

Background: Covalent coupling of SUMO by the E2 and E3 enzymes confers repression activity to transcriptional regulators.
Results: Identification of a non-covalent E2·SUMO·E3 complex that can also function in transcriptional repression.
Conclusion: SUMO participates in repression as both a covalent modification and through non-covalent interactions with E2 and E3 enzymes.
Significance: Similar interaction interfaces in other ubiquitin-like proteins and their cognate enzymes suggest they form analogous ternary complexes.
Keywords: Bioluminescence Resonance Energy Transfer (BRET), Calorimetry, Gene Regulation, Nuclear Magnetic Resonance, Phosphorylation, Protein Complexes, Sumoylation, E2 SUMO Conjugating Enzyme, E3 SUMO Ligase, SUMO Interacting Motif
Abstract
Post-translational modifications with ubiquitin-like proteins require three sequentially acting enzymes (E1, E2, and E3) that must unambiguously recognize each other in a coordinated fashion to achieve their functions. Although a single E2 (UBC9) and few RING-type E3s (PIAS) operate in the SUMOylation system, the molecular determinants regulating the interactions between UBC9 and the RING-type E3 enzymes are still not well defined. In this study we use biochemical and functional experiments to characterize the interactions between PIAS1 and UBC9. Our results reveal that UBC9 and PIAS1 are engaged both in a canonical E2·E3 interaction as well as assembled into a previously unidentified non-covalent ternary complex with SUMO as evidenced by bioluminescence resonance energy transfer, nuclear magnetic resonance spectroscopy, and isothermal titration calorimetry studies. In this ternary complex, SUMO functions as a bridge by forming non-overlapping interfaces with UBC9 and PIAS1. Moreover, our data suggest that phosphorylation of serine residues adjacent to the PIAS1 SUMO-interacting motif favors formation of the non covalent PIAS1·SUMO·UBC9 ternary complex. Finally, our results also indicate that the non-covalent ternary complex is required for the known transcriptional repression activities mediated by UBC9 and SUMO1. Taken together, the data enhance our knowledge concerning the mode of interaction of enzymes of the SUMOylation machinery as well as their role in transcriptional regulation and establishes a framework for investigations of other ubiquitin-like protein systems.
Introduction
Post-translational modifications by ubiquitin-like proteins (UBLs)5 occur on a large number of target proteins, altering their functions and subsequently cell response to stimuli. The covalent coupling of UBL family members (ubiquitin, NEDD8, SUMO, ISG15) to specific substrate proteins is achieved through a series of sequential steps involving three distinct enzymatic activities (E1 activating enzyme, E2 conjugating enzyme, and E3 ligase) (1). The ubiquitination machinery has been extensively studied, and it is currently known that the human genome encodes 2 E1 enzymes, >20 E2 enzymes, and ∼500 RING-type E3 ligases (2–4). The appropriate pairing of E2 and E3 enzymes is an important determinant for specificity of target recognition and ubiquitination (5). Considering the numerous possible combinations of E2 and E3 enzymes, it remains challenging to identify the functional ubiquitin E2·E3 pair required for ubiquitination of a given substrate and to understand the molecular basis that control specific interactions by UBLs.
Human cells possess at least three functional SUMO proteins (SUMO1, -2, and -3) that adopt a three-dimensional structure similar to ubiquitin despite sharing only 20% sequence identity (6–15). In comparison to other known UBL systems, the SUMOylation machinery appears to be the simplest in terms of possible E2·E3 combinations (16). In the SUMOylation pathway, there is a single E1 activating enzyme (SAE1/SAE2 heterodimer), a single E2 conjugating enzyme (UBC9), and a limited number of RING-type E3 ligases (the Siz/PIAS (yeast/human) family members) (17–25). Like ubiquitin, SUMO proteins must be activated before their conjugation with target proteins (26). This maturation step, performed by the SUMO-specific proteases (SENPs), results in the cleavage of the carboxyl-terminal residues that exposes a di-glycine motif of SUMO required for their subsequent adenylation by the E1 enzyme SAE1·SAE2 (20). Similar to the activation step for ubiquitin, the adenylated SUMO is then attacked by the catalytic cysteine residue of the E1 enzyme, resulting in the formation of a thioester bond (27). The thioester-bound SUMO is then transferred to the catalytic cysteine residue of the sole SUMO E2 conjugating enzyme, UBC9, before being covalently linked to a target lysine residue on the substrate protein (20, 22, 28). Unlike what is observed with ubiquitination sites, a large proportion of SUMO-modified lysine residues are found within consensus motifs corresponding to the sequence ψKX(E/D) (ψ is a hydrophobic residue, and X corresponds to any residues) (29). This is consistent with the fact that a single E2-conjugating enzyme functions in the SUMOylation pathway and to the uniqueness of the catalytic cleft of UBC9 (30).
The SUMO E2 conjugating enzyme UBC9 shares a similar catalytic core and tertiary structure (the UBC fold) with all other known UBL E2 conjugating enzymes (4). However, the electrostatic potentials of the UBC9 interfaces involved in interactions with the E1 enzyme and SUMO differ considerably from those found in other UBL E2s (15, 31). These differences may allow UBC9 to form specific interactions with various components of the SUMOylation machinery including the E3 ligases. To date, several structurally unrelated classes of proteins appear to act as SUMO E3 ligases in mammalian cells. These include the Siz/PIAS family members, RanBP2, PC2, and Topors (32–37). Among them, the members of the Siz/PIAS family (PIAS1–4, Siz1 and -2) belong to the SP-RING-type (for Siz/PIAS-RING) SUMO E3 ligases that share several characteristics with the RING-type ubiquitin E3 ligases. In particular, RING E3 ligases function as scaffold proteins that position both the UBL-loaded E2 conjugating enzyme and the substrate to allow modification of the target lysine residue (38). Moreover, a unifying theme among the SUMO E3 ligases is their ability to interact non-covalently with SUMO proteins via their SUMO interacting motifs (SIMs), which appears to be required for the activity of select SUMO E3 ligases (24, 33, 39–43). These non-covalent interactions between SUMO proteins and their cognate E3 ligases seem to be shared among other UBL E3 ligases, as several ubiquitin E3 ligases display ubiquitin binding domains that contribute to the specificity of the ubiquitination process (44–46).
To better understand the mechanisms that govern the correct selection of E2·E3 combinations in SUMOylation, we attempted to identify key determinants that regulate interactions between the SUMO E3 ligase PIAS1 and the SUMO E2 conjugating enzyme UBC9. Based on structure- and sequence-guided mutational analysis, we performed a series of in vivo and in vitro experiments to help define the interaction interfaces formed between SUMO proteins, PIAS1 and UBC9. Based on our results, we conclude that PIAS1 and UBC9 engage in a canonical E2·E3 RING interaction with each other, where specific residues of the PIAS1 SP-RING domain are involved in interaction with the L4 loop of UBC9. In addition, we provide evidence that PIAS1, UBC9, and SUMO proteins form a ternary complex through a series of non-covalent interactions, where SUMO functions as a bridge to link PIAS1 and UBC9. Although covalent modification of UBC9 by SUMO is not required for formation of the ternary complex, the non-covalent interaction of UBC9 and SUMO appears to enhance the ability of UBC9 to interact with PIAS1 within the ternary complex. Moreover, our data support a model in which the PIAS1·SUMO·UBC9 ternary complex functions in transcriptional repression and its formation is regulated by the phosphorylation state of the SIM module of PIAS1.
EXPERIMENTAL PROCEDURES
Expression Vectors
Bioluminescence Resonance Energy Transfer (BRET) Vectors
Human UBC9 cDNA was isolated from a human fetal brain MATCHMAKER cDNA library (Clontech) and cloned as a blunted NotI-SpeI fragment into a blunted BamHI-digested pBluescript SK+ vector. A SK+-UBC9-ΔSTOP codon construct was generated using site-directed mutagenesis (Stratagene). UBC9-ΔSTOP was cloned as a SacI-ApaI fragment into the pGFP2N2 vector (PerkinElmer Life Sciences) and as a blunted SacI-ApaI fragment into a EcoRV-ApaI-digested pHRLucN2 vector (PerkinElmer Life Sciences). All the UBC9 mutants (UBC9-R13A, UBC9-K14R, UBC9-R17A, UBC9-H20D, UBC9-F22A, UBC9-P69A, UBC9-C93S, UBC9-C93A, UBC9-D100A-K101A, UBC9-P105A, UBC9-P128A, UBC9-Y134A) were generated by site-directed mutagenesis. Human PIAS1 cDNA was isolated from a human fetal brain MATCHMAKERcDNA library and cloned as a EcoRI-XhoI fragment into the pBluescript SK+ vector. PIAS1 was subcloned as a EcoRI-KpnI fragment into the pHRLucC1 vector (PerkinElmer Life Sciences). All the PIAS1 mutants (PIAS1-L337A, PIAS1-SIMmt (PIAS1-V457A/V459A/I460A/L462A/I464A), PIAS1–3SA (PIAS1-S466A/S467A/S468A) and PIAS1–3SD (PIAS1-S466D/S467D/S468D)) were generated by site-directed mutagenesis. PIAS1–5EA (PIAS1-E470A/E471A/E472A/E473A/E474A)was ordered (Bio Basic) as a XbaI-KpnI fragment and subcloned into pHRLuc-PIAS1 in replacement of the wild-type fragment. The non-conjugate-able versions of human SUMO1 and human SUMO2 fused to pGFP10 were generated by site-directed mutagenesis from pGFP10-SUMO1 and pGFP10-SUMO2 (47), respectively, mutating the SUMO di-glycine motif to two alanine residues. All the SUMO1 (SUMO1mt (SUMO1-F36A/K37A/K39A/K45A/K46A) and SUMO1-E67R) and SUMO2 (SUMO2-D63R) mutants were generated by site-directed mutagenesis starting from the cDNAs encoding the non-conjugate-able forms of SUMO1 and SUMO2, respectively. All clones were verified by DNA sequencing. A detailed description of the protein variants used in this study is provided in Table 1.
TABLE 1.
Protein mutations and interactions tested
| Mutation in | Mutant tested | Tested for interaction with | Assaya | Figure |
|---|---|---|---|---|
| PIAS1 SP-RING | PIAS1 L337 | UBC9 | BRET (↓) | 1B |
| PIAS1 SIM | PIAS1 SIMmtb | UBC9 | BRET (↓) | 3B, Not shown |
| SUMO1 or SUMO2 | ||||
| PIAS1 serines adjacent to the SIM | PIAS1 3SD | UBC9 | BRET (↑) | 3C and D |
| PIAS1 3SD peptide encompassing the SIMc | SUMO1 SUMO2 UBC9 UBC9 and SUMO1 |
ITC (↑), NMR ITC (↑) NMR NMR |
3F, 4, A–C and F Not shown Not shown 6 |
|
| PIAS1 3SA | UBC9 | BRET (↓) | 3C and D | |
| PIAS1 acidic region recognized by CK2 and adjacent to the SIM | PIAS1 5EA | UBC9 | BRET (↓) | 3D |
| UBC9 loops predicted to interact with PIAS1 SP-RING | UBC9 P69A | PIAS1 | BRET (↓) | 1D |
| UBC9 P105A | PIAS1 | BRET (n.c) | 1D | |
| UBC9 catalytic site | UBC9 C93S or C93A | PIAS1 | BRET( n.c) | 2B |
| UBC9 SUMOylation site | UBC9 K14A | PIAS1 | BRET (n.c) | 2D, Not shown |
| SUMO1 | ||||
| UBC9 backside region forming an interface with β1, 4 and 5 strands of SUMO | UBC9 R13A | PIAS1 | BRET (↓) | 2G, Not shown |
| SUMO1 | ||||
| UBC9 R17A | PIAS1 | BRET (↓) | 2E, Not shown | |
| SUMO1 or SUMO2 | ||||
| UBC9 H20D | PIAS1 | BRET (↓) | 2E, Not shown | |
| SUMO1 or SUMO2 | ||||
| UBC9 F22A | PIAS1 | BRET (↓) | 2F | |
| SUMO surface interacting with UBC9 backside | SUMO1 E67R | UBC9 | BRET (↓) | Not shown |
| SUMO2 D63R | UBC9 | BRET (↓) | Not shown | |
| SUMO1 surface interacting with PIAS SIM sequence | SUMO1mt (Phe-36, Lys-37, Lys-39, Lys-45, Lys-46 mutated to Ala) | PIAS1 | BRET (↓) | Not shown |
a The results relative to wild-type controls in the BRET or ITC signal are indicated in the parentheses: increase (↑), decrease (↓), or no significant change (n.c.).
b SIMmt amino acids 457–464: VEVIDLTI → AEAADATA.
c PIAS1 phosphomimetic peptide amino acids 456–480: KVEVIDLTIDDDDDEEEEEPSAKRT (the SIM is underlined, and the serines mutated to aspartic acid are in bold).
Recombinant Protein Expression Vectors
Oligonucleotides encoding for the peptide sequences of the human PIAS1-SIM (residues 456–480) and PIAS1-SIM-3SD (PIAS1-SIM with Ser-466, Ser-467, and Ser-468 mutated to aspartic acid) were synthesized with BamHI and EcoRI restriction sites (Integrated DNA Technologies), 5′-phosphorylated, annealed, and cloned as BamHI-EcoRI fragments into the pGEX-2T vector (GE Healthcare). SUMO1 (residues 2–97 of human SUMO1) cDNA was PCR amplified from pGFP10-SUMO1 and cloned as a XbaI fragment into the pGEX-4T3 vector (GE Healthcare). A SUMO1-C52A point mutant was generated using site-directed mutagenesis. SUMO2 (residues 1–93 of human SUMO2) cDNA was PCR-amplified from pGFP10-SUMO2 and cloned as a BamHI-EcoRI fragment into the pGEX-2T vector. UBC9 (residues 1–158 of human UBC9) cDNA was PCR amplified and cloned as a BamHI-EcoRI fragment into the pGEX-2T vector. All clones were verified by DNA sequencing.
Transcriptional Assay Vectors
For expression of the Gal4DBD fusion proteins (Gal4DBD-UBC9 and Gal4DBD-SUMO1), human UBC9 cDNA sequence was cloned as a BsrGI-BamHI fragment in the pcDNA3.1-Gal4DBD vector, and human SUMO1 with the di-glycine motif mutated to alanine residues was cloned as a XbaI-BamHI blunt fragment in the pRSV-Gal4DBD vector. All the Gal4DBD-UBC9 mutants (UBC9-K14R, UBC9-R17A, UBC9-H20D, UBC9-P69A, UBC9-C93S, UBC9-D100A-K101A, UBC9-P128A, UBC9-Y134A) and Gal4DBD-SUMO1 mutants (SUMO1-F36A and SUMO1-E67R) were generated by site-directed mutagenesis and verified by DNA sequencing.
Transient Transfections
Human embryonic kidney 293T cells (HEK 293T) maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum (Wisent), 100 μg/ml penicillin and streptomycin, and 1 mm l-glutamine were seeded at a density of ∼1 × 106 cells per 100-mm dish for BRET experiments and ∼ 2 × 105 cells per well in 6-well plates for transcriptional assays. Transient transfections of plasmids were performed on the following day using the calcium phosphate precipitation method except for transcription assays where FuGENE transfection reagent (3 μl/1 μg of DNA) (ThermoFisher) was used. The total amount of transfected DNA was kept constant (10 μg for 100-mm dishes and 1 μg per well for 6-well plates).
BRET Experiments
The BRET assays were conducted as previously described (48). Briefly, cells transiently transfected with plasmids encoding fusion proteins of the Luciferase donor (2 or 3 μg depending the construction) and GFP acceptor (from 0.125–7 μg) were resuspended and distributed in 96-well plates. Upon the addition of the cell-permeant Luciferase substrate coelenterazine deep blue (PerkinElmer Life Sciences), the bioluminescence signal resulting from its degradation was detected using a 370–450-nm band pass filter (donor emission peak at 400 nm). The energy transferred resulting in a fluorescence signal emitted by the GFP acceptor (excitation peak at 400 nm, emission peak at 510 nm) was detected using a 500–530-nm band pass filter. The BRET signal (BRET ratio) was quantified by calculating the acceptor fluorescence/donor bioluminescence ratio as previously reported, (49) using a modified Top- count apparatus (BRET count, Packard Instrument Co.). The expression level of each fusion protein was determined by direct measurements of total fluorescence or luminescence on aliquots of transfected cell samples. The GFP total fluorescence was measured using a Fusion Alpha FP (Packard) with excitation at 425 nm and emission at 515 nm. The total luminescence was measured with the same cells incubated with coelenterazine H (Molecular Probes) for 10 min (emission peak at 485 nm) using a Fusion Alpha FP instrument (Packard). The BRET ratios (% BRET) were plotted as a function of the GFP/Luc fusion protein expression ratio to take into account the potential variations in the expression of individual fusion proteins for a given transfection.
Transcriptional Assays
Transient transfections of reporter and effector plasmids were performed as described above. Briefly, 200 ng per well of the Firefly Luciferase reporter plasmid, pGL3–5xGal4, which corresponds to pGL3 vector under the control of the SV40 promoter (Promega) with five repeats of the Gal4-DNA binding sequence (AGGGTATATAATG), was used. The Renilla Luciferase vector phRLuc-C1 (PerkinElmer Life Sciences) was co-transfected (20 ng/well) to normalize for transfection efficiency. Up to 400 ng of effector plasmid that corresponds to pcDNA3.1 (Invitrogen) in which Gal4DBD-(1–147) was cloned and fused to UBC9 and mutants or to pRSV-Gal4DBD fused to SUMO1 and mutants (see “Transcriptional Assay Vectors” above) were transfected. The total amount of transfected DNA was kept constant by the addition of the pcDNA3.1 empty vector. Cell lysates were prepared 48 h after transfection and split into two samples for determination of the Luciferase activity and assessment of protein expression levels by Western blotting. The equivalent of 35 μg of cell lysates was processed for the Luciferase activity using the Dual GloTM Luciferase assay kit (Promega). A mouse monoclonal anti-Gal4DBD antibody (sc-510, Santa Cruz Biotechnology) was used to monitor the expression level of Gal4DBD fusion proteins.
Induction and Purification of Glutathione S-Transferase (GST) Fusion Proteins
SUMO1, SUMO2, UBC9, PIAS1-SIM, and PIAS1-SIM-3SD were expressed as GST fusion proteins in Escherichia coli host strain TOPP2 (Stratagene). The cells were grown at 37 °C in Luria broth media, and protein expression was induced for 4 h at 30 °C with 0.7 mm isopropyl-β-d-thiogalactopyranoside (Inalco). The cells were harvested by centrifugation and resuspended in lysis buffer (20 mm Tris-HCl (pH 7.4), 1 m NaCl, 0.2 mm EDTA, and 1 mm DTT). The cells were then lysed in a French press and centrifuged at 35,000 × g for 1 h at 4 °C. The supernatant was then collected and incubated for 1 h with glutathione (GSH)-Sepharose resin (GE Healthcare) at 4 °C. After incubation, the resin was collected by centrifugation and washed with lysis buffer and phosphate-buffered saline (PBS; 10 mm Na2HPO4, 2 mm KH2PO4 (pH 7.4), 140 mm NaCl, and 3 mm KCl). The resin bound proteins were incubated 2 h with 100 units of thrombin (Calbiochem) to cleave the GST tag from proteins. The SUMO and UBC9 proteins were then eluted in PBS and dialyzed against sodium phosphate buffer (20 mm sodium phosphate, 1 mm EDTA, 1 mm DTT) at an appropriate pH. For further purification, a Q-Sepharose High-performance column (GE Healthcare) was used for SUMO proteins, and a SP-Sepharose High-performance column (GE Healthcare) was used for UBC9. After elution, the PIAS peptides were dialyzed against 5% acetic acid and purified over a C4-reversed phase HPLC column (Vydac). An additional purification step on a Sephadex-75 gel-filtration column (GE Healthcare) was performed for SUMO and UBC9 proteins. Proteins and peptides were then desalted, lyophilized (for SUMOs and SIM-containing peptides), and kept at −80 °C until being processed for isothermal titration calorimetry (ITC) or nuclear magnetic resonance (NMR) experiments. 15N-Labeled proteins were prepared as described, but the E. coli host strain was grown in M9 minimal media containing 15NH4Cl (Sigma) as the sole nitrogen source.
ITC Experiments
For ITC experiments, lyophilized proteins were resuspended in water and dialyzed overnight at 4 °C against Tris buffer (20 mm Tris-HCl (pH 7.4)). The protein concentrations were determined from absorbance at 280 nm. ITC measurements were performed at 25 °C using a Microcal VP-ITC calorimeter (GE Healthcare). For each titration experiment, the concentration of the protein or peptide in the syringe was 10 times higher than in the sample cell. All titration experiments were performed at least two times. The base-line-corrected data were fit to a single binding site interaction with 1:1 stoichiometry using the Origin 7 software.
NMR Spectroscopy
NMR experiments were carried out at 300 K on Varian Unity Inova 500- and 600-MHz spectrometers. For the NMR chemical shift perturbation experiments of SUMO1, 0.5 mm 15N-labeled SUMO1 was used in 20 mm sodium phosphate (pH 6.5), 90% H2O, and 10% D2O. To map the PIAS1-SIM peptide binding sites on SUMO1, unlabeled PIAS1-SIM or PIAS1-SIM-3SD peptide was titrated to a final ratio of 1:1.5 (15N-SUMO1·PIAS-SIM peptide). To map the UBC9 binding sites on SUMO1, unlabeled UBC9 protein was sequentially added to a final ratio of 1:1 (15N-SUMO1·UBC9). For the NMR chemical shift perturbation experiments of the PIAS1-SIM-3SD peptide, 0.5 mm 15N-labeled PIAS1-SIM-3SD was used in 20 mm sodium phosphate (pH 6.5), 90% H2O, and 10% D2O. The 15N-PIAS1-SIM-3SD·SUMO1 complex was obtained by titration of unlabeled SUMO1 to a final ratio of 1:2 (15N-PIAS1-SIM-3SD·SUMO1). The PIAS1-SIM-3SD·UBC9 complexes were prepared by titration of either unlabeled UBC9 to 0.5 mm of 15N-labeled PIAS1-SIM-3SD peptide to a final ratio of 1:3 (15N-PIAS1-SIM-3SD·UBC9) or unlabeled PIAS1-SIM-3SD peptide in 0.5 mm 15N-labeled UBC9 to a final ratio of 1:4 (15N-UBC9·PIAS1-SIM-3SD). For preparation of the PIAS1-SIM-3SD·SUMO1·UBC9 ternary complex, two methods were employed. First, 0.5 mm 15N-labeled SUMO1 in 20 mm sodium phosphate (pH 6.5), 90% H2O, and 10% D2O was supplemented with 0.75 mm unlabeled PIAS1-SIM-3SD peptide and then unlabeled UBC9 to a final concentration of 0.5 mm. Alternatively, 0.5 mm 15N-labeled SUMO1 in 20 mm sodium phosphate (pH 6.5), 90% H2O, and 10% D2O was supplemented with 0.5 mm unlabeled UBC9 and then unlabeled PIAS1-SIM-3SD peptide to a final concentration of 0.75 mm. The backbone assignments of SUMO1 and UBC9 were obtained from the Biological Magnetic Resonance Data Bank (accession numbers 6304 and 4132, respectively). The NMR data were processed with NMRPipe/NMRDraw (50) and analyzed with CCPNMR (51).
RESULTS
A Conserved Hydrophobic Residue in PIAS1 Is Required for Interaction with UBC9
The SUMO E3 ligases from the Siz/PIAS family possess a SP-RING domain that displays both sequence and structure homology with the RING and U-box domains from ubiquitin E3 ligases (38) (Fig. 1A and data not shown). Several in vitro binding studies have shown that SP-RING domains are capable of directly interacting with UBC9, but very little is known regarding the determinants of this interaction (24, 25, 33, 35). To better understand the role of the SP-RING domain of PIAS proteins in binding to UBC9, we searched for highly conserved residues in known RING-type domains of E3 ligases that could mediate this interaction. Based on sequence alignments (data not shown), we identified several residues that are either identical or homologous in RING-type domains. Among these, we chose to mutate a conserved hydrophobic residue (Leu-337) to assess its role in the binding of PIAS1 to UBC9 in a cellular context. This particular residue was chosen because a similar residue in Siz1 (Ile-363) has been shown to be essential for in vitro ligase activity (38).
FIGURE 1.
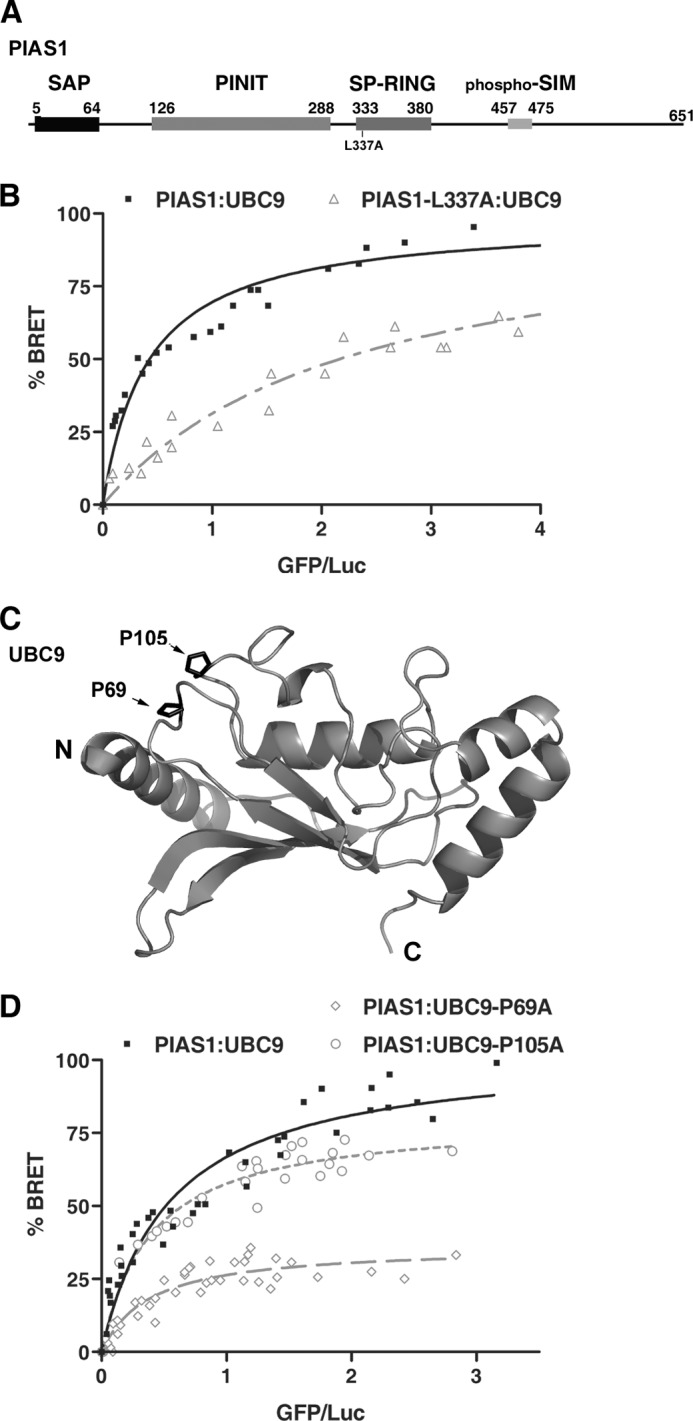
Conserved residues within PIAS1 and UBC9 play a key role in the PIAS1·UBC9 complex formation. A, schematic representation of the full-length human PIAS1. Key regions of the protein are highlighted with boxes including the SAP (SAF-A/B, Acinus, and PIAS) domain, PINIT (proline-isoleucine-asparagine-isoleucine-threonine) motif-containing domain, SP-RING (Siz/PIAS-Really Interesting New Gene) domain, and the phospho-SIM module. The L337A mutation in the PIAS1 SP-RING domain is indicated. B, BRET titration curves obtained with wild-type UBC9 and either wild-type PIAS1 or the PIAS1-L337A mutant. The experiments were performed with a fixed amount of either RLuc-PIAS1 (■) or RLuc-PIAS1-L337A (▵) and increasing amounts of UBC9-GFP in HEK293T cells. For each curve the data of two independent experiments (40 independent transfections) were pooled. 100% BRET corresponds to the maximum BRET signal (as defined under “Experimental Procedures”) value of 0.55 measured with wild-type PIAS1 and UBC9. C, ribbon representation of human UBC9 (PDB code 1U9A) showing proline residues (Pro-69 and Pro-105) subjected to mutational analysis. N and C correspond respectively to the amino and carboxyl termini of UBC9. D, BRET titration curves obtained with wild-type PIAS1 and either wild-type UBC9 or UBC9 loop mutants. The experiments were performed using a fixed amount of wild-type RLuc-PIAS1 and increasing amounts of UBC9-GFP (■), UBC9-P105A-GFP (L7 loop mutant) (○), or UBC9-P69A-GFP (L4 loop mutant) (%#x25CA;). For each curve, the data from at least two independent experiments were pooled. 100% BRET corresponds to the maximum BRET signal value of 0.56 measured with wild-type PIAS1 and UBC9.
The interaction between PIAS1-L337A and UBC9 was monitored in HEK293T cells using a BRET assay (48, 52). HEK293T cells were co-transfected with a fixed amount of a DNA construct coding for either the wild-type PIAS1 or the PIAS1-L337A mutant fused to Renilla Luciferase (RLuc-PIAS1or RLuc-PIAS1-L337A) along with increasing amounts of a DNA construct coding for UBC9 fused to the green fluorescent protein (UBC9-GFP) (Fig. 1B). For wild-type RLuc-PIAS1, the BRET ratio increases as a function of UBC9-GFP concentration and reaches a maximum when the UBC9-GFP expression levels (BRET acceptor) are no longer limiting relative to the RLuc-PIAS1 expression levels (BRET donor). The saturation curve is indicative of a specific interaction between PIAS1 and UBC9 under these experimental conditions. In contrast, a much lower BRET signal is obtained for the interaction between UBC9-GFP and the RLuc-PIAS1-L337A mutant. This is indicative of a weaker interaction and suggests that Leu-337 within the SP-RING domain of PIAS1 is an important determinant for the interaction with UBC9. This result demonstrates that Leu-337 is required for efficient PIAS1·UBC9 association in a cellular context and is consistent with the fact that an equivalent residue is essential for the in vitro SUMO E3 ligase activity of Siz1 (38). Moreover, this result suggests that different classes of RING-type E3 ligases use similar molecular determinants to recognize their cognate E2-conjugating enzyme (53–56).
A Conserved Proline Residue in UBC9 Is Required for Binding to PIAS1
Next, we attempted to identify the region of UBC9 involved in the interaction with the SP-RING domain of PIAS1. Structural studies indicate that UBC9 shares a common three-dimensional structure with ubiquitin E2 enzymes (31, 57). Furthermore, the integrity of the L4 and/or L7 loops of several ubiquitin E2 enzymes have been shown to mediate their interaction with the RING domain of their ubiquitin E3 ligases (58–62). In an attempt to identify residues in either the L4 or L7 loop of UBC9 that could mediate interaction with PIAS1, we aligned UBC9 sequences from several species along with various E2-conjugating enzymes (data not shown). Based on the alignments, we identified two highly conserved proline residues that correspond to Pro-69 in loop 4 and Pro-105 in loop 7 of human UBC9. Interestingly, these two proline residues are solvent-exposed in all UBC9 crystal structures (Fig. 1C) and thus are available to form interactions with the SP-RING domain of PIAS1. To validate this hypothesis, we generated UBC9-P69A and UBC9-P105A mutants to test them for association with PIAS1 using our BRET assay. In comparison to wild-type UBC9, only a slight decrease in the BRET signal is obtained with the UBC9-P105A (Fig. 1D). In contrast, a significant reduction in the BRET signal is observed for the interaction between PIAS1 and the UBC9-P69A (Fig. 1D). Based on these results, it appears that the conserved Pro-69 residue in the L4 loop of UBC9 is crucial for the interaction with PIAS1. Furthermore, superpositions of the three-dimensional structures of Ubc9 and Siz1 onto the structures of several ubiquitin E2·E3 RING or U-box complexes indicates that the conserved proline residue in the L4 loop of UBC9 should be positioned at the interface with the SP-RING domain of PIAS1 (data not shown). Taken together, these data indicate that PIAS1·UBC9 complex shares a similar type of canonical interface with complexes formed by ubiquitin RING-type E3 and E2 enzymes, where specific residues within the SP-RING domain of PIAS1 (E3) contact the L4 loop of UBC9 (E2).
PIAS1 Is Capable of Recruiting Non-SUMO-modified UBC9
Several in vitro studies indicate that ubiquitin E2-conjugating enzymes are usually charged with ubiquitin before interacting with their E3 ligases (63, 64). To explore the role of the SUMO moiety conjugated to the catalytic cysteine residue of UBC9 in the interaction with PIAS1, two catalytically inactive UBC9 mutants (UBC9-C93S, UBC9-C93A) were generated, and their respective ability to associate with PIAS1 were measured using the BRET assay (Fig. 2A). Surprisingly, the BRET ratio obtained between PIAS1 and the two catalytically inactive UBC9 mutants is similar to the one observed with wild-type UBC9, suggesting that SUMO thioester-linked to UBC9 is dispensable for PIAS1·UBC9 association (Fig. 2, B and C). In addition, SUMOylation of UBC9 at Lys-14 (65) is not required for the PIAS1·UBC9 binding because the UBC9-K14R mutant appears to interact to the same level with PIAS1 in the BRET assay as wild-type UBC9 (Fig. 2D). These results demonstrate that neither SUMO modification of UBC9 nor the formation of a thioester linkage between SUMO and UBC9 is essential for PIAS1 to bind UBC9 in a cellular context. These results suggest that PIAS proteins are able to specifically interact with free UBC9 in a non-covalent fashion.
FIGURE 2.

The non-covalent interaction between SUMO and UBC9 stabilize the PIAS1·UBC9 complex. A, ribbon representation of human UBC9 (PDB code 1U9A) showing residues Arg-13, Lys-14, Arg-17, His-20, Phe-22, and Cys-93 subjected to mutational analysis. N and C correspond, respectively, to the amino and carboxyl termini of UBC9. B, BRET titration curves obtained with wild-type PIAS1 and either wild-type UBC9 or a catalytically inactive UBC9-C93S mutant. The experiments were performed with a fixed amount of RLuc-PIAS1 and increasing amounts of either UBC9-GFP (■) or UBC9-C93S-GFP (%#x25CA;). For each curve the data from two independent experiments were pooled. 100% BRET corresponds to the maximum BRET signal value of 0.69 measured with the wild-type PIAS1 and UBC9. C, bar graph showing a comparison of the BRET ratios obtained with wild-type PIAS1 and either wild-type UBC9, UBC9-C93S, or the UBC9-C93A mutant at similar GFP acceptor/Luc donor expression ratios. D, BRET titration curves obtained with wild-type PIAS1 and either wild-type UBC9 or the UBC9-K14R mutant. The experiments were performed with a fixed amount of RLuc-PIAS1 and increasing amounts of either UBC9-GFP (■) or UBC9-K14R-GFP (%#x25CA;). 100% BRET corresponds to the maximum BRET signal value of 0.56 measured with wild-type PIAS1 and UBC9. E, BRET titration curves obtained with wild-type PIAS1 and either wild-type UBC9, the UBC9-R17A mutant, or the UBC9-H20D mutant. The experiments were performed with a fixed amount of wild-type RLuc-PIAS1 and increasing amounts of wild-type UBC9-GFP (■), UBC9-R17A-GFP (×), or UBC9-H20D-GFP (%#x25CA;). For each curve the data of at least three independent experiments were pooled, and 100% BRET corresponds to the maximum BRET signal value of 0.50 measured with wild-type PIAS1 and UBC9. F, BRET titration curves obtained with wild-type PIAS1 and either the wild-type UBC9 or the UBC9-F22A mutant. The experiments were performed with a fixed amount of wild-type RLuc-PIAS1 with increasing amounts of wild-type UBC9-GFP (■) or UBC9-F22A-GFP (○). 100% BRET represents the maximum BRET signal value of 0.50 in wild-type curve. G, BRET titration curves obtained with wild-type PIAS1 and either the wild-type UBC9 or the UBC9-R13A mutant. The experiment was performed with a fixed amount of wild-type RLuc-PIAS1 with increasing amounts of wild-type UBC9-GFP (■) or UBC9-R13A-GFP (□). 100% BRET represents a maximum BRET signal value of 0.52 measured with wild-type PIAS1 and UBC9.
In Vivo Evidence for a Non-covalent PIAS1·SUMO·UBC9 Ternary Complex
Previous in vitro studies have characterized the formation of non-covalent binary complexes between SUMO proteins and UBC9 (SUMO·UBC9) as well as between the SIM of PIAS2 and SUMO1 (PIAS2-SIM·SUMO1) (15, 66–68). In the non-covalent complex between SUMO and UBC9, the backside of UBC9 spanning from the end of the first α-helix (Arg-13) to the second loop (Lys-30) (human numbering) forms an interface with a region encompassing the first, the fourth, and the fifth β-strands (β1, β4, and β5) of SUMO proteins (residues Lys-25–Ser-31, Arg-63–Gln69, and Glu-83–Tyr-91 in human SUMO1). In the PIAS2·SUMO1 non-covalent complex, the SIM of PIAS2 (V467-I474 in human PIAS2) forms the interface with the region of SUMO1 commencing at the first residues of the β2-strand (Glu-33 in human SUMO1) and ending at the last residues of the α-helix (Arg-54 in human SUMO1). Because, the residues of SUMO that form the interfaces with UBC9 and PIAS proteins are located on non-overlapping surfaces, we posited that SUMO mediates the formation of a non-covalent ternary complex by specifically bridging UBC9 and PIAS proteins.
To address the possibility of a non-covalent PIAS1·SUMO·UBC9ternary complex, we first confirmed the SUMO·UBC9 non-covalent interaction in the cell-based BRET assay (data not shown). Next, we tested several UBC9 mutants, including R13A, R17A, H20D, and F22A for binding to PIAS1 by BRET (Fig. 2, A and E–G). When compared with the BRET signal obtained for the wild-type PIAS1·UBC9 complex, a significant decrease in BRET signal was obtained with the UBC9-R17A, UBC9-H20D, and UBC9-F22A mutants (Fig. 2, E and F). In addition, a less dramatic, but significant effect was also observed with the UBC9-R13A mutant (Fig. 2G). These UBC9 mutants were designed because the UBC9 residues are highly conserved in several eukaryotic species and are not found in other human UBL E2-conjugating enzymes (data not shown). Moreover, they have been found to alter UBC9 binding to SUMO proteins in vitro (57, 69) and are confirmed here in a cellular context with our BRET assay (data not shown). Taken together, these results strongly suggest that the SUMO·UBC9 non-covalent interaction is required for the in vivo formation of the PIAS1·UBC9 non-covalent complex and thus support the existence of a PIAS1·SUMO·UBC9 ternary complex.
Recently, PIAS1 was found to form a non-covalent interaction with SUMO proteins through its SIM sequence (70) (Fig. 3A). This interaction between PIAS1 and SUMO proteins was verified in our cell-based BRET assay (data not shown). To further define the role of the PIAS1 SIM in the formation of the PIAS1·SUMO·UBC9 ternary complex, the interaction between a SIM mutant of the PIAS1 protein (PIAS1-SIMmt) and UBC9 was assessed in the BRET assay. As expected, a significant decrease was observed in the BRET signal for the PIAS1-SIMmt·UBC9 complex in comparison to the signal with the wild-type PIAS1·UBC9 complex (Fig. 3B). This result strongly argues that the non-covalent recruitment of SUMO proteins to PIAS1 is required for stabilization and/or formation of the PIAS1·UBC9 interaction, further supporting the existence of a non-covalent PIAS1·SUMO·UBC9 ternary complex.
FIGURE 3.

The phospho-SIM module of PIAS1 positively regulates the PIAS1·UBC9 interaction. A, schematic representation of the full-length human PIAS1 and amino acid sequence alignment of the SUMO interacting motifs for PIAS SUMO E3 ligase family members. Conserved hydrophobic residues within the minimal SIM core (SIM) and serine residues (▾) from human PIAS1 subjected to mutational analysis are indicated. B, BRET titration curves obtained with wild-type UBC9 and either wild-type PIAS1 or the PIAS1-SIMmt mutant. The experiments were performed using a fixed amount of either RLuc-PIAS1 (■) or RLuc-PIAS1-SIMmt (Δ) with increasing amounts of UBC9-GFP. For each curve, the data of two independent experiments were pooled. 100% BRET represents the maximum BRET signal value of 0.50 measured with wild-type PIAS1 and UBC9. C, BRET titration curves obtained with wild-type UBC9 and either wild-type PIAS1, PIAS1–3SD, or PIAS1–3SA. The experiments were performed using increasing amounts of UBC9-GFP with a fixed amount of either wild-type RLuc-PIAS1 (■), RLuc-PIAS1–3SD (▵), or RLuc-PIAS1–3SA (%#x25CA;). For each curve, the data of two independent experiments were pooled. 100% BRET represents a maximum BRET signal value of 0.56 measured with wild-type PIAS1 and UBC9. D, bar graph showing a comparison of the BRET ratios obtained with UBC9-GFP and either RLuc-PIAS1 or RLuc-PIAS1 mutants at similar GFP acceptor/Luc donor expression ratios. E and F, representative ITC thermograms for the interaction between either the wild-type PIAS1-SIM (E) or PIAS1-SIM-3SD (F) peptides and SUMO1. The PIAS1-SIM peptide binds to SUMO1 with an apparent dissociation constant (KD) of 2.0 ± 0.5 μm, whereas the PIAS1-SIM-3SD peptide binds to SUMO1 with a KD of 0.50 ± 0.15 μm.
Phosphorylation of the PIAS1 SIM Enhances Formation of the Ternary Complex
Previous studies have shown that CK2-dependent phosphorylation of residues immediately adjacent to the SIM sequences in DAXX (death-associated protein 6) and PML (promyelocytic leukemia protein) play an important role in their function (71, 72). More recently, three conserved serine residues immediately adjacent to the SIM sequence of PIAS1 have also been shown to be phosphorylated by CK2 and to influence the PIAS1·SUMO interaction (Fig. 3A) and (70). Based on these results, we postulated that phosphorylation of these serine residues of PIAS1 might enhance the formation of a PIAS1·SUMO·UBC9 ternary complex. To test this possibility, we generated PIAS1 mutants where all three serine residues were modified to either a phosphomimetic aspartic acid residue (PIAS1–3SD) or a non-phosphorylatable alanine residue (PIAS1–3SA). Interestingly, BRET saturation curves indicate that UBC9 binds more efficiently to the PIAS1–3SD mutant than to the PIAS1–3SA mutant (Fig. 3, C and D). However, only a small increase in binding efficiency was observed for UBC9 binding to the PIAS1–3SD mutant compared with the wild-type PIAS1 (Fig. 3, C and D). This small difference between the PIAS1–3SD mutant and the wild-type protein suggests that PIAS1 may be constitutively phosphorylated in our assay conditions. To explore this possibility, a PIAS1 mutant containing five alanine substitutions for five glutamic acid residues (PIAS1–5EA) was generated. This mutant was designed because it alters the CK2 consensus sites without mutating the serine residues that undergo phosphorylation (70). In addition, previous NMR and ITC studies have shown that this stretch of acidic residues plays no role in PIAS binding to SUMO1 (67). In agreement with what is observed with the PIAS1–3SA mutant, a decrease in the BRET ratio was observed for the association between the PIAS1–5EA mutant and UBC9 in comparison to wild-type PIAS1 (Fig. 3D). Taken together, these results support a model where the formation of the PIAS1·SUMO·UBC9 ternary complex is positively regulated by CK2-dependent phosphorylation of serine residues adjacent to the PIAS1 SIM.
Phosphomimetic Substitutions in the PIAS1 SIM Enhance in Vitro Binding to SUMO
To quantitatively assess the potential role of phosphorylation in PIAS1 binding to SUMO1, we determined the apparent dissociation constant (KD) for SUMO1 binding to a 25-residue peptide encompassing the SIM of PIAS1 (PIAS1-SIM; residues 456–480) (Fig. 3A) and compared it to a phosphomimetic PIAS1-SIM peptide (PIAS1-SIM-3SD) using ITC. By ITC, the PIAS1-SIM peptide binds to SUMO1 with a KD of 2.0 ± 0.5 μm, whereas the PIAS1-SIM-3SD peptide binds with a KD of 0.50 ± 0.15 μm (Fig. 3, E and F). Thus, the phosphomimetic PIAS1-SIM-3SD peptide binds SUMO1 with approximately a 4-fold higher affinity. The binding of the PIAS1-SIM peptide is similar to what was previously observed in ITC studies examining the binding of a PIAS2-SIM peptide to SUMO1 (67). A similar increase in affinity was also obtained when comparing the PIAS1-SIM·SUMO2 (KD of 2.6 ± 0.3 μm) and PIAS1-SIM-3SD·SUMO2 (KD of 0.53 ± 0.05 μm) interactions (data not shown). Thus, these ITC studies are consistent with our BRET results and suggest that phosphorylation of CK2 sites adjacent to the SIM of PIAS1 enhances the interaction between SUMO proteins and PIAS1.
To more specifically address the potential impact of the PIAS1 phosphorylation status on the formation of the PIAS1·SUMO·UBC9 ternary complex in vitro, chemical shift perturbations studies were performed using NMR spectroscopy. In these studies we mapped the binding sites of the PIAS1-SIM and PIAS1-SIM-3SD peptides on SUMO1. 1H,15N HSQC experiments were conducted using 15N-labeled SUMO1 titrated with either the native PIAS1-SIM peptide or the PIAS1–3SD peptide. As expected, the addition of either the PIAS1-SIM-3SD or the PIAS1-SIM peptides resulted in significant chemical shift changes (Δδ (ppm) >0.2) for specific signals of SUMO1 (Fig. 4, A, B, and D and data not shown). Interestingly, superposition of the 1H,15N HSQC spectra from the two titrations allowed us to identify the signals of SUMO1 that are specifically changed due to the presence of the three phosphomimetic residues (data not shown). The SUMO1 signals displaying phosphomimetic-dependent changes are associated with residues located mainly in a region spanning from the end the β2-strand to the start of the α-helix of SUMO1. Within this region, His-43 and Lys-46 undergo the most dramatic differences in chemical shift changes due to the presence of the three aspartic acid residues (Δδ (ppm) >0.07) (Fig. 4, C and D).
FIGURE 4.

Characterization of the PIAS1-SIM-3SD·SUMO1 interaction by NMR spectroscopy. A, histogram of the variation in chemical shifts (Δδ (ppm)) observed in the 1H,15N HSQC spectra of 15N-SUMO1 upon the addition of the unlabeled PIAS1-SIM-3SD peptide. The chemical shift variations were calculated with the formula Δδ = [(0.17ΔNH)2 + (ΔHN)2]1/2 and are given in parts per million. SUMO1 residues significantly shifting [Δδ (ppm) > 0.2] are identified according to the human SUMO1 numbering. B, overlay from the two-dimensional 1H,15N HSQC spectra of 15N-labeled SUMO1 (0.5 mm) in the free form (black) and in presence of either 0.25 mm (red) or 0.75 mm (orange) of unlabeled PIAS1-SIM-3SD peptide. Arrows depict the direction of the change (free → bound). C, histogram of the differences in chemical shifts (CSP) between 15N-labeled SUMO1·PIAS1-SIM-3SD and 15N labeled SUMO1·PIAS1-SIM. The differences were calculated with the formula Δδ = [(0.17ΔNH)2 + (ΔHN)2]1/2 and are given in parts per million. SUMO1 residues significantly shifting (Δδ (ppm) >0.07) are identified according to the human SUMO1 numbering. D, ribbon representation of human SUMO1 (PDB code 1A5R; residues 19–95) indicating residues significantly shifting (cyan) with either the PIAS1-SIM or the PIAS1-SIM-3SD peptides. The arrows (magenta) indicate residues specifically shifting upon binding to PIAS1-SIM-3SD. N and C indicate, respectively, the amino and carboxyl termini of SUMO1. E and F, two-dimensional 1H,15N HSQC spectra of 15N-labeled PIAS1-SIM-3SD (0.5 mm) in the free form (black) (E) and in the presence of unlabeled SUMO1 (1 mm; purple) (F).
To confirm that the PIAS1-SIM peptide binds only to SUMO1 and not to UBC9, a second set of NMR chemical shift perturbation studies was conducted using 15N-labeled PIAS1-SIM-3SD peptide with either unlabeled SUMO1 or unlabeled UBC9. As expected, chemical shift changes were observed for signals of the 15N-labeled PIAS1-SIM-3SD peptide upon the addition of SUMO1 (Fig. 4, E and F) but not after the addition of UBC9 (data not shown). Together, these results confirm that the PIAS1 SIM directly binds SUMO1 and that the region of SUMO1 encompassing His-43 and Lys-46 is important for the recognition of the phosphomimetic PIAS1 SIM peptide.
Two Distinct Interfaces of SUMO Are Required for the Non-covalent Ternary Complex
To confirm that PIAS1 and UBC9 form a non-covalent complex bridged by SUMO, we performed NMR chemical shift mapping studies using 15N-labeled SUMO1, unlabeled UBC9, and unlabeled PIAS1-SIM-3SD peptide. Two dimensional 1H,15N HSQC spectra were collected to follow the addition of unlabeled UBC9 or unlabeled PIAS1-SIM-3SD to 15N-labeled SUMO1 (data not shown). As anticipated, upon formation of the PIAS1-SIM-3SD·SUMO1 and SUMO1·UBC9 complexes, the SUMO1 residues exhibiting significant 1H and 15N chemical shift changes are located within two distinct regions of the SUMO1 surface (Δδ (ppm) >0.2) (data not shown). Superposition of the resultant two-dimensional 1H,15N HSQC spectra reveals differences in numerous chemical shifts for several signals of SUMO1, confirming that non-overlapping SUMO1 surfaces are involved in the formation of the two binary complexes (Fig. 5A). To form the ternary complexes, PIAS1-SIM-3SD was added to the sample containing the 15N-labeled SUMO1·UBC9 proteins and, reciprocally, UBC9 was added to the 15N-labeled SUMO1·PIAS1–3SD complex. As expected, the two resultant two-dimensional 1H,15N HSQC spectra obtained were very similar (Fig. 5, B and C, and data not shown), supporting formation of identical ternary complexes. These results clearly demonstrate that a non-covalent ternary complex can form between PIAS1-SIM-3SD, SUMO1, and UBC9, where SUMO1 acts as a bridge that is specifically recognized by both the SIM module of the PIAS1 E3 ligase and the backside of the UBC9 E2-conjugating enzyme.
FIGURE 5.
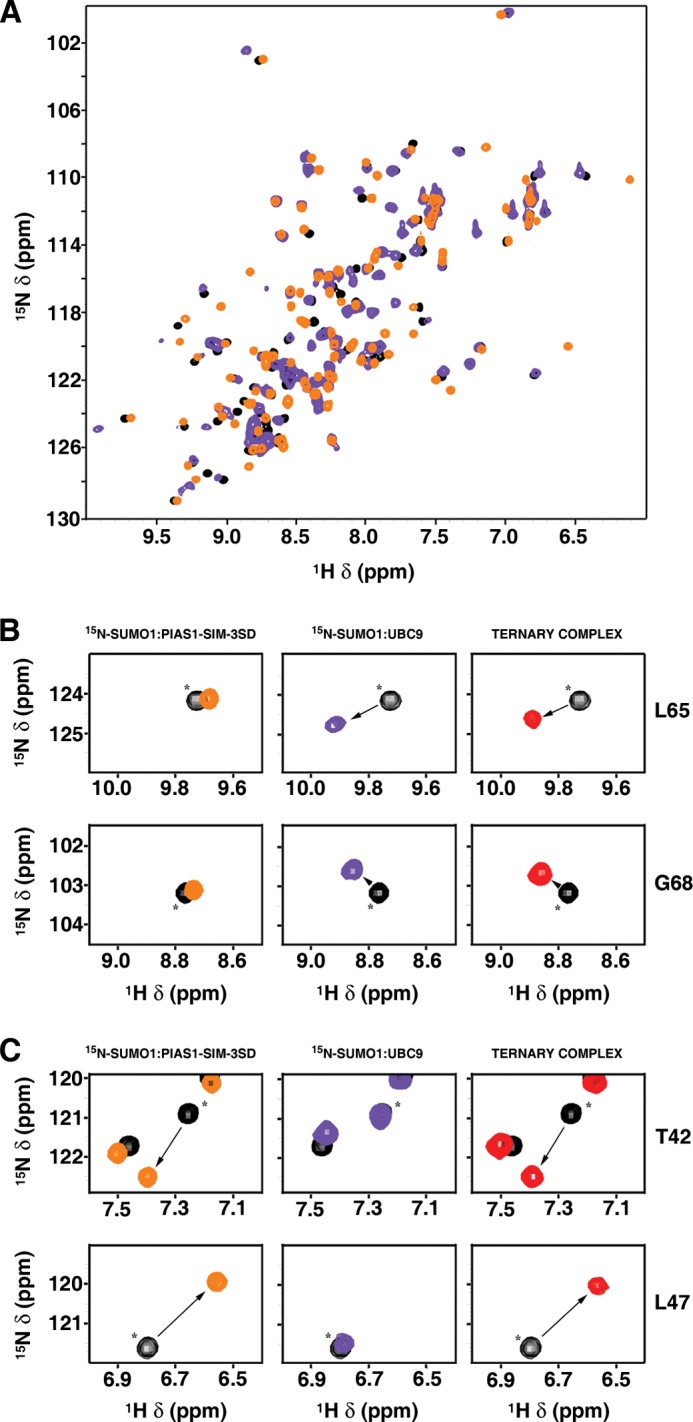
Characterization of a ternary complex between PIAS1, SUMO1, and UBC9 by NMR spectroscopy. A, overlay of the two-dimensional 1H,15N HSQC spectra of 15N-labeled SUMO1 (0.5 mm) in the free form (black) and in the presence of either unlabeled PIAS1-SIM-3SD peptide (0.75 mm; orange) or unlabeled UBC9 (0.75 mm, purple). B and C, selected regions of two-dimensional 1H,15N HSQC spectra of 15N-labeled SUMO1 (0.5 mm; black) illustrating perturbations to specific SUMO1 residues upon formation of either binary (PIAS1-SIM-3SD·15N-SUMO1 (left panels) and 15N-SUMO1·UBC9 (middle panels)) or ternary complexes (PIAS1-SIM-3SD·15N-SUMO1·UBC9; right panels). Asterisks (*) indicate the position of free 15N-labeled SUMO1 signals, and arrows depict the direction of the change (free → bound). B, the signals from Leu-65 and Gly-68 of SUMO1 are significantly shifted upon the addition of unlabeled UBC9 (0.75 mm; purple; middle panels), but they are unaffected upon the addition of unlabeled PIAS1-SIM-3SD peptide (0.75 mm; orange; left panels). C, the signals from Thr-42 and Leu-47 of SUMO1 are significantly shifted upon the addition of unlabeled PIAS1-SIM-3SD peptide (0.75 mm; orange; left panels), but they are unaffected upon the addition of unlabeled UBC9 (0.75 mm; purple; middle panels). In the formation of the ternary complex (ternary complex; red; right panels), the four SUMO1 residues (Thr-42, Leu-47, Leu-65, and Gly-68) are all perturbed, indicating simultaneous binding of unlabeled UBC9 and PIAS1-SIM-3SD peptide to 15N-labeled SUMO1.
The Ternary Complex Is Required for the Transcriptional Repression Activities of UBC9 and SUMO1
Previous studies have shown that PIAS1, UBC9, and SUMO have transcriptional regulatory properties, and we were interested in determining if the PIAS·SUMO·UBC9 ternary complex played a role in these activities (70, 73, 74). We first assessed the role of the PIAS·SUMO·UBC9 ternary complex in UBC9-mediated transcriptional repression. For this purpose UBC9 was fused to a heterologous DNA binding domain (Gal4DBD-UBC9), and its effect on transcription was determined by monitoring the expression of a Luciferase reporter gene located downstream from a Gal4 response element (5xGal4UAS) (Fig. 6A). HEK293T cells were co-transfected with a vector encoding for the Gal4DBD-UBC9 fusion protein (or Gal4DBD alone) along with the Luciferase reporter gene plasmid under the control of the SV40 promoter. In this assay the Gal4DBD-UBC9 fusion, but not the Gal4DBD alone, significantly repressed the expression of the Luciferase reporter in a dose-dependent manner (up to 5-fold) (Fig. 6B). In contrast, a complete loss of the repressive activity was observed with both the UBC9-R17A and the UBC9-H20D mutants (Fig. 6C), two mutants that alter non-covalent binding of UBC9 to SUMO (data not shown and Fig. 2E). In addition, the UBC9-P69A mutant displayed a reduced repressive activity when compared with wild-type UBC9, indicating that the non-covalent association between the PIAS1 SP-RING domain and UBC9 may further stabilize the repressive complex. These results suggest that, like E3 ligases of the SUMO pathway, the SUMO E2-conjugating enzyme has a repressive activity on transcription under our experimental conditions and that non-covalent interaction between UBC9 and SUMO is required for the repressive activity of UBC9. These data also indicate that stabilization of the PIAS·SUMO·UBC9 ternary complex through an interaction between PIAS and UBC9 is required for UBC9-repressive activity.
FIGURE 6.

Regulation of UBC9- and SUMO1-dependent repression by SUMO non-covalent interactions. A, schematic representation of the firefly Luciferase reporter plasmid and Gal4-effector constructs used in the repression assay. The Luciferase reporter is under the control of the strong SV40 promoter and SV40 enhancer sequence and contains five consensus Gal4 UAS (5xGal4UAS). Effectors proteins UBC9 and SUMO1 and mutants fused to Gal4DBD are shown. B, transcriptional activity of Gal4DBD and Gal4DBD-UBC9 proteins directly recruited on the Luciferase reporter plasmid. Increasing amounts (100, 200, and 400 ng) of the effector constructs were used. C, transcriptional activity of Gal4DBD and Gal4DBD fusion proteins (UBC9, UBC9-C93S, UBC9-K14R, UBC9-P69A, UBC9-R17A, and UBC9-H20D) directly recruited on the Luciferase reporter plasmid. Increasing amounts (200 and 400 ng) of the effector constructs were used. The expression of Gal4DBD-UBC9 and Gal4DBD-UBC9 mutants (200 ng) was monitored by immunoblotting with an anti-Gal4DBD antibody. D, transcriptional activity of Gal4DBD and Gal4DBD fusion proteins (UBC9, UBC9-D100A-K101A, UBC9-P128A, and UBC9-Y134A) directly recruited on the Luciferase reporter plasmid. Increasing amounts (100, 200, and 400 ng) of the effector constructs were used. The expression of Gal4DBD-UBC9 and Gal4DBD-UBC9 mutants (200 ng) was monitored by immunoblotting with an anti-Gal4DBD antibody. In B–D, the -fold repression is the ratio of Luciferase activity measured in the presence of Gal4DBD control divided by the activity measured in the presence of the Gal4DBD-UBC9 fusions constructs. In each case, the -fold repression was normalized to account for transfection efficiency. Error bars represent the S.D. for at least three independent experiments performed with duplicate or triplicate samples. IB, immunoblot. E, transcriptional activity of Gal4DBD fused to the non-conjugate-able form of SUMO1 and SUMO1 mutants (SUMO1-F36A and SUMO1-E67R) directly recruited on the Luciferase reporter plasmid. 200 ng of the effector constructs were used. The percentage change in -fold repression of the Gal4DBD-SUMO1 constructs is expressed relative to the Luciferase activity of the Gal4DBD control fixed to 1-fold repression; thus, the activity of Gal4DBD control corresponds to 0% change in -fold repression. Error bars represent S.D. for at least two independent experiments performed with duplicate samples. The expression of Gal4DBD-SUMO1 constructs was monitored by immunoblotting with an anti-Gal4DBD antibody.
Because SUMOylation of several transcriptional regulatory factors leads to repression of their target genes (74–77), we were also curious to determine if UBC9 catalytic activity contributes to UBC9-mediated repression. To test this we first assessed the repressive activity of the catalytically inactive UBC9 mutant (UBC9-C93S). Intriguingly, UBC9-C93S exerted almost the same repressive activity as the wild-type UBC9 (Fig. 6C). In addition, UBC9 proteins with mutations of specific residues near the catalytic cysteine residue that are known to prevent and/or alter the recognition of the SUMOylation consensus sequence of substrates all retain full repressive activity (78, 79) (Fig. 6D). Thus, covalent modification of cellular transcriptional regulatory factors with SUMO is unlikely required for the observed UBC9-dependent transcriptional repression. Moreover, SUMOylation of UBC9 on lysine residue 14 is not required for this activity as the UBC9-K14R mutant also retains full activity (Fig. 6C). Noticeably, both UBC9-C93S and UBC9-K14R mutants retain the ability to non-covalently interact with SUMO proteins in our BRET assay (data not shown), and consequently with PIAS1. Next, we investigated whether or not the repressive activity of SUMO1 depends on its ability to bridge a non-covalent ternary complex with UBC9 and PIAS proteins. To test this we used SUMO1 and two mutants fused to the Gal4DBD and compared their relative repressive activity. The SUMO1 mutants have been shown to disrupt the interaction with either PIAS1 (SUMO1-F36A) (66) or UBC9 (SUMO1-E67R) (69). As previously reported, Gal4DBD-SUMO1 induces an increase in the repression of the Luciferase reporter relative to the Gal4DBD control (Fig. 6E) (74). In contrast, a loss in the repressive activity was observed with both SUMO1 mutants (Fig. 6E). Taken together, these results suggest that the repressive activity of SUMO1 depends on its ability to bridge the PIAS1·SUMO1·UBC9 ternary complex, and this confirms the functional role of the PIAS1·SUMO1·UBC9 complex in transcriptional repression.
DISCUSSION
Although the enzymes of the SUMOylation pathway were identified more than a decade ago, the molecular determinants regulating the interactions between the E2·E3 enzymes have not been well defined. In this study we used a combination of cellular and in vitro experiments to establish that the interaction between PIAS1 and UBC9 is stabilized by their ability to non-covalently bind distinct surfaces on SUMO1 leading to the formation of a functional ternary complex. In the PIAS1·SUMO1·UBC9 ternary complex, SUMO1 bridges its cognate E2 and E3 enzymes by concomitantly binding to the SIM of PIAS1 and the backside surface of UBC9. Our data also suggests that phosphorylation of serine residues adjacent to the SIM of PIAS1 facilitates the formation of the ternary complex by increasing the affinity between PIAS1 and SUMO1. Furthermore, our data suggest that PIAS1 and UBC9 can engage in canonical E2·E3 RING interactions that require conserved residues located in the PIAS1 SP-RING domain and the L4 loop of UBC9. This canonical UBC9·PIAS1 interaction also appears to increase the stabilization of the ternary complex. In addition, we show that formation of the covalent thioester bond between SUMO1 and UBC9 does not appear to be a prerequisite for this complex formation. Moreover, we provide evidence that UBC9- and SUMO-dependent transcriptional repression requires formation of the PIAS1·SUMO·UBC9 ternary complex.
SUMO Bridges PIAS1 and UBC9 in the Ternary Complex
Although the functions of UBLs including SUMO, NEDD8, or ubiquitin as modifiers of protein targets has been intensively studied, less efforts have been devoted to unraveling the roles that these proteins play as non-covalent interacting partners in macromolecular complexes assembly. SUMO2/3 are mainly found in their un-conjugated form in unstressed cells, and a recent study reports significant levels of free ubiquitin in different cell lines (80, 81). Here, we present convincing evidence that SUMO proteins function in part by bridging a non-covalent interaction between its E2 and E3 enzymes. The simplest model, supported by our in vivo BRET data using full-length proteins as well as our in vitro ITC and NMR results involves formation of a ternary complex formed between PIAS1, SUMO1, and UBC9. In this ternary complex the SIM of PIAS1 and the backside of UBC9 bind to two distinct surfaces of SUMO1. This suggests that SUMO1 operates as a hub bridging together its own E3 ligase and E2-conjugating enzyme. Because both the SIM of PIAS1 and the backside of UBC9 have been shown to bind to different SUMO paralogs, all SUMO family members could, in principle, form a similar ternary complex with PIAS1 and UBC9. Moreover, because other PIAS proteins contain a SIM sequence analogous to the one of PIAS1, all PIAS proteins could participate in a similar ternary complex with SUMO proteins and UBC9. This result is equivalent with what has been recently found for the in vitro interaction between ubiquitin and either the ZNF216·p62 or ZNF4·UBE2D1 complexes. In the ZNF216·p62 and ZNF4·UBE2D1 complexes, the isolated A20 zinc-finger domain of either ZNF216 or ZNF4 contacts the ubiquitin polar patch, whereas the ubiquitin hydrophobic surface binds to either the UBA (ubiquitin-associated) domain of p62 or the backside of UBE2D1 (82, 83). Thus, both SUMO and ubiquitin are highly versatile proteins that can function both as posttranslational modifying factors and as scaffolding proteins in non-covalent macromolecular assemblies.
Non-SUMO-modified UBC9 Interacts with PIAS1
It has been generally accepted that to form an active E2·E3 complex capable of posttranslationally modifying a given substrate, the E3 ligase must interact with its E2-conjugating enzyme after it has been charged with its cognate UBL. However, few other studies have reported direct interactions between the E3 ligase and free E2-conjugating enzymes (84, 85). Using our in vivo BRET assay, we show that PIAS1 can form complexes with free UBC9. Using catalytically inactivated forms of UBC9, we clearly demonstrate that uncharged UBC9 is able to specifically interact with PIAS1. Overall, this non-covalent recruitment of UBC9 to PIAS1 by SUMO is likely to be a conserved process among the PIAS family proteins.
PIAS1 and UBC9 Also Engage in Canonical E2·E3 Interactions
In addition to the non-covalent interaction mediated by SUMO1, our BRET studies suggest that UBC9 can directly bind to PIAS1 through a canonical E2·E3 RING interaction similar to the one used by the E2·E3 enzymes of the ubiquitination pathway. We demonstrate that Leu-337 within the SP-RING domain of PIAS1 and Pro-69 within the L4 loop of UBC9 are required for the interaction between PIAS1 and UBC9. Interestingly, equivalent residues are conserved in the RING E3 and E2 enzymes of the ubiquitination pathway, and structural models demonstrate that these residues are crucial for E2·E3 interactions (54, 56, 58, 62, 86). This suggests that PIAS1 and UBC9 engage in interactions similar to the one employed by E2·E3 enzymes of the ubiquitination pathway. Moreover, our results also suggest that the interaction interface used by UBC9 (L4 loop) to bind the PIAS1 SP-RING domain overlaps with the one used to bind the IR1 motif of RanBP2 (40). Interestingly, the RanBP2 IR1 motif and the PIAS/Siz SP-RING domain have no sequence or structural similarities. This indicates that UBC9 can recognize two structurally distinct SUMO E3 ligases using the same interface. A similar phenomenon has been observed for enzymes of the ubiquitination pathway, where the E2 conjugating enzyme UbcH7 (UBE2L3) uses a similar surface to contact two structurally distinct E3 ligases (54, 87).
Phosphorylation of PIAS1 Enhances Formation of the PIAS1·SUMO·UBC9 Complex
All members of the PIAS family of SUMO E3 ligases possess a SIM that is adjacent to a cluster of serine residues located within CK2 consensus phosphorylation sites (70). The phosphorylation of these serine residues in PIAS1 has been proposed to dictate its binding to SUMO-family proteins (70). However, we detect a relatively strong binding between SUMO1 and the native PIAS1-SIM peptide by ITC, and this is in agreement with a previous study examining the interaction between the SIM of PIAS2 and SUMO1 (67). Interestingly, the affinity of SUMO1 for the PIAS1-SIM peptide is enhanced when three serine residues are changed to phosphomimetic aspartic acid residues in the PIAS-SIM-3SD peptide. These results are further supported by NMR data where more pronounced chemical shift changes are observed for signals of SUMO1 residues with the PIAS1-SIM-3SD peptide in comparison with the PIAS1-SIM peptide. In particular, we observe significant changes for the signals corresponding to His-43 and Lys-46 of SUMO1 in the interaction with the PIAS1-SIM-3SD peptide. This suggests that although CK2-dependent phosphorylation is not a prerequisite for the PIAS1·SUMO interaction, it significantly increases its affinity. This regulation of SUMO binding by CK2-dependent phosphorylation is analogous with what has been observed for DAXX (88). Moreover, these in vitro studies are consistent with our cellular BRET data, suggesting that the phosphorylation status of the PIAS1 SIM regulates the PIAS1·UBC9 interaction in a SUMO-dependent manner. Altogether, these results provide strong evidence suggesting that the phosphorylation status of the PIAS1 SIM helps regulate the formation of the PIAS1·SUMO·UBC9 ternary complex. Because analogous phospho-SIM sequences are present in all PIAS proteins, similar phospho-regulate-able ternary complexes might form with other PIAS proteins.
UBC9- and SUMO-mediated Transcriptional Repression Require Formation of Ternary Complexes
So far the functional roles of SUMOylation have been largely connected to transcriptional repression, with SUMO mainly acting as covalent protein modifier that specifically recruits SIM-containing repressor complexes (89). Here we demonstrate that SUMO-mediated repression depends on its capacity to non-covalently bridge PIAS1 and UBC9 in a ternary complex. Interestingly, members of the SUMO E3 PIAS family have been found to repress the activity of several transcriptional activators such as p53, p73α, and the nuclear androgen receptor (35, 90, 91). Of note, PIAS2-induced transcriptional repression requires both the integrity of its SP-RING domain and phospho-SIM sequence (35, 70). These observations are consistent with our data showing that both the PIAS1 SP-RING domain and phospho-SIM sequence are implicated in the formation of a non-covalent repressive E3·SUMO·E2 ternary complex. Similar to the PIAS proteins, UBC9 induces transcriptional repression when directly tethered to DNA (73). Interestingly, we demonstrate that this repressive activity is independent of UBC9 SUMOylation at Lys-14. Moreover, we show that neither the UBC9 recognition of the SUMOylation consensus site nor its catalytic activity is required for UBC9-dependent repression. Thus, as shown for the PIAS proteins (92, 93), the UBC9-induced repression can occur independently of its catalytic activity (94). In addition, we establish that altering the formation of the PIAS1·SUMO·UBC9 ternary complex by disrupting either the SUMO·UBC9 non-covalent binding or the PIAS1 SP-RING·UBC9 L4 loop interaction interferes with the ability of UBC9 to repress transcription. Altogether, our results suggest that SUMO-mediated repression depends on its non-covalent interaction with PIAS and UBC9 proteins, and this repression occurs independently of E2-E3 catalytic activities. It is thus tempting to speculate that the SUMO·UBC9 non-covalent complex may function in additional ternary complexes with other phospho-SIM containing proteins (Fig. 7).
FIGURE 7.
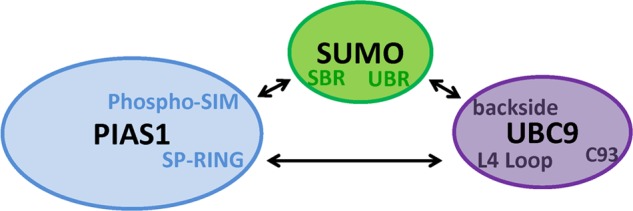
Summary model of interactions involved in the formation of the non-covalent PIAS E3·SUMO·UBC9 E2 ternary complex. In our model SUMO bridges the non-covalent ternary complex by simultaneously interacting with the phospho-SIM motif of PIAS1 via its SIM binding region (SBR) and with the backside of UBC9 through its UBC9 binding region (UBR) domain. In addition, this ternary complex appears to be reinforced through interactions between PIAS1 SP-RING domain and UBC9 L4 loop.
Acknowledgment
We thank Pascale Legault for critical review of the manuscript.
This work was supported by a grant from the Canadian Cancer Society (to J. G. O.), the Canadian Institutes for Health Research (to J. G. O.; MOP-74739), and the Natural Sciences and Engineering Research Council of Canada (to M. A.).
- UBL
- ubiquitin-like protein
- SIM
- SUMO interacting motif
- BRET
- bioluminescence resonance energy transfer
- ITC
- isothermal titration calorimetry
- HSQC
- heteronuclear single quantum correlation.
REFERENCES
- 1. Schulman B. A. (2011) Twists and turns in ubiquitin-like protein conjugation cascades. Protein Sci. 20, 1941–1954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Deshaies R. J., Joazeiro C. A. (2009) RING domain E3 ubiquitin ligases. Annu. Rev. Biochem. 78, 399–434 [DOI] [PubMed] [Google Scholar]
- 3. Schulman B. A., Harper J. W. (2009) Ubiquitin-like protein activation by E1 enzymes. The apex for downstream signalling pathways. Nat. Rev. Mol. Cell Biol. 10, 319–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. van Wijk S. J., Timmers H. T. (2010) The family of ubiquitin-conjugating enzymes (E2s). Deciding between life and death of proteins. FASEB J. 24, 981–993 [DOI] [PubMed] [Google Scholar]
- 5. Christensen D. E., Klevit R. E. (2009) Dynamic interactions of proteins in complex networks. Identifying the complete set of interacting E2s for functional investigation of E3-dependent protein ubiquitination. FEBS J. 276, 5381–5389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Boddy M. N., Howe K., Etkin L. D., Solomon E., Freemont P. S. (1996) PIC 1, a novel ubiquitin-like protein that interacts with the PML component of a multiprotein complex that is disrupted in acute promyelocytic leukaemia. Oncogene 13, 971–982 [PubMed] [Google Scholar]
- 7. Matunis M. J., Coutavas E., Blobel G. (1996) A novel ubiquitin-like modification modulates the partitioning of the Ran-GTPase-activating protein RanGAP1 between the cytosol and the nuclear pore complex. J. Cell Biol. 135, 1457–1470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Okura T., Gong L., Kamitani T., Wada T., Okura I., Wei C. F., Chang H. M., Yeh E. T. (1996) Protection against Fas/APO-1- and tumor necrosis factor-mediated cell death by a novel protein, sentrin. J. Immunol. 157, 4277–4281 [PubMed] [Google Scholar]
- 9. Shen Z., Pardington-Purtymun P. E., Comeaux J. C., Moyzis R. K., Chen D. J. (1996) UBL1, a human ubiquitin-like protein associating with human RAD51/RAD52 proteins. Genomics 36, 271–279 [DOI] [PubMed] [Google Scholar]
- 10. Mahajan R., Delphin C., Guan T., Gerace L., Melchior F. (1997) A small ubiquitin-related polypeptide involved in targeting RanGAP1 to nuclear pore complex protein RanBP2. Cell 88, 97–107 [DOI] [PubMed] [Google Scholar]
- 11. Chen A., Mannen H., Li S. S. (1998) Characterization of mouse ubiquitin-like SMT3A and SMT3B cDNAs and gene/pseudogenes. Biochem. Mol. Biol. Int. 46, 1161–1174 [DOI] [PubMed] [Google Scholar]
- 12. Kamitani T., Kito K., Nguyen H. P., Fukuda-Kamitani T., Yeh E. T. (1998) Characterization of a second member of the sentrin family of ubiquitin-like proteins. J. Biol. Chem. 273, 11349–11353 [DOI] [PubMed] [Google Scholar]
- 13. Saitoh H., Pu R. T., Dasso M. (1997) SUMO-1. Wrestling with a new ubiquitin-related modifier. Trends Biochem. Sci. 22, 374–376 [DOI] [PubMed] [Google Scholar]
- 14. Bayer P., Arndt A., Metzger S., Mahajan R., Melchior F., Jaenicke R., Becker J. (1998) Structure determination of the small ubiquitin-related modifier SUMO-1. J. Mol. Biol. 280, 275–286 [DOI] [PubMed] [Google Scholar]
- 15. Liu Q., Jin C., Liao X., Shen Z., Chen D. J., Chen Y. (1999) The binding interface between an E2 (UBC9) and a ubiquitin homologue (UBL1). J. Biol. Chem. 274, 16979–16987 [DOI] [PubMed] [Google Scholar]
- 16. Gareau J. R., Lima C. D. (2010) The SUMO pathway. Emerging mechanisms that shape specificity, conjugation, and recognition. Nat. Rev. Mol. Cell Biol. 11, 861–871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Desterro J. M., Thomson J., Hay R. T. (1997) Ubch9 conjugates SUMO but not ubiquitin. FEBS Lett. 417, 297–300 [DOI] [PubMed] [Google Scholar]
- 18. Gong L., Kamitani T., Fujise K., Caskey L. S., Yeh E. T. (1997) Preferential interaction of sentrin with a ubiquitin-conjugating enzyme, Ubc9. J. Biol. Chem. 272, 28198–28201 [DOI] [PubMed] [Google Scholar]
- 19. Johnson E. S., Blobel G. (1997) Ubc9p is the conjugating enzyme for the ubiquitin-like protein Smt3p. J. Biol. Chem. 272, 26799–26802 [DOI] [PubMed] [Google Scholar]
- 20. Johnson E. S., Schwienhorst I., Dohmen R. J., Blobel G. (1997) The ubiquitin-like protein Smt3p is activated for conjugation to other proteins by an Aos1p/Uba2p heterodimer. EMBO J. 16, 5509–5519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Johnson E. S., Gupta A. A. (2001) An E3-like factor that promotes SUMO conjugation to the yeast septins. Cell 106, 735–744 [DOI] [PubMed] [Google Scholar]
- 22. Okuma T., Honda R., Ichikawa G., Tsumagari N., Yasuda H. (1999) In vitro SUMO-1 modification requires two enzymatic steps, E1 and E2. Biochem. Biophys. Res. Commun. 254, 693–698 [DOI] [PubMed] [Google Scholar]
- 23. Kahyo T., Nishida T., Yasuda H. (2001) Involvement of PIAS1 in the sumoylation of tumor suppressor p53. Mol. Cell 8, 713–718 [DOI] [PubMed] [Google Scholar]
- 24. Takahashi Y., Kahyo T., Toh-E A., Yasuda H., Kikuchi Y. (2001) Yeast Ull1/Siz1 is a novel SUMO1/Smt3 ligase for septin components and functions as an adaptor between conjugating enzyme and substrates. J. Biol. Chem. 276, 48973–48977 [DOI] [PubMed] [Google Scholar]
- 25. Takahashi Y., Toh-e A., Kikuchi Y. (2001) A novel factor required for the SUMO1/Smt3 conjugation of yeast septins. Gene 275, 223–231 [DOI] [PubMed] [Google Scholar]
- 26. Hay R. T. (2007) SUMO-specific proteases. A twist in the tail. Trends Cell Biol. 17, 370–376 [DOI] [PubMed] [Google Scholar]
- 27. Olsen S. K., Capili A. D., Lu X., Tan D. S., Lima C. D. (2010) Active site remodelling accompanies thioester bond formation in the SUMO E1. Nature 463, 906–912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Desterro J. M., Rodriguez M. S., Kemp G. D., Hay R. T. (1999) Identification of the enzyme required for activation of the small ubiquitin-like protein SUMO-1. J. Biol. Chem. 274, 10618–10624 [DOI] [PubMed] [Google Scholar]
- 29. Rodriguez M. S., Dargemont C., Hay R. T. (2001) SUMO-1 conjugation in vivo requires both a consensus modification motif and nuclear targeting. J. Biol. Chem. 276, 12654–12659 [DOI] [PubMed] [Google Scholar]
- 30. Sampson D. A., Wang M., Matunis M. J. (2001) The small ubiquitin-like modifier-1 (SUMO-1) consensus sequence mediates Ubc9 binding and is essential for SUMO-1 modification. J. Biol. Chem. 276, 21664–21669 [DOI] [PubMed] [Google Scholar]
- 31. Giraud M. F., Desterro J. M., Naismith J. H. (1998) Structure of ubiquitin-conjugating enzyme 9 displays significant differences with other ubiquitin-conjugating enzymes which may reflect its specificity for sumo rather than ubiquitin. Acta Crystallogr. D Biol. Crystallogr. 54, 891–898 [DOI] [PubMed] [Google Scholar]
- 32. Sachdev S., Bruhn L., Sieber H., Pichler A., Melchior F., Grosschedl R. (2001) PIASy, a nuclear matrix-associated SUMO E3 ligase, represses LEF1 activity by sequestration into nuclear bodies. Genes Dev. 15, 3088–3103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kotaja N., Karvonen U., Jänne O. A., Palvimo J. J. (2002) PIAS proteins modulate transcription factors by functioning as SUMO-1 ligases. Mol. Cell. Biol. 22, 5222–5234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pichler A., Gast A., Seeler J. S., Dejean A., Melchior F. (2002) The nucleoporin RanBP2 has SUMO1 E3 ligase activity. Cell 108, 109–120 [DOI] [PubMed] [Google Scholar]
- 35. Schmidt D., Müller S. (2002) Members of the PIAS family act as SUMO ligases for c-Jun and p53 and repress p53 activity. Proc. Natl. Acad. Sci. U.S.A. 99, 2872–2877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kagey M. H., Melhuish T. A., Wotton D. (2003) The polycomb protein Pc2 is a SUMO E3. Cell 113, 127–137 [DOI] [PubMed] [Google Scholar]
- 37. Weger S., Hammer E., Heilbronn R. (2005) Topors acts as a SUMO-1 E3 ligase for p53 in vitro and in vivo. FEBS Lett. 579, 5007–5012 [DOI] [PubMed] [Google Scholar]
- 38. Yunus A. A., Lima C. D. (2009) Structure of the Siz/PIAS SUMO E3 ligase Siz1 and determinants required for SUMO modification of PCNA. Mol. Cell 35, 669–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Weger S., Hammer E., Engstler M. (2003) The DNA topoisomerase I binding protein topors as a novel cellular target for SUMO-1 modification. Characterization of domains necessary for subcellular localization and sumolation. Exp. Cell Res. 290, 13–27 [DOI] [PubMed] [Google Scholar]
- 40. Reverter D., Lima C. D. (2005) Insights into E3 ligase activity revealed by a SUMO-RanGAP1-Ubc9-Nup358 complex. Nature 435, 687–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tatham M. H., Kim S., Jaffray E., Song J., Chen Y., Hay R. T. (2005) Unique binding interactions among Ubc9, SUMO and RanBP2 reveal a mechanism for SUMO paralog selection. Nat. Struct. Mol. Biol. 12, 67–74 [DOI] [PubMed] [Google Scholar]
- 42. Merrill J. C., Melhuish T. A., Kagey M. H., Yang S. H., Sharrocks A. D., Wotton D. (2010) A role for non-covalent SUMO interaction motifs in Pc2/CBX4 E3 activity. PLoS ONE 5, e8794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Werner A., Flotho A., Melchior F. (2012) The RanBP2/RanGAP1*SUMO1/Ubc9 complex is a multisubunit SUMO E3 ligase. Mol. Cell 46, 287–298 [DOI] [PubMed] [Google Scholar]
- 44. Gyrd-Hansen M., Darding M., Miasari M., Santoro M. M., Zender L., Xue W., Tenev T., da Fonseca P. C., Zvelebil M., Bujnicki J. M., Lowe S., Silke J., Meier P. (2008) IAPs contain an evolutionarily conserved ubiquitin-binding domain that regulates NF-κB as well as cell survival and oncogenesis. Nat. Cell Biol. 10, 1309–1317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Davies G. C., Ettenberg S. A., Coats A. O., Mussante M., Ravichandran S., Collins J., Nau M. M., Lipkowitz S. (2004) Cbl-b interacts with ubiquitinated proteins. Differential functions of the UBA domains of c-Cbl and Cbl-b. Oncogene 23, 7104–7115 [DOI] [PubMed] [Google Scholar]
- 46. Smit J. J., Monteferrario D., Noordermeer S. M., van Dijk W. J., van der Reijden B. A., Sixma T. K. (2012) The E3 ligase HOIP specifies linear ubiquitin chain assembly through its RING-IBR-RING domain and the unique LDD extension. EMBO J. 31, 3833–3844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mascle X. H., Germain-Desprez D., Huynh P., Estephan P., Aubry M. (2007) Sumoylation of the transcriptional intermediary factor 1β (TIF1β), the co-repressor of the KRAB Multifinger proteins, is required for its transcriptional activity and is modulated by the KRAB domain. J. Biol. Chem. 282, 10190–10202 [DOI] [PubMed] [Google Scholar]
- 48. Germain-Desprez D., Bazinet M., Bouvier M., Aubry M. (2003) Oligomerization of transcriptional intermediary factor 1 regulators and interaction with ZNF74 nuclear matrix protein revealed by bioluminescence resonance energy transfer in living cells. J. Biol. Chem. 278, 22367–22373 [DOI] [PubMed] [Google Scholar]
- 49. Mercier J. F., Salahpour A., Angers S., Breit A., Bouvier M. (2002) Quantitative assessment of β1- and β2-adrenergic receptor homo- and heterodimerization by bioluminescence resonance energy transfer. J. Biol. Chem. 277, 44925–44931 [DOI] [PubMed] [Google Scholar]
- 50. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., Bax A. (1995) NMRPipe. A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 51. Vranken W. F., Boucher W., Stevens T. J., Fogh R. H., Pajon A., Llinas M., Ulrich E. L., Markley J. L., Ionides J., Laue E. D. (2005) The CCPN data model for NMR spectroscopy. Development of a software pipeline. Proteins 59, 687–696 [DOI] [PubMed] [Google Scholar]
- 52. Angers S., Salahpour A., Joly E., Hilairet S., Chelsky D., Dennis M., Bouvier M. (2000) Detection of β2-adrenergic receptor dimerization in living cells using bioluminescence resonance energy transfer (BRET). Proc. Natl. Acad. Sci. U.S.A. 97, 3684–3689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Brzovic P. S., Keeffe J. R., Nishikawa H., Miyamoto K., Fox D., 3rd, Fukuda M., Ohta T., Klevit R. (2003) Binding and recognition in the assembly of an active BRCA1/BARD1 ubiquitin-ligase complex. Proc. Natl. Acad. Sci. U.S.A. 100, 5646–5651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zheng N., Wang P., Jeffrey P. D., Pavletich N. P. (2000) Structure of a c-Cbl-UbcH7 complex. RING domain function in ubiquitin-protein ligases. Cell 102, 533–539 [DOI] [PubMed] [Google Scholar]
- 55. Albert T. K., Hanzawa H., Legtenberg Y. I., de Ruwe M. J., van den Heuvel F. A., Collart M. A., Boelens R., Timmers H. T. (2002) Identification of a ubiquitin-protein ligase subunit within the CCR4-NOT transcription repressor complex. EMBO J. 21, 355–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Dominguez C., Bonvin A. M., Winkler G. S., van Schaik F. M., Timmers H. T., Boelens R. (2004) Structural model of the UbcH5B/CNOT4 complex revealed by combining NMR, mutagenesis, and docking approaches. Structure 12, 633–644 [DOI] [PubMed] [Google Scholar]
- 57. Capili A. D., Lima C. D. (2007) Structure and analysis of a complex between SUMO and Ubc9 illustrates features of a conserved E2-Ubl interaction. J. Mol. Biol. 369, 608–618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Christensen D. E., Brzovic P. S., Klevit R. E. (2007) E2-BRCA1 RING interactions dictate synthesis of mono- or specific polyubiquitin chain linkages. Nat Struct Mol Biol 14, 941–948 [DOI] [PubMed] [Google Scholar]
- 59. Yin Q., Lin S. C., Lamothe B., Lu M., Lo Y. C., Hura G., Zheng L., Rich R. L., Campos A. D., Myszka D. G., Lenardo M. J., Darnay B. G., Wu H. (2009) E2 interaction and dimerization in the crystal structure of TRAF6. Nat. Struct. Mol. Biol. 16, 658–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Bentley M. L., Corn J. E., Dong K. C., Phung Q., Cheung T. K., Cochran A. G. (2011) Recognition of UbcH5c and the nucleosome by the Bmi1/Ring1b ubiquitin ligase complex. EMBO J. 30, 3285–3297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Mace P. D., Linke K., Feltham R., Schumacher F. R., Smith C. A., Vaux D. L., Silke J., Day C. L. (2008) Structures of the cIAP2 RING domain reveal conformational changes associated with ubiquitin-conjugating enzyme (E2) recruitment. J. Biol. Chem. 283, 31633–31640 [DOI] [PubMed] [Google Scholar]
- 62. Xu Z., Kohli E., Devlin K. I., Bold M., Nix J. C., Misra S. (2008) Interactions between the quality control ubiquitin ligase CHIP and ubiquitin conjugating enzymes. BMC Struct. Biol. 8, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Spratt D. E., Wu K., Kovacev J., Pan Z. Q., Shaw G. S. (2012) Selective recruitment of an E2∼ubiquitin complex by an E3 ubiquitin ligase. J. Biol. Chem. 287, 17374–17385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Levin I., Eakin C., Blanc M. P., Klevit R. E., Miller S. I., Brzovic P. S. (2010) Identification of an unconventional E3 binding surface on the UbcH5 ∼ Ub conjugate recognized by a pathogenic bacterial E3 ligase. Proc. Natl. Acad. Sci. U.S.A. 107, 2848–2853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Knipscheer P., Flotho A., Klug H., Olsen J. V., van Dijk W. J., Fish A., Johnson E. S., Mann M., Sixma T. K., Pichler A. (2008) Ubc9 sumoylation regulates SUMO target discrimination. Mol. Cell 31, 371–382 [DOI] [PubMed] [Google Scholar]
- 66. Song J., Zhang Z., Hu W., Chen Y. (2005) Small ubiquitin-like modifier (SUMO) recognition of a SUMO binding motif. A reversal of the bound orientation. J. Biol. Chem. 280, 40122–40129 [DOI] [PubMed] [Google Scholar]
- 67. Song J., Durrin L. K., Wilkinson T. A., Krontiris T. G., Chen Y. (2004) Identification of a SUMO-binding motif that recognizes SUMO-modified proteins. Proc. Natl. Acad. Sci. U.S.A. 101, 14373–14378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Tatham M. H., Kim S., Yu B., Jaffray E., Song J., Zheng J., Rodriguez M. S., Hay R. T., Chen Y. (2003) Role of an N-terminal site of Ubc9 in SUMO-1, -2, and -3 binding and conjugation. Biochemistry 42, 9959–9969 [DOI] [PubMed] [Google Scholar]
- 69. Knipscheer P., van Dijk W. J., Olsen J. V., Mann M., Sixma T. K. (2007) Noncovalent interaction between Ubc9 and SUMO promotes SUMO chain formation. EMBO J. 26, 2797–2807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Stehmeier P., Muller S. (2009) Phospho-regulated SUMO interaction modules connect the SUMO system to CK2 signaling. Mol. Cell 33, 400–409 [DOI] [PubMed] [Google Scholar]
- 71. Lin D. Y., Huang Y. S., Jeng J. C., Kuo H. Y., Chang C. C., Chao T. T., Ho C. C., Chen Y. C., Lin T. P., Fang H. I., Hung C. C., Suen C. S., Hwang M. J., Chang K. S., Maul G. G., Shih H. M. (2006) Role of SUMO-interacting motif in Daxx SUMO modification, subnuclear localization, and repression of sumoylated transcription factors. Mol. Cell 24, 341–354 [DOI] [PubMed] [Google Scholar]
- 72. Scaglioni P. P., Yung T. M., Cai L. F., Erdjument-Bromage H., Kaufman A. J., Singh B., Teruya-Feldstein J., Tempst P., Pandolfi P. P. (2006) A CK2-dependent mechanism for degradation of the PML tumor suppressor. Cell 126, 269–283 [DOI] [PubMed] [Google Scholar]
- 73. Shiio Y., Eisenman R. N. (2003) Histone sumoylation is associated with transcriptional repression. Proc. Natl. Acad. Sci. U.S.A. 100, 13225–13230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Ross S., Best J. L., Zon L. I., Gill G. (2002) SUMO-1 modification represses Sp3 transcriptional activation and modulates its subnuclear localization. Mol. Cell 10, 831–842 [DOI] [PubMed] [Google Scholar]
- 75. Perdomo J., Verger A., Turner J., Crossley M. (2005) Role for SUMO modification in facilitating transcriptional repression by BKLF. Mol. Cell. Biol. 25, 1549–1559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Hsu Y. H., Sarker K. P., Pot I., Chan A., Netherton S. J., Bonni S. (2006) Sumoylated SnoN represses transcription in a promoter-specific manner. J. Biol. Chem. 281, 33008–33018 [DOI] [PubMed] [Google Scholar]
- 77. Rytinki M. M., Palvimo J. J. (2008) SUMOylation modulates the transcription repressor function of RIP140. J. Biol. Chem. 283, 11586–11595 [DOI] [PubMed] [Google Scholar]
- 78. Yunus A. A., Lima C. D. (2006) Lysine activation and functional analysis of E2-mediated conjugation in the SUMO pathway. Nat. Struct. Mol. Biol. 13, 491–499 [DOI] [PubMed] [Google Scholar]
- 79. Tatham M. H., Chen Y., Hay R. T. (2003) Role of two residues proximal to the active site of Ubc9 in substrate recognition by the Ubc9.SUMO-1 thiolester complex. Biochemistry 42, 3168–3179 [DOI] [PubMed] [Google Scholar]
- 80. Kaiser S. E., Riley B. E., Shaler T. A., Trevino R. S., Becker C. H., Schulman H., Kopito R. R. (2011) Protein standard absolute quantification (PSAQ) method for the measurement of cellular ubiquitin pools. Nat. Methods 8, 691–696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Saitoh H., Hinchey J. (2000) Functional heterogeneity of small ubiquitin-related protein modifiers SUMO-1 versus SUMO-2/3. J. Biol. Chem. 275, 6252–6258 [DOI] [PubMed] [Google Scholar]
- 82. Garner T. P., Strachan J., Shedden E. C., Long J. E., Cavey J. R., Shaw B., Layfield R., Searle M. S. (2011) Independent interactions of ubiquitin-binding domains in a ubiquitin-mediated ternary complex. Biochemistry 50, 9076–9087 [DOI] [PubMed] [Google Scholar]
- 83. Bosanac I., Wertz I. E., Pan B., Yu C., Kusam S., Lam C., Phu L., Phung Q., Maurer B., Arnott D., Kirkpatrick D. S., Dixit V. M., Hymowitz S. G. (2010) Ubiquitin binding to A20 ZnF4 is required for modulation of NF-κB signaling. Mol. Cell 40, 548–557 [DOI] [PubMed] [Google Scholar]
- 84. Purbeck C., Eletr Z. M., Kuhlman B. (2010) Kinetics of the transfer of ubiquitin from UbcH7 to E6AP. Biochemistry 49, 1361–1363 [DOI] [PubMed] [Google Scholar]
- 85. Bailly V., Lamb J., Sung P., Prakash S., Prakash L. (1994) Specific complex formation between yeast RAD6 and RAD18 proteins. A potential mechanism for targeting RAD6 ubiquitin-conjugating activity to DNA damage sites. Genes Dev. 8, 811–820 [DOI] [PubMed] [Google Scholar]
- 86. Zhang M., Windheim M., Roe S. M., Peggie M., Cohen P., Prodromou C., Pearl L. H. (2005) Chaperoned ubiquitylation-crystal structures of the CHIP U box E3 ubiquitin ligase and a CHIP-Ubc13-Uev1a complex. Mol. Cell 20, 525–538 [DOI] [PubMed] [Google Scholar]
- 87. Huang L., Kinnucan E., Wang G., Beaudenon S., Howley P. M., Huibregtse J. M., Pavletich N. P. (1999) Structure of an E6AP-UbcH7 complex. Insights into ubiquitination by the E2-E3 enzyme cascade. Science 286, 1321–1326 [DOI] [PubMed] [Google Scholar]
- 88. Chang C. C., Naik M. T., Huang Y. S., Jeng J. C., Liao P. H., Kuo H. Y., Ho C. C., Hsieh Y. L., Lin C. H., Huang N. J., Naik N. M., Kung C. C., Lin S. Y., Chen R. H., Chang K. S., Huang T. H., Shih H. M. (2011) Structural and functional roles of Daxx SIM phosphorylation in SUMO paralog-selective binding and apoptosis modulation. Mol. Cell 42, 62–74 [DOI] [PubMed] [Google Scholar]
- 89. Garcia-Dominguez M., Reyes J. C. (2009) SUMO association with repressor complexes, emerging routes for transcriptional control. Biochim. Biophys. Acta 1789, 451–459 [DOI] [PubMed] [Google Scholar]
- 90. Gross M., Yang R., Top I., Gasper C., Shuai K. (2004) PIASy-mediated repression of the androgen receptor is independent of sumoylation. Oncogene 23, 3059–3066 [DOI] [PubMed] [Google Scholar]
- 91. Munarriz E., Barcaroli D., Stephanou A., Townsend P. A., Maisse C., Terrinoni A., Neale M. H., Martin S. J., Latchman D. S., Knight R. A., Melino G., De Laurenzi V. (2004) PIAS-1 is a checkpoint regulator which affects exit from G1 and G2 by sumoylation of p73. Mol. Cell. Biol. 24, 10593–10610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Tolkunova E., Malashicheva A., Parfenov V. N., Sustmann C., Grosschedl R., Tomilin A. (2007) PIAS proteins as repressors of Oct4 function. J. Mol. Biol. 374, 1200–1212 [DOI] [PubMed] [Google Scholar]
- 93. Zhou S., Si J., Liu T., DeWille J. W. (2008) PIASy represses CCAAT/enhancer-binding protein δ (C/EBPδ) transcriptional activity by sequestering C/EBPδ to the nuclear periphery. J. Biol. Chem. 283, 20137–20148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Kobayashi S., Shibata H., Kurihara I., Yokota K., Suda N., Saito I., Saruta T. (2004) Ubc9 interacts with chicken ovalbumin upstream promoter-transcription factor I and represses receptor-dependent transcription. J. Mol. Endocrinol. 32, 69–86 [DOI] [PubMed] [Google Scholar]