

Background: The acetyl-CoA biosynthetic pathways of the malaria parasite are unclear.
Results: 13C-Labeling experiments in parasites lacking a functional pyruvate dehydrogenase (PDH) complex show that the PDH does not contribute significantly to the acetyl-CoA pool.
Conclusion: The majority of acetyl-CoA biosynthesis in the parasite derives from a PDH-like enzyme and acetyl-CoA synthetase.
Significance: The two routes for acetyl-CoA synthesis appear to have separate functions.
Keywords: Acetyl Coenzyme A, Glycolysis, Malaria, Phosphoenolpyruvate Carboxykinase, Plasmodium, Pyruvate Dehydrogenase Complex, Tricarboxylic Acid (TCA) Cycle, Acetate
Abstract
The malaria parasite Plasmodium falciparum depends on glucose to meet its energy requirements during blood-stage development. Although glycolysis is one of the best understood pathways in the parasite, it is unclear if glucose metabolism appreciably contributes to the acetyl-CoA pools required for tricarboxylic acid metabolism (TCA) cycle and fatty acid biosynthesis. P. falciparum possesses a pyruvate dehydrogenase (PDH) complex that is localized to the apicoplast, a specialized quadruple membrane organelle, suggesting that separate acetyl-CoA pools are likely. Herein, we analyze PDH-deficient parasites using rapid stable-isotope labeling and show that PDH does not appreciably contribute to acetyl-CoA synthesis, tricarboxylic acid metabolism, or fatty acid synthesis in blood stage parasites. Rather, we find that acetyl-CoA demands are supplied through a “PDH-like” enzyme and provide evidence that the branched-chain keto acid dehydrogenase (BCKDH) complex is performing this function. We also show that acetyl-CoA synthetase can be a significant contributor to acetyl-CoA biosynthesis. Interestingly, the PDH-like pathway contributes glucose-derived acetyl-CoA to the TCA cycle in a stage-independent process, whereas anapleurotic carbon enters the TCA cycle via a stage-dependent phosphoenolpyruvate carboxylase/phosphoenolpyruvate carboxykinase process that decreases as the parasite matures. Although PDH-deficient parasites have no blood-stage growth defect, they are unable to progress beyond the oocyst phase of the parasite mosquito stage.
Introduction
Over the course of its 48-h intraerythrocytic developmental cycle the human malaria parasite, Plasmodium falciparum, matures and replicates. This rapid development necessitates a constant supply of exogenous nutrients, with the parasite primarily dependent upon glucose to sustain its energy demands (for review, see Refs. 1–3). Parasite-infected erythrocytes consume up to 100-fold more glucose than uninfected erythrocytes via glycolysis (4–6). Most of this glucose is metabolized to pyruvate that is subsequently reduced to lactate and excreted (7–11). However, P. falciparum possess several metabolic pathways that are typically supplied by pyruvate-derived acetyl-CoA including the mitochondrial tricarboxylic acid cycle (TCA), cytosolic fatty acid elongation, and nuclear histone acetylation (12–16). In most organisms acetyl-CoA is predominantly synthesized through the action of the pyruvate dehydrogenase (PDH)3 complex. In Plasmodium, however, the PDH is localized within the apicoplast, a plastid-like organelle enclosed by four membranes (17, 18). Although it is clear that P. falciparum uses glucose-derived acetyl-CoA to supply the mitochondrial TCA cycle (14), it remains unclear to what extent PDH-derived acetyl-CoA is utilized by the parasite. In particular, transport of acetyl-CoA out of the apicoplast (via an unknown transport mechanism) would be required, which is highly unlikely.
PDH is a large multienzyme complex that converts pyruvate and co-enzyme A into acetyl-CoA using thiamine, NAD, and lipoic acid as cofactors. PDH is comprised of the E1 pyruvate dehydrogenase (which exists as a heteromer of E1α and E1β), E2 dihydrolipoamide acetyltransferase, and E3 dihydrolipoamide dehydrogenase subunits (19). The E1 subunit mediates the covalent attachment of pyruvate to thiamine pyrophosphate, decarboxylating pyruvate to an acetyl group, and then transfers the acetyl group to lipoic acid. The acetyl group is then transferred to co-enzyme A (CoA) via the E2 subunit, and dihydrolipoic acid is oxidized back to lipoic acid by the E3 subunit. Because of its localization to the apicoplast in malaria parasites, the E3 subunit is not shared between other dehydrogenase complexes (17).
In the rodent malaria Plasmodium yoelii, disrupting the E1α or E3 subunits of PDH has no effect on blood-stage development but prevents parasites from developing into liver-stage exo-erythrocytic merozoites (20). Because this phenotype is consistent with the fatty acid synthase II knock-out parasites, PDH is thought to be involved in de novo fatty acid synthesis during liver-stage development (17, 21, 22). The catalytically active domain of the E2 subunit was demonstrated to have acetyl-CoA synthetic activity (17), but it has not been determined whether the PDH complex significantly contributes to acetyl-CoA metabolism or if it is essential for blood stages of the human malaria parasite P. falciparum.
Herein, we have applied a rapid stable-isotope labeling technique in combination with high resolution mass spectrometry to study blood-stage acetyl-CoA metabolism and determine the contribution of the PDH to central carbon metabolism. In a comparative analysis of wild type versus PDH-deficient (pdh e1α-) parasites, we come to the surprising conclusion that the PDH does not appreciably contribute to the acetyl-CoA pool or metabolic pathways downstream of this central precursor. Moreover, we observe significant fluxes through acetyl-CoA-dependent pathways despite the absence of a functional PDH. We propose that acetyl-CoA is predominantly synthesized through the mitochondrial branched-chain keto acid dehydrogenase complex (BCKDH), an enzyme complex typically associated with amino acid degradation. We show that this PDH-like metabolism can account for the majority of acetyl-CoA synthesis and that direct synthesis of acetyl-CoA from acetate can also contribute to the acetyl-CoA pool when parasites are supplied with an abundant acetate source. These findings demonstrate that blood-stage parasites use a PDH-like enzyme and acetate fixation for supplying acetyl-CoA pools.
To further investigate central carbon metabolism of P. falciparum, we measured the relative contribution of glucose- and glutamine-derived carbon to the TCA cycle throughout the intraerythrocytic developmental cycle. We demonstrate that as the parasite matures, it reduces the incorporation of anapleurotic carbon from glucose and increases incorporation via glutamine. This finding highlights the parasite's ability to restructure metabolism to meet its developmental requirements.
EXPERIMENTAL PROCEDURES
P. falciparum in Vitro Cultivation
P. falciparum NF54 and pdh e1α-G2/B9 clones were cultured and synchronized by standard methods (23, 24) with the modification that culture flasks were maintained at 37 °C in an atmospherically controlled incubator set at 5% CO2, 6% O2. Parasitemia and synchronicity was monitored using microscopy and Giemsa-stained thin-blood smear slides. Uninfected erythrocytes (from the same donor) were cultured for 48-h in parallel before any experimentation. Mycoplasma testing was performed routinely to ensure contamination-free cultures. Cell counts were taken for all experiments with a Neubauer hemocytometer.
P. falciparum Sporozoite Production
Anopheles stephensi mosquitoes (originating from the Walter Reed Army Institute of Research) were maintained at 27 °C and 75% humidity on a 12-h light/dark cycle. Larval stages were reared after standard protocols as described in the MR4 manual with larval stages maintained on finely ground Tetramin fish food and adult mosquitoes maintained on 8% dextrose in 0.05% para-aminobenzoic acid water.
In vitro P. falciparum NF54 blood stage cultures were maintained in RPMI 1640 (25 mm HEPES, 2 mm l-glutamine) supplemented with 50 μm hypoxanthine and 10% A+ human serum in an atmosphere of 5% CO2, 5% O2, and 90% N2. Cells were subcultured into O+ erythrocytes. Gametocyte cultures were initiated at 5% hematocrit and 0.8–1% parasitemia (mixed stages) and maintained for up to 17 days with daily media changes.
Non blood-fed adult female mosquitoes (3–7 days post-emergence) were fed on gametocyte cultures. Gametocyte cultures were quickly spun down, and the pelleted infected erythrocytes were diluted to a 40% hematocrit with fresh A+ human serum and O+ erythrocytes. Mosquitoes were allowed to feed through Parafilm for up to 20 min. After blood feeding, mosquitoes were maintained for up to 19 days at 27 °C, 75% humidity and provided with 8% dextrose solution in PABA water. Infection prevalence was checked at days 7–10 by examining dissected midguts under light microscopy for the presence of oocysts with salivary gland dissections performed at days 14–19.
Infected Erythrocyte Enrichment
Parasite cultures were enriched using a modified magnetic enrichment method. A custom-built magnetic-separation apparatus was designed using Google Sketch and produced by Pokono via high density plastic three-dimensional printing. 1.5 in3 magnets (pull force 220 lbs) were purchased, and metal inserts cut on-site. The apparatus allowed the simultaneous use of four CS VarioMacs columns (Miltenyi Biotech) and the design can be downloaded from 3dwarehouse. Enrichment was performed by resuspending 1 ml of packed cell culture (10% parasitemia) in 15 ml (∼8% hematocrit) and passed through a single CS column with constant addition of complete RPMI until the eluent was transparent. Infected erythrocytes were then eluted separate from the magnetic apparatus, yielding 85–98% parasitemia. Separate strains were enriched simultaneously to maintain consistency between cell lines and allowed to recover at 37 °C for 1 h before experimentation.
Isotope Labeling
For [13C]glucose labeling experiments, complete RPMI was made using glucose-free RPMI powder (Sigma), and an appropriate amount of [6-13C]glucose (all stable-isotope compounds were acquired from Cambridge Isotopes) was added to give a final concentration of 11 mm. For thiamine-free/oxythiamine experiments, RPMI was reconstituted using 50× RPMI amino acid solution (Sigma) and individual vitamins, salts, and oxythiamine purchased from Sigma and Fischer. Fully reconstituted RPMI was always made in parallel and used as a control condition to ensure there was no variation in parasite growth between batches. The IC50 of oxythiamine was determined, and a low dose long-term incubation strategy was required to allow conversion of oxythiamine to oxythiamine pyrophosphate. [5-13C]Glutamine was added to glutamine-free RPMI at a final concentration of 2 mm for [5-13C]glutamine labeling experiments. [6-13C,15N]leucine labeling experiments were performed in complete custom-made RPMI without leucine and supplemented with [6-13C,15N]leucine at the RPMI concentration.
[2-13C]Acetate and [1-13C]pyruvate (label in the 2-C position) labeling experiments were performed using complete RPMI with an appropriate amount of each compound added to give a concentration of 2 and 10 mm, respectively. Media were pH-titrated and added as a 1:1 ratio to cell suspensions containing label-free RPMI, giving a final concentration of 1 mm (acetate) and 5 mm (pyruvate). 11 mm unlabeled glucose was present in all experiments.
Metabolite Extraction
Four methods were employed to extract metabolites. Steady-state incubations were extracted using the methods described previously (25) with slight modifications. Briefly, infected erythrocytes were transferred to microcentrifuge tubes and spun at 14,000 × g for 30 s. The supernatant was rapidly aspirated, and the cell pellet was extracted with 1 ml of 90% ice-cold methanol (containing the internal standard [4-13C,15N]aspartate). Samples were dried under nitrogen flow and stored at −80 °C until analysis. Isolated parasites were extracted from 10% parasitemia cultures by saponin lysis (0.08%) in Eppendorf tubes. Samples were rapidly washed 3 times with ice-cold PBS, and the parasite pellet was extracted with 90% methanol.
The rapid-labeling and extraction method was adapted from previously described radiolabeled flux techniques (26). 2-ml microcentrifuge tubes were loaded with 300 μl of 30% trifluoroacetic acid with 700 μl of an oil mixture (5:4 dibutyl phthalate:dibutyl octanol, density 1.02) layered above. Time courses were initiated with the 1:1 addition of cell suspensions to pre-warmed isotope-labeled media (1–2% final hematocrit) and incubated at 37 °C. At the appropriate time points, 1 ml of cell suspension was layered on top of the oil mixture and immediately centrifuged at 14,000 × g for 30 s. The supernatant and oil layer was removed, and the acid layer was mixed with 700 μl of 90% ice-cold methanol (containing the internal standard [4-13C,15N]aspartate) and centrifuged. The metabolite extracts were transferred to a fresh microcentrifuge tube and immediately dried under nitrogen flow. Extracts were resuspended in 100 μl of H2O pH neutralized with 5–10 μl of 1 m ammonia bicarbonate, transferred to a fresh microcentrifuge tube, dried under nitrogen flow, and stored at −80 °C until analysis. Validation of this method was achieved by comparison to the standard 90% methanol extraction method.
Fatty acid extractions were performed as described previously (27) with processing blanks extracted in parallel. [16-13C]Palmitate was used as an internal standard and used to normalize all signals. After data acquisition, processing blanks were subtracted from biological samples, providing the total ion counts associated with each condition.
Mass Spectrometry
For all LC-MS analysis, samples were reconstituted in 100 μl of H2O and analyzed on an Exactive Orbitrap mass spectrometer as previously described (28). The data presented in the glutamine panel of Fig. 5C were collected on a Finnigan TSQ Quantum Ultra triple quadrupole mass spectrometer equipped with an electrospray ionization source, operating in positive mode.
FIGURE 5.

Stage-dependent changes in [6-13C]glucose and [5-13C]glutamine labeling. At 12, 26, and 40 h post invasion, bulk cultures were incubated for 2 h in RPMI containing either 11 mm [6-13C]glucose or 2 mm [5-13C]glutamine. Cultures were saponin-isolated and rapidly washed with ice-cold PBS and extracted. A, schematic of the IDC and the proposed metabolic architecture of central-carbon metabolism in P. falciparum. The transcriptional profile of PEPC and PEPCK are presented as the log2 ratio relative to the total RNA pool from (45). DHAP, dihydroxyacetone phosphate, PEP, phosphoenolpyruvate. B, [6-13C]glucose labeling of central carbon intermediates across the IDC. C, [5-13C]glutamine labeling of the TCA cycle across the IDC. Data are presented as percent of total metabolite pool as mean ± S.E. from n = 4 experiments.
Heteronuclear Single Quantum Correlation Nuclear Magnetic Resonance (NMR) Acquisition
Uninfected and enriched infected erythrocytes were incubated for 2 h in [6-13C]glucose RPMI and subsequently extracted with 90% methanol. Samples were dried down under N2 gas, then resuspended in 99.9% D2O and titrated to pH 7.40 (±0.01; uncorrected glass electrode reading). Two-dimensional 13C,1H heteronuclear single quantum correlation NMR spectra were collected on a 500-MHz Bruker spectrometer equipped with a 1H-optimized triple resonance cryoprobe. Heteronuclear single quantum correlations were acquired in 4 transients, with 8192 directly acquired points and 1028 increments. Spectra were Fourier transform with shifted sine bell window function, zero-filled, and phased in TopSpin. Spectra were analyzed using rNMR (29), and metabolites were identified and quantified using established methods (30).
MS Data Processing and Analysis
Thermo Fisher mass spectrometry RAW files were converted from profile mode into centroid mode using the ReAdW program (31) and loaded into MAVEN, a publicly available analysis program (32). Correct assignment was confirmed via the addition of pure standard for all metabolites presented. Isotope-labeled forms were identified using the expected mass shifts given by 13C and 15N. Peaks below an ion count of 1000 were excluded from analysis. Raw signals were normalized to the internal control, corrected for natural abundance where appropriate, and adjusted to ion count/108 cells using cells counts taken for each sample.
For quantification of glycolytic intermediates the signal ratio of the 13C-labeled metabolite (complete pool labeling was achieved with [13C]glucose and [13C]acetate for 2 h) to its unlabeled internal standard was used and to convert ion count/108 cells into intracellular concentration as previously described (33), assuming the intracellular volume of an infected erythrocyte is 75 fl (34). For TCA cycle intermediates, pure 13C-labeled standards were added to unlabeled parasite extracts and quantified as described above.
Graphical representations and statistical analysis were performed in GraphPad. A paired two-tail Student's t test was performed for pairwise statistical comparisons, and a one-way analysis of variance was performed for multiple statistical comparison. See supplemental Methods for further experimental details.
RESULTS
Dynamic Isotope Labeling in P. falciparum-infected Erythrocytes
To investigate central carbon metabolism of P. falciparum-infected erythrocytes, we used stable-isotope labeling. Unfortunately, parasite biology is not amenable to existing rapid-labeling techniques utilized in bacterial metabolomics studies, which use filter paper-adhered cells to rapidly transfer samples between labeled media to extraction solution (35, 36). Therefore, we developed a rapid stable-isotope labeling technique to measure dynamic glycolytic flux in Plasmodium (see “Experimental Procedures”). Magnet-enriched uninfected and infected erythrocytes (trophozoite stage) suspended in [12C]glucose RPMI were supplemented with [6-13C]glucose (final concentration 11 mm at an isotope ratio of 1:1 12C:13C). Turnover of intracellular glycolytic intermediates was monitored over a 1-h period via LC-MS (Fig. 1A and supplemental Fig. S1). The total metabolite pool (presented as intracellular concentration; Fig. 1A, top panel) was unchanged across the time course, indicating that the conditions used did not perturb parasite metabolism. Isotopic labeling kinetics in glycolytic intermediates indicated differential labeling rates between the upper and lower halves of glycolysis; fructose 1,6-bisphosphate was completely labeled in less than 2 min, whereas phosphoenolpyruvate, pyruvate, and lactate required an hour to reach isotopic equilibrium (Fig. 1A). Infected erythrocytes (NF54) showed a significant increase in the total pool of most metabolic intermediates and in the glycolytic flux relative to uninfected erythrocytes (supplemental Table S1; all p < 0.01). Surprisingly, unlike glycolytic intermediates, the acetyl-CoA pool did not completely label in the conditions tested, labeling only 42 ± 9% of the total pool after 1 h and 77 ± 3% after 3 h (Fig. 1B).
FIGURE 1.

A, dynamic [6-13C]glucose labeling of P. falciparum-infected erythrocytes. RPMI containing 11 mm [6-13C]glucose was added to enriched P. falciparum-infected and uninfected erythrocyte suspensions at a 1:1 ratio. Rapid quenching and metabolite extraction was performed, and metabolic flux was assessed via LC-MS. Percent labeling was adjusted to the maximal theoretical enrichment for equal 12C:13C glucose mixing and natural abundance. Data are presented as intracellular concentration [IC] (top panel) and percent labeling of the total metabolite pool %[13C] (bottom panel) as the mean ± S.E. from n = 4 experiments. B, steady-state [6-13C]glucose labeling of P. falciparum-infected erythrocytes. Enriched P. falciparum-infected and uninfected erythrocytes (uRBC) were incubated in RPMI containing 11 mm [6-13C]glucose for 3 h, extracted with 90% methanol, and measured via LC-MS. Isotope labeling was adjusted for natural abundance and presented as the fraction of the total intracellular concentration as the mean ± S.E. from n = 4 experiments. Isotope labels were combined for clarity, the individual isotopes detected and combined were as follows: acetyl-CoA M+2, M+3, M+4, M+7 and citrate M+2, M+3, M+5. The error above each bar represents the S.E. of the total ion signal converted to intracellular concentration, whereas the error below the labeled fraction reflects the S.E. of the combined isotope label. PEP, phosphoenolpyruvate.
PDH-disrupted Parasites Possess Normal Acetyl-CoA Metabolism
The E1 subunit of the PDH complex mediates the thiamine pyrophosphate-dependent decarboxylation of pyruvate and is considered critical for normal PDH function. To ensure that P. falciparum produce the E1 subunit and all of the additional subunits of the PDH complex, we first used MS/MS proteomics to identify the subunits using the NF54 strain during the blood stage (supplemental Fig. S2 and Table S2). Knowing that the complex was expressed, we generated a pdh e1α- line by homologous recombination to investigate acetyl-CoA metabolism in the parasite (supplemental Fig. S3). Surprisingly, PDH-disrupted parasites had no signification alterations in glycolytic flux (Fig. 1A). Moreover, PDH disrupted parasites converted [6-13C]glucose into +2 atomic mass unit-shifted acetyl-CoA (consistent with the incorporation of two 13C atoms, referred to henceforth as M+2) and M+2 citrate (the first product of acetyl-CoA metabolism by the TCA cycle) (Fig. 1, A and B). The citrate labeling was composed of M+2, M+3, and M+5, indicating that both acetyl-CoA and oxaloacetate derived from glucose were incorporated into the TCA cycle. Neither the acetyl-CoA nor the citrate labeling was significantly different between the pdh e1α- and the wild type NF54 parasite lines. These observations were consistent with our finding that disruption of the PDH had no effect on parasite growth over 15 days (supplemental Fig. S4).
Disruption of the PDH Complex Does Not Affect the Fatty Acid Profile of P. falciparum-infected Erythrocytes
Given the lack of growth rate phenotype, the negligible impact of PDH disruption on total acetyl-CoA synthesis, its localization to the apicoplast, and its likely role in type 2 fatty acid synthesis, we hypothesized that PDH disruption might alter the fatty acid profiles of blood-stage parasites. Several fatty acid species were more abundant in infected erythrocytes in comparison to uninfected erythrocytes; these included C16:0, C18:0, and C18:1 and the longer unsaturated fatty acid species C18:2, C18:3, C20:3, C20:5, and C22:5 (Fig. 2). However, disruption of the PDH complex did not perturb the levels of fatty acid species of infected erythrocytes. Similarly, global transcriptome analysis failed to identify any significant change in the transcription of metabolic genes when PDH is disrupted (supplemental Table S3 and Methods).
FIGURE 2.
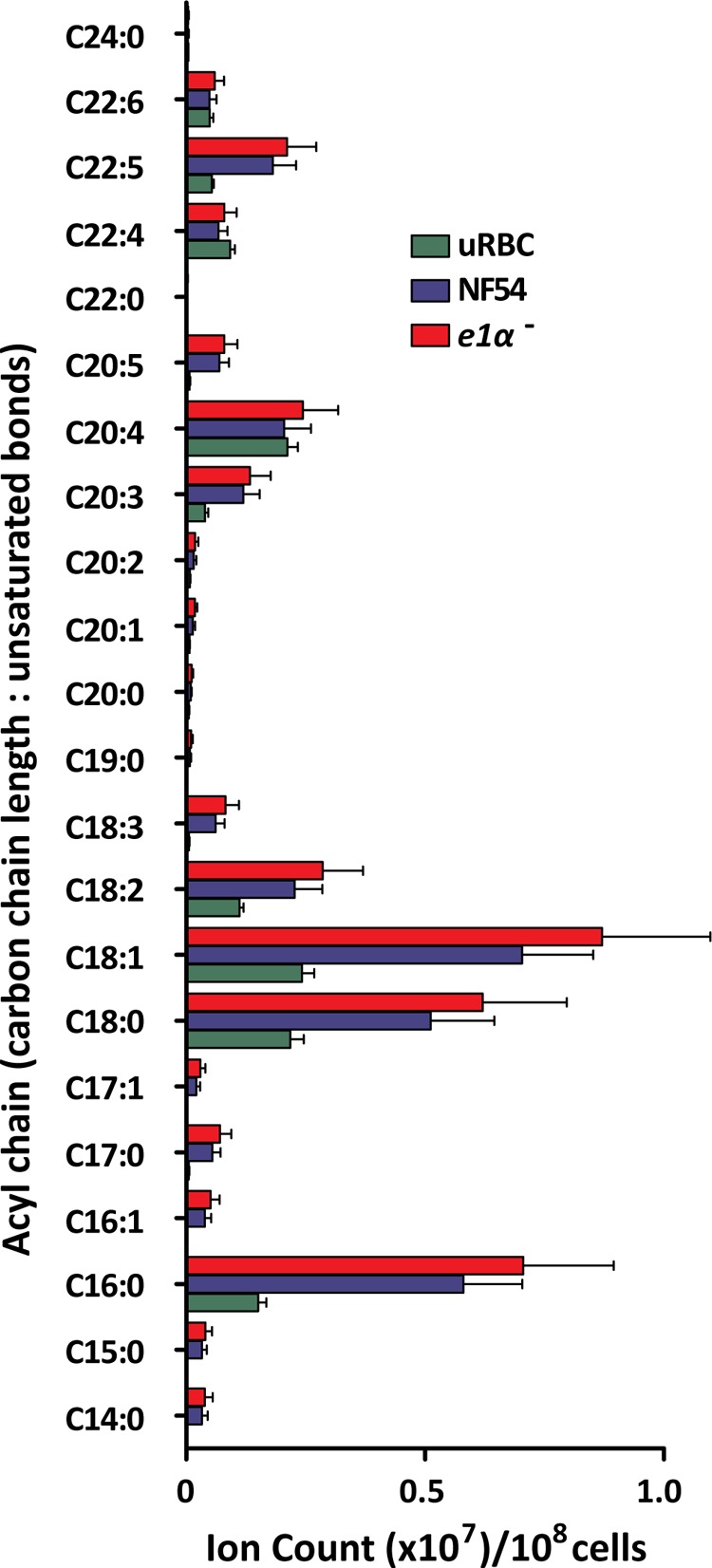
Fatty acid profile of NF54 and pdh e1α- infected erythrocytes in comparison to uninfected erythrocytes (uRBC). Processing blanks were measured and subtracted from the total ion count and signals normalized to the internal standard ([16-13C]palmitate). Data are presented as total ion counts/108 cells mean ± S.E. from n = 3 experiments.
pdh e1α- Parasites Cannot Form Sporozoites during the Mosquito Stage
To complete the lifecycle, Plasmodium parasites must differentiate into sexual gametocytes that fuse within the mosquito midgut to produce an oocyte that develops into an oocyst, which results in 1000s of infectious motile sporozoite parasites for delivery back into another human (37). Because disruption of PDH had no effect on metabolism during intraerythocytic development, we examined the progression of the pdh e1α- parasite line through the sexual and mosquito stages. Disruption of PDH showed no significant impact on gametocytogenesis, exflagellation, or development into oocysts (Table 1). However, oocyst development was aberrant including the following; 1) a large number of pdh e1α- oocysts were seen 14 days post blood meal, whereas most of the wild type parasites had matured into midgut oocyst sporozoites; 2) the observation of wild type but not pdh e1α- sporozoites in mosquito midguts; 3) the total absence of sporozoites in salivary glands of mosquitoes infected with pdh e1α- parasites despite an average of 78,000 ± 21,000 sporozoites in salivary glands of mosquitoes infected with wild type parasites (Table 1). These observations suggest that PDH-disrupted parasites are unable to progress beyond the oocyst stage and fail to produce sporozoites.
TABLE 1.
Enumeration of midgut oocyst density and salivary gland sporozoites in wild type NF54 and the pdh e1α- parasite, clones B9 and D2
| Parasite line | Day 7a | Day 9 | Day 11 | Day 14 | Day 19 | Midgut spzsb | Spzsc |
|---|---|---|---|---|---|---|---|
| Wild type NF54 | 33 (0–83) | 45 (0–157) | 10 (0–70) | 2 (0–9) | 0 | Yes | 78,000 ± 21,000 |
| pdh e1α-B9 | 37 (0–134) | 32 (0–120) | 34 (0–116) | 14 (0–35) | 0 | No | 0 |
| pdh e1α-G2 | 38 (0–142) | 26 (0–97) | 8 (0–47) | 10 (0–44) | 0 | No | 0 |
a Mean of 20 mosquitoes (10 from each of 2 cages taken from independent infectious blood meals). Data are days after the infectious blood meal.
b Mosquito midguts (10 from each of 2 cages taken from independent infectious blood meals) were analyzed for the presence of mature midgut oocyst sporozoites (Midgut spzs) every other day from day 9 until day 19 after the infectious blood meal.
c Salivary gland sporozoites (Spzs) were counted from 20 mosquitoes (10 from each of two cages taken from independent infectious blood meals) on days 14, 15, and 16 after the infectious blood meal.
Glucose-derived Acetyl-CoA Synthesis Is Mediated by a PDH-like Enzyme
As there was no significant difference in glucose-derived acetyl-CoA turnover between NF54 and the pdh e1α- line throughout blood-stage development, we sought to determine how the parasite was able to convert [6-13C]glucose (M+6) into [2-13C]acetyl-CoA (M+2). The PDH synthesis of acetyl-CoA is a thiamine pyrophosphate-dependent reaction that can be inhibited by oxythiamine (which is converted to oxythiamine pyrophosphate by the parasite), a broad-spectrum inhibitor of thiamine pyrophosphate-dependent reactions. We measured the effect of oxythiamine when added to thiamine-free RPMI for 48 h (at an IC20 concentration of 200 μm; data not shown) compared with identical cultures maintained in standard RPMI or thiamine-free RPMI. After 48 h of exposure, infected erythrocytes were enriched and incubated in their respective RPMI, to which was added [6-13C]glucose (Fig. 3A). In the presence of oxythiamine, the synthesis of [2-13C]acetyl-CoA was inhibited by 70 and 80% in both pdh e1α- and NF54 parasite lines, respectively (Fig. 3A; p < 0.05). Comparable inhibition was also observed in the 13C-labeling of the downstream metabolites citrate and acetyl glutamate (supplemental Fig. S5, A and B). The 13C-labeling of pyruvate in thiamine-free oxythiamine-containing RPMI was not significantly different from the thiamine-free control (p > 0.1), indicating that the oxythiamine does not inhibit glycolysis (Fig. 3A). In addition to PDH, P. falciparum contains only three additional thiamine pyrophosphate-dependent enzymes, transketolase (PFF0550w), ketoglutarate dehydrogenase (KDH; E1, PF08_0045; E2, PF13_0121; E3, PFL1550w), and the BCKDH (E1α, PF13_0070; E1β, PFE0225w; E2, PFC0170c; E3, PFL1550w), which can all be inhibited by oxythiamine. In P. falciparum, transketolase and KDH have well established functions, and oxythiamine elicited clear inhibitory effects on both (supplemental Fig. S5, C and D), causing a reduction of the downstream products AMP (transketolase) and succinate (KDH).
FIGURE 3.
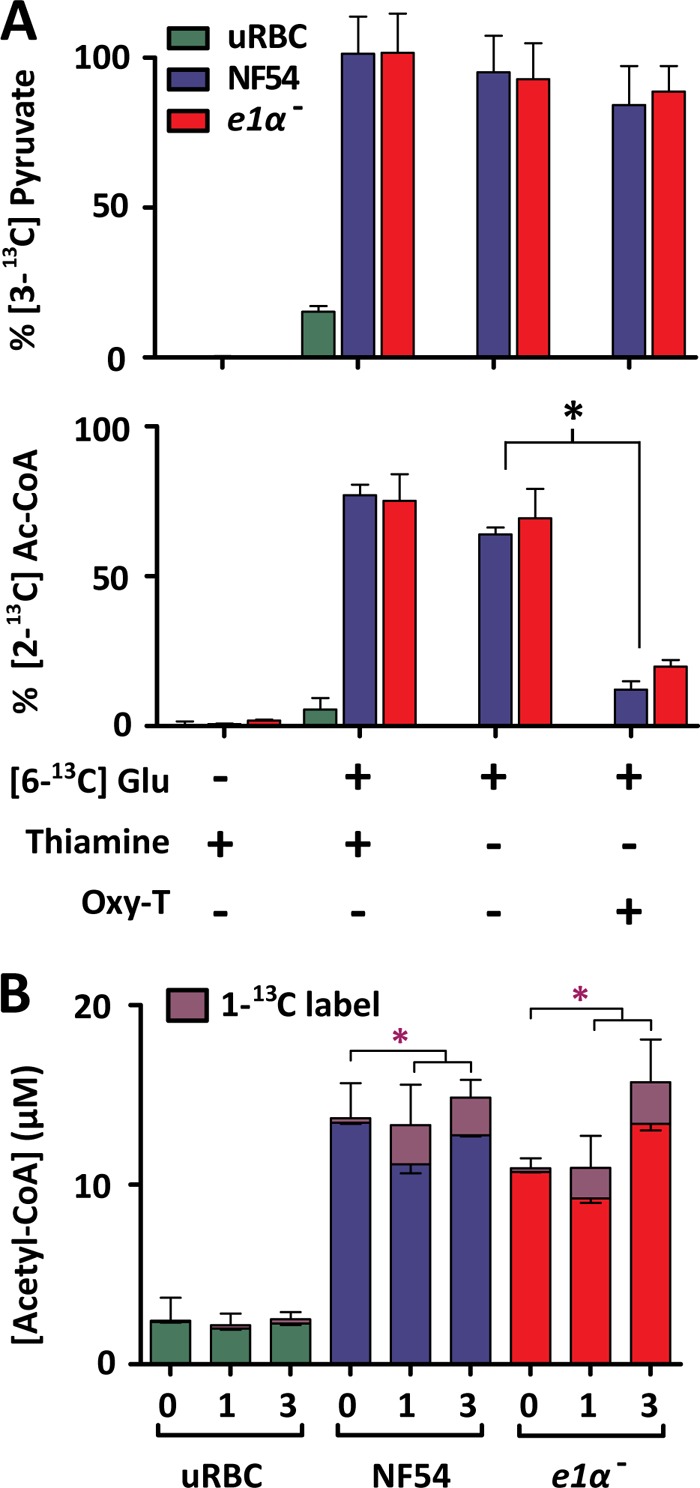
A, the effect of thiamine depletion and oxythiamine exposure on long-term [6-13C]glucose labeling of P. falciparum-infected and uninfected erythrocytes (uRBC). NF54 and pdh e1α- parasite strains were grown under standard culturing conditions for 48 h in standard RPMI, thiamine-free RPMI, and thiamine-free RPMI with 200 μm oxythiamine. Enriched infected erythrocytes were then incubated for 3 h in equivalent media containing 1:1 [6-12C]glucose:[6-13C]glucose. The top panel represents the fractional isotope labeling of pyruvate, and the bottom panel represents acetyl-CoA isotope labeling. Signals were corrected for natural abundance and maximal theoretical enrichment for equal 12C:13C labeling. The percentages of 3-13C or 2-13C label (pyruvate and acetyl-CoA, respectively) of the total metabolite pools are presented for each condition as the mean ± S.E. from n = 3. B, 3 h [1-13C]pyruvate labeling of P. falciparum-infected erythrocytes. Enriched P. falciparum-infected and uninfected erythrocyte suspensions were added to RPMI containing 10 mm [1-13C]pyruvate in a 1:1 ratio (or standard RPMI for time = 0) giving a final concentration of 5 mm [1-13C]pyruvate. Acetyl-CoA labeling was measured by LC-MS presented as the percent of 1-13C label of the total acetyl-CoA pool (corrected for natural abundance). The purple asterisk denotes statistical significance of [1-13C]acetyl-CoA between time = 0 and 1 and 2 h of [1-13C]pyruvate incubation. Data are presented as the mean ± S.E. from n = 3 experiments.
Further experiments were carried out to determine whether pyruvate, the substrate utilized by PDH for acetyl-CoA production, was still converted to acetyl-CoA when the PDH complex was disrupted. Both NF54 and pdh e1α–infected erythrocytes were able to incorporate [1-13C]pyruvate into [1-13C]acetyl-CoA (Fig. 3B, in the presence of 11 mm unlabeled glucose) at equivalent rates, 14 ± 1 and 15 ± 2% of the total acetyl-CoA pool after 3 h (NF54 and pdh e1α-, respectively). The labeled acetyl-CoA pool was attributed to the parasite itself as opposed to the host cells, as evidenced by the smaller acetyl-CoA pool and absence of labeling via [1-13C]pyruvate in uninfected erythrocytes (Fig. 3B). The utilization of pyruvate for acetyl-CoA production in pdh e1α- parasites indicates the presence of an alternate oxythiamine-sensitive PDH-like reaction in the parasite.
The Orphan BCKDH Complex and Its Putative Role in Acetyl-CoA Synthesis
In light of the role of transketolase in the pentose phosphate pathway and KDH in the TCA cycle (supplemental Fig. S5), we examined the role of the sole remaining thiamine pyrophosphate-dependent enzyme BCKDH during intraerythrocytic development. The BCKDH complex is expressed during the asexual blood stage and predicted to localize to the mitochondrion (38). BCKDH usually participates in the multienzyme branched-chain amino acid degradation pathway.
To investigate whether the parasite possesses a functional branched-chain amino acid degradation pathway, P. falciparum-infected and uninfected erythrocytes were incubated with 1 mm [6-13C,15N]leucine over multiple time points and analyzed via LC-MS. As illustrated in Table 2, the percent 2-13C label of acetyl-CoA was unchanged over a 2-h period, remaining at natural abundance levels throughout. The first step in the branched-chain amino acid degradation pathway is the conversion of leucine into methyl-3-oxo-valerate via the branched-chain aminotransferase enzyme. The reaction involves the transfer of the amine group from leucine to α-ketoglutarate, forming glutamate. [15N]Glutamate formation was also monitored and did not change over the time course measured (Table 2). Last, the intermediates immediately upstream and downstream of BCKDH (methyl-3-oxo-valerate and isovaleryl-CoA, respectively) were determined to be below the limit-of-detection (intracellular concentration of 34 ± 12 and 215 ± 23 nm, respectively). These results indicate that the parasite does not possess a functional branched-chain amino acid degradation pathway, leaving BCKDH without a recognized function. Considering BCKDH mediates a similar reaction to PDH (including the cofactors required) and previous work in archaea and mammals indicates that BCKDH can convert pyruvate to acetyl-CoA (39–42), we propose that BCKDH is responsible for the PDH-like activity observed.
TABLE 2.
Percent labeling into the branched-chain amino acid degradation pathway
[15N ,6-13C]Leucine labeling indicates an absence of branched-chain amino acid degradation in P. falciparum-infected erythrocytes. Detection of intermediates of the branched-chain amino acid degradation was monitored via LC-MS, and the proportion of isotopic species detected is presented as % of total pool. Data are presented as the mean ± S.E. from n = 3. [15N]Glutamate percent labeling was monitored as an indicator of the transamination reaction immediately upstream of BCKDH. [2-13C]Acetyl-CoA percent labeling was monitored as an indicator of BCKDH activity in P. falciparum-infected erythrocytes.
| Metabolite | Natural abundance | NF54 | pdh e1α- | ||||
|---|---|---|---|---|---|---|---|
| h | h | ||||||
| 0.5 | 1 | 2 | 0.5 | 1 | 2 | ||
| [15N]Glutamate | 0.36 | 0.36 ± 0.05 | 0.56 ± 0.04 | 0.68 ± 0.02 | 0.36 ± 0.03 | 0.38 ± 0.06 | 0.63 ± 0.03 |
| [2-13C]Acetyl-CoA | 2.40 | 3.3 ± 0.3 | 3.0 ± 0.1 | 3.7 ± 0.14 | 2.7 ± 0.1 | 2.0 ± 0.4 | 2.4 ± 0.9 |
Acetate Flux through acetyl-CoA Synthetase Is Rapid and a Major Contributor to Acetyl-CoA Metabolism in P. falciparum
The incomplete labeling of acetyl-CoA from glucose via the PDH-like pathway (Fig. 1, A and B) may be due to slow acetyl-CoA utilization or the result of another unlabeled carbon source contributing to acetyl-CoA generation. The parasite possesses an acetyl-CoA synthetase (ACS; PFF1350c), which bears the most similarity to members of the AMP-forming subgroup of the acetyl-CoA synthetase family. This subgroup mediates the synthesis of acetyl-CoA from acetate and ATP via the formation of an acetyl-AMP intermediate. To test whether this single enzyme reaction contributes to acetyl-CoA metabolism, rapid labeling of infected and uninfected erythrocytes with [2-13C]acetate (1 mm) was performed (in the presence of 11 mm unlabeled glucose). 72 ± 3% of the acetyl-CoA pool became 2-13C-labeled within 30 s, reaching 82 ± 1% after 1 h (Fig. 4A). Labeled acetyl-CoA was actively metabolized, with the 13C-labeled acetyl group being incorporated into acetylated glutamate and acetylated alanine but not into the TCA cycle intermediate α-ketoglutarate (or any other TCA cycle/glycolytic intermediate). Importantly, the conversion of [2-13C]acetate into [2-13C]acetyl-CoA only occurred in infected erythrocytes, whereas uninfected erythrocytes possessed minimal [2-13C]acetyl-CoA labeling (Fig. 4B).
FIGURE 4.
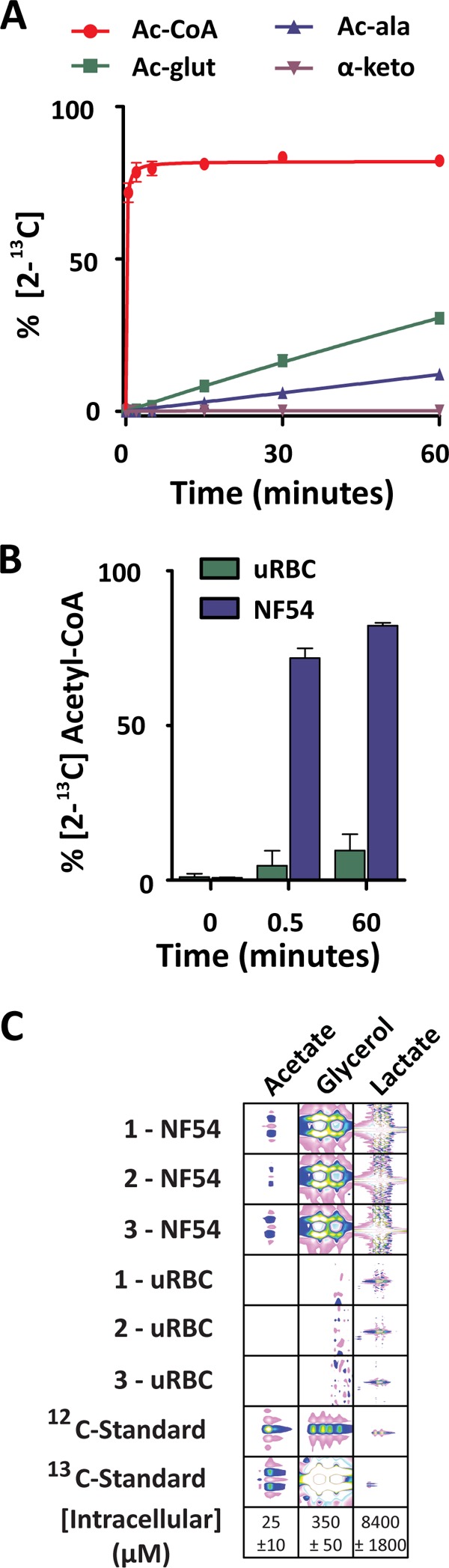
Acetyl-CoA and downstream metabolite labeling via [2-13C]acetate. Enriched infected erythrocyte and uninfected erythrocyte suspensions were added 1:1 with RPMI containing 2 mm acetate (pH adjusted to 7.4), giving a final concentration of 1 mm. Rapid metabolite extraction was performed, and the natural abundance-corrected percent labeling into acetyl-CoA and downstream acetyl-metabolites in NF54 (A) and acetyl-CoA in uninfected erythrocytes (uRBC) and NF54 (B) is shown. Data are presented as the mean ± S.E. from n = 4. C, two-dimension 13C,1H NMR identification and quantification of non-ionizing metabolites from enriched P. falciparum-infected erythrocytes (NF54) and uninfected erythrocytes after a 2-h incubation in RPMI containing [6-13C]glucose. 12C and 13C standards were run in a dilution series to provide identification and quantification. Unlabeled acetate and glycerol were not detectable. The data are presented from three individual experiments collected on separate days as signal intensity. Quantification is presented as intracellular concentration mean ± S.E. Peak intensity is presented using the following colors; pink = negative, blue-white = increasingly positive.
Although the parasite has the capacity to convert acetate into acetyl-CoA, how the intracellular acetate pool (600 μm as estimated by Teng et al. (43)) is maintained remains poorly understood. Acetate is usually found at low concentrations in human plasma (30 μm) (44) but is absent from RPMI. We used two-dimension 13C,1H NMR spectroscopy to investigate [6-13C]glucose labeling of glucose-derived metabolites in trophozoite-stage P. falciparum-infected erythrocytes (Fig. 4C and supplemental Fig. S6). Carbon coupling indicated measurable 13C-labeling of acetate and glycerol pools in infected erythrocytes. The quantification of each metabolite indicated that although glycerol labeling was substantial (350 ± 50 μm), the amount of acetate 13C-labeled was only 25 ± 10 μm, and the unlabeled pool of acetate was below the limit of detection (12C = 200 μm; 13C = 2 μm), preventing any assessment of the fraction of the total acetate pool labeled. Nonetheless, these data indicate that parasites possess a dynamic intracellular pool of acetate, which is partially derived from glucose and can be utilized for acetyl-CoA generation.
Glucose-derived Carbon Flux Shifts Away from the TCA Cycle and into the Pentose Phosphate Pathway during Asexual Development
The evidence presented indicates acetyl-CoA metabolism in the parasite is mediated by ACS and a PDH-like enzyme. We also found that although [6-13C]glucose was incorporated into TCA cycle intermediates, [2-13C]acetate incorporation into the TCA cycle was not detectable (Figs. 1 and 4). We, therefore, asked how different carbon sources contribute to the TCA cycle across different stages of the intraerythrocytic developmental cycle (IDC) (Fig. 5A). Infected erythrocyte cultures were incubated for 2 h in either RPMI containing [6-13C]glucose or [5-13C]glutamine (at 12, 26, and 40 h post invasion) and extracted at 14, 28, and 42 h post invasion (Fig. 5, B and C). Cultures were saponin-isolated before extraction to obtain pure parasite extracts. This technique modifies the intracellular metabolite pool through efflux during the wash procedure (43); however, the percent label is internally controlled because different isotopes of the same metabolite will efflux at the same rate, maintaining the isotope ratios.
The 6-13C-labeling of glucose 6-phosphate (the first step of glycolysis) was unchanged throughout the IDC (Fig. 5B), indicating that changes downstream were a consequence of altered metabolism and not differences in glucose transport during parasite development. The flux through the pentose phosphate pathway progressively increased during parasite development, as indicated by a significant increase in M+5 ribose phosphate (Fig. 5B). Ribose incorporation into AMP followed a similar trend (M+5). The extent of glucose labeling into acetyl-CoA (M+2, acetyl-group labeling; M+7, acetyl-group plus ribose-group labeling) did not change significantly during parasite development, consistent with the stage-independent turnover of pyruvate (Fig. 5B). As previously reported, acetyl-CoA incorporates into the TCA cycle, which combined with oxaloacetate produces citrate (Fig. 1B) (14). Although oxaloacetate is not detectable by the methods used, the immediate downstream products malate and aspartate were used as proxies. The percent isotope labeling of malate and aspartate after 2 h of incubation with [6-13C]glucose at 28 h post invasion (HPI) was 26 ± 1 and 27 ± 3%, respectively (for NF54). The extent of 13C-labeling of the malate and aspartate pools was stage-dependent, decreasing to only 6 ± 1 and 6 ± 2% of the total pool at 42 HPI, respectively (p < 0.05). The reduction in the M+5 species of citrate (derived from M+2 acetyl-CoA and M+3 oxaloacetate) and the comparable increase in M+2 species of citrate were consistent with the stage-dependent 3-13C labeling of oxaloacetate via [6-13C]glucose. The significant decrease in M+2 (derived from either acetyl-CoA or oxaloacetate) and M+4 succinate confirmed a stage-dependent incorporation of glucose into the TCA cycle via the anapleurotic phosphoenolpyruvate carboxylase (PEPC)/phosphoenolpyruvate carboxykinase (PEPCK) route (Fig. 5B).
The parasite possesses both a PEPC (PF14_0246) and PEPCK (PF13_0234)). These enzymes mediate the forward and backward reaction of phosphoenolpyruvate to oxaloacetate, respectively. Parasites do not possess pyruvate carboxylase (metabolizing pyruvate to oxaloacetate). Previous transcriptional analysis of PEPC/PEPCK suggests that these reactions are used at non-coincidental times during parasite development (45).
To further investigate how the parasite alters carbon utilization during development, [5-13C]glutamine labeling was conducted in an identical manner as previously described for glucose (Fig. 5C). A progressive increase of labeling in downstream intermediates and incorporation into the TCA cycle was observed; however, the increased glutamine uptake indicates that this result is partially due to altered transport properties of the mature parasite. Nonetheless, the increased glutamine commitment to the TCA cycle was commensurate to the decrease in anapleurotic glucose incorporation at 42 HPI.
Our data suggest that the two pathways for glucose-derived carbon entering the TCA cycle differ in their contributions during parasite development. The labeling of the acetyl group in acetyl-CoA via the PDH-like reaction is constant throughout blood stage parasite development. In contrast, the anapleurotic oxaloacetate synthesis via the PEPC/PEPCK reaction is stage-dependent and decreases during trophozoite-schizont development.
DISCUSSION
The apicoplast localization of PDH has presented a conundrum for parasite biology as it provides no rationale for how the acetyl-CoA pool is maintained outside of this organelle. Our analysis of PDH-deficient parasites demonstrates that the PDH does not appreciably contribute to the bulk acetyl-CoA pool, TCA metabolism (Fig. 1), fatty acid profiles (Fig. 2), or viability of blood stage parasites (supplemental Fig. S4). In short, PDH-deficient parasites have no detectable phenotypes until the mosquito stage of the parasite lifecycle. Moreover, we provide evidence that two enzymes, a PDH-like enzyme (which we propose is the BCKDH) and ACS, are responsible for the metabolic activities formerly ascribed to the PDH. Our data indicate that the PDH-like enzyme supplies acetyl-CoA to the TCA cycle, whereas ACS contributes to amino acid acetylation reactions (Fig. 6).
FIGURE 6.

Schematic of the proposed metabolic architecture of P. falciparum. Pertinent enzymes are highlighted in red. Metabolite nomenclature is as follows: PEP, phosphoenolpyruvate; Pyr, pyruvate; Oxa, oxaloacetate; Mal, malate; Cit, citrate; Iso, isocitrate; α-Kg, α-ketoglutarate; Suc-CoA, succinyl-CoA; Suc, succinate; Fum, fumarate; Gln, glutamine; Glu, glutamate; GABA, γ-aminobutyric acid. The gene IDs of the relevant metabolic enzymes are as follows; PEPC (PF14_0246), PEPCK (PF13_0234), citrate synthase I (CS-I, PF10_0218), citrate synthase II (CS-II, PFF0455w), ACS (PFF1350c), histone deacetylases (KDAC, PFI1260c, PF14_069, PF10_0078), BCKDH E1α (PF13_0070), BCKDH E1β (PFE0225w), BCKDH E2 (PFC0170c), BCKDH E3 (PFL1550w), PDH E1α (PF11_0256), PDH E1β (PF14_0441), PDH E2 (PF10_0407), PDH E3 (PF08_0066), fatty acid biosynthesis type II (FAS II).
The question, therefore, remains, What is the function of PDH during the blood stage? The absence of a change in the fatty acid profile (Fig. 2) and growth phenotype in pdh e1α- (supplemental Fig. S4) suggests that blood-stage parasites can meet their fatty acid requirements using exogenous sources (e.g. serum). Recent work has shown that type II fatty acid metabolism is functional in blood-stage parasites grown under minimal fatty acid conditions (C16:0 and C18:1 alone) (12). Given this finding, PDH may serve a more important role under starvation conditions. This interpretation could explain why PDH and FAS II enzymes are expressed during the blood stage despite their apparent dispensability in rich media (21, 22).
Despite its equivocal function in blood stages, we show that PDH is essential for oocyst sporozoite development in P. falciparum (Table 1). This finding disagrees with similar analyses conducted with the mouse malaria parasite P. yoelii, where PDH is only essential for complete liver-stage development (20). This result underscores the fact that the metabolic architecture and nutritional requirement of a rodent malaria species does not necessarily predict that of a human malaria species. Because Pf pdh e1α- parasites arrest at the mosquito stage, we were unable to determine if PDH is also essential for P. falciparum liver-stage development.
To gain a more comprehensive understanding of parasite acetyl-CoA metabolism, rapid labeling was used to measure the kinetic flux through glycolysis (supplemental Table S1). We demonstrated that there were no dynamic (Fig. 1A), steady-state (Fig. 1B), or developmental-specific (Fig. 5) metabolic phenotypes when the PDH subunit E1α was disrupted. These findings, in conjunction with our pyruvate labeling data, show that another enzyme must be supplying the acetyl-CoA pool. This activity can be inhibited by oxythiamine (Fig. 3) suggesting that the unknown enzyme uses thiamine as a cofactor. At present, there are only four established thiamine-dependent enzymes expressed in blood stage parasites: 1) PDH, 2) transketolase, 3) keto-dehydrogenase, and 4) BCKDH. Because we have knocked out PDH and both transketolase and keto-dehydrogenase have clear functions in the pentose phosphate pathway and TCA cycle (supplemental Fig. S5, C and D), the only remaining known oxythiamine-sensitive enzyme is BCKDH.
Although the complete BCKDH complex is expressed during asexual development and predicted to be localized to the mitochondrion (38), we find that there is no detectable BCAA degradation (Table 2). Conversion of pyruvate into acetyl-CoA by the BCKDH would simplify the mitochondrial carbon requirement, as BCKDH could provide acetyl-CoA directly to the TCA cycle without necessitating the transport of acetyl-CoA out of the apicoplast (Fig. 6). It has previously been reported that the parasite does not possess any of the remaining genes in the branched-chain amino acid degradation pathway (2, 38, 46, 47), leaving the function of the enzyme complex an open question. Considering the BCKDH utilizes the same cofactors and mediates a stepwise reaction comparable to PDH, it is plausible that BCKDH is responsible for the PDH-like activity observed. This hypothesis is supported by the observation that the BCKDH of other organisms can convert pyruvate to acetyl-CoA (39–42).
This study indicates that BCKDH may not be the only significant contributor to the parasite acetyl-CoA pool. P. falciparum has a putative ACS gene, and our [13C]acetate labeling data show that acetyl-CoA can be actively synthesized from this precursor (Fig. 4, A and B). It remains unclear how the intracellular pool of acetate is maintained, but here we provide evidence it is partially derived from glucose (Fig. 4C). The isotope-labeled pool we observed may have resulted from the bidirectional activity of ACS, as previously reported by Yoshii et al. (48, 49) or through an indirect mechanism such as histone deacetylation (Fig. 6). These two mechanisms are the most likely, as there is no evidence for an acetyl-CoA hydrolase in the P. falciparum genome (50). The half-life for histone acetylation can be on the order of minutes, which may well contribute to the [2-13C]acetate detected in Fig. 4C (51). Acetylation of enzymes is also an important means to regulate cellular metabolism, and therefore, the intracellular acetate/acetyl-CoA balance is crucial to development and viability (16, 52–54). Although it is unlikely that ACS is a significant contributor to total acetyl-CoA biosynthesis, its role in regulating this sensitive balance may prove to be essential.
In addition to elucidating acetyl-CoA metabolism, we observed significant developmental changes in parasite carbon metabolism. Most dramatically, glucose-derived carbon entering the TCA cycle is dramatically diminished between 28 and 42 HPI (Fig. 5B). This stage dependence was only via the anapleurotic oxaloacetate-forming PEPC reaction, whereas acetyl-CoA formation and commitment to the TCA cycle was unchanged throughout the IDC. Diminished anapleurotic contributions from glucose were offset by increased reliance on [5-13C]glutamine as a TCA carbon source (Fig. 5C). The reduction in [13C]glucose labeling into aspartate and TCA cycle intermediates between 28 and 42 HPI is likely controlled by the PEPC/PEPCK conversion of phosphoenolpyruvate to oxaloacetate (as pyruvate carboxylase is absent from the genome, Fig. 5). These findings indicate that the parasite restructures metabolic flux to meet the changing environment during intraerythrocytic development. As the parasite progressively requires glucose-derived carbon for other uses (nucleotide/membrane synthesis) (55–57) and glutamine becomes increasingly abundant (as more hemoglobin is catabolized into free amino acids and via exogenous import), the source of carbon for TCA anapleurosis is switched.
In summary, we have demonstrated that 1) PDH is neither essential nor a substantial contributor to acetyl-CoA metabolism of blood stage parasites, 2) that parasites have two independent mechanisms for synthesizing acetyl-CoA and that these capacities are likely attributable to BCKDH and ACS, and 3) parasites undergo significant stage-dependent metabolic alterations over the course of their intraerythrocytic developmental cycle. These findings explain how P. falciparum circumvents the paradoxical apicoplast localization of PDH and may inform future drug development efforts.
Acknowledgments
We thank Kiaran Kirk for his thoughtful comments on the manuscript, the insectary staff at Seattle BioMed for dedication to gametocyte production and mosquito rearing, and the Red Cross Blood Service (Melbourne, Australia) for erythrocytes.
This work was supported, in whole or in part, by National Institutes of Health Director's New Innovators Award 1DP2OD001315-01) (to M. L.) and Grants R56 AI080685 (to S. H. I. K.) and P50 GM071508 (Center for Quantitative Biology; to M. L.). This work was also supported through generous support from the Burroughs Welcome Fund (to M. L.) and National Health and Medical Research Council (NHMRC) of Australia and Victorian State Government Operational Infrastructure Support and Australian Government NHMRC Independent Research Institutes Infrastructure Support Scheme (to A. F. C.).

This article contains supplemental Methods, Tables S1–S3, and Figs. S1–S6.
- PDH
- pyruvate dehydrogenase
- BCKDH
- branched-chain keto acid dehydrogenase complex
- KDH
- ketoglutarate dehydrogenase
- ACS
- acetyl-CoA synthetase
- IDC
- intraerythrocytic developmental cycle
- HPI
- hours post invasion
- PEPC
- phosphoenolpyruvate carboxylase
- PEPCK
- phosphoenolpyruvate carboxykinase.
REFERENCES
- 1. Olszewski K. L., Llinás M. (2011) Central carbon metabolism of Plasmodium parasites. Mol. Biochem. Parasitol. 175, 95–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Polonais V., Soldati-Favre D. (2010) Versatility in the acquisition of energy and carbon sources by the Apicomplexa. Biol. Cell 102, 435–445 [DOI] [PubMed] [Google Scholar]
- 3. Mogi T., Kita K. (2010) Diversity in mitochondrial metabolic pathways in parasitic protists Plasmodium and Cryptosporidium. Parasitol. Int. 59, 305–312 [DOI] [PubMed] [Google Scholar]
- 4. Mehta M., Sonawat H. M., Sharma S. (2006) Glycolysis in Plasmodium falciparum results in modulation of host enzyme activities. J. Vector Borne Dis. 43, 95–103 [PubMed] [Google Scholar]
- 5. Roth E. F., Jr., Raventos-Suarez C., Perkins M., Nagel R. L. (1982) Glutathione stability and oxidative stress in P. falciparum infection in vitro. Responses of normal and G6PD-deficient cells. Biochem. Biophys. Res. Commun. 109, 355–362 [DOI] [PubMed] [Google Scholar]
- 6. Zolg J. W., Macleod A. J., Scaife J. G., Beaudoin R. L. (1984) The accumulation of lactic acid and its influence on the growth of Plasmodium falciparum in synchronized cultures. In Vitro 20, 205–215 [DOI] [PubMed] [Google Scholar]
- 7. Cranmer S. L., Conant A. R., Gutteridge W. E., Halestrap A. P. (1995) Characterization of the enhanced transport of l- and d-lactate into human red blood cells infected with Plasmodium falciparum suggests the presence of a novel saturable lactate proton cotransporter. J. Biol. Chem. 270, 15045–15052 [DOI] [PubMed] [Google Scholar]
- 8. Elliott J. L., Saliba K. J., Kirk K. (2001) Transport of lactate and pyruvate in the intraerythrocytic malaria parasite, Plasmodium falciparum. Biochem. J. 355, 733–739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kanaani J., Ginsburg H. (1991) Transport of lactate in Plasmodium falciparum-infected human erythrocytes. J. Cell Physiol. 149, 469–476 [DOI] [PubMed] [Google Scholar]
- 10. Pfaller M. A., Krogstad D. J., Parquette A. R., Nguyen-Dinh P. (1982) Plasmodium falciparum. Stage-specific lactate production in synchronized cultures. Exp. Parasitol. 54, 391–396 [DOI] [PubMed] [Google Scholar]
- 11. Vander Jagt D. L., Hunsaker L. A., Campos N. M., Baack B. R. (1990) d-Lactate production in erythrocytes infected with Plasmodium falciparum. Mol. Biochem. Parasitol. 42, 277–284 [DOI] [PubMed] [Google Scholar]
- 12. Botté C. Y., Yamaryo-Botté Y., Rupasinghe T. W., Mullin K. A., MacRae J. I., Spurck T. P., Kalanon M., Shears M. J., Coppel R. L., Crellin P. K., Maréchal E., McConville M. J., McFadden G. I. (2013) Atypical lipid composition in the purified relict plastid (apicoplast) of malaria parasites. Proc. Natl. Acad. Sci. U.S.A. 110, 7506–7511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cui L., Miao J. (2010) Chromatin-mediated epigenetic regulation in the malaria parasite Plasmodium falciparum. Eukaryot. Cell 9, 1138–1149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. MacRae J. I., Dixon M. W., Dearnley M. K., Chua H. H., Chambers J. M., Kenny S., Bottova I., Tilley L., McConville M. J. (2013) Mitochondrial metabolism of sexual and asexual blood stages of the malaria parasite Plasmodium falciparum. BMC Biol. 11, 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fritzler J. M., Millership J. J., Zhu G. (2007) Cryptosporidium parvum long-chain fatty acid elongase. Eukaryot. Cell 6, 2018–2028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Miao J., Lawrence M., Jeffers V., Zhao F., Parker D., Ge Y., Sullivan W. J., Jr., Cui L. (2013) Extensive lysine acetylation occurs in evolutionarily conserved metabolic pathways and parasite-specific functions during Plasmodium falciparum intraerythrocytic development. Mol. Microbiol. 89, 660–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Foth B. J., Stimmler L. M., Handman E., Crabb B. S., Hodder A. N., McFadden G. I. (2005) The malaria parasite Plasmodium falciparum has only one pyruvate dehydrogenase complex, which is located in the apicoplast. Mol. Microbiol. 55, 39–53 [DOI] [PubMed] [Google Scholar]
- 18. Ralph S. A. (2005) Strange organelles. Plasmodium mitochondria lack a pyruvate dehydrogenase complex. Mol. Microbiol. 55, 1–4 [DOI] [PubMed] [Google Scholar]
- 19. Zhou Z. H., McCarthy D. B., O'Connor C. M., Reed L. J., Stoops J. K. (2001) The remarkable structural and functional organization of the eukaryotic pyruvate dehydrogenase complexes. Proc. Natl. Acad. Sci. U.S.A. 98, 14802–14807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pei Y., Tarun A. S., Vaughan A. M., Herman R. W., Soliman J. M., Erickson-Wayman A., Kappe S. H. (2010) Plasmodium pyruvate dehydrogenase activity is only essential for the parasite's progression from liver infection to blood infection. Mol. Microbiol. 75, 957–971 [DOI] [PubMed] [Google Scholar]
- 21. Vaughan A. M., O'Neill M. T., Tarun A. S., Camargo N., Phuong T. M., Aly A. S., Cowman A. F., Kappe S. H. (2009) Type II fatty acid synthesis is essential only for malaria parasite late liver stage development. Cell Microbiol. 11, 506–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yu M., Kumar T. R., Nkrumah L. J., Coppi A., Retzlaff S., Li C. D., Kelly B. J., Moura P. A., Lakshmanan V., Freundlich J. S., Valderramos J. C., Vilcheze C., Siedner M., Tsai J. H., Falkard B., Sidhu A. B., Purcell L. A., Gratraud P., Kremer L., Waters A. P., Schiehser G., Jacobus D. P., Janse C. J., Ager A., Jacobs W. R., Jr., Sacchettini J. C., Heussler V., Sinnis P., Fidock D. A. (2008) The fatty acid biosynthesis enzyme FabI plays a key role in the development of liver-stage malarial parasites. Cell Host Microbe 4, 567–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lambros C., Vanderberg J. P. (1979) Synchronization of Plasmodium falciparum erythrocytic stages in culture. J. Parasitol. 65, 418–420 [PubMed] [Google Scholar]
- 24. Trager W., Jensen J. B. (1976) Human malaria parasites in continuous culture. Science 193, 673–675 [DOI] [PubMed] [Google Scholar]
- 25. Olszewski K. L., Llinás M. (2013) Extraction of hydrophilic metabolites from Plasmodium falciparum-infected erythrocytes for metabolomic analysis. Methods Mol. Biol. 923, 259–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cobbold S. A., Martin R. E., Kirk K. (2011) Methionine transport in the malaria parasite Plasmodium falciparum. Int. J. Parasitol. 41, 125–135 [DOI] [PubMed] [Google Scholar]
- 27. Kamphorst J. J., Fan J., Lu W., White E., Rabinowitz J. D. (2011) Liquid chromatography-high resolution mass spectrometry analysis of fatty acid metabolism. Anal. Chem. 83, 9114–9122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Xu Y. F., Létisse F., Absalan F., Lu W., Kuznetsova E., Brown G., Caudy A. A., Yakunin A. F., Broach J. R., Rabinowitz J. D. (2013) Nucleotide degradation and ribose salvage in yeast. Mol. Syst. Biol. 9, 665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lewis I. A., Schommer S. C., Markley J. L. (2009) rNMR. Open source software for identifying and quantifying metabolites in NMR spectra. Magn. Reson. Chem. 47, S123–S126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lewis I. A., Schommer S. C., Hodis B., Robb K. A., Tonelli M., Westler W. M., Sussman M. R., Markley J. L. (2007) Method for determining molar concentrations of metabolites in complex solutions from two-dimensional 1H-13C NMR spectra. Anal. Chem. 79, 9385–9390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Keller A., Eng J., Zhang N., Li X. J., Aebersold R. (2005) A uniform proteomics MS/MS analysis platform utilizing open XML file formats. Mol. Syst. Biol. 1, 0017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Clasquin M. F., Melamud E., Rabinowitz J. D. (2012) LC-MS data processing with MAVEN. A metabolomic analysis and visualization engine. Curr. Protoc. Bioinformatics, Chapter 14, Unit 14.11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bennett B. D., Yuan J., Kimball E. H., Rabinowitz J. D. (2008) Absolute quantitation of intracellular metabolite concentrations by an isotope ratio-based approach. Nat. Protoc. 3, 1299–1311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Saliba K. J., Horner H. A., Kirk K. (1998) Transport and metabolism of the essential vitamin pantothenic acid in human erythrocytes infected with the malaria parasite Plasmodium falciparum. J. Biol. Chem. 273, 10190–10195 [DOI] [PubMed] [Google Scholar]
- 35. Amador-Noguez D., Brasg I. A., Feng X. J., Roquet N., Rabinowitz J. D. (2011) Metabolome remodeling during the acidogenic-solventogenic transition in Clostridium acetobutylicum. Appl. Environ. Microbiol. 77, 7984–7997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Xu Y. F., Amador-Noguez D., Reaves M. L., Feng X. J., Rabinowitz J. D. (2012) Ultrasensitive regulation of anapleurosis via allosteric activation of PEP carboxylase. Nat. Chem. Biol. 8, 562–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Beier J. C., Vanderberg J. P. (1998) in Malaria: Parasite Biology, Pathogenesis, and Protection (Sherman I. ed.) pp. 49–61, American Society for Microbiology, Washington, D. C [Google Scholar]
- 38. Seeber F., Limenitakis J., Soldati-Favre D. (2008) Apicomplexan mitochondrial metabolism. A story of gains, losses, and retentions. Trends Parasitol. 24, 468–478 [DOI] [PubMed] [Google Scholar]
- 39. Heath C., Posner M. G., Aass H. C., Upadhyay A., Scott D. J., Hough D. W., Danson M. J. (2007) The 2-oxoacid dehydrogenase multienzyme complex of the archaeon Thermoplasma acidophilum. Recombinant expression, assembly, and characterization. FEBS J. 274, 5406–5415 [DOI] [PubMed] [Google Scholar]
- 40. Paxton R., Harris R. A. (1982) Isolation of rabbit liver branched chain α-ketoacid dehydrogenase and regulation by phosphorylation. J. Biol. Chem. 257, 14433–14439 [PubMed] [Google Scholar]
- 41. Paxton R., Scislowski P. W., Davis E. J., Harris R. A. (1986) Role of branched-chain 2-oxo acid dehydrogenase and pyruvate dehydrogenase in 2-oxobutyrate metabolism. Biochem. J. 234, 295–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pettit F. H., Yeaman S. J., Reed L. J. (1978) Purification and characterization of branched chain α-keto acid dehydrogenase complex of bovine kidney. Proc. Natl. Acad. Sci. U.S.A. 75, 4881–4885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Teng R., Junankar P. R., Bubb W. A., Rae C., Mercier P., Kirk K. (2009) Metabolite profiling of the intraerythrocytic malaria parasite Plasmodium falciparum by 1H NMR spectroscopy. NMR Biomed. 22, 292–302 [DOI] [PubMed] [Google Scholar]
- 44. Lentner C. (1992) Geigy Scientific Tables (Lentner C., ed.) 8th Revised Ed., vol. 1, pp. 167–177, Basel, Switzerland [Google Scholar]
- 45. Bozdech Z., Llinás M., Pulliam B. L., Wong E. D., Zhu J., DeRisi J. L. (2003) The transcriptome of the intraerythrocytic developmental cycle of Plasmodium falciparum. PLoS Biol. 1, E5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gardner M. J., Hall N., Fung E., White O., Berriman M., Hyman R. W., Carlton J. M., Pain A., Nelson K. E., Bowman S., Paulsen I. T., James K., Eisen J. A., Rutherford K., Salzberg S. L., Craig A., Kyes S., Chan M. S., Nene V., Shallom S. J., Suh B., Peterson J., Angiuoli S., Pertea M., Allen J., Selengut J., Haft D., Mather M. W., Vaidya A. B., Martin D. M., Fairlamb A. H., Fraunholz M. J., Roos D. S., Ralph S. A., McFadden G. I., Cummings L. M., Subramanian G. M., Mungall C., Venter J. C., Carucci D. J., Hoffman S. L., Newbold C., Davis R. W., Fraser C. M., Barrell B. (2002) Genome sequence of the human malaria parasite Plasmodium falciparum. Nature 419, 498–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ginsburg H. (2006) Progress in in silico functional genomics. The malaria metabolic pathways database. Trends Parasitol. 22, 238–240 [DOI] [PubMed] [Google Scholar]
- 48. Yoshii Y., Waki A., Furukawa T., Kiyono Y., Mori T., Yoshii H., Kudo T., Okazawa H., Welch M. J., Fujibayashi Y. (2009) Tumor uptake of radiolabeled acetate reflects the expression of cytosolic acetyl-CoA synthetase. Implications for the mechanism of acetate PET. Nucl. Med. Biol. 36, 771–777 [DOI] [PubMed] [Google Scholar]
- 49. Yoshii Y., Furukawa T., Yoshii H., Mori T., Kiyono Y., Waki A., Kobayashi M., Tsujikawa T., Kudo T., Okazawa H., Yonekura Y., Fujibayashi Y. (2009) Cytosolic acetyl-CoA synthetase affected tumor cell survival under hypoxia. The possible function in tumor acetyl-CoA/acetate metabolism. Cancer Sci. 100, 821–827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tielens A. G., van Grinsven K. W., Henze K., van Hellemond J. J., Martin W. (2010) Acetate formation in the energy metabolism of parasitic helminths and protists. Int. J. Parasitol. 40, 387–397 [DOI] [PubMed] [Google Scholar]
- 51. Waterborg J. H. (2002) Dynamics of histone acetylation in vivo. A function for acetylation turnover? Biochem. Cell Biol. 80, 363–378 [DOI] [PubMed] [Google Scholar]
- 52. Lee S., Son H., Lee J., Min K., Choi G. J., Kim J. C., Lee Y. W. (2011) Functional analyses of two acetyl coenzyme A synthetases in the ascomycete Gibberella zeae. Eukaryot. Cell 10, 1043–1052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Rivière L., Moreau P., Allmann S., Hahn M., Biran M., Plazolles N., Franconi J. M., Boshart M., Bringaud F. (2009) Acetate produced in the mitochondrion is the essential precursor for lipid biosynthesis in procyclic trypanosomes. Proc. Natl. Acad. Sci. U.S.A. 106, 12694-12699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Starai V. J., Escalante-Semerena J. C. (2004) Acetyl-coenzyme A synthetase (AMP forming). Cell. Mol. Life Sci. 61, 2020–2030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Atamna H., Pascarmona G., Ginsburg H. (1994) Hexose-monophosphate shunt activity in intact Plasmodium falciparum-infected erythrocytes and in free parasites. Mol. Biochem. Parasitol. 67, 79–89 [DOI] [PubMed] [Google Scholar]
- 56. Déchamps S., Shastri S., Wengelnik K., Vial H. J. (2010) Glycerophospholipid acquisition in Plasmodium. A puzzling assembly of biosynthetic pathways. Int. J. Parasitol. 40, 1347–1365 [DOI] [PubMed] [Google Scholar]
- 57. Mi-Ichi F., Kita K., Mitamura T. (2006) Intraerythrocytic Plasmodium falciparum utilize a broad range of serum-derived fatty acids with limited modification for their growth. Parasitology 133, 399–410 [DOI] [PubMed] [Google Scholar]