

Background: The tetraspanins peripherin and ROM1 interact and localize to the rim structures of vertebrate photoreceptor disc membranes.
Results: Native peripherin-ROM1 complex displays a tetrameric structure with a 2-fold symmetry and induces membrane curvature.
Conclusion: Biochemical and structural analyses confirm the tetrameric state of the complex.
Significance: Analyses provide evidence for the role of the peripherin-ROM1 complex in the rim structure.
Keywords: Retina, Retinal Degeneration, Retinal Metabolism, Vision, Vitamin A
Abstract
Peripherin and its homologue ROM1 are retina-specific members of the tetraspanin family of integral membrane proteins required for morphogenesis and maintenance of photoreceptor outer segments, regions that collect light stimuli. Over 100 pathogenic mutations in peripherin cause inherited rod- and cone-related dystrophies in humans. Peripherin and ROM1 interact in vivo and are predicted to form a core heterotetrameric complex capable of creating higher order oligomers. However, structural analysis of tetraspanin proteins has been hampered by their resistance to crystallization. Here we present a simplified methodology for high yield purification of peripherin-ROM1 from bovine retinas that permitted its biochemical and biophysical characterization. Using size exclusion chromatography and blue native gel electrophoresis, we confirmed that the core native peripherin-ROM1 complex exists as a tetramer. Peripherin, but not ROM1, is glycosylated and we examined the glycosylation site and glycan composition of ROM1 by liquid chromatographic tandem mass spectrometry. Mass spectrometry was used to analyze the native complex in detergent micelles, demonstrating its tetrameric state. Our electron microscopy-generated structure solved to 18 Å displayed the tetramer as an elongated structure with an apparent 2-fold symmetry. Finally, we demonstrated that peripherin-ROM1 tetramers induce membrane curvature when reconstituted in lipid vesicles. These results provide critical insights into this key retinal component with a poorly defined function.
Introduction
The vertebrate retina contains a host of distinct cell types but none more specialized than rod and cone photoreceptor cells. Each of these post-mitotic cells is composed of four distinct parts; an axon that connects the cell to other neurons, an inner segment containing the nucleus and other organelles, an outer segment (OS),2 which collects light stimuli and a ciliary structure that connects the inner and outer segments. Rod outer segments (ROS) are cylindrical structures that envelop stacks of independent membranous discs that contain the light-sensing pigment, rhodopsin. Disruption of these discs within the OS, either through genetic mutation or injury, can result in progressive retinal degeneration leading to blindness. Throughout the lifetime of an animal these discs are continually produced at the base of the OS and shed at the distal end or tip, a process of renewal that assures their constant function (1). Just how these structures are produced and maintained remains an area of intense research.
Tetraspanins are a family of glycoproteins linked to diverse cellular functions such as development, signal transduction, and motility (2). They often self-assemble into oligomers thought to be central regulators of complex protein networks, also referred to as “tetraspanin webs” (3). Primary sequence analysis has characterized tetraspanins as consisting of four transmembrane helices, short intracellular N and C termini, and two extracellular loops with one such loop (EC2) representing most of the soluble portion of this protein (4). Based on sequence analysis, the EC2 domain also is predicted to be the portion of each tetraspanin that determines its family identity and specificity (5). Each EC2 can be subdivided into a conserved and a variable domain (4). Although exhibiting some sequence variation among family members, the conserved domain appears to have a structurally conserved fold, whereas the variable domain is highly diverse in size, secondary structure, and folding (2). The EC2 domain also contains a number of conserved residues including invariant cysteine residues that form important intramolecular disulfide bridges (5).
Peripherin and its homologue ROM1, members of the tetraspanin family of integral membrane proteins, are two of the most highly expressed proteins in the OS other than rhodopsin (6). These two proteins, exclusively expressed in the retina and localized to the rim regions of the outer segment discs, have been implicated in disc morphogenesis and stability (7). Most pathogenic mutations in peripherin and ROM1, as with other tetraspanins, are localized to the EC2 domain (8). Loss-of-function or null alleles of peripherin cause an absence of disc formation in the ROS with accompanying blindness. Interestingly, similar mutations in ROM1 do not result in loss of vision unless accompanied by a mutation in one allele of peripherin, suggesting a more limited role for ROM1 (9).
Structural analysis of tetraspanins has been hampered by their resistance to crystallization. Moreover, that tetraspanins are integral membrane proteins with a complex disulfide network in the EC2 domain presents problems for proper folding of heterologously expressed proteins. The only crystal structure for any portion of a tetraspanin is the EC2 domain of the ubiquitously expressed CD81, expressed in Escherichia coli as an IgG fusion protein and solved to 1.6 Å (10). A cryoelectron crystallographic structure of the tetraspanin uroplakin demonstrated a tightly packed elongated structure capped by the EC2 domain (11). Uroplakin is found as a two-dimensional crystalline array on the urothelial surface, providing a suitable native source from which to isolate this protein for analysis (12).
When isolated from native sources, primarily bovine retina, peripherin, and ROM1 proteins co-purify in what was believed to be a heterotetrameric complex (13). Peripherin has a predicted molecular mass of 39.1 kDa, whereas ROM1 is 37.4 kDa, making the tetramer a predicted 153 kDa. Analysis of detergent-solubilized ROS membranes under reducing conditions identified a core complex of peripherin-ROM1 that associated non-covalently in the predicted tetramer (14). Disulfide-linked dimers were present under non-reducing conditions and displayed sedimentation coefficients consistent with higher order oligomers, suggesting that disulfide bonds naturally occur between but not within the core complexes (14–16).
Peripherin and ROM1 present an interesting opportunity to study tetraspanins produced and assembled in their native environment. Their relatively high expression in the eye and the ready accessibility of bovine retinas make this feasible. Previous work on peripherin was performed with both native protein and heterologously-expressed protein from mammalian cell lines (14, 17–19). But former purification procedures did not offer a simple, high yield method for isolating the native proteins (19). Additionally, whereas a number of sedimentation analyses were carried out to ascertain the size and oligomeric state of the complex, no definitive evidence was provided to confirm these indirect results (13, 16).
Here we present a simple purification procedure for peripherin-ROM1 from bovine retinas, which yielded a significant quantity of highly purified peripherin-ROM1 complex. We also provide evidence for the existence of peripherin homotetramers isolated independently of peripherin-ROM1 tetramers. Use of native blue gel electrophoresis and size exclusion chromatography (SEC) confirmed that the complex size is consistent with a tetrameric state. Previous work done by several laboratories has suggested the presence of a glycan moiety at Asn-229 (20, 21) and here we present an analysis of the composition of this glycan by mass spectrometry. Our work demonstrates that this glycan contains a mixture of α-mannose and N-acetylglucosamine residues. We used a newly developed mass spectrometry method capable of analyzing intact detergent-solubilized membrane proteins (20) to confirm the oligomeric state of the peripherin-ROM1 tetrameric complex. As opposed to two assembled homodimers, this complex was found to consist of two peripherin monomers and two ROM1 monomers assembled together in an a1b1/a2b2 organization. Detergent-solubilized tetramers were analyzed by single particle electron microscopy and an 18-Å structure is presented, the first for the peripherin-ROM1 complex. Last, we demonstrate the ability of isolated peripherin-ROM1 tetramers to force membrane curvature when they are reconstituted in lipid vesicles by cyclodextrin sequestration of detergent. This new purification procedure and related analyses provide a more detailed glimpse into the structure of this notoriously difficult complex.
EXPERIMENTAL PROCEDURES
Chemicals
Chemicals used for peripherin and ROM1 purification were purchased from Sigma. Amphipol, A8-35, as well as detergents including n-dodecyl-β-d-maltopyranoside (DDM) and lauryl maltose neopentyl glycol (LMNG), were purchased from Affymetrix (Cleveland, OH).
Purification of Rod Outer Segments Containing Peripherin and ROM1
Fresh bovine eyes obtained from a local slaughterhouse (Mahan Packing) were dissected under dim red light and retinas were removed and frozen at −80 °C. Rod outer segments were isolated from retinas as previously reported (21). Isolated ROS membranes were immediately re-suspended in 20 ml of isotonic buffer (20 mm HEPES, pH 7.5, 5 mm MgCl2, 1 mm DTT, 100 mm NaCl). The suspension was centrifuged at 27,000 × g for 25 min at 4 °C. ROS membranes were re-suspended in 40 ml of hypotonic buffer (5 mm HEPES, pH 7.5, 0.1 mm EDTA, 1 mm DTT) and homogenized with 10 strokes in a glass-glass homogenizer. The homogenate was then centrifuged at 35,000 × g for 30 min at 4 °C. Both the hypotonic buffer wash and homogenization steps were repeated. The resulting pellet containing washed ROS membranes was wrapped in aluminum foil and frozen at −80 °C until further use.
Purification of Peripherin and ROM1 from Washed ROS Membranes
Frozen washed ROS membranes were thawed on ice and re-suspended in solubilization buffer (20 mm BisTris propane, pH 8.0, 1 mm DTT, 25 mm DDM) and incubated at room temperature for 1 h with constant agitation. The lysate then was centrifuged at 150,000 × g for 1 h at 4 °C. To prevent reactions between free cysteine residues, the crude protein lysate was treated with 3 mm iodoacetamide for 1 h at room temperature. Insoluble material and aggregated proteins were cleared from the sample by centrifugation at 150,000 × g for 1 h at 4 °C. The sample then was immediately loaded onto a 5-ml DEAE MacroPrep Cartridge (Bio-Rad) pre-equilibrated with 20 mm BisTris propane, pH 8.0, and 0.35 mm DDM (2× CMC)) attached to a BioLogic LP System (Bio-Rad) at 4 °C. The cartridge was washed with 100 ml of low salt buffer (20 mm BisTris propane, pH 8.0, 0.35 mm DDM, 50 mm NaCl) to remove residual rhodopsin. Elution of the remaining protein was accomplished with high salt buffer (20 mm BisTris propane, pH 8.0, 0.35 mm DDM, 500 mm NaCl). Protein-containing fractions were combined and concentrated to 2.5 ml with a 100-kDa cutoff Amicon Ultra Centrifugal Filter (EMD Millipore, Billerica, MA). Excess salt was removed by running the protein sample through a PD-10 desalting column (GE Healthcare) pre-equilibrated with 20 mm BisTris propane, pH 8.0, 0.35 mm DDM, 10 mm NaCl. Protein purified for MS analysis was promptly loaded onto a pre-equilibrated SOURCE 15Q anion exchange column (GE Healthcare) and washed with 5 column volumes of equilibration/wash buffer (20 mm BisTris propane, pH 8.0, 0.35 mm DDM, 10 mm NaCl). Proteins were eluted with a linear gradient of 10–550 mm NaCl in 20 mm BisTris propane, pH 8.0, 0.35 mm DDM, and 1-ml fractions were collected. When LMNG detergent was used for protein purification, the 4 ml of protein collected from the PD-10 column was immediately added to 16 ml of 20 mm BisTris propane, pH 8.0, 10 mm NaCl, 1 mm LMNG (100× CMC) and incubated at room temperature for 1 h. This protein sample then was loaded onto a SOURCE 15Q column pre-equilibrated with 20 mm BisTris propane, pH 8.0, 20 μm LMNG (2× CMC), 10 mm NaCl. The column was washed with 5 column volumes of equilibration/wash buffer. Proteins were eluted with a linear gradient of 10–550 mm NaCl in 20 mm BisTris propane, pH 8.0, 20 μm LMNG. Protein isolated in either DDM or LMNG was analyzed by SDS-PAGE and fractions containing peripherin and ROM1 or peripherin alone were combined and concentrated to 500 μl with a 100-kDa cutoff Amicon Ultra Centrifugal Filter (EMD Millipore). Samples were centrifuged at 100,000 × g to remove aggregated protein. Protein purified in DDM was run on a Superdex 200 10/300 GL gel filtration column (GE Healthcare) pre-equilibrated with 25 mm BisTris propane, pH 8.0, 100 mm NaCl, 0.35 mm DDM, and 0.5-ml fractions were collected. Protein purified in LMNG was run on a Superdex 10/300 GL gel filtration column pre-equilibrated with 25 mm BisTris propane, pH 8.0, 100 mm NaCl, 10 μm LMNG (1× CMC), with 0.5-ml fractions collected. All protein samples were analyzed by SDS-PAGE and fractions containing peripherin were combined and concentrated. Gel filtration standards used to determine the molecular weights of the peripherin complex and its components were purchased from Bio-Rad.
Protein Immunoblot Analyses
Isolated proteins were resolved on 10% acrylamide SDS-PAGE gels and transferred to PVDF membranes with Bio-Rad Mini-PROTEAN Tetra Cell. Membranes were blocked with 5% nonfat milk for 1 h at room temperature. Anti-peripherin primary monoclonal antibody (custom) and anti-ROM1 primary rabbit polyclonal antibody (Abcam, Cambridge, MA) were diluted as indicated in 5% nonfat milk and incubated with membranes for 2 h at room temperature. Membranes were washed in PBS 3 times for 10 min each and incubated with HRP-conjugated secondary antibodies (1:5000; Jackson ImmunoResearch Laboratories, West Grove, PA) in 5% nonfat milk for 1 h at room temperature. Membranes then were washed in PBS 3 times for 10 min each. Bands were visualized by using the ThermoScientific SuperSignal West Pico Chemiluminescent Substrate with subsequent exposure to CL-XPosure Film (Waltham, MA).
Lectin Binding Assays and Mass Spectrometric Analysis of Glycosylation
Purified peripherin-ROM1 was separated by SDS-PAGE and proteins were transferred to PVDF membranes (Bio-Rad). Membranes were blocked by preincubation in a blocking solution from a DIG glycan differentiation kit (Roche Applied Science) according to the manufacturer's instructions. Following blocking, membranes were incubated for 1 h in the dark with various fluorescein-labeled lectins (Vector Labs, Burlingame, CA) in Tris-buffered saline, pH 7.4, supplemented with 1 mm MgCl2, 1 mm MnCl2, 1 mm CaCl2. After incubation, blots were washed three times with the same buffer, dried, and imaged by a Typhoon 9410 scanner (GE Healthcare) with excitation set at 488 nm and emission through a 526-nm filter.
For carbohydrate analyses, purified proteins were first either treated with a 10 m excess of PNGase F or left untreated overnight at room temperature. Coomassie-stained SDS-PAGE gel bands containing intact and PNGase F-treated bovine peripherin were subjected to in-gel digestion with sequencing grade modified chymotrypsin (Roche Applied Science) according to a formerly described procedure (22). Briefly, pieces excised from a SDS-PAGE were first destained with 50% acetonitrile in 100 mm ammonium bicarbonate, and then dehydrated with acetonitrile. Before an overnight proteolytic digestion, proteins were first reduced with 20 mm DTT at room temperature for 1 h and then alkylated with 50 mm iodoacetamide in 50 mm ammonium bicarbonate for 30 min in the dark. After proteolytic digestion, peptides were extracted from the gel with 5% formic acid in 50% acetonitrile and then resuspended in 0.1% formic acid after being dried completely with a Speed-Vac. Liquid chromatography-tandem mass spectrometry analysis of the resulting peptides was performed with a LTQ Orbitrap XL linear ion trap mass spectrometer (Thermo Fisher Scientific, Waltham, MA) coupled with an Ultimate 3000 HPLC system (Dionex, Sunnyvale, CA). Spectra were acquired by data-dependent MS to MS/MS switching methods. A full MS scan was performed with 60,000 resolution in the Orbitrap detector, whereas the MS/MS scan in the ion trap detector was carried out on the top five most abundant precursor ions in the CID mode at a normalized collision energy of 30%. Obtained data were submitted to Mascot Daemon (version 2.2.0, Matrix Science, Boston, MA) against a customized peripherin-2 database. Candidate N-glycosylation sites were initially identified with sugar-free peptides based on conversion of Asn to Asp residues by PNGase F, and then further confirmed with glycopeptides. Sugar compositions of glycopeptides were verified by manual interpretation of the obtained MS/MS spectra.
Native Blue Gel Electrophoresis
Native blue gel electrophoresis of isolated peripherin complexes was performed with the NativePAGE BisTris Gel System at 4 °C (Invitrogen). Proteins were resolved on 4–16% polyacrylamide gels and stained with Commassie G-250 added to the cathode buffer. For sodium dodecyl sulfate and dithiothreitol treatments, chemicals were added just prior to loading of the gels. Molecular weights of resolved proteins were estimated by comparison with NativeMark Unstained Protein Standards (Invitrogen).
Transmission Electron Microscopy and Single Particle Analyses
Protein purified in DDM was incubated with Amphipol A8-35 at a 5 times molar eq for 1 h on ice. To remove excess amphipol, protein was purified on a Superdex 200 column with 50 mm BisTris propane, pH 7.2, 100 mm NaCl. Peripherin-ROM1 samples were adsorbed onto carbon film-coated copper microscope grids and stained with 1% uranyl acetate following the procedure described earlier (23). Micrographs were taken at a magnification of ×70,400 with a FEI Tecnai F20 microscope (FEI, Eindhoven, Netherlands) operated at 200 kV and equipped with a Gatan US4000 UHS CCD camera (4K × 4K). The data set contained 50 untilted micrographs and 15 micrographs recorded at a 45° tilt. The pixel size was 2.13 Å/pixel. To account for defocus variations in the tilted images, micrographs were split in slices that were then processed independently (23). The defocus range of the whole data set was 0.2 to 1.6 μm under focus. A total of 20,096 particles were selected by using the ”swarm“ mode of e2boxer from the EMAN2 software package (24). Initial models were calculated with EMAN2 from 50 two-dimensional class averages assuming no symmetry was present. The best of these models was used for the reference-based projection alignment and three-dimensional reconstruction procedure implemented in SPIDER (25). The resulting map was contrast transfer function-corrected and subjected to multiple rounds of angular refinement. The resulting three-dimensional structure and its two-dimensional projections suggested the presence of a 2-fold rotational symmetry axis along the longest dimension of the peripherin-ROM1 complex. Thus, the SPIDER reconstruction/refinement procedure was repeated with imposed C2 symmetry. The resolution of the final structure was estimated by using the Fourier shell correlation method with a cut-off of 0.5. The amount of Amphipol bound to the transmembrane region of peripherin-ROM1 was estimated based on the known value for bacteriorhodopsin (54 kDa of Amphipol bound to seven transmembrane helices (26)) and by assuming a linear relationship between the length of the transmembrane region circumference (considered circular for this purpose) and the amount of Amphipol required to cover it. The final model was visualized with UCSF Chimera (27).
Reconstitution of Peripherin-ROM1 and Rhodopsin in Lipid Vesicles by Cyclodextrin
Peripherin-ROM1 protein was purified using DDM throughout the process. Fifteen μl of purified protein (1 mg/ml) were dispensed to wells of a 384-conical well plate, together with different salts, buffers, and lipids. The last were either E. coli polar lipids (Avanti Polar Lipids Inc., Alabaster, AL) or a mixture of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine, 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1′-rac-glycerol), and 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (Avanti Polar Lipids) that mimicked the native lipid membrane. Lipids delivered in chloroform were dried under a stream of nitrogen, desiccated overnight, and solubilized at a concentration of 5 mg/ml in 4% DM. Lipid to protein ratios were varied from 0.1 to 1. Detergent concentrations of protein solutions were measured with the sessile drop method (28). Plates were placed in a custom-made robot that gradually added methyl-β-cyclodextrin at a controlled rate and used light scattering to monitor the reconstitution process. Plates were held at 23 °C for 7 h, when the estimated amount of cyclodextrin required to sequester the DDM was added. Once the CMC was reached, the temperature was raised to 37 °C for 7 h and then reduced back to 23 °C for an additional 7 h. Samples were harvested after about 24 h. Rhodopsin dimers in DDM purified according to Jastrzebska et al. (29) were reconstituted with or without the peripherin-ROM1 protein under identical conditions, but kept in the dark throughout the experiment. Samples were collected after 24 h, adsorbed onto carbon-coated EM grids, stained with uranyl acetate, and imaged at a magnification of ×50,000 with a FEI Tecnai F20 microscope operated at 200 kV equipped with a Gatan US4000 UHS CCD camera (4K × 4K). Image analyses of regular lipid protein assemblies were performed with the SEMPER image processing software (30).
Mass Spectrometry of Native Peripherin-ROM1 Complexes
Peripherin-ROM1 complexes were isolated in the presence of DDM as described above except that the alkylation step with iodoacetamide was omitted to obtain an accurate mass measurement. Mass spectrometry experiments were performed with a quadrupole time-of-flight MS instrument (Synapt, Waters Inc., Milford, MA) equipped with a nanoelectrospray ionization source using metal coated borosilicate glass capillaries (nanoflow probe tips, long thin walled, Waters Corporation). Samples were buffer exchanged via a Bio-Rad P6 column into 100 mm ammonium acetate containing 1 mm DDM. Solution flow rates of ∼75 nl/min and an electrospray ionization capillary voltage of ∼1.3 kV were used for all experiments. The source temperature was 50 °C and the pressure of the vacuum/backing region was 3.5 mbar. The signal was typically averaged for about 10–60 min. Source conditions were used to minimize gas phase unfolding of the protein (sample cone, 50–150 V; trap voltage, 10–150 V; transfer lens, 12 V; bias, 25; cone gas, 30 liter/h; PTRAP(Ar), 0.0175 mbar). Sulfur hexafluoride (SF6) was used as carrier gas in the trap and transfer cells. The instrument was externally mass calibrated with a 50 μg/μl CsI solution. Waters' raw data files were translated to Matlab binary files with in-house developed software. Spectral deconvolution was performed using either Waters' maximum entropy algorithm or an algorithm based on minimum l(1)-norm reconstruction (31) developed in-house at Amgen Inc. (M. Toupiko, I. D. Campuzano, and P. D. Schnier).
RESULTS
Purification of Peripherin and ROM1 from Native Eye Tissues
Isolated bovine retinal ROS contain a highly complex mixture of proteins, with the visual pigment rhodopsin constituting >90% of the membrane protein fraction by weight. Prior to solubilization with detergent, isolated ROS were washed and homogenized in both isotonic and hypotonic buffers to remove soluble and loosely bound membrane proteins. Previous work in our laboratory indicated that rhodopsin, unlike many retinal proteins, does not bind readily to DEAE anion exchange resin so most rhodopsin would pass through the column and any loosely bound rhodopsin could be gently washed from the resin with a low salt wash. This strategy was used to pre-purify peripherin-ROM1 from bovine ROS and remove >95% of rhodopsin (Fig. 1A). Integral membrane proteins were released from membranes by solubilizing washed bovine ROS in 25 mm DDM. The resulting sample was reduced with dithiothreitol (DTT) and alkylated with iodoacetamide to prevent protein oligomerization and then purified on a commercial DEAE cartridge. Subsequent analysis by SDS-PAGE confirmed the nearly complete removal of rhodopsin and retention of peripherin-ROM1 (Fig. 1A). A detergent screen identified LMNG, which functions as an amphiphile with an extremely low CMC (10 μm), as an ideal detergent to continue peripherin-ROM1 purification. After an exchange into LMNG the resulting preparation was further purified on a SOURCE 15Q column and analyzed by SDS-PAGE (Fig. 1B). The first two major peaks eluted from this column contained a significant amount of peripherin-ROM1 with few other contaminating proteins (Fig. 1C). Interestingly, the first peak contained only peripherin, whereas the second peak contained both peripherin and ROM1. Fractions containing peripherin alone or peripherin-ROM1 were further purified on a Superdex 200 GL SEC column. Chromatograms of both samples displayed a sharp peak which, based on SDS-PAGE analysis, contained >95% of the targeted protein (Fig. 1D). Immunoblot analysis confirmed that the first peak from the SOURCE 15Q column contained only peripherin, whereas the second contained both proteins (Fig. 1E). This highly reproducible purification method also provided significant yields. Three consecutive purifications from ROS isolated from 100 retinas each produced 1.2, 1.3, and 1.6 mg of purified protein, respectively.
FIGURE 1.

Peripherin-ROM1 complex purification. A, SDS-PAGE gel of partially purified Per/rds:ROM1 on a weak anion exchange column (DEAE-cellulose) to remove rhodopsin. B, FPLC chromatogram from a SOURCE 15Q anion exchange column purification of peripherin and ROM1. C, SDS-PAGE gel from the SOURCE 15Q column purification. Fractions labeled Peripherin correspond to Peak 1 in the chromatogram and primarily contain peripherin. Fractions labeled Peripherin-ROM1 correspond to Peak 2 and contain both peripherin and ROM1. The third peak primarily contains rhodopsin as determined by immunoblot analysis (data not shown). D, chromatograms derived from purification of the Per/Per homotetramer and Per/ROM1 heterotetramer on a Superdex 200 GL 10/300 size exclusion column with corresponding gel images of the protein-containing fractions (right) are shown. E, immunoblot analyses of both complexes with an anti-Per mAb and an anti-ROM1 pAb confirm the presence of peripherin in both complexes but ROM1 only in the heterotetramer.
Analysis of Peripherin Glycosylation by Lectin-binding Assays and Mass Spectrometry
It has been established first that peripherin exists as a glycoprotein in vivo, whereas ROM1 lacks the conserved asparagines of peripherin (Fig. 2, A and B) (32) and second that glycosylation does not participate in peripherin-ROM1 complex formation or function (33). However, the composition of this glycan has never been reported because a highly purified peripherin-ROM1 complex was not available. Here, treatment of the purified native peripherin-ROM1 complex with PNGase F showed a shift in the molecular weight of peripherin but not ROM1, confirming previous results (Fig. 2A). The identity of the conjugated glycans was then determined by the use of fluorescein-labeled lectins and mass spectrometry. Purified peripherin-ROM1 was first resolved by SDS-PAGE electrophoresis and then transferred to a PVDF membrane. Individual lanes were probed with a series of fluorescein-labeled lectins and then analyzed with a fluorescence scanner. Fig. 2B shows that two classes of lectins were bound to purified peripherin: the wheat germ agglutinins WGA and SuWGA, which recognize N-acetylglucosamine, and concanavalin A, LCA, and PSA which recognize α-linked mannose residues. To further investigate the position and identity of these glycans, we analyzed our samples by high sensitivity LC MS/MS. Our results confirm that the glycosylation of peripherin occurred at Asn-229 and that the adduct is a mixture of N-acetylglucosamine and α-linked mannose residues (Fig. 2, C and D).
FIGURE 2.
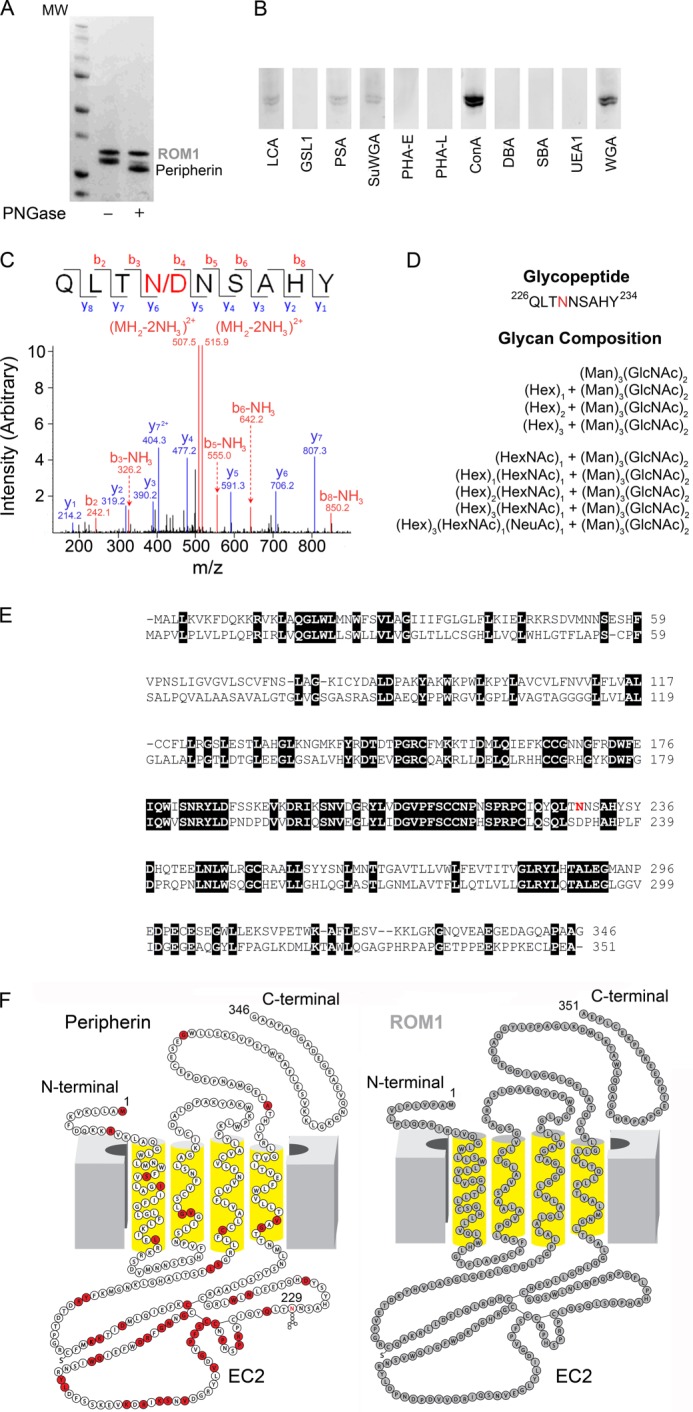
Identification of glycosylation species by mass spectrometry and comparison of peripherin and ROM1 sequences and two-dimensional structures. A, SDS-PAGE gel of untreated and PNGase F-treated purified Per/ROM1 complex demonstrates a shift in peripherin but not ROM1. B, staining of PVDF membranes containing purified Per/ROM1 complex with various fluorescein-conjugated lectins. C, representative MS/MS spectrum of de-glycopeptide QLTNNSAHY after PNGase F treatment. D, list of glycans detected for Asn-229 by mass spectrometry. Abbreviations: Hex, hexose; Man, mannose; HexNAc, N-acetylhexosamine; GlcNAc, N-acetylglucosamine; NeuAc, N-acetylneuraminic acid. E, sequence alignment of bovine peripherin and ROM1, transmembrane domains are underlined with broad black lines and numbered TM1–4. Peripherin glycosylation site Asn-229 is highlighted in red. F, peripherin and ROM1 two-dimensional structures displaying topology within the membrane, Asn-229 is highlighted in the peripherin structure. Red residues: missense mutation or in-frame deletion. Abbreviations: LCA, Lens culinaris agglutinin; GSLI, Griffonia (Bandeiraea) simplicifolia lectin I; PSA, Pisum sativum agglutinin; SuWGA, succinylated WGA; PHA-E, Phaseolus vulgaris agglutinin E; PHA-L, P. vulgaris agglutinin L; ConA, concanavalin A; DBA, Dolichos biflorus agglutinin; SBA, soybean agglutinin; UEAI, Ulex europaeus agglutinin I.
Analysis of the Peripherin-ROM1 Complex Molecular Weight by SEC and Native Blue Gel Electrophoresis
Tetraspanin proteins are known to be central regulators of complex interaction networks, often referred to as tetraspanin webs (3). The complexity of these networks makes it difficult to ascertain their true molecular weight in vivo. Previous work relied mainly on centrifugal sedimentation to determine the molecular weight of this complex (13). Based on SEC analysis, the molecular mass of our purified protein was estimated to be ∼150 kDa, in good agreement with the predicted molecular mass of the tetrameric complex previously described (Fig. 1D) (13). In agreement with this result, native blue gel electrophoresis also showed a molecular mass of ∼150 kDa for our highly purified complex, confirming its tetrameric structure (Fig. 3A). Although peripherin can be found in a dimeric state in native membranes (14), exactly where the disulfide bonds form has not been clear. Loewen and Molday (14) demonstrated that under non-reducing conditions peripherin from solubilized ROS was found as monomers and dimers that sedimented as both tetramers and higher order oligomers. When the same samples were analyzed by SDS-PAGE under reducing conditions they found only monomers. This was consistent with a tetrameric state and suggested that disulfide bonds did not contribute to the basic oligomeric state of the complex. To determine whether our purified native protein evidenced a similar behavior in the presence of reducing agents, we treated the complex with 5 mm DTT, 2% SDS or both before loading it on a gel (Fig. 3A). Our results confirmed that this core complex of peripherin-ROM1 does not associate via disulfide bonds but instead associates through a non-covalent interaction that can be disrupted by SDS treatment.
FIGURE 3.
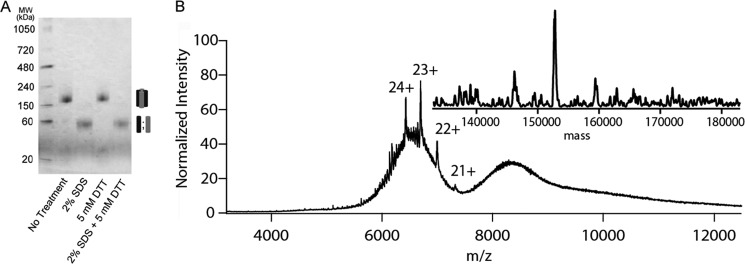
Analysis of the native peripherin-ROM1 complex by blue native gel electrophoresis and mass spectrometry. A, purified peripherin-ROM1 complex was resolved on a 4–16% native PAGE gel and stained by Commassie G-250 supplied in the cathode buffer. Protein was treated with 2% SDS, 5 mm DTT or both just prior to loading on the gel. B, native nanoelectrospray mass spectrometric analysis of peripherin-ROM1 complex displayed over m/z range 4000–12,000. The spectrum was smoothed using wavelet thresholding. A deconvoluted “zero charge” spectrum resulting in a mass of ∼153 kDa consistent with a tetrameric complex is displayed in the inset.
Analysis of Native Peripherin-ROM1 Complex by Mass Spectrometry
Recent work by several groups has revolutionized analysis of native intact membrane proteins and complexes (20, 34, 35). The technique involves the release of proteins, solubilized in a nonionic detergent micelle after collision activation in the gas phase of the mass spectrometer. The resultant apoprotein/complex is then detected without the interference of bound detergent. Upon MS and/or MS/MS and spectral smoothing using wavelet thresholding, a series of multiply charged peaks were observed in the m/z range of 6,000–7,000. Upon deconvolution an intact mass of 153 kDa was inferred to represent the intact peripherin-ROM1 complex (Fig. 3B). The intact mass of 153 kDa would be consistent with the subunit stoichiometry of two peripherin and two ROM1 monomers. These data provide direct evidence that the complex is indeed present as a tetramer.
Peripherin-ROM1 Complex Structure Determined by Electron Microscopy
Although expression systems and purification methods have improved substantially, structural descriptions of full-length tetraspanin proteins remain rare with only one available to date (11). Recently our laboratory successfully employed electron microscopy to determine the structures of previously uncharacterized proteins and complexes (23, 36). To determine the molecular architecture of peripherin-ROM1, we used an electron microscope to image purified samples negatively stained with uranyl acetate (Fig. 4A). Consistent with the composition of the complex, examination of individual particles as well as two-dimensional class averages suggested the presence of a 2-fold symmetry. An 18.0-Å resolution map was obtained from ∼20,000 particles (Fig. 4B). The iso-surface level of this map was chosen such that it encompassed a volume of 280 nm3, which corresponds to 82 kDa of Amphipol bound to the 153-kDa protein complex. The final model measured 11 × 9 × 6 nm, with the larger side roughly triangular in shape.
FIGURE 4.

EM model of the peripherin-ROM1 complex. A, a micrograph illustrating negatively stained particles of peripherin-ROM1 enclosed in white boxes (left). Examples of two-dimensional class averages are shown on the right. B, angular views of the three-dimensional map of peripherin-ROM1 obtained by single particle analysis. A comparison of selected forward projections of this model (top) with the corresponding experimental averages (bottom) is shown below the model. C, the hypothetical location of the Amphipol belt near the bottom of the model is shown with a darker color. Arrowheads indicate positions where the horizontal cross-sections were taken to calculate the cross-sectional areas shown below.
Lack of a structural description of peripherin-ROM1 and related tetraspannins makes it difficult to identify structural elements unequivocally in the EM model. However, a reasonable suggestion can be made based on the membrane topology of this protein complex and certain known properties of Amphipol. In particular, the soluble part of peripherin-ROM1 is largely composed of intradiscal domains that constitute about 47% of the complex. Because the N- and C-terminal fragments located across the membrane from these domains are small, it can be inferred that the transmembrane region occupies either the top or bottom part of the EM model. Several studies of membrane proteins found that Amphipol forms an ∼4-nm high belt wrapped around the hydrophobic region (23, 37, 38). Such a belt can be observed near the bottom of the present model (Fig. 4C). The area of the horizontal cross-section taken in the middle of this hypothetical belt was 41 nm2, whereas the average sectional area of a single transmembrane helix was estimated to be 1.4 nm2 (39). Thus, the bottom portion of the model is sufficiently large to accommodate 16 transmembrane helices (22.4 nm2) and the 1.5 ± 0.2-nm thick Amphipol (40). In contrast, the area of the horizontal cross-section at the top of the model was only 29 nm2, which makes it a less likely candidate to host the transmembrane region. Based on these considerations, we hypothesize that the bottom portion of the complex represents the membrane-spanning helices, whereas the top conical part is occupied by the soluble intradiscal domains.
Forced Membrane Curvature by Reconstitution of the Peripherin-ROM1 Complex in Lipid Vesicles
The ability of ROS disc membranes to maintain their “pancake-like” structure has confounded researchers for decades. The localization of peripherin and ROM1 exclusively to the rim regions of the discs has been presented as evidence for their possible role in this process. Here we present reconstitution experiments over a wide range of conditions that demonstrate that incorporation of purified peripherin-ROM1 complex into lipid vesicles forces membrane curvature. The peripherin-ROM1 complex was reconstituted in the presence of lipids by cyclodextrin sequestration of detergent with a custom built robot (41, 42). One experiment presented here was conducted at pH 7, but additional experiments were performed at pH 6.5, 8, and 9 (data not shown). To compare the behavior of the peripherin-ROM1 complex alone and in the presence of other disc proteins, co-reconstitution experiments with rhodopsin were carried out. The light scattering signal measured by our robot allowed the progress of reconstitution to be monitored during cyclodextrin addition (Fig. 5A). A striking difference was observed between reconstitutions of peripherin-ROM1 alone, rhodopsin alone, and co-reconstitution of peripherin-ROM1 with rhodopsin over all conditions assessed. Whereas a low scattering signal indicates the presence of mostly small structures for peripherin-ROM1 (Fig. 5A, left panel), the onset of larger vesicles or aggregates is indicated by a sigmoid increase of the scattering signal when rhodopsin was present (Fig. 5A, middle and right panels). In all three cases, exogenous lipid was added at a lipid to protein ratio of 0.1.
FIGURE 5.

Peripherin-ROM1 and rhodopsin incorporation into lipid vesicles. Purified peripherin-ROM1 complexes and/or rhodopsin were incorporated into protein-lipid aggregates by slow addition of methyl-β-cyclodextrin. A, light scattering is used to monitor incorporation of protein into vesicles. The transition from low to high scattering indicates formation of larger structures and is only observed in the presence of rhodopsin. B, micrographs of negatively stained reconstitution products. B, upon detergent removal, peripherin-ROM1 forms cylindrical rings. Scale bar: 200 nm. Upon addition of rhodopsin to the peripherin-ROM1-lipid mixture, larger discs with clear rims and rings are found. Scale bar, 200 nm. Rhodopsin alone reconstitutes into larger vesicles than in the presence of Peripherin-ROM1. Pronounced rims are missing. Scale bar, 1 μm. C, histogram showing distribution of vesicle diameters for the indicated proteins. Reconstitution conditions: peripherin-ROM1 (1 mg/ml), peripherin-ROM1-rhodospin (1:1, 1 mg/ml), or rhodopsin (1 mg/ml) were mixed with lipids at a lipid to protein ratio of 0.1 (w/w) in 100 mm NaCl, 5 mm MgCl2, 10 mm Bis-tris propane, pH 7. Fifty μm 11-cis-retinal was present in wells containing rhodopsin.
In excellent agreement, negatively stained reconstituted protein lipid structures revealed a strikingly different morphology (Fig. 5B). Most of the peripherin-ROM1-lipid complexes had a cylindrical shape as documented by top and side views. These cylindrical discs were quite uniform in size (38 ± 6 nm (n = 49); Fig. 5C, left panel) and had a pronounced border of ∼6 nm width. The discs exhibited a thickness of 7 ± 1 nm (n = 12). Few larger protein-containing vesicles up to a size of ∼100 nm were also present. Larger circular disc-like structures, together with the small cylindrical discs, assembled when peripherin-ROM1 and rhodopsin were co-reconstituted (Fig. 5B, middle panel). These discs had a pronounced rim and a diameter of 57 ± 22 nm (n = 54). Vesicles up to a size of 0.5 μm were also present, but infrequent (Fig. 5B, middle panel). In the presence of lipids and rhodopsin alone, small vesicles were reconstituted as well, but they exhibited a larger average size of 157 ± 56 nm (n = 26), and lacked the rim observed when peripherin-ROM1 was present. A much wider distribution of vesicle diameters emerged upon reconstitution of rhodopsin alone, extending up to several μm (Fig. 5, B and C, right panels). The sample shown was prepared under close to physiological conditions, but the interaction of peripherin-ROM1 with lipids was observed for all conditions tested (data not shown).
At a lipid-to-protein ratio of 1, larger and more diverse assembled products were observed. The first products were comprised of S- and U-shaped structures as well as discs of different sizes (Fig. 6A). Their characteristic features were a membrane region with a thickness of 6.5 ± 0.9 nm (n = 20) and a distinct regular protein structure extending from the membrane region at an average distance of 4.4 ± 0.6 nm (n = 20). Averaging 11 U-shaped segments of similar sizes enhanced the visibility of the extended peripherin-ROM1 domains, which had a length of 4.6 ± 0.5 nm (n = 10) (Fig. 6B). The other kind of product consisted of two disc-like structures with distinct peripheral borders connected by a membrane-enclosed ribbon-like structure (Fig. 7A). The borders had a thickness of 6.1 ± 0.9 nm (n = 24). Upon closer inspection, some regularity was recognized, which was enhanced by averaging 36 ribbon segments (Fig. 7B, inset bottom right). The step size of the ladder-like arrangement was 4.6 ± 0.3 nm (n = 8), and extensions that appeared to interact in the middle of the ribbon had a length of 5.4 ± 0.7 nm (n = 9).
FIGURE 6.

Micrographs of peripherin-ROM1-lipid structures. A, representative images of negatively stained peripherin-ROM1-containing reconstituted products exhibiting characteristic S- and U-shaped structures. These exhibited a weaker curvature than the discs obtained at a lipid to protein ratio of 0.1. Micrographs were recorded at ×62,000 magnification with a FEI Tecnai F20 microscope running at 200 kV. Scale bar, 100 nm. B, average of 11 U-shaped images used for quantification. Scale bar, 20 nm. Reconstitution conditions: peripherin-ROM1 (1 mg/ml) was mixed with lipids at a lipid-to-protein ratio of 1.0 (w/w) in 100 mm NaCl, 5 mm MgCl2, 10 mm Bis-tris propane, pH 7.
FIGURE 7.

Micrographs of peripherin-ROM1-lipid ribbons. A, representative images of disk-like structures connected by ribbons formed during peripherin-ROM1 reconstitution. Scale bar: 100 nm. B, correlation average from 36 ribbon segments revealed a distinct periodicity (bottom left inset). Scale bar: 20 nm. The unprocessed ribbon segment exhibited a weak periodic pattern (top left inset) revealed in its power spectrum (top right inset). Scale bar: (5 nm)−1. Reconstitution conditions: peripherin-ROM1 (1 mg/ml) was mixed with lipids at a lipid to protein ratio of 1.0 (w/w) in 100 mm NaCl, 5 mm MgCl2, 10 mm Bis-tris propane, pH 7.
DISCUSSION
The recalcitrance of tetraspanins to crystallize when purified from heterologous hosts required a different approach to study these important proteins. The purification and crystallization of natively expressed proteins was once the bedrock of this field but has been largely supplanted by the ease of protein purification and high yields of recombinant techniques. Although the latter techniques can produce milligrams of proteins, these often lack post-translational modifications, proper disulfide bond formation, and appropriate intermolecular interactions with their native partners. The use of native proteins for biochemical and structural analysis may again be gaining support for the study of individual proteins in their cellular contexts.
Peripherin and ROM1 present an example of proteins that, when expressed in heterologous hosts, lack some critical attributes including glycosylation, proper disulfide bond formation, and the potential formation of higher order oligomers. Their identity as integral membrane proteins dictates a low success rate for efficient recombinant expression. Fortunately, both peripherin and ROM1 represent two of the most highly expressed proteins in the vertebrate eye. The availability of bovine eye tissue, along with its relatively high expression of these proteins, make this an ideal source for isolation of peripherin-ROM1 proteins. Here we present a highly simplified purification scheme employing anion exchange and size exclusion chromatography. Previously published purification methods for these proteins involved more harsh techniques such as hydroxyapatite chromatography and chromatofocusing, which expose proteins to very low pH (19). Our technique consistently yielded >1 mg of highly purified complex from ∼100 retinas, a very reasonable amount of starting material. The complex remained quite stable in a number of detergents and required harsh treatment with chemicals like SDS to break apart. Consistent with previous reports, the complex purified as a tetramer with a mass of ∼150 kDa, confirmed by both native PAGE and SEC. Most of the isolated complex was composed of both peripherin and ROM1. Although, we did manage to purify the peripherin-only tetrameric complex by using the neopentyl glycol detergent, LMNG, this complex represented only ∼10% of the total isolated complex suggesting that the dominant species is the heterotetramer. Our treatment of the purified complex with reducing agents also confirmed previous findings (14) that the core complex does not associate through intermolecular disulfide bonds but likely through non-covalent hydrophobic interactions. Peripherin, but not ROM1, is known to be glycosylated at Asn-229 (33) but the nature of the glycan has not been reported. Moreover, the role of this glycosylation remains undefined, as it was shown that a glycosylation impaired mutant was still able to interact with ROM1 and also rescue rds mutant mice (33). Our analysis by mass spectrometry confirmed the location of glycosylation on Asn-229 and identified the residues as a mixture of α-mannose and N-acetylglucosamine. The latter finding was also confirmed by lectin-binding assays in which only lectins that bind these two residues were able to recognize the purified protein.
Recent advances in mass spectrometry have enabled routine MS analyses of intact proteins or complexes that are considerably larger than those previously thought possible. Moreover, these include integral membrane proteins present in detergent micelles. Pioneered by the Robinson group (20, 34, 35), such analyses involve the release of proteins from micelles after collision activation in the gas phase. Removal (by collision-induced dissociation) of the detergent leaves the protein and lipid interactions intact while separating any loosely associated small molecules. Structural studies of tetraspanin proteins have been sparse, impeding our acquisition of knowledge at an atomic level. Structural studies of other tetraspanins to date have involved either crystallization of soluble portions of the protein (EC2) for x-ray crystallography or electron crystallography of purified native proteins (10, 11). Ultimately a full-length structure of a tetraspanin at an atomic resolution is required. We used single particle three-dimensional EM to determine the structure of the peripherin-ROM1 complex to a resolution of 18 Å, similar to studies of the pentameric rhodopsin/G-protein complex and the ABC transporter ABCA4 (23, 36). The peripherin-ROM1 complex exhibited an elongated cylindrical structure when the detergent belt was omitted. This structural layout is consistent with the only other known full-length tetraspanin structure, the 6-Å cryoelectron crystallographic structure of uroplakin composed of a tightly bunched group of α-helices. The bulkier domain likely represents the transmembrane helices wrapped in an Amphipol belt of 1.5 nm thickness, because this is the only area of the structure with sufficient density to accommodate 16 α-helices. There also is evidence of a 2-fold symmetry within the model, consistent with the heterotetrameric nature of the complex. Moreover, the 2-fold symmetry displayed in Fig. 4B presents some insights into the nature of the tetramer. The bulging protrusions seen on opposing sides of the structure when looking down the long axis of the protein suggest that the complex is set up in an a1b1/a2b2 configuration with each peripherin or ROM1 monomer being juxtaposed to the other, as opposed to right next to each other. These results, along with our native mass spectrometry, provide convincing evidence as to the organization of this tetramer.
The prevailing theory of peripherin-ROM1 function within the OS has been that its strict localization to the rim region of the disc suggests a role in maintaining the curved disc structure. Maintenance of membranes curved at such an angle would require considerable force. It is known that the C-terminal domain of peripherin is capable of fusogenic activity in the presence of lipids, suggesting that it may play a key role in the morphogenesis of disc membranes (43). It was also shown that when peripherin was integrated into vesicles in vitro, the vesicles displayed a flattened structure (44, 45). One disadvantage with interpreting these studies is that the investigators used in vitro translated protein or heterologously expressed truncated proteins. In addition to the peripherin-ROM1 complex there are also other proteins that show localization to this region of the OS disc, including the ABC transporter ABCA4. We recently presented an EM structure of this protein indicating an intra-discal region that protrudes roughly 10 nm into the lumen (23). Here we provide a structure of our purified peripherin-ROM1 complex indicating a 4–5-nm region localized to the intra-discal lumen. Several studies utilizing cryoelectron tomography have carefully evaluated the dimensions of OS disc membranes, providing disc thicknesses as well as diameters for the disc rim regions (46, 47). These measurements show a 5-nm disc membrane thickness with a 26-nm diameter for the rim region, including the disc membrane. Measurements of distances between membranes in the flattened portion of the disc are about 4 nm. The corresponding area would likely be too small for either peripherin-ROM1 or ABCA4, forcing them into the rim regions of the disc, which by our calculations should have sufficient space to accommodate these proteins.
We reconstituted peripherin-ROM1 by very slow addition of cyclodextrin to a solution containing membrane proteins in detergent micelles and solubilized lipids using a custom-built robot (42). This instrument allowed us to screen many conditions with small amounts of protein and it monitored progress of the reconstitution process by light scattering. When peripherin-ROM1 was reconstituted at a lipid to protein ratio of 0.1, we consistently observed homogeneous cylindrical lipid protein structures that had a diameter of 38 ± 6 nm (Fig. 5A, left panel) and a height of 7 ± 1 nm, as documented by the top and side views in Fig. 5B. Their pronounced rims exhibited a thickness of ∼6 nm, often revealing an inner fainter ring. Larger proteoliposomes emerged when peripherin-ROM1 was co-reconstituted with rhodopsin. A major fraction of these structures had diameters of 57 ± 22 nm (Fig. 5, B and C, middle panel). These proteoliposomes exhibited a ∼6 nm rim similar to that of peripherin-ROM1-lipid cylinders. Rimless vesicles assembled under identical conditions when rhodopsin alone was reconstituted (Fig. 5B, right panel). These had a much greater size variation (diameters of 491 ± 475 nm; Fig. 5C, right panel) than proteoliposomes comprising peripherin-ROM1. This demonstrates the capability of peripherin-ROM1 to induce a strong curvature of the lipid membrane. It is interesting to note that the curvature of the membrane at the periphery of native discs (13 nm)−1 (47) is similar to that of the smallest peripherin-ROM1-lipid cylinders with curvatures of >(15 nm)−1, and not much different to the average curvature in such cylinders ((19 nm)−1; Fig. 5C, left panel).
Upon reconstitution of peripherin-ROM1 at a lipid to protein ratio of 1, a larger variety of protein-lipid aggregates formed. S- or U-shaped structures and ribbons with cylindrical ends were found. These structures allowed the packing of peripherin-ROM1 complexes to be resolved (Figs. 6 and 7). Peripherin-ROM1 extensions were distinct in S- and U-shaped assemblies, consistently forming an inner ring in curved membranes (Fig. 6). Although curvature variations and poor regularity of the packing arrangement hindered achieving an average that reveals the individual protein complexes, micrographs such as that shown in Fig. 6 allowed the average distance between extensions (d = 4.4 ± 0.6 nm) and their length (n = 4.5 ± 0.5 nm) to be estimated. Taking into account the thickness of the rim of the structures (t = 6.5 ± 0.5 nm), we estimated the length of the peripherin-ROM1 complex to be ∼11 nm. These results are in excellent agreement with the 18-Å three-dimensional structure of negatively stained peripherin-ROM1 complexes kept in solution by Amphipol (Fig. 4). The conical domain interpreted to be intra-discal had a shape and dimensions that promote the packing arrangement of peripherin-ROM1 complexes, as indicated in Fig. 8. Ribbon-like assemblies were not sufficiently straight and ordered to allow elucidation of the precise packing arrangement of peripherin-ROM1 (Fig. 7). Nevertheless, the average distance between extensions was 4.6 ± 0.3 nm, identical to that in S- and U-shaped assemblies within experimental error. How these ribbon-like structures assemble remains to be determined, although the images suggest an interaction between the tips of the intradiscal peripherin-ROM1 domains. Human patients with mutations in peripherin display a wide range of pathologies including macular dystrophy, rod and cone-rod dystrophy, and retinitis pigmentosa. A majority of the mutations identified to date are located in the EC2 domain, long thought to be the site of interaction between the monomers in the complex (Fig. 2F). The model presented here would suggest that these mutations, many of which are transmitted in a dominant inheritance pattern, would disrupt the formation of the core tetramer and thus affect its ability to properly form the rim structures of the disc membranes, leading to a disease phenotype.
FIGURE 8.
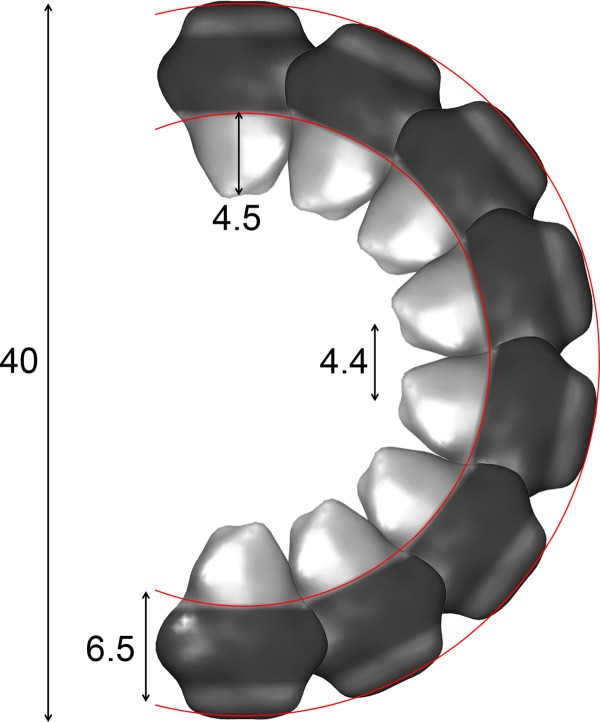
Model of peripherin-ROM1 packing in the disc rim. Packing of the peripherin-ROM1 complex is depicted using the EM structure presented in Fig. 4. Dimensions presented were determined empirically using both the EM structure and reconstitution assays.
Acknowledgments
We thank Dr. Leslie T. Webster Jr., and members of the Palczewski laboratory for helpful comments on the manuscript. We thank Dr. Benlian Wang for glycan mass spectral analyses.
This work was supported, in whole or in part, by National Institutes of Health Grants EY008061 and EY019478. This work was also supported by the Transcontinental EM Initiative for Membrane Protein Structure, which is a center for membrane protein structure determination funded by the National Institutes of Health Protein Structure Initiative under Grant U54GM094598.
- OS
- outer segment
- ROM1
- retinal outer segment membrane protein 1
- EC2
- extracellular loop 2
- ROS
- rod outer segment
- EM
- electron microscopy
- DDM
- n-dodecyl-β-d-maltoside
- LMNG
- lauryl maltose neopentyl glycol
- SEC
- size exclusion chromatography
- BisTris propane
- 1,3-bis[tris(hydroxymethyl)methylamino]propane
- EC
- extracellular
- CMC
- critical micellular concentration
- PNGase F
- peptide N-glycosidase F
- WGA
- wheat germ agglutinin.
REFERENCES
- 1. Kevany B. M., Palczewski K. (2010) Phagocytosis of retinal rod and cone photoreceptors. Physiology 25, 8–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hemler M. E. (2005) Tetraspanin functions and associated microdomains. Nat. Rev. Mol. Cell Biol. 6, 801–811 [DOI] [PubMed] [Google Scholar]
- 3. Rubinstein E. (2011) The complexity of tetraspanins. Biochem. Soc. Trans. 39, 501–505 [DOI] [PubMed] [Google Scholar]
- 4. Stipp C. S., Kolesnikova T. V., Hemler M. E. (2003) Functional domains in tetraspanin proteins. Trends Biochem. Sci. 28, 106–112 [DOI] [PubMed] [Google Scholar]
- 5. Seigneuret M., Delaguillaumie A., Lagaudrière-Gesbert C., Conjeaud H. (2001) Structure of the tetraspanin main extracellular domain. A partially conserved fold with a structurally variable domain insertion. J. Biol. Chem. 276, 40055–40064 [DOI] [PubMed] [Google Scholar]
- 6. Mustafi D., Kevany B. M., Genoud C., Okano K., Cideciyan A. V., Sumaroka A., Roman A. J., Jacobson S. G., Engel A., Adams M. D., Palczewski K. (2011) Defective photoreceptor phagocytosis in a mouse model of enhanced S-cone syndrome causes progressive retinal degeneration. FASEB J. 25, 3157–3176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Farjo R., Naash M. I. (2006) The role of Rds in outer segment morphogenesis and human retinal disease. Ophthalmic Genet. 27, 117–122 [DOI] [PubMed] [Google Scholar]
- 8. Boon C. J., den Hollander A. I., Hoyng C. B., Cremers F. P., Klevering B. J., Keunen J. E. (2008) The spectrum of retinal dystrophies caused by mutations in the peripherin/RDS gene. Prog. Retin. Eye Res. 27, 213–235 [DOI] [PubMed] [Google Scholar]
- 9. Dryja T. P., Hahn L. B., Kajiwara K., Berson E. L. (1997) Dominant and digenic mutations in the Peripherin/RDS and ROM1 genes in retinitis pigmentosa. Invest. Ophthalmol. Vis. Sci. 38, 1972–1982 [PubMed] [Google Scholar]
- 10. Kitadokoro K., Bordo D., Galli G., Petracca R., Falugi F., Abrignani S., Grandi G., Bolognesi M. (2001) CD81 extracellular domain 3D structure. Insight into the tetraspanin superfamily structural motifs. EMBO J. 20, 12–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Min G., Wang H., Sun T. T., Kong X. P. (2006) Structural basis for tetraspanin functions as revealed by the cryo-EM structure of uroplakin complexes at 6-Å resolution. J. Cell Biol. 173, 975–983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wu X. R., Manabe M., Yu J., Sun T. T. (1990) Large-scale purification and immunolocalization of bovine uroplakin-I, uroplakin-II, and uroplakin-III, molecular markers of urothelial differentiation. J. Biol. Chem. 265, 19170–19179 [PubMed] [Google Scholar]
- 13. Goldberg A. F., Molday R. S. (1996) Subunit composition of the peripherin/rds-rom-1 disk rim complex from rod photoreceptors. Hydrodynamic evidence for a tetrameric quaternary structure. Biochemistry 35, 6144–6149 [DOI] [PubMed] [Google Scholar]
- 14. Loewen C. J., Molday R. S. (2000) Disulfide-mediated oligomerization of peripherin/Rds and Rom-1 in photoreceptor disk membranes. Implications for photoreceptor outer segment morphogenesis and degeneration. J. Biol. Chem. 275, 5370–5378 [DOI] [PubMed] [Google Scholar]
- 15. Chakraborty D., Ding X. Q., Conley S. M., Fliesler S. J., Naash M. I. (2009) Differential requirements for retinal degeneration slow intermolecular disulfide-linked oligomerization in rods versus cones. Hum. Mol. Genet. 18, 797–808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chakraborty D., Ding X. Q., Fliesler S. J., Naash M. I. (2008) Outer segment oligomerization of Rds. Evidence from mouse models and subcellular fractionation. Biochemistry 47, 1144–1156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Goldberg A. F., Loewen C. J., Molday R. S. (1998) Cysteine residues of photoreceptor peripherin/rds. Role in subunit assembly and autosomal dominant retinitis pigmentosa. Biochemistry 37, 680–685 [DOI] [PubMed] [Google Scholar]
- 18. Muller-Weeks S., Boesze-Battaglia K., Fitzgerald C. (2002) Deletional analysis of the rod photoreceptor cell peripherin/RDS carboxy-terminal region. Exp. Eye Res. 75, 143–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Boesze-Battaglia K., Kong F., Lamba O. P., Stefano F. P., Williams D. S. (1997) Purification and light-dependent phosphorylation of a candidate fusion protein, the photoreceptor cell peripherin/rds. Biochemistry 36, 6835–6846 [DOI] [PubMed] [Google Scholar]
- 20. Zhou M., Morgner N., Barrera N. P., Politis A., Isaacson S. C., Matak-Vinković D., Murata T., Bernal R. A., Stock D., Robinson C. V. (2011) Mass spectrometry of intact V-type ATPases reveals bound lipids and the effects of nucleotide binding. Science 334, 380–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Papermaster D. S. (1982) Preparation of retinal rod outer segments. Methods Enzymol. 81, 48–52 [DOI] [PubMed] [Google Scholar]
- 22. Tsybovsky Y., Wang B., Quazi F., Molday R. S., Palczewski K. (2011) Posttranslational modifications of the photoreceptor-specific ABC transporter ABCA4. Biochemistry 50, 6855–6866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tsybovsky Y., Orban T., Molday R. S., Taylor D., Palczewski K. (2013) Molecular organization and ATP-induced conformational changes of ABCA4, the photoreceptor-specific ABC transporter. Structure 21, 854–860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tang G., Peng L., Baldwin P. R., Mann D. S., Jiang W., Rees I., Ludtke S. J. (2007) EMAN2. An extensible image processing suite for electron microscopy. J. Struct. Biol. 157, 38–46 [DOI] [PubMed] [Google Scholar]
- 25. Shaikh T. R., Gao H., Baxter W. T., Asturias F. J., Boisset N., Leith A., Frank J. (2008) SPIDER image processing for single-particle reconstruction of biological macromolecules from electron micrographs. Nat. Protoc. 3, 1941–1974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gohon Y., Dahmane T., Ruigrok R. W., Schuck P., Charvolin D., Rappaport F., Timmins P., Engelman D. M., Tribet C., Popot J. L., Ebel C. (2008) Bacteriorhodopsin/amphipol complexes. Structural and functional properties. Biophys. J. 94, 3523–3537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E. (2004) UCSF Chimera. A visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]
- 28. Kaufmann T. C., Engel A., Rémigy H. W. (2006) A novel method for detergent concentration determination. Biophys. J. 90, 310–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jastrzebska B., Orban T., Golczak M., Engel A., Palczewski K. (2013) Asymmetry of the rhodopsin dimer in complex with transducin. FASEB J. 27, 1572–1584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Saxton W. O. (1996) Semper. Distortion compensation, selective averaging, 3-D reconstruction, and transfer function correction in a highly programmable system. J. Struct. Biol. 116, 230–236 [DOI] [PubMed] [Google Scholar]
- 31. Donoho D. L., Johnstone I. M., Hoch J. C., Stern A. S. (1992) Maximum entropy and the nearly black object. J. R. Stat. Soc. B 54, 41–81 [Google Scholar]
- 32. Travis G. H., Sutcliffe J. G., Bok D. (1991) The retinal degeneration slow (rds) gene-product is a photoreceptor disk membrane-associated glycoprotein. Neuron 6, 61–70 [DOI] [PubMed] [Google Scholar]
- 33. Kedzierski W., Bok D., Travis G. H. (1999) Transgenic analysis of rds/peripherin N-glycosylation. Effect on dimerization, interaction with rom1, and rescue of the rds null phenotype. J. Neurochem. 72, 430–438 [DOI] [PubMed] [Google Scholar]
- 34. Barrera N. P., Di Bartolo N., Booth P. J., Robinson C. V. (2008) Micelles protect membrane complexes from solution to vacuum. Science 321, 243–246 [DOI] [PubMed] [Google Scholar]
- 35. Barrera N. P., Isaacson S. C., Zhou M., Bavro V. N., Welch A., Schaedler T. A., Seeger M. A., Miguel R. N., Korkhov V. M., van Veen H. W., Venter H., Walmsley A. R., Tate C. G., Robinson C. V. (2009) Mass spectrometry of membrane transporters reveals subunit stoichiometry and interactions. Nat. Methods 6, 585–587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jastrzebska B., Ringler P., Lodowski D. T., Moiseenkova-Bell V., Golczak M., Müller S. A., Palczewski K., Engel A. (2011) Rhodopsin-transducin heteropentamer. Three-dimensional structure and biochemical characterization. J. Struct. Biol. 176, 387–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Althoff T., Mills D. J., Popot J. L., Kühlbrandt W. (2011) Arrangement of electron transport chain components in bovine mitochondrial supercomplex I1III2IV1. EMBO J. 30, 4652–4664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cvetkov T. L., Huynh K. W., Cohen M. R., Moiseenkova-Bell V. Y. (2011) Molecular architecture and subunit organization of TRPA1 ion channel revealed by electron microscopy. J. Biol. Chem. 286, 38168–38176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Eskandari S., Wright E. M., Kreman M., Starace D. M., Zampighi G. A. (1998) Structural analysis of cloned plasma membrane proteins by freeze-fracture electron microscopy. Proc. Natl. Acad. Sci. U.S.A. 95, 11235–11240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Vahedi-Faridi A., Jastrzebska B., Palczewsk K., Engel A. (2013) 3D imaging and quantitative analysis of small solubilized membrane proteins and their complexes by transmission electron microscopy. Microscopy 62, 95–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Iacovache I., Biasini M., Kowal J., Kukulski W., Chami M., van der Goot F. G., Engel A., Rémigy H. W. (2010) The 2DX robot. A membrane protein 2D crystallization Swiss Army knife. J. Struct. Biol. 169, 370–378 [DOI] [PubMed] [Google Scholar]
- 42. Signorell G. A., Kaufmann T. C., Kukulski W., Engel A., Rémigy H. W. (2007) Controlled 2D crystallization of membrane proteins using methyl-β-cyclodextrin. J. Struct. Biol. 157, 321–328 [DOI] [PubMed] [Google Scholar]
- 43. Boesze-Battaglia K., Lamba O. P., Napoli A. A., Jr., Sinha S., Guo Y. (1998) Fusion between retinal rod outer segment membranes and model membranes. A role for photoreceptor peripherin/rds. Biochemistry 37, 9477–9487 [DOI] [PubMed] [Google Scholar]
- 44. Wrigley J. D., Ahmed T., Nevett C. L., Findlay J. B. (2000) Peripherin/rds influences membrane vesicle morphology. Implications for retinopathies. J. Biol. Chem. 275, 13191–13194 [DOI] [PubMed] [Google Scholar]
- 45. Khattree N., Ritter L. M., Goldberg A. F. (2013) Membrane curvature generation by a C-terminal amphipathic helix in peripherin-2/rds, a tetraspanin required for photoreceptor sensory cilium morphogenesis. J. Cell Sci. 126, 4659–4670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gilliam J. C., Chang J. T., Sandoval I. M., Zhang Y., Li T., Pittler S. J., Chiu W., Wensel T. G. (2012) Three-dimensional architecture of the rod sensory cilium and its disruption in retinal neurodegeneration. Cell 151, 1029–1041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Nickell S., Park P. S., Baumeister W., Palczewski K. (2007) Three-dimensional architecture of murine rod outer segments determined by cryoelectron tomography. J. Cell Biol. 177, 917–925 [DOI] [PMC free article] [PubMed] [Google Scholar]