the high SDS region (5–100 mm), where UCP1 tight dimers could be further dissociated into their loose dimeric and monomeric forms. All values were normalized, and the data were fitted to the Hill equation. The K1/2 for the first and second transitions were 1.3 ± 0.1 and 23 ± 1 mm, respectively. a.u., absorbance units. B, plot of the maximum emission wavelength (λmax) versus SDS revealed no significant change in the maximum Trp emission wavelength up to ∼20 mm SDS, where the fluorophores became more exposed to the polar environments and exhibited a red shift in its maximum emission wavelength.
Taken together, the spectroscopic measurements, analytical ultracentrifugation, and semi-native SDS-PAGE data suggest that UCP1 self-associates in lipid bilayers. Dissociation of UCP1 by SDS revealed two distinct types of interaction in the associated forms of UCP1. The tetrameric unit of UCP1 is held together by a relatively weak dimer-dimer interaction that can be disrupted at low concentrations of SDS near the CMC. The dimeric unit of UCP1, on the other hand, is more tightly packed and becomes only partially dissociated in the presence of high SDS concentration (∼40 × CMC). It is important to note that CD spectroscopy monitors the change in the average secondary structure of the protein, whereas fluorescence spectroscopy gives a microscopic view of the local environment of fluorophores in the protein. The combination of these techniques used in this study provides a more complete picture of the oligomeric states of the protein in a lipid environment and the dissociation process.
CL Interacts with UCP1 and Enhances Its Proton Transport Activity
To study the effect of the mitochondrial lipid CL on the structure and function of UCP1, the protein was reconstituted in liposomes supplemented with increasing concentrations of CL (0–50% mol). The secondary structure and proton transport activity of reconstituted UCP1 were examined in each lipid system. The data provide evidence that CL interacts directly with UCP1 and influences its structure and proton transport function.
Addition of CL to the lipid bilayers induced a change in the secondary structure of reconstituted UCP1 and increased its helicity (Table 1). As shown in Fig. 7, A and B, increasing the CL content in the phospholipid bilayers from 0 to 50% mol induced a gradual increase of the θ208/θ222 ratio, indicating the protein adopted a less associated conformation at higher CL concentrations. This θ208/θ222 ellipticity ratio remained less than 1 in all CL-containing lipid vesicles (Fig. 7B). In addition, the highly helical character of UCP1 was preserved in all lipid systems, as shown by the 2:1 ellipticity ratio of the positive maximum at ∼195 nm (π → π* ⊥ transition) to negative maximum at ∼222 nm (π → π* ‖ transition) (Fig. 7A). The weakening of association in the tetrameric form of UCP1 can be detected by the gradual increase in the θ208/θ222 ratio with the increase in CL/UCP1 molar ratio (Fig. 7B). The conformation of the UCP1 tetramer remained unchanged after the CL/UCP1 molar ratio reached ∼800. Semi-native SDS-PAGE of UCP1 in CL-containing liposomes revealed a single UCP1 tetramer band comparable to that in the CL-free proteoliposomes when the liposomes were not pretreated with SDS before loading the semi-native SDS-PAGE gel.
FIGURE 7.

Effect of CL on the conformation, stability and proton transport activity of associated UCP1 in liposomes. A, normalized far-UV CD spectra of UCP1 reconstituted in POPC vesicles in the absence and presence of CL. The molar ellipticity of UCP1 in different liposome systems was normalized at 222 nm. B, plot of θ208/θ222 versus CL/UCP1 molar ratio. C, plot of θ208/θ222 versus SDS concentration revealed a cooperative unfolding of reconstituted UCP1 tetramers in POPC supplemented with CL. The θ208/θ222 ratio was normalized, and the K1/2 in each lipid system was calculated using the Hill fitting. D, plot of UCP1 tetramer dissociation constants (K1/2) versus CL/UCP1 molar ratio. E, average corrected proton transport rate mediated by UCP1 in l-α-lecithin vesicles supplemented with various concentrations of CL. PA and LA were used as proton transport activators. A one-way analysis of variance statistical test was performed to determine the statistical significance of data, and p values were obtained. p < 0.05 when comparing the proton transport mediated by UCP1 in the CL-supplemented vesicles to that in l-α-lecithin vesicles (*).
The effect of CL on the stability of UCP1 tetramers and dimers (and monomers) was studied using the same SDS titration experiment as described above for UCP1 reconstituted in CL-free liposomes (Fig. 7, C and D). Assuming that the predominant form of the UCP1 in liposomes was tetrameric, as with the SDS titration of the proteoliposomes shown in Fig. 4B, the SDS titration of CL-containing liposomes was consistent with the dissociation of tetramers to dimers. In this case, in comparison to the CL-free system (K1/2 = 1.4 mm), higher K1/2 values (from 1.7 mm (for [CL]/[UCP] = 62.5) to 2.3 mm (for [CL]/[UCP] = 833)) for the tetramer-dimer transition curve indicated a more stable tetrameric form of UCP1 in the presence of CL (Fig. 7, C and D). As shown in Fig. 7D, a higher content of CL in proteoliposomes increased the dissociation K1/2 values of the UCP1 tetramer. Similar to the CL-free case, evidence from semi-native SDS-PAGE also supported the presence of predominantly dimeric UCP1 at high SDS concentration in the end of the SDS titration curve (data not shown). Collectively, these results suggest that CL stabilizes UCP1 tetramers in liposomes (Fig. 7, B and D, and Table 1).
Finally, the proton transport activity of UCP1 was examined in different CL-containing liposomes. PA and LA were used as the proton transport activators, and the UCP1-mediated proton transport rate was calculated in each lipid system. As shown in Fig. 7E, in the l-α-lecithin liposome system (and other examined liposomes) a consistently higher proton flux was observed when using PA as the activator. Interestingly, both the LA- and PA-activated UCP1-mediated proton transport activities were enhanced in CL-supplemented vesicles (Fig. 7E). It is noteworthy that a previous study on the effect of CL on AAC substrate transport function reported a similar enhancement of transport rate when AAC was reconstituted in the CL-supplemented vesicles (8). It is also important to note that at 500 μm ATP, a complete inhibition of UCP1-mediated proton transport was obtained in CL-containing liposomes. Overall, it appears that the presence of CL in phospholipid vesicles has a stabilizing role by forming complexes with the protein subunits. This associated UCP1-CL complex has a higher proton transport activity compared with self-associated UCP1 in the absence of CL.
DISCUSSION
This study describes a novel method for obtaining native-like recombinant UCP1 in high yield from a bacterial expression system. A combination of protein engineering, a modified induction/expression method, and detergent screening for protein extraction has allowed us to obtain folded UCP1 that is suitable for both structural and functional studies. Of particular interest is the evidence for the existence of self-associated forms of UCP1 extracted from the bacterial membranes. The associated state of UCP1 was preserved through the course of membrane isolation, extraction of the protein from membranes, purification, and reconstitution into phospholipid bilayers. Finally, the study provides new insights into the effect of the mitochondrial lipid CL on the structure and function of UCP1. These findings are essential for understanding the structure and ion transport activity of UCP1 (and likely, by extension, other human UCPs) in the IMM.
Bacterial Membrane Extracted UCP1 Is Suitable for Structural and Functional Studies
To date the majority of molecular studies on UCP1 have relied on the reconstitution of extracted proteins from bacterial inclusion bodies (18, 19, 34). This method allows for the production of large quantities of high purity protein from inclusion bodies that can be reconstituted into liposomes. The reconstituted proteins in these cases are typically only partially refolded from the inclusion bodies (6). Other disadvantages of the method include the heterogeneity of protein conformations that are obtained and the limited number of detergents suitable for the solubilization of inclusion bodies and subsequent refolding of the protein. Protein expression in yeast and mammalian cell systems usually offers low yields of recombinant proteins as compared with E. coli-based systems, which are inadequate for structural studies (40, 41). In the current study we used a modified bacterial expression system as an inexpensive and efficient source of recombinant UCP1 that was targeted to the bacterial membrane rather than accumulating exclusively in inclusion bodies. The PelB leader sequence was fused to the N terminus of UCP1 to achieve targeting to the bacterial membranes (42). The PelB leader sequence is a periplasmic targeting sequence that is cleaved by signal peptidase in the periplasm (42). Presumably, when fused to UCP1, the leader sequence still reaches the periplasm to be cleaved, but the remainder of the protein remains associated with the membrane rather than being completely transported to the periplasm because of the six hydrophobic transmembrane α-helices. The mild auto-induction expression method was also a key factor in optimizing protein expression in the E. coli system. Presumably, this approach avoided potential cell toxicity due to protein overexpression and/or accumulation in inclusion bodies, as has been observed when using IPTG-based induction (6, 17, 18, 34). Under our experimental conditions, OG was shown to be the best detergent for UCP1 extraction from bacterial membranes. UCP1 extracted in 1% OG had a high helical content, was thermally stable, and interacted with the proton transport inhibitor ATP (Figs. 2, D and E, and Table 1). Reconstitution of UCP1 from OG into phospholipid bilayers further enhanced the folding of the protein and its helical content (Fig. 3A and Table 1). The estimated helical content of UCP1 in lipid bilayers (75–85% helix) is the highest yet reported for this protein (6, 17, 19, 40). The expression and reconstitution protocol described in this study is appropriate for preparing highly pure and native-like folded proteins that can be used for obtaining high resolution structures of UCP1 and other UCPs.
Associated State of UCP1; Structure and Function
The native molecular state of mitochondrial carriers, including UCP1, has long been a matter of debate. The functional unit of UCP1 has been argued to be either dimeric or monomeric (43, 44). In this study UCP1 existed in three forms: tetramer, dimer, and monomer. All these forms were detected for the protein extracted from bacterial membranes, where UCP1 was overexpressed (Fig. 2A). The tetrameric form of the protein was further stabilized during extraction and purification processes from bacterial membranes (Figs. 2A, 4C, and 5). This tetrameric form of UCP1 in liposomes was able to transport protons across the liposome membranes, and its activity was stimulated by fatty acids and inhibited by purine nucleotides (Fig. 3D). When reconstituted into liposomes including the mitochondrial lipid CL, UCP1 tetramers transported protons at a higher rate (Fig. 7E). Our study has, therefore, provided new evidence for a functional tetrameric unit of UCP1. The monomeric form of UCP1, refolded from inclusion bodies, also exhibited proton transport activity (17, 18). Interestingly, based on the evidence provided in the current study, it appears that depending on the levels of protein expression and the composition of membranes, all three forms of functional UCP1 can coexist, with the possible increase in the abundance of associated forms at higher levels of protein expression (or concentration) and/or different compositions of mitochondrial membranes under different cellular conditions (Figs. 2–5).
If UCP1 can adopt a tetrameric conformation, questions arise regarding how UCP1 monomers self-associate and what the overall architecture of the oligomeric UCP1 is. Data from the current study suggest two types of UCP1 association, a monomer-monomer interaction to form a homodimer and a dimer-dimer interaction to form a tetramer (Fig. 8A). When titrated with SDS (CMC ∼ 2 mm), UCP1 tetramers were fully dissociated into predominantly dimers and some monomers (Fig. 4C). UCP1 dimers, which we have named “tight dimers,” appeared immediately after the formation of SDS micelles (Fig. 4, B and C). Excess SDS (∼40× CMC) did not completely dissociate UCP1 dimers but did appear to weaken the binding between monomers in the homodimer, resulting in a mixture of dimers and monomers (Figs. 4, B and C, and 6); we have termed the dimers that persist at this high concentration of SDS “loose dimers.” These data suggest a strong monomer-monomer interaction and a stable dimeric unit as well as a weaker dimer-dimer interaction within the tetrameric unit. Fluorescence data support the existence of different interaction surfaces between monomer-monomer association in the dimeric unit and dimer-dimer association in the tetrameric unit (Fig. 6). Despite the dissociation of tetramers to dimers and monomers in the presence of SDS, Trp residues remained in a weakly polar environment until the dissociation of homodimers to monomers was initiated (Fig. 6). Based on these observations, we propose that intermolecular UCP1 interactions leading to oligomerization can occur at two distinct sites, with different affinities, leading to the loose binding of UCP1 dimers to form tetramers and the tight binding of UCP1 monomers to form homodimers (Fig. 8A).
FIGURE 8.

Proposed models for UCP1 associated states and its structure. A, UCP1 tetramer is composed of two loosely associated homodimers. The tetramer can be dissociated into homodimers at low SDS concentrations (SDS ∼ CMC). In each dimer UCP1 monomers interact and tightly associate with each other through direct protein-protein interactions and/or protein interactions mediated by lipids, to form a tight association. These tight dimers retain their quaternary structures and can be partially unfolded/dissociated at higher concentrations of SDS to form loose dimers and/or monomers. Loose dimers may be fully separated to monomers at much higher concentrations of SDS. B, three-dimensional homology model of UCP1, built on the basis of sequence alignment (T-Coffee) using the crystal structure of AAC. The nucleotide binding pockets are located in the middle of the funnel-shaped UCP1 model. Basic amino acid residues (blue spheres) are mostly clustered in the cavity close to the matrix loops. On the other hand, acidic amino acid residues (red spheres) are distributed evenly throughout the protein. The number of basic amino acid residues is much higher than that of acidic amino acid residues, resulting in a +10 overall charge for UCP1 at physiological pH (refer to “Experimental Procedures” for detailed methods).
As mentioned above, the association of UCP1 could occur through direct protein interactions and/or protein interactions mediated by lipids. We have shown that the mitochondrial lipid CL strongly interacts with UCP1. The presence of CL in liposomes affects both the structure and function of UCP1. First, UCP1 tetramers exhibited a more relaxed helical conformation in CL-supplemented liposomes (Fig. 7A). This could be due to the specific interaction of CL with UCP1, transforming the protein-protein tetramer (in the absence or very low concentrations of CL) to a protein-lipid-protein tetramer complex (Fig. 7, A and B). In addition, CL enhances the stability of the tetrameric conformation (Fig. 7D). Second, UCP1 transports protons at a much higher rate in the CL-supplemented vesicles (Fig. 7E). Interestingly, the proton transport rate and tetrameric stability both reached their optimal values at the CL/UCP1 molar ratio of ∼800. At this CL/UCP1 molar ratio, the UCP1 tetramer is most stable (K1/2 = 2.3 mm; Fig. 7, C and D) and is at its most “relaxed” conformation, as indicated by the maximum value of θ208/θ222 of UCP1 far-UV CD spectra. Collectively, these data provide strong evidence for the specific interaction of UCP1 and CL and its direct relationship to the UCP1 proton transport function.
Homology three-dimensional modeling of UCP1, based on the crystal structure of AAC, exhibits a high helical content; ∼66%, as compared with an estimation of 67–85% in the CD analysis of the reconstituted protein in detergents and lipid vesicles (Fig. 8B and Table 1). The three conserved Arg residues (located on transmembrane domains 2, 4, and 6) involved in the nucleotide binding of UCP1 are located in the middle of the funnel-shaped structure, dividing the protein into two regions, cytoplasmic and matrix sides (Fig. 8B) (2, 5). The results from this study (UCP1 tetramers) (Figs. 2D and 3A) and previous work (UCP1 monomers), both, indicate a minor change in the overall conformation of UCP1 and other UCPs upon binding to ATP (6, 19). Based on the spectroscopic data of this study and the molecular model presented in Fig. 8B, it is plausible that ATP interacts with UCP1 in the middle of the funnel-shaped structure, causing a local conformational change (as detected by near-UV CD) leading to inhibition of ion (proton) transport, whereas the overall structure of the protein remained less affected (as detected by far-UV CD). Given the high pI (9.26) value of UCP1 and the negative charge present in the CL head group at physiological pH, it is possible that electrostatic attraction is among the initial driving forces for UCP1-CL interaction followed by shorter range intermolecular and hydrophobic interactions. In support of this idea, the UCP1 model shows an accumulation of positively charged residues on the matrix side of the funnel-shaped structure (Fig. 8B). Thus, it is possible that the negatively charged head group of CL interacts with UCP1 at the monomer interface situated toward the inner leaflet of the mitochondrial inner membrane and stabilizes the associated forms of UCP1. In fact, three CL molecules in the AAC crystals were found at the interfaces between monomers, where their phosphate head groups point toward the matrix interface (16). Based on our results, it is concluded that the presence of CL stabilizes UCP1-associated forms and could also release the stress resulting from the protein-protein interactions between UCP1 monomers, leading to a higher proton transport activity mediated by the associated forms of the protein. The interaction of CL with UCP1 and other mitochondrial carriers is thought to be important in minimizing the energy cost of protein conformational changes during substrate/ion transport (45). It is also noteworthy that bacterial membranes contain ∼5% CL (46). Therefore, it is plausible that the association of UCP1 in bacterial membranes was driven by the CL-UCP1 interaction when UCP1 was targeted toward the bacterial membranes. Based on these arguments, it is expected that UCP1 associated states would be different in CL-free bacteria.
In conclusion, we have shown that UCP1 can exist in different functional monomeric and associated states when targeted to and extracted from bacterial membranes. These associated forms can be closely related to the native form(s) of UCP1 in mitochondria. The dimeric form of UCP1 is very stable, even at low concentrations of the protein, implying that it is likely that this form has a role in the function of UCP1, including proton transport. Finally, the mitochondrial lipid CL likely plays an essential role as a mediator of UCP1 oligomerization and serves as an enhancer of the proton transport function of the protein.
Acknowledgments
We thank Dr. Frances Sharom, Dr. Joel Weadge, Dr. Yoshito Kakihara, Adam Clay, Miljan Kuljanin, Joseph Chu, Tyler Auld, James Campbell, Nguyen Vo, and Kyle Reinhart for technical support.
This work was supported by grants from the Canada Foundation for Innovation (CFI) and the Natural Sciences and Engineering Research Council of Canada (NSERC) CFI 6786 and NSERC 250119 (to M. J. N.) and CFI 11292 and NSERC 312143 (to M. D. S.).
This work is dedicated to the memory of Professor Grikor Terpoghossian.
- IMM
- inner mitochondrial membrane
- AAC
- ADP/ATP carrier
- CL
- cardiolipin
- CMC
- critical micelle concentration
- C8E4
- octyltetraoxyethylene
- DDM
- dodecyl maltoside
- FA
- fatty acid
- IPTG
- isopropyl-β-thiogalactopyranoside
- LA
- lauric acid
- LDAO
- lauryldimethylamine-N-oxide
- OG
- octyl glucoside
- PA
- palmitic acid
- POPC
- 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
- SPQ
- 6-methoxy-N-(3-sulfopropyl)quinolinium
- TES
- N-[tris(hydroxymethyl)methyl]-2-aminoethane-sulfonic acid
- UCP
- uncoupling protein
- EB
- extraction buffer.
REFERENCES
- 1. Nicholls D. G., Bernson V. S., Heaton G. M. (1978) The identification of the component in the inner membrane of brown adipose tissue mitochondria responsible for regulating energy dissipation. Experientia Suppl. 32, 89–93 [DOI] [PubMed] [Google Scholar]
- 2. Krauss S., Zhang C.-Y., Lowell B. B. (2005) The mitochondrial uncoupling-protein homologues. Nat. Rev. Mol. Cell Biol. 6, 248–261 [DOI] [PubMed] [Google Scholar]
- 3. Kozak L. P., Anunciado-Koza R. (2008) UCP1. Its involvement and utility in obesity. Int. J. Obes. (Lond) 32, S32–S38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cederberg A., Grønning L. M., Ahrén B., Taskén K., Carlsson P., Enerbäck S. (2001) FOXC2 is a winged helix gene that counteracts obesity, hypertriglyceridemia, and diet-induced insulin resistance. Cell 106, 563–573 [DOI] [PubMed] [Google Scholar]
- 5. Echtay K. S. (2007) Mitochondrial uncoupling proteins. What is their physiological role? Free Radic. Biol. Med. 43, 1351–1371 [DOI] [PubMed] [Google Scholar]
- 6. Ivanova M. V., Hoang T., McSorley F. R., Krnac G., Smith M. D., Jelokhani-Niaraki M. (2010) A comparative study on conformation and ligand binding of the neuronal uncoupling proteins. Biochemistry 49, 512–521 [DOI] [PubMed] [Google Scholar]
- 7. Berardi M. J., Shih W. M., Harrison S. C., Chou J. J. (2011) Mitochondrial uncoupling protein 2 structure determined by NMR molecular fragment searching. Nature 476, 109–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Klingenberg M. (2009) Cardiolipin and mitochondrial carriers. Biochim. Biophys. Acta 1788, 2048–2058 [DOI] [PubMed] [Google Scholar]
- 9. Haines T. H. (2009) A new look at cardiolipin. Biochim. Biophys. Acta 1788, 1997–2002 [DOI] [PubMed] [Google Scholar]
- 10. Pfeiffer K., Gohil V., Stuart R. A., Hunte C., Brandt U., Greenberg M. L., Schägger H. (2003) Cardiolipin stabilizes respiratory chain supercomplexes. J. Biol. Chem. 278, 52873–52880 [DOI] [PubMed] [Google Scholar]
- 11. Chicco A. J., Sparagna G. C. (2007) Role of cardiolipin alterations in mitochondrial dysfunction and disease. Am. J. Physiol. Cell Physiol. 292, C33–C44 [DOI] [PubMed] [Google Scholar]
- 12. Schlame M., Towbin J. A., Heerdt P. M., Jehle R., DiMauro S., Blanck T. J. (2002) Deficiency of tetralinoleoyl-cardiolipin in Barth syndrome. Ann. Neurol. 51, 634–637 [DOI] [PubMed] [Google Scholar]
- 13. Beyer K., Klingenberg M. (1985) ADP/ATP carrier protein from beef heart mitochondria has high amounts of tightly bound cardiolipin, as revealed by 31P nuclear magnetic resonance. Biochemistry 24, 3821–3826 [DOI] [PubMed] [Google Scholar]
- 14. Kadenbach B., Mende P., Kolbe H. V., Stipani I., Palmieri F. (1982) The mitochondrial phosphate carrier has an essential requirement for cardiolipin. FEBS Lett. 139, 109–112 [DOI] [PubMed] [Google Scholar]
- 15. Noël H., Pande S. V. (1986) An essential requirement of cardiolipin for mitochondrial carnitine acylcarnitine translocase activity. Lipid requirement of carnitine acylcarnitine translocase. Eur. J. Biochem. 155, 99–102 [DOI] [PubMed] [Google Scholar]
- 16. Nury H., Dahout-Gonzalez C., Trézéguet V., Lauquin G., Brandolin G., Pebay-Peyroula E. (2005) Structural basis for lipid-mediated interactions between mitochondrial ADP/ATP carrier monomers. FEBS Lett. 579, 6031–6036 [DOI] [PubMed] [Google Scholar]
- 17. Hoang T., Smith M. D., Jelokhani-Niaraki M. (2012) Toward understanding the mechanism of ion transport activity of neuronal uncoupling proteins UCP2, UCP4, and UCP5. Biochemistry 51, 4004–4014 [DOI] [PubMed] [Google Scholar]
- 18. Jaburek M., Garlid K. D. (2003) Reconstitution of recombinant uncoupling proteins UCP1, -2, and -3 have similar affinities for ATP and are unaffected by coenzyme Q10. J. Biol. Chem. 278, 25825–25831 [DOI] [PubMed] [Google Scholar]
- 19. Jelokhani-Niaraki M., Ivanova M. V., McIntyre B. L., Newman C. L., McSorley F. R., Young E. K., Smith M. D. (2008) A CD study of uncoupling protein-1 and its transmembrane and matrix-loop domains. Biochem. J. 411, 593–603 [DOI] [PubMed] [Google Scholar]
- 20. Zhou Q.-J., Sun J., Zhai Y.-J., Sun F. (2010) Prokaryotic expression of active mitochondrial uncoupling protein 1. Prog. Biochem. Biophys. 37, 56–62 [Google Scholar]
- 21. Douette P., Navet R., Gerkens P., Galleni M., Lévy D., Sluse F. E. (2005) Escherichia coli fusion carrier proteins act as solubilizing agents for recombinant uncoupling protein 1 through interactions with GroEL. Biochem. Biophys. Res. Commun. 333, 686–693 [DOI] [PubMed] [Google Scholar]
- 22. Knyght I., Laila K., Zhai C., Parsons R., Lawrence M. J., Panaretou B., Barlow D. J. (2012) Fusion protein expression and insertion of human mitochondrial uncoupling protein 1 in the inner membrane of Escherichia coli. J. Biol. Res. (Hong Kong) 1, 12–16 [Google Scholar]
- 23. Blesneac I., Ravaud S., Juillan-Binard C., Barret L.-A., Zoonens M., Polidori A., Miroux B., Pucci B., Pebay-Peyroula E. (2012) Production of UCP1 a membrane protein from the inner mitochondrial membrane using the cell free expression system in the presence of a fluorinated surfactant. Biochim. Biophys. Acta 1818, 798–805 [DOI] [PubMed] [Google Scholar]
- 24. Studier F. W. (2005) Protein production by auto-induction in high-density shaking cultures. Protein Expr. Purif. 41, 207–234 [DOI] [PubMed] [Google Scholar]
- 25. Osborn M. J., Gander J. E., Parisi E., Carson J. (1972) Mechanism of assembly of the outer membrane of Salmonella typhimurium. Isolation and characterization of cytoplasmic and outer membrane. J. Biol. Chem. 247, 3962–3972 [PubMed] [Google Scholar]
- 26. Voulhoux R., Bos M. P., Geurtsen J., Mols M., Tommassen J. (2003) Role of a highly conserved bacterial protein in outer membrane protein assembly. Science 299, 262–265 [DOI] [PubMed] [Google Scholar]
- 27. Laue T. M., Shah B. D., Ridgeway T. M., Pelletier S. L. (1992) Theory of computer aided interpretation of sedimentation data, pp 90–125, Royal Society of Chemistry, Cambridge, England [Google Scholar]
- 28. Schuck P. (2000) Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys. J. 78, 1606–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Whitmore L., Wallace B. A. (2004) DICHROWEB, an online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic Acids Res. 32, W668–W673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lees J. G., Miles A. J., Wien F., Wallace B. A. (2006) A reference database for circular dichroism spectroscopy covering fold and secondary structure space. Bioinformatics 22, 1955–1962 [DOI] [PubMed] [Google Scholar]
- 31. Notredame C., Higgins D. G., Heringa J. (2000) T-Coffee. A novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 302, 205–217 [DOI] [PubMed] [Google Scholar]
- 32. Sali A., Potterton L., Yuan F., van Vlijmen H., Karplus M. (1995) Evaluation of comparative protein modeling by MODELLER. Proteins 23, 318–326 [DOI] [PubMed] [Google Scholar]
- 33. DeLano W. L. (2002) PyMOL, DeLano Scientific, San Carlos, CA [Google Scholar]
- 34. Echtay K. S., Winkler E., Frischmuth K., Klingenberg M. (2001) Uncoupling proteins 2 and 3 are highly active H+ transporters and are highly nucleotide sensitive when activated by coenzyme Q (ubiquinone). Proc. Natl. Acad. Sci. 98, 1416–1421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rupprecht A., Sokolenko E. A., Beck V., Ninnemann O., Jaburek M., Trimbuch T., Klishin S. S., Jezek P., Skulachev V. P., Pohl E. E. (2010) Role of the transmembrane potential in the membrane proton leak. Biophys. J. 98, 1503–1511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Greenfield N. J., Hitchcock-DeGregori S. E. (1993) Conformational intermediates in the folding of a coiled-coil model peptide of the N terminus of tropomyosin and α-tropomyosin. Protein Sci. 2, 1263–1273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cooper T. M., Woody R. W. (1990) The effect of conformation on the CD of interacting helices. A theoretical study of tropomyosin. Biopolymers 30, 657–676 [DOI] [PubMed] [Google Scholar]
- 38. Salom D., Hill B. R., Lear J. D., DeGrado W. F. (2000) pH-dependent tetramerization and amantadine binding of the transmembrane helix of M2 from the influenza A virus. Biochemistry 39, 14160–14170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Heerklotz H., Seelig J. (2000) Titration calorimetry of surfactant-membrane partitioning and membrane solubilization. Biochim. Biophys. Acta 1508, 69–85 [DOI] [PubMed] [Google Scholar]
- 40. Douette P., Navet R., Bouillenne F., Brans A., Sluse-Goffart C., Matagne A., Sluse F. E. (2004) Secondary-structure characterization by far-UV CD of highly purified uncoupling protein 1 expressed in yeast. Biochem. J. 380, 139–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jastroch M. (2012) Expression of uncoupling proteins in a mammalian cell culture system (HEK293) and assessment of their protein function. Methods Mol. Biol. 810, 153–164 [DOI] [PubMed] [Google Scholar]
- 42. Lei S. P., Lin H. C., Wang S. S., Callaway J., Wilcox G. (1987) Characterization of the Erwinia carotovora pelB gene and its product pectate lyase. J. Bacteriol. 169, 4379–4383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lin C. S., Hackenberg H., Klingenberg E. M. (1980) The uncoupling protein from brown adipose tissue mitochondria is a dimer. A hydrodynamic study. FEBS Lett. 113, 304–306 [DOI] [PubMed] [Google Scholar]
- 44. Kunji E. R., Crichton P. G. (2010) Mitochondrial carriers function as monomers. Biochim. Biophys. Acta 1797, 817–831 [DOI] [PubMed] [Google Scholar]
- 45. Claypool S. M. (2009) Cardiolipin, a critical determinant of mitochondrial carrier protein assembly and function. Biochim. Biophys. Acta 1788, 2059–2068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Dowhan W. (1997) Molecular basis for membrane phospholipid diversity. Why are there so many lipids? Annu. Rev. Biochem. 66, 199–232 [DOI] [PubMed] [Google Scholar]