

Background: A large GTPase movement on the signal recognition particle (SRP) RNA activates GTP hydrolysis to complete protein targeting.
Results: GTPase activation requires distinct primary and secondary docking sites and catalytic bases, optimally positioned on the SRP RNA.
Conclusion: The SRP RNA forms a preorganized surface to mediate specific GTPase activation.
Significance: Study of the SRP RNA broadens our understanding of RNA-protein recognition and protein targeting.
Keywords: G Proteins, Nuclear Magnetic Resonance, Protein Targeting, Protein-Nucleic Acid Interaction, RNA, Single Molecule Biophysics
Abstract
The signal recognition particle (SRP) RNA is a universally conserved and essential component of the SRP that mediates the co-translational targeting of proteins to the correct cellular membrane. During the targeting reaction, two functional ends in the SRP RNA mediate distinct functions. Whereas the RNA tetraloop facilitates initial assembly of two GTPases between the SRP and SRP receptor, this GTPase complex subsequently relocalizes ∼100 Å to the 5′,3′-distal end of the RNA, a conformation crucial for GTPase activation and cargo handover. Here we combined biochemical, single molecule, and NMR studies to investigate the molecular mechanism of this large scale conformational change. We show that two independent sites contribute to the interaction of the GTPase complex with the SRP RNA distal end. Loop E plays a crucial role in the precise positioning of the GTPase complex on these two sites by inducing a defined bend in the RNA helix and thus generating a preorganized recognition surface. GTPase docking can be uncoupled from its subsequent activation, which is mediated by conserved bases in the next internal loop. These results, combined with recent structural work, elucidate how the SRP RNA induces GTPase relocalization and activation at the end of the protein targeting reaction.
Introduction
The signal recognition particle (SRP,3 Fig. 1A) is a ribonucleoprotein complex responsible for targeting proteins to their proper membrane destinations (1–3). SRP recognizes the signal sequence emerging from the ribosomal exit tunnel (termed ribosome-nascent chain complex or RNC; Fig. 1A, step 1) (4–8). The RNC-SRP complex is delivered to the membrane through the interaction between the SRP and SRP receptor (SR; Fig. 1A, step 2) (9–12). At the membrane, when the RNC-SRP-SR ternary complex localizes near the SecYEG translocon, RNC dissociates from the SRP-SR complex and is loaded onto SecYEG (Fig. 1A, step 3), where the nascent protein is integrated into or translocated across the membrane (13, 14). The SRP-SR complex then disassembles to enter another round of protein targeting (Fig. 1A, step 4).
FIGURE 1.
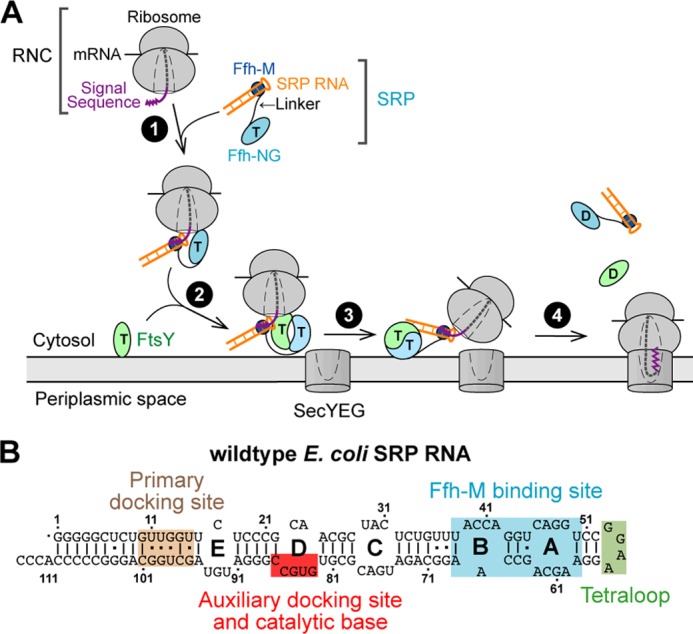
Scheme depicting the function of SRP RNA during co-translational protein targeting. A, working model of co-translational protein targeting by the SRP. B, secondary structure of the E. coli SRP RNA. The four sites on the RNA that mediate different functions are noted with different colors.
In prokaryotic cells, SRP consists of a SRP54 protein (Ffh in bacteria) and a 4.5S SRP RNA (Fig. 1A, SRP). Ffh contains a methionine-rich M domain, which binds the SRP RNA and the signal sequence on the translating ribosome. In addition, an NG domain in Ffh, comprising a GTPase G domain and a four-helix bundle N domain, forms a tight complex with a highly homologous NG domain in the SRP receptor (called FtsY in bacteria) in the presence of GTP (9, 10). GTP hydrolysis at the end of the SRP cycle drives the disassembly of the Ffh-FtsY GTPase complex (15, 16). The assembly of the SRP-FtsY GTPase complex and its GTPase activation require discrete conformational rearrangements in the SRP (see next paragraph) that are regulated by the RNC and the target membrane (17–21), respectively, thus ensuring the spatial and temporal precision of these molecular events during protein targeting.
In addition to the SRP and SRP receptor GTPases, the SRP RNA is the only other universally conserved and essential component of the cytosolic SRP system (Fig. 1B). Extensive previous work showed that the SRP RNA provides an active scaffold that regulates protein-protein interactions and protein conformational rearrangements during the targeting reaction. The bacterial 4.5S SRP RNA adopts a hairpin structure containing five internal loops, capped by a highly conserved GGAA tetraloop (Fig. 1B). Two internal loops, A and B, mediate binding of the SRP RNA to the Ffh M domain with picomolar affinity (22, 23). The tetraloop of the SRP RNA mediates a transient electrostatic interaction with FtsY (24) during its initial recruitment to the SRP, thus accelerating the assembly of the stable NG domain complex between Ffh and FtsY by 102–103 -fold (15, 25, 26). More recently, crystallographic and single molecule analyses showed that GTPase activation requires the Ffh-FtsY GTPase complex to interact with a distal docking site at the 5′,3′-distal end of the SRP RNA (Fig. 1B) (27, 28). Together with biochemical studies, these data demonstrate a global relocalization of the Ffh-FtsY GTPase complex, from the tetraloop end of the SRP RNA during initial complex assembly to the distal end where the GTPase complex hydrolyzes GTP (Fig. 1A, step 3).
The global rearrangement along the SRP RNA is important for multiple reasons. First, it provides temporal regulation of GTP hydrolysis. On the one hand, rapid GTP hydrolysis is desired at the end of the SRP cycle to regenerate free SRP. On the other hand, GTP hydrolysis should be delayed to minimize premature complex disassembly before the RNC is productively delivered to the SecYEG translocation machinery. The spatial separation of the sites on the SRP RNA that mediate GTPase assembly and activation resolves this dilemma: the Ffh-FtsY GTPase complex only migrates to the distal end of the RNA and hydrolyzes GTP at the end of the SRP cycle, after encounter with the SecYEG machinery (17, 28). Second, the global rearrangement of SRP enables coordinated handover of RNC from targeting to the translocation machinery. In the RNC-SRP complex, Ffh binds the L23 protein on the large ribosomal subunit (5, 7, 8). Strikingly, the same site is used by SecYEG to interact with the ribosome (Fig. 1A) (29, 30). Relocalizing the GTPase complex to the distal end of the SRP RNA ensures the productive exchange of the targeting and translocation machineries at the L23 binding site and thus efficient co-translational protein targeting.
Despite the biological importance of the GTPase relocalization on the SRP RNA, its underlying molecular mechanisms remain incompletely understood. First, how the Ffh-FtsY GTPase complex interacts specifically with the distal docking site remains obscure, especially given the low degree of sequence conservation at the distal docking site and the predominantly electrostatic nature of the GTPase interactions with this site observed in the crystal structure (27). Indeed, nonspecific docking of the Ffh-FtsY GTPase complex on the SRP RNA has been observed in single molecule experiments that represent “trial-and-error” searches of the complex for the correct docking site (28). However, little is known about the information that specifies the correct distal docking site. Second, different crystal structures suggest two different bases, G83 and C86, as potential catalytic moieties that could insert into the GTPase active site (27, 48). Third, it is unclear whether activation of GTP hydrolysis is coupled to, or independent of, the GTPase relocalization on the SRP RNA.
In this work, we utilized biochemical, single molecule, and NMR tools to investigate the molecular mechanism of GTPase docking and activation at the SRP RNA distal end. We show that two docking sites mediate the interaction of the Ffh-FtsY GTPase complex with the RNA distal end. A highly conserved internal loop controls the correct orientation and positioning of these two sites and plays crucial roles in generating a preformed surface for stable docking of the GTPase complex. This docking event can be uncoupled from subsequent GTPase activation, which is mediated by conserved bases in the next internal loop. These results, combined with recent structural work, elucidate how the SRP RNA induces GTPase relocalization and activation at the end of the protein targeting reaction.
EXPERIMENTAL PROCEDURES
Plasmids
Plasmids for in vivo expression of Ffh, full-length FtsY, and SRP RNA have been described in previous studies (15). Plasmids for SRP RNA mutants were constructed using the QuikChange mutagenesis protocol (Stratagene) following the manufacturer's instructions. The plasmid for in vitro transcription of Hammerhead-SRP RNA-HDV was a gift from Adrian Ferre-D'Amare. The hammerhead ribozyme was cleaved from the 5′-end of the RNA sequence during in vitro transcription, and the SRP RNA was extended with an overhang sequence to fuse with the DNA splint handle for single molecule experiments (28).
Protein and RNA Preparations
Ffh, FtsY, and SRP RNA were expressed in vivo as described in previous studies. SRP RNAs for smFRET experiments (smRNA) were prepared by in vitro transcription using T7 polymerase based on the Megascript protocol (Ambion) (28).
Fluorescence Labeling
Single cysteine mutants of Ffh-C153 were labeled with Cy3-maleimide (GE Healthcare) using a protocol similar to that of previous studies (28). The labeling reaction was carried out with a 1:5 protein:dye molar ratio at room temperature for 2 h. Unconjugated dyes were removed by gel filtration chromatography using Sephadex G-25 resin (Sigma).
Single Molecule Instrument
A home-built objective-type total internal reflection fluorescence microscope was used to carry out all the single molecule experiment. Green (532-nm) and red (635-nm) lasers were introduced in a 100× oil immersed objective and focused on the coverslip where SRP was immobilized. Scattering light was removed by a 560-nm and a 660-nm long pass filter (Chroma) for the green and red lasers, respectively. Cy3 and Quasar670 signals were split by a DV2 Dualview (Photometrics) and focused onto the Ixon 897 camera (Andor). Data were recorded at 30-ms time resolution.
Single Molecule Assay and Data Analysis
All protein samples were ultracentrifuged at 100,000 rpm (Optima TLX; Beckman Coulter) for 1 h immediately before use to remove aggregates during freeze-thaw cycle. PEGylated slides and coverslips were assembled to a flowing chamber (31). Neutravidin was applied to the chamber and incubated for 10 min before flowing in fluorescent molecules of interest.
SRP complexes were assembled in SRP buffer (50 mm K-HEPES, pH 7.5, 150 mm KOAc, 2 mm Mg(OAc)2, 2 mm DTT) supplemented with 0.01% Nikkol. The samples were diluted to a final concentration of 50 pm in imaging buffer (SRP buffer supplemented with 0.4% glucose and 1% Gloxy in Trolox), flowed into the sample chamber, and incubated for 5 min before imaging. Movies were recorded at 30-ms intervals until most fluorescent molecules were photobleached. Data were analyzed by home-written scripts in IDL and Matlab. Hidden Markov Modeling (HMM) of the fluorescence resonance energy transfer (FRET) trajectory and binning of FRET efficiencies were described previously (32).
GTPase Assay
GTPase rate constants were determined as described previously (15). In general, reactions contained 100 nm Ffh and 200 nm SRP RNA (wild type or mutants), and varying concentrations of FtsY were incubated with 100 μm GTP (doped with [γ-32P]GTP). Reactions were quenched by 0.75 m KH2PO4, pH 3.3, at different time points, separated by thin layer chromatography (TLC), and quantified by autoradiography.
NMR Sample Preparation
Unlabeled, uniformly 13C,15N-labeled WT, E−1, and E+1 samples, and base-specifically G/C-13C,15N-labeled WTx, E−1x, and E+1x samples were prepared by in vitro transcription using T7 polymerase (P266L mutant) (33) with synthetic DNA templates as described previously (34). After in vitro transcription, the RNA samples were ethanol-precipitated, purified using 15% denaturing polyacrylamide gels, electroeluted with Elutrap system (Whatman), and purified by anion exchange with a 5-ml Hi-Trap Q column (GE Healthcare). All RNA samples were then desalted and exchanged into water using an Amicon filtration system with 3000 molecular weight cut-off membranes (Millipore). The RNA samples were diluted with water to concentrations of ∼10 μm, heated at 95 °C for 3 min, and snap-cooled on ice for 30 min. All of the WT, E−1, E+1, WTx, E−1x, and E+1x samples were then exchanged into 10 mm potassium phosphate, pH 6.3, 50 mm KCl, and 50 μm EDTA and concentrated to ∼1 mm using an Amicon filtration system. For H2O samples, 10% D2O was added. For D2O samples, the RNA solutions were repeatedly lyophilized and redissolved in the same volume of 99.996% D2O (Sigma).
NMR Spectroscopy
All NMR experiments were carried out on Bruker Avance 800 MHz spectrometers equipped with 5-mm triple resonance cryogenic probes. Exchangeable proton NOESY spectra were recorded using H2O samples at 283K with a 200-ms mixing time, and nonexchangeable proton NOESY spectra were recorded on D2O samples at 298K with a 200-ms mixing time. NMR spectra were processed and analyzed with XWINNMR 3.5 (Bruker), NMRPipe (35), and Sparky 3.110 (University of California, San Francisco, CA).
The assignments were obtained using two-dimensional NOESY, two-dimensional total correlation spectroscopy, 1H-15N HSQC, 1H-13C HSQC experiments on the unlabeled and uniformly 13C,15N-labeled RNA samples (WT, E+1, and E−1) (34, 36). The secondary structure was determined by analyzing the two-dimensional H2O NOESY, two-dimensional D2O NOESY, and 1H-15N HSQC. Imino proton resonances were assigned from sequential connectivities in H2O NOESY spectra.
For analysis of nucleotide dynamics on the pico- to nanosecond time scale, the resonance intensities in 1H-13C HSQC spectra of each type of C-H spin (C1′H1′, C5H5, C6H6, and C8H8) were normalized against the lowest intensity from the helix to the reference value of 0.1 (see Fig. 14). To obtain information on interhelical orientation and relative motion between the distal docking and ED stems, C-H (C1′H1′, C5H5, C6H6, and C8H8) and N-H (imino) residual dipolar couplings (RDCs) were measured on base-specifically GC-13C,15N-labeled WTx, E−1x, and E+1x samples in the presence and absence of ∼6 mg/ml Pf1 phage (ASLA Biotech, Ltd.) at 298K on a 800-MHz spectrometer (supplemental Table S1). One-bond C-H RDCs were measured from the splittings of 1H-13C doublets along the 1H dimension using the transverse relaxation-optimized spectroscopy (TROSY)-based two-dimensional 1H-13C S3CT-HSQC (37), and one-bond H-N RDCs were determined by using standard 1H-15N HSQC experiments without decoupling in the indirect or direct dimension (38) (supplemental Table S1). NMR spectra for RDCs were processed and analyzed using NMRPipe/NMRDraw (35). These measured RDCs were then subjected to order tensor analysis (39) using the RAMAH program (40), where idealized A-form helices were used as input coordinates (41). The idealized A-form helices were constructed using Insight II (Molecular Simulations) with propeller twist angles corrected from +14.5° to −14.5° (41). To account for different degrees of alignment due to internal motions, the RDCs from each stem were analyzed independently. Excellent fits were obtained (supplemental Table S2), further supporting the validity of using idealized A-form helices in the order tensor analysis of RDCs. The resultant principal order tensor parameter and the generalized degree of order (GDO, or theta),
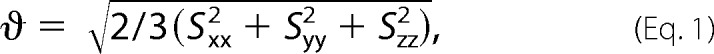 |
where Sxx, Syy, and Sz are the order tensor frames, describe the degree of alignment of each stem in the alignment media. The resultant Euler angles (α, β, γ) were used to generate a structural model that represents the average relative orientation between the two stems, by superimposing their order tensor frames (39, 42). The average interhelical bend was obtained from analyzing these structural models using Curves 5.3 (43).
FIGURE 14.
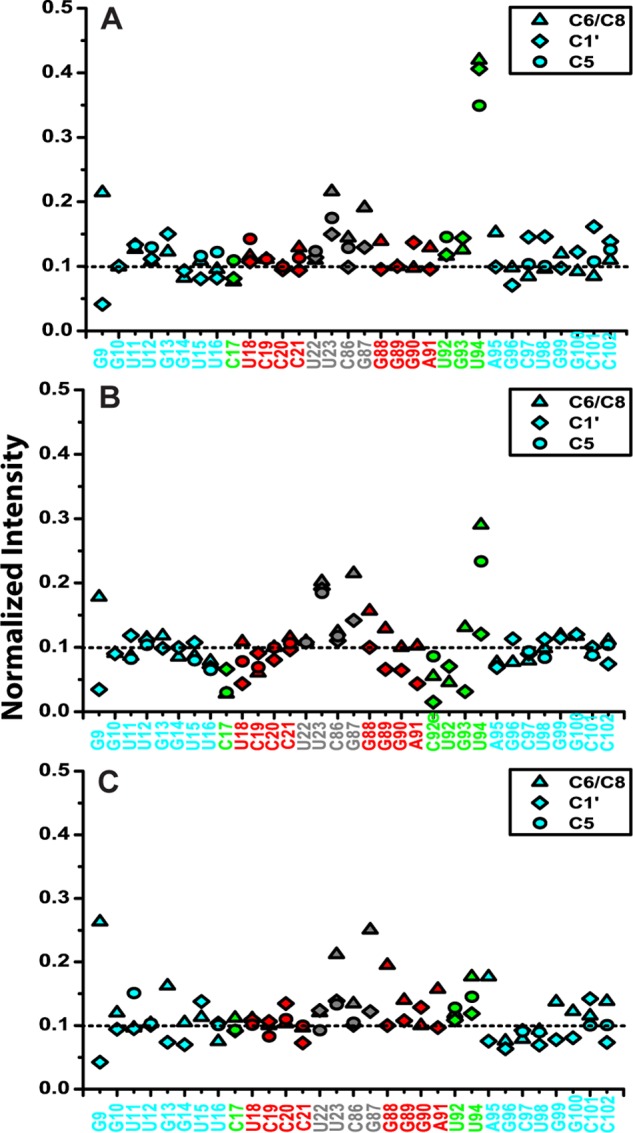
Normalized resonance intensities of WT (A), E+1 (B), and E−1 (C) from non-constant-time two-dimensional 1H-13C HSQC experiments. The nucleotides are colored by subdomain as in the inset of Fig. 11B.
RESULTS
An Intact Primary Docking Site Is Required for GTPase Activation at the RNA Distal End
To provide mechanistic details on how the SRP RNA distal end triggers GTP hydrolysis in the Ffh-FtsY GTPase complex, we revisited the 92-mer SRP RNA (nucleotides 10–101 of the wild-type SRP RNA), the minimal RNA construct that can stimulate GTP hydrolysis (27). We systematically truncated individual nucleotides from the 3′-end of the 92-mer (Fig. 2A, 91-mer to 87-mer) and determined the activity of these mutants using a well established GTPase assay (Fig. 2A) (15). In this assay, the value of kcat/Km is rate-limited by, and thus equal to, the rate constant of SRP-FtsY complex formation (15). The observed GTPase rate constant at saturating FtsY concentrations (kcat) includes both GTPase relocalization and activated GTP hydrolysis at the SRP RNA distal end (15, 28). Deletion of every nucleotide beyond C101 reduced the kcat value of the SRP-FtsY complex to levels in the absence of the SRP RNA (Fig. 2, A and B), even though single mutations of most of these nucleotides exhibited no significant defect (Fig. 3). These observations suggest that an intact docking site at the distal end of SRP RNA is required to stimulate GTPase activation.
FIGURE 2.
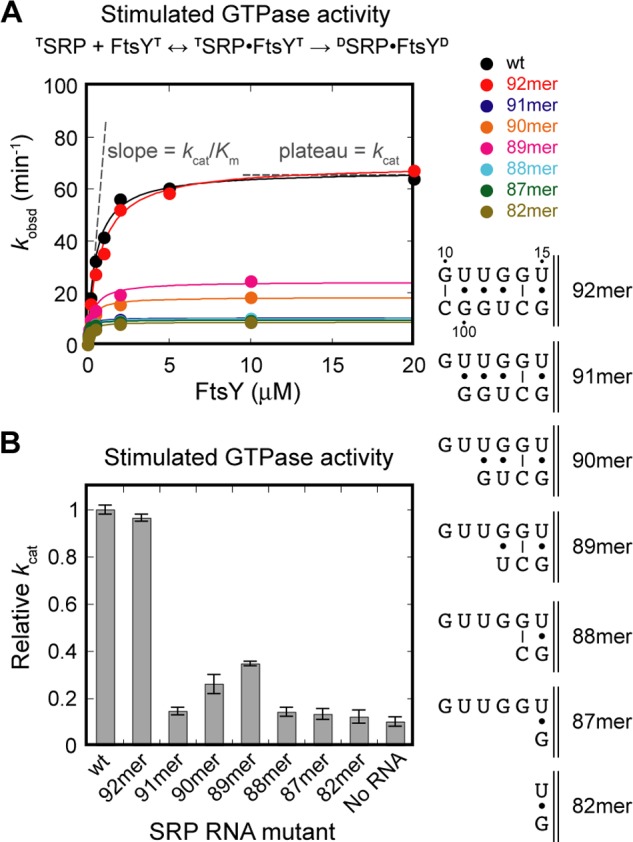
An intact SRP RNA distal end is required for efficient GTPase activation. A, GTPase assay showing the function of the SRP RNA truncation mutants. B, summary of the GTPase rate constant for single nucleotide truncation mutants of 92-mer. All kcat values are reported relative to that of wild-type SRP RNA.
FIGURE 3.
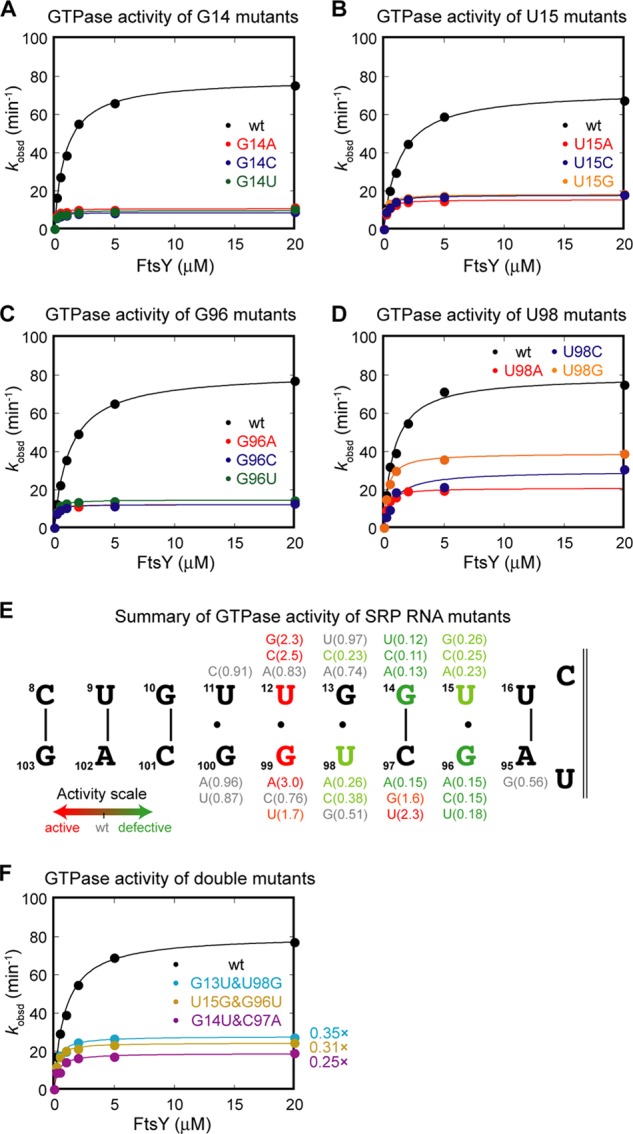
Specific nucleotides at the SRP RNA distal end play crucial roles in GTPase activation. A–D, site-directed mutagenesis of specific bases at the SRP RNA distal end and their GTPase activity. The four nucleotides whose mutations cause the most deleterious effects are shown: G14 (A), U15 (B), G96 (C), and U98 (D). E, summary of the GTPase activity of the point mutants at the distal end docking site. Values in parentheses denote the kcat values of the mutant relative to that of wild-type RNA. F, GTPase activity of base pair-switched mutants.
To probe the nucleotide specificity of the interactions between the distal docking site on the SRP RNA and the Ffh-FtsY GTPase complex, we mutated the individual bases in and surrounding this site. Several nucleotides stood out in this analysis (Fig. 3, A–D; summarized in 3E): G14, U15, G96, and U98. Mutation of G14 (Fig. 3A) or G96 (Fig. 3C) to any other bases lowered the GTPase rate to the level in the absence of SRP RNA. Mutation of U15 (Fig. 3B) or U98 (Fig. 3D) also severely impaired GTPase activation, although to a lesser extent than G96 and G14. The defects in GTPase activation by mutants G14, G96, and U98 were not rescued by compensatory mutations that restore base-pairing interactions at these sites (Fig. 3F), indicating that specific bases, rather than the base-paired structure, are important. Besides these four nucleotides, mutation of other nucleotides does not lead to defective GTPase activation (Fig. 3E). These results indicate that four specific nucleotides are crucial for driving GTPase docking and activation at the distal site. Combined with the results of deletional analyses (Fig. 2), we define the base-pairing region (10–15:96–101) as the primary docking site.
Interestingly, several mutants show higher GTPase activity than wild-type SRP RNA, most notably mutations at G99, U12, and C97 (Fig. 3E, red nucleotides). Mutant G99A has been shown to prolong GTPase docking at the distal end, which correlates with its faster GTP hydrolysis rate (28). Thus, by modifying the GTPase docking interface, the efficiency of activation of the Ffh-FtsY GTPase complex can be specifically tuned.
Conserved Bases in Loop D Specifically Catalyze GTP Hydrolysis
Once the GTPase complex docks at the SRP RNA distal end, GTP hydrolysis is activated >100-fold (28). In two crystallographic analyses, two distinct nucleotides in loop D, C86 or G83, have each been observed to insert into the composite active site formed at the interface between the Ffh and FtsY NG domains (Fig. 4, A and B) (27, 48). Biochemical studies demonstrated the importance of these two bases: deletion or substitution of G83 by any other nucleotide completely abolishes the stimulatory effect of the SRP RNA on GTP hydrolysis (48). Mutations at C86 yield a more complex pattern: whereas C86A and C86U completely abolish GTPase activation by the RNA, ΔC86 and C86G reduce GTPase activity by only 50% (27). In additional, when G83 is mutated, substitution of C86 with guanine rescues the SRP RNA-mediated stimulation of GTPase activity to 50% of wild-type rate (48). These results suggest that a guanine at residue 86 could compete with and substitute for G83 as a catalytic base.
FIGURE 4.

Catalytic bases in loop D specifically contribute to catalysis. A, crystal structure of the SRP-FtsY complex in which the GTPase complex is docked at the distal end (Protein Data Bank ID 2XXA (27). Shown in yellow is the protruding base (C86) that inserts into the Ffh-FtsY NG domain interface. B, secondary structure of the SRP RNA loop D. C, single molecule setup to observe the migration of the Ffh-FtsY NG complex along the SRP RNA. Ffh-C153 is labeled with Cy3. The 3′-end of the SRP RNA is labeled with Quasar670. D, fluorescence signals (upper panel) and FRET trajectory (lower panel) of the SRP-FtsY complex in GppNHp. HMM of the FRET trajectory is shown in navy. E, single molecule traces (left panel) and FRET histograms (right panel) of G83A and C86G mutants.
To distinguish whether these nucleotides are responsible for docking of the GTPase complex at the distal end or for enhancing GTP hydrolysis, we carried out single molecule fluorescence-total internal reflection fluorescence microscopy analyses with SRP RNA mutants G83A and C86G immobilized on the microscope slide (Fig. 4C) (28). We labeled the Ffh protein with a donor (Cy3) dye and the distal end of the SRP RNA with an acceptor (Quasar670). Successful docking of the GTPase complex at the distal end brings these two dyes into close proximity, resulting in a high efficiency of FRET (see Fig. 4) (28). In previous work, we showed that wild-type SRP RNA mediates dynamic and reversible movement of the NG domain complex on the SRP RNA in the presence of the nonhydrolyzable GTP analog GMPPNP (Fig. 4D). The probability to reach the high FRET state correlates directly with the observed GTPase rate constant, indicating that stable docking is necessary for GTPase activation (28). This assay allows us to specifically monitor the movement of the GTPase complex to the distal end, regardless of the catalytic function of these nucleotides.
Despite defective GTP hydrolysis, neither the G83A nor C86G mutant shows any detectable defect in the efficiency of GTPase docking at the distal end (Fig. 4E). The Ffh-FtsY GTPase complex assembled with both RNA mutants exhibits stable high FRET states, and their frequency of reaching the high FRET state is the same, within experimental error, as that of wild-type SRP RNA (Fig. 4, D versus E; summarized in Fig. 5A). These mutants lie far away from the linear correlation between observed GTPase rate and the frequency of attaining high FRET established for mutants that affect GTPase docking at the distal site (Fig. 5A), indicating that they uncouple GTP hydrolysis from the movement of the GTPase complex to the RNA distal end. Thus, a guanine at position 83 or 86 serves as a catalytic base that specifically triggers GTP hydrolysis.
FIGURE 5.
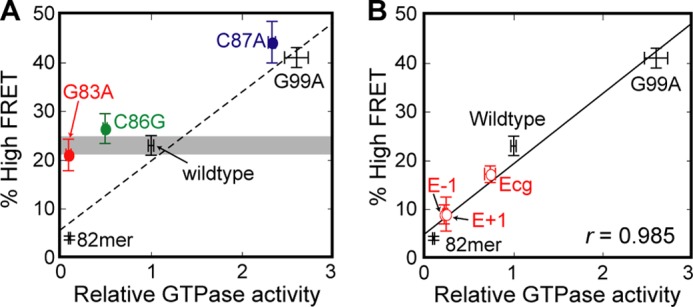
Correlation between the probability of attaining the high FRET state and the observed GTPase activity of the SRP-FtsY complex. Standard curve (dashed line in A and solid line in B) is the linear fit of the six data points with WT RNA and distal site docking mutants (B). The data for G99A and 82mer RNA are from Ref. 28 and were included in the linear regression. The data points for G83A, C86G, and C87A (colored circles) are not included in the fit.
An Extruded Base in Loop D Provides an Additional Site to Facilitate GTPase Docking
Strikingly, mutation of C87, an extruded base in loop D, also modulates GTPase activity (Fig. 6A). For example, mutant C87A triggers GTP hydrolysis ∼2.5-fold faster than wild type, whereas deletion of C87 reduces the observed GTPase rate (Fig. 6A). This suggests that C87 participates in the docking or activation of the GTPase complex. To distinguish between these possibilities, we tested mutant C87A using the single molecule assay. Mutant C87A displays an even higher efficiency of GTPase docking at the distal end than wild-type SRP RNA (Fig. 6B). The data with mutant C87A are consistent with the linear correlation obtained with other distal site docking mutants (Fig. 5A), suggesting that the major function of this nucleotide is to provide an additional site that assists in the stable docking of the GTPase complex at the RNA distal end.
FIGURE 6.

C87 provides an auxiliary docking site for the GTPase complex. A, GTPase activity of C87 mutants. B, single molecule trace (left panel) and FRET histogram (right panel) of the C87A mutant. C, C87 acting independently of the distal docking site. GTPase activity of the SRP RNA contains a combination of activating mutations. C97U and G99A are activating mutations at the primary docking site. C87A is the activating mutation at the auxiliary docking site.
To determine whether C87 and the previously identified primary distal docking site act independently or cooperatively, we combined the C87A with the G99A or C97U mutation, which also improves docking of the GTPases at the distal end (cf. red nucleotides in Fig. 3E). If the two docking sites are independent of one another, these double mutants will have an additive effect. This was indeed observed (Fig. 6C). Combining either G99A or C97U with C87A generated “superactive” SRP RNA double mutants that hydrolyze GTP 5.5- and 4.6-fold faster than wild-type SRP RNA, respectively. The observed enhancement in GTPase rate with these double mutants is consistent with an additive effect from the individual mutations. In contrast, combining the G99A and C97U mutations in the primary distal docking site did not further enhance GTPase activity compared with the single mutations (Fig. 6C). Together with the single molecule data, these results strongly suggest that nucleotide C87 provides an additional docking site that further stabilizes the interaction of the GTPase complex with the SRP RNA distal end. We therefore define C87 as the auxiliary docking site.
Loop E Controls the Action of the Distal End Docking Sites
The results above show that stable docking of the Ffh-FtsY GTPase complex at the SRP RNA distal end requires a bidentate interaction with the primary (nucleotides 10–15:96–101) and auxiliary (C87 in loop D) docking sites. These two sites are bridged by the asymmetric loop E (C-UGU in Escherichia coli, Fig. 1B). In the crystal structure, loop E is located on the opposite side of the distal docking site and hence could not make direct contacts with the GTPase complex. However, this asymmetric internal loop is conserved across species in the SRP RNA, implicating it in an important function. We therefore probed the structure and function of this loop.
Two roles could be envisioned for loop E. It could be crucial for accurately positioning and orienting the primary and auxiliary docking sites, optimizing them for stable interaction with the GTPase complex. Alternatively or in addition, loop E could introduce flexibility that enables more efficient search and docking by the GTPase complex. To test these hypotheses, we measured the GTPase activity of several mutant RNAs in which loop E is either replaced by base pairs (Fig. 7A, ΔE, ΔE+1, cE) or systematically varied in size (Fig. 7A, E−1, E+1, E+2). All of the mutations that removed loop E completely abolished the stimulatory effect of the SRP RNA on GTP hydrolysis (Fig. 7, B and C). All of the mutants that alter the size of loop E reduced the GTPase rate constant to ∼20% of that of wild-type SRP, only 2-fold higher than that in the absence of the SRP RNA (Fig. 7, B, upper panel, and C). This indicates that the size of loop E is critical for GTPase docking and activation at the RNA distal end. Finally, when the flanking AU pairs are replaced with more stable GC pairs, which may reduce interhelical flexibility as has been observed in other RNAs (44), GTPase activation by the SRP RNA is reduced by ∼30% (Fig. 7B, lower panel). These observations suggest that the major function of loop E is to accurately control the orientation of the distal end docking sites, whereas the flexibility introduced by loop E contributes only marginally to the function of the RNA distal end.
FIGURE 7.
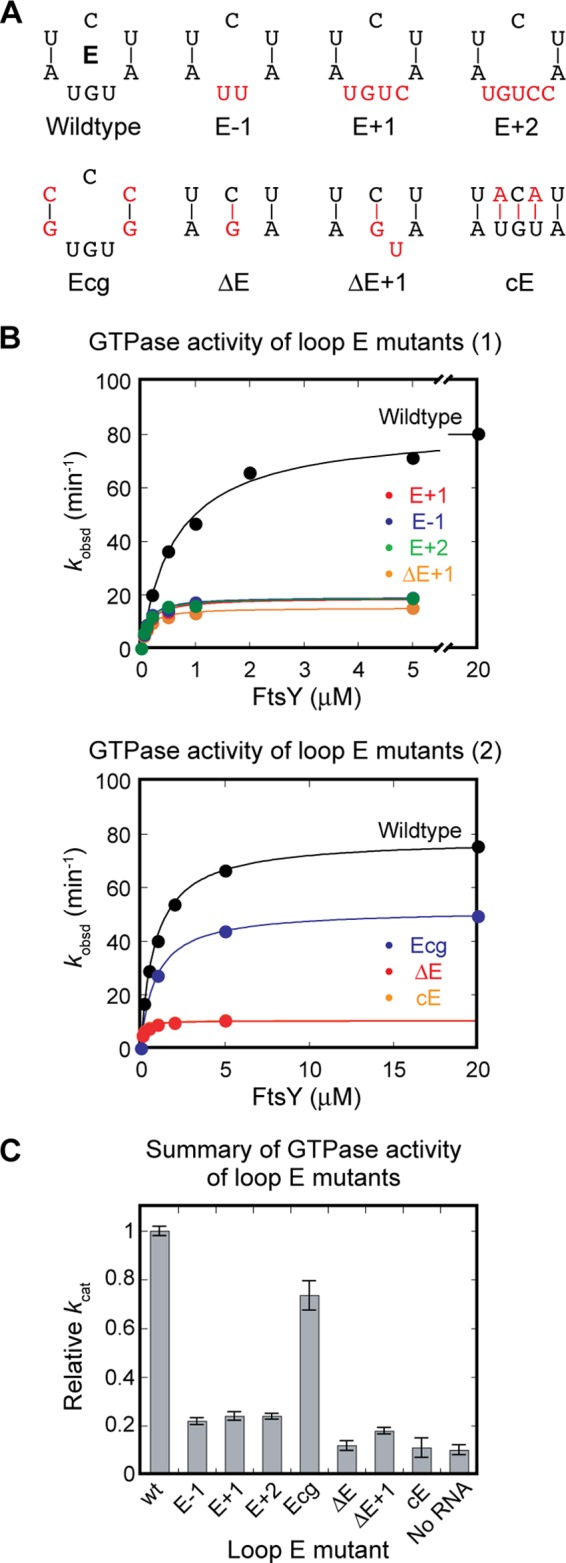
Loop E plays a crucial role in GTPase activation by the SRP RNA. A, loop E mutants characterized in this work. E−1, E+1, and E+2 alter the size of loop E. Ecg reduces potential dynamics of loop E by replacing the UA pairs with CG pairs. ΔE, ΔE+1, and cE eliminate loop E. B, GTPase activity of the loop E mutants in A. C, summary of the relative GTPase rate constant (kcat) of the loop E mutants relative to wild-type RNA.
To test directly the role of loop E in mediating the docking of the Ffh-FtsY GTPase complex at the RNA distal end, we used the single molecule assay (Fig. 8). Mutants E−1 and E+1 exhibit a much lower frequency of attaining the high FRET state and a much shorter dwell time in this state compared with wild-type SRP RNA (cf. Figs. 8 and 4D). The probability at which each mutant attains the high FRET state directly correlates with its observed GTPase rate constant, falling on the same linear correlation generated by wild type, G99A, and 82-mer (Fig. 5B). This indicates that loop E affects observed GTPase activity by tuning the efficiency of GTPase docking at the RNA distal end.
FIGURE 8.
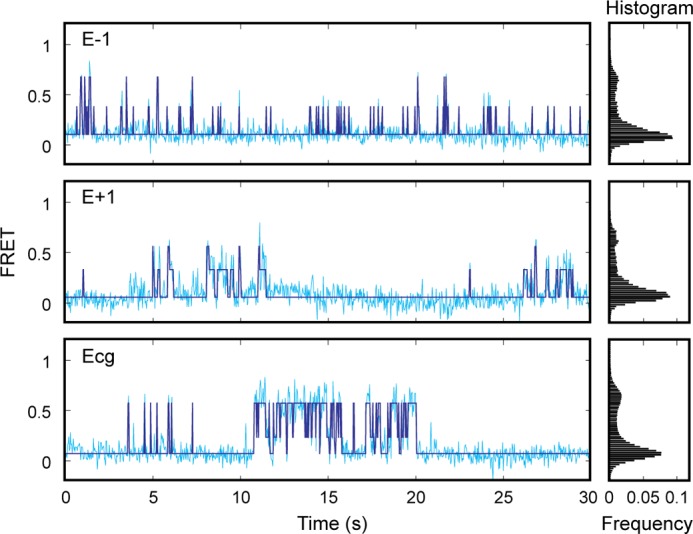
Loop E mutants disrupt correct docking of the GTPase complex at the distal end of the SRP RNA. Sample FRET trajectories (cyan) and HMM simulation (navy) of E−1, E+1, and Ecg SRP RNA mutants (left panel) are shown. The FRET histograms for each mutant are shown in the right panel.
Although loop E mutants do not maintain the GTPase complex in the high FRET state as well as wild-type SRP RNA, they allow the GTPase complex to nonspecifically visit intermediate FRET states (Fig. 8). However, molecules that reached these intermediate states primarily return to the low FRET state, rather than proceeding to the high FRET state. We reasoned that these intermediate states represent nonspecific binding modes of the GTPase complex on the SRP RNA, which do not allow stable docking of the GTPase complex and are not conducive to GTPase activation. Thus, loop E controls the specific docking of the GTPase complex at the correct distal end docking sites.
Loop E Conformation and Dynamics Determined by NMR
To gain further insights into the proposed function of loop E in the docking process, we applied solution NMR spectroscopy for structural analysis, together with a domain elongation strategy to measure the amplitude of interhelical motions (45), to investigate the structure and dynamics of the SRP RNA distal end. The RNA construct includes the distal end docking stem extended by one G-C base pair at the distal end, loop E, and the ED stem (the stem between loops D and E) (nucleotides 10–21:88–101) capped by a UUCG tetraloop. The distal end docking stem was elongated using a stretch of unlabeled A-U base pairs with otherwise 13C,15N-labeled G/C nucleotides for the WT, E+1, and E−1 sequences (WTx, E+1x, and E−1x, Fig. 9A). The elongated distal end docking stem dominates the overall molecular tumbling and therefore serves as the internal reference frame for accurate NMR characterization of interhelical bending and conformational flexibility of the RNA (45). The excellent agreement between the NMR spectra of the elongated (WTx) and nonelongated wild-type (WT) SRP RNAs indicates that the WTx construct faithfully recapitulates the structure and dynamics properties of the nonelongated RNA (Fig. 10).
FIGURE 9.
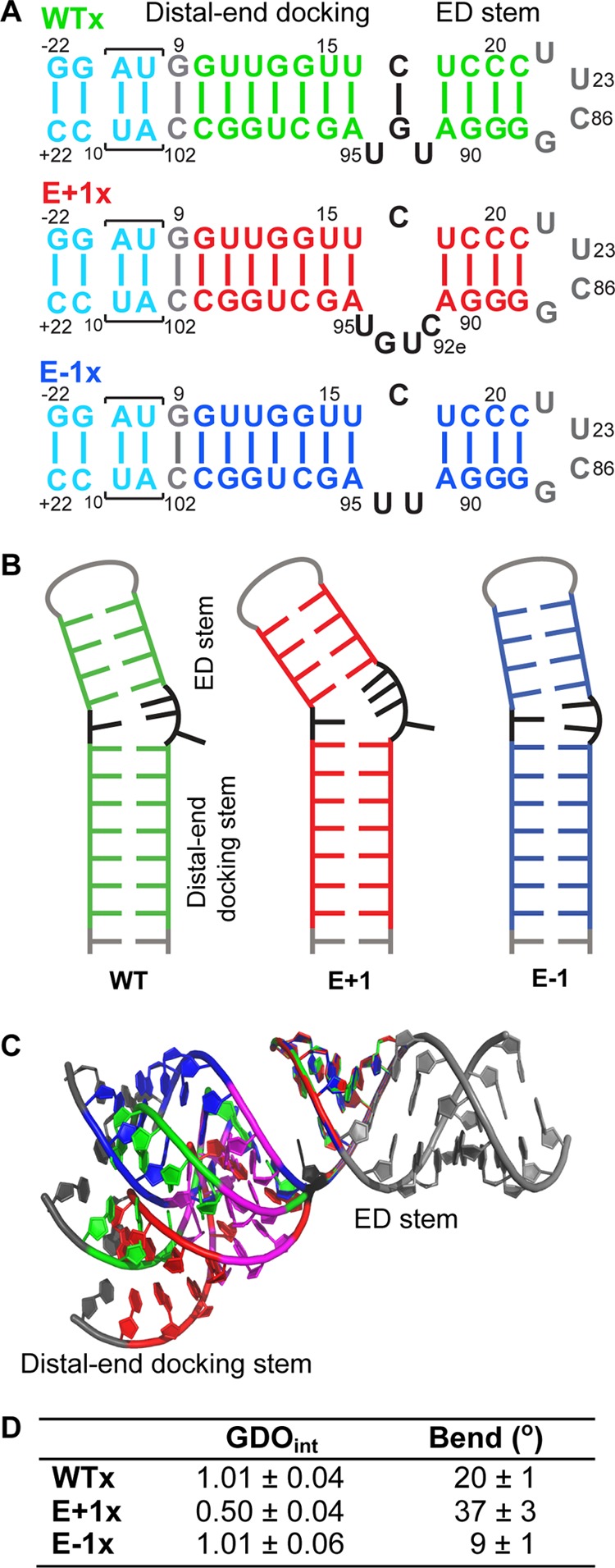
NMR study directly visualizes the orientation and flexibility of loop E region. A, sequence and secondary structures of WTx, E+1x, and E−1x constructs. B, schematic representation of the secondary structures of WT, E+1, and E−1 derived from NMR data. C, computational modeling of the NMR-derived structures of WT (green), E+1 (red), and E−1 (blue) superimposed on ED stem. G14, U15, G96, and U98 are colored magenta. The elongated gray nucleotides on ED stem are guides for visualization. D, RDC analysis results of WTx, E+1x, and E−1x.
FIGURE 10.

Comparison of elongated WT SPR (WTx) and WT SRP. A, sequence and secondary structures of WT SPR and WTx constructs. B–D, overlapping spectra of two-dimensional 1H-13C HSQC of the C6H6/C8H8 (B), sugar C1′H1′ (C), and C5H5 (D) from WT (orange) and WTx (green). The asterisks denote resonances that belong to two terminal guanine (G−21 and G−22) and cytosine (C+21 and C+22) residues in WTx.
We first investigated whether loop E induces flexibility between the two stems, by measuring NMR RDCs of WTx using Pf1 phage as an alignment medium (supplemental Table S1). Order tensor analysis of the RDCs allowed us to characterize interhelical motion that is faster than the millisecond time scale by comparing the degree of order (ϑ) for individual stems (supplemental Table S2) (39). The ratio between the degree of order for each stem, defined as the interhelical generalized degree of order (ϑint = ϑED/ϑDistal), provides a measure of interhelical motions with ϑint = 1 corresponding to entirely rigid and ϑint = 0 to maximum interhelical motion. Consistent with the biochemical assays, yet surprising for a 1–3 (C-UGU) asymmetric loop, there is almost no interhelical motion across loop E in WTx SRP RNA (ϑint = 1.01 ± 0.04). RDC analysis also shows that there is a well defined bend angle (20 ± 1°) between the distal end docking and ED stems (Fig. 9D). To explain the unexpected rigidity across loop E, we analyzed its secondary/tertiary structure using NMR. Analysis of the imino proton resonances and cross-peaks in 1H-15N HSQC spectra of WTx, and imino NOESY of WT showed that G93 forms a base pair with C17 within loop E (Figs. 11 and 12). An imino proton resonance for this base pair is observed in the NOESY spectrum (Fig. 11), and its assignment is confirmed in the 1H-15N HSQC spectrum (Fig. 12). Furthermore, analysis of the D2O NOESY spectrum showed that residue U92 is stacked between the U18-A91 and C17-G93 base pairs, C17-G93 is stacked on U16-A95, and U94 is flipped out of loop E (Fig. 13). In addition, in 1H-13C HSQC spectra, U94 has abnormally high resonance intensities compared with other residues in WT, consistent with conformational flexibility that would be expected for a flipped out base (Fig. 14A). Higher intensity, as a result of motional narrowing, is indicative of internal motion that is faster than the overall molecular tumbling rate (45). Therefore, the 1–3 asymmetric loop E adopts a conformation that has a stable G93-C17 base pair and a stacked-in U92 residue, which results in formation of a rigid bend between the distal end docking and ED stems.
FIGURE 11.
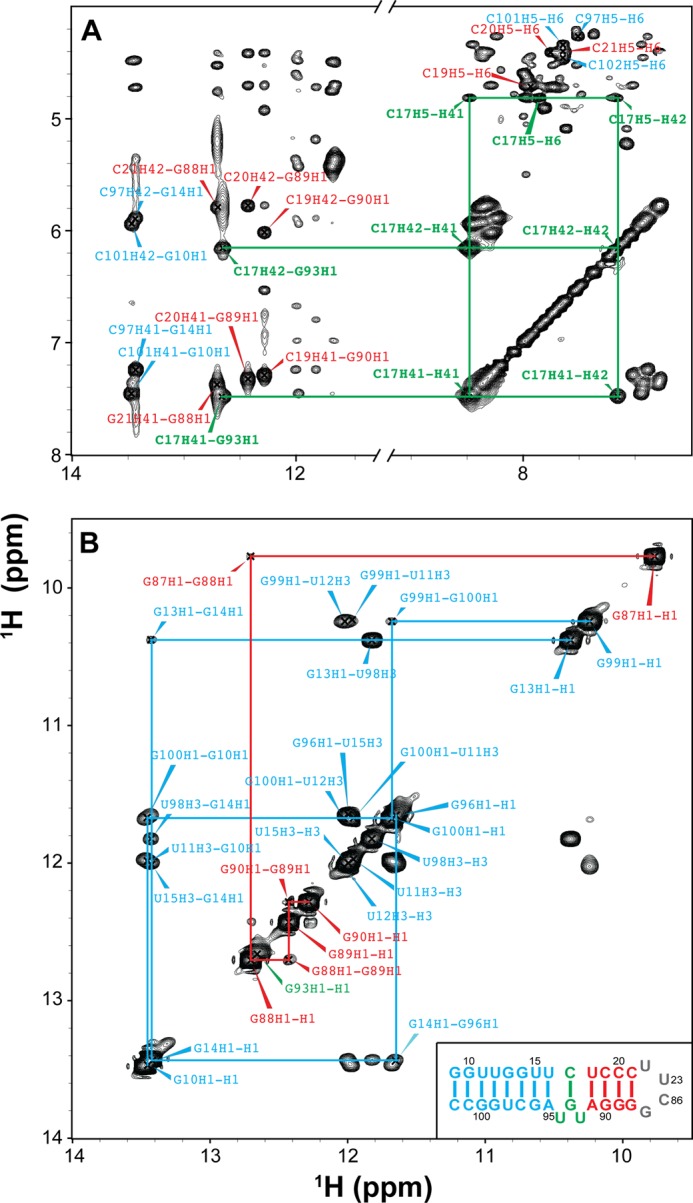
Identification of base pairing of the wild-type SRP RNA by NMR spectra. A, two-dimensional NOESY of wild-type SRP RNA in H2O. Selected assignments are labeled in the spectrum. The NOEs (C17H41-G93H1 and C17H42-G93H1) provide direct evidence that C17 and G93 form a canonical GC base pair. B, imino-imino region in two-dimensional NOESY of WT SRP RNA in H2O. The lines indicate sequential imino proton connectivities. The inset in B shows the sequence and secondary structure of wild-type SRP RNA. The assignments and lines are colored by subdomain as in the inset.
FIGURE 12.

1H-15N HSQC spectra of WTx (A), E+1x (B), and E−1x (C). The cross-peaks represent base-paired guanines. Base-paired G93 can be observed in WTx, but not in E+1x and E−1x.
FIGURE 13.

Identification of base stacking of the wild-type SRP RNA by two-dimensional NOESY in D2O. The cross-peaks are between the neighboring aromatic protons (H2, H6, or H8). No NOE connecting with U94H6 was observed. NOE (G93H8-A95H8) indicates that the base of G93 is stacked on A95 and U94 is flipped out of loop E. The assignments are colored by subdomain as in the inset of Fig. 11B.
Next, to investigate the effect of the size of loop E on the interhelical motion and orientation, we acquired RDC data on both E+1x and E−1x constructs. The RDC analysis showed that E+1x has an average interhelical angle of 37°, approximately 17° larger than WTx (Fig. 9D). Moreover, E+1x has a significant interhelical motion (ϑint = 0.50 ± 0.04). Secondary structure information obtained from NMR NOESY, total correlation spectroscopy (see “Experimental Procedures”; data not shown), and HSQC spectra explained this observation. NMR 1H-15N HSQC spectra showed that in the presence of the extra cytosine (C92e) in the E+1x loop E, the C17-G93 base pair does not form (Fig. 12B), resulting in a less ordered and therefore more mobile loop E. A flexible U94, which is probably flipped out, is also observed in E+1, based on the normalized intensities in a 1H-13C HSQC spectrum (Fig. 14B). In contrast, E−1x showed a limited interhelical motion similar to that in WTx and a negligible (9 ± 1°) interhelical bend angle (Fig. 9D and supplemental Table S2). In NOESY spectra of E−1 (data not shown) sequential NOEs are observed from A91-U92-U94-A95, indicating that, in the absence of G93, U92 and U94 are stacked on each other and into the helix opposite C17, and therefore E−1 forms an almost linear stem (Fig. 9B). Together with GTPase activity results, these data suggest that a defined orientation between the primary and auxiliary docking sites is important for efficient docking of the Ffh-FtsY GTPase complex and its activity. We further propose that the C17-G93 base pair in loop E might play an essential role in stabilizing the overall interhelical conformation and orienting the docking sites.
DISCUSSION
During co-translational protein targeting, a global conformational rearrangement of the Ffh-FtsY GTPase complex from the tetraloop to the distal end of the SRP RNA has been demonstrated by crystallographic, biochemical, and single molecule studies (27, 28). However, the molecular basis underlying the specificity of GTPase docking and activation at the RNA distal end has been puzzling, given the low sequence conservation at the RNA distal end and the largely electrostatic nature of the interaction of the GTPase complex with this end. In this work, we combined biochemical and biophysical assays to investigate how GTP hydrolysis is specifically and productively triggered at the RNA distal end.
Using mutagenesis screening, we identified four critical bases, G14, U15, G96, and U98, whose mutations severely disrupt docking of the GTPase complex at the distal end. In the crystal structure, these nucleotides form a contiguous minor groove that docks against FtsY (Fig. 15, orange). Nevertheless, all of these nucleotide bases point inward into the RNA duplex and do not directly interact with FtsY. Instead, FtsY contacts this groove primarily via electrostatic interactions with the backbone phosphate groups (27). Thus, it is most likely that the specific base composition is important for providing an optimal conformation and electrostatic potential in this minor groove (46), allowing it to provide a primary docking site for the GTPase complex.
FIGURE 15.
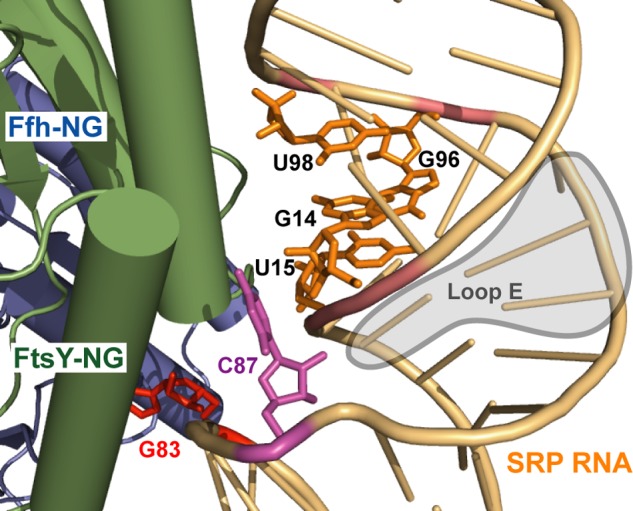
Bidentate interaction between the Ffh-FtsY GTPase complex and the distal end of the SRP RNA (Protein Data Bank ID 4C7O) (48). The four critical nucleotides that form the minor groove in the primary docking site are shown in orange. C87, the protruding base that forms the auxiliary docking site, is in magenta. The catalytic base G83 is in red.
In addition to interactions with this primary docking site, we identified an auxiliary interaction mediated by nucleotide C87 in loop D. C87 is an extruded base in the crystal structure (Fig. 15; Ref. 48), and previous chemical probing experiments revealed that two nucleotides in loop D, C87 and G83, are exposed and sensitive to modification by small molecules (47). In agreement with these observations, we found that the identity of the base at position 87 modulates the docking of the GTPase complex at the RNA distal end, and further, mutations at C87 exhibit additive effects with those in the primary docking site to regulate the stability and frequency of GTPase docking at the RNA distal end. These results suggest that C87 provides an additional interaction site to enable stable docking of the Ffh-FtsY GTPase complex and that correct GTPase docking at the SRP RNA distal end requires a bidentate interaction with both sites.
Successful docking at the distal end is necessary but insufficient for activation of GTP hydrolysis. Previous biochemical and structural work identified the critical guanine base at position 83, which inserts into the Ffh-FtsY NG domain interface and repositions active site residues and the water network, and thus could be responsible for stimulating GTP hydrolysis (48). An additional nucleotide, C86, could serve as an alternative catalytic base when replaced by G (48). In this study, single molecule experiments further allowed us to distinguish the role of distal end nucleotides in the docking step from that in GTPase activation. We found that neither G83 nor C86 contributes to the docking of the GTPase complex at the SRP RNA distal end, but instead are specifically responsible for activating GTP hydrolysis. Thus, docking of the GTPase complex and its activation at the RNA distal end involve distinct sets of nucleotides and can be conceptually and experimentally uncoupled from one another.
The two sites necessary for GTPase docking at the RNA distal end are bridged by the internal loop E, whose size and position relative to the two docking sites are evolutionarily conserved. Here, biochemical, single molecule, and NMR analyses characterized the properties of loop E and for the first time provided clues for its function. For the wild-type SRP RNA, we found that loop E forms a rigid structure, with a C17-G93 base pair and stacked-in U92, which induces a defined bend between the distal end docking and ED stems. Perturbations to the size or composition of loop E induce structural changes that are strongly correlated with the defects of the loop E mutants in mediating GTPase docking and activation at the RNA distal end. We propose that for these loop E mutants, the GTPase complex spends most of the time in nonproductive searches, during which it likely makes contacts with one or the other distal docking sites but not both. Finally, we note that the structure of loop E and the interhelical bend introduced by it are highly similar for the free RNA, determined here by solution NMR, and for the RNA bound to the Ffh-FtsY observed in two crystal structures. Together, the results suggest that a major function of this conserved internal loop is to correctly orient the relative positioning of the primary docking site with respect to the auxiliary docking site and to the catalytic base. This generates a preformed interaction surface complementary in shape and electrostatic potential to that on the GTPase complex, thereby enabling productive and stable interaction of the GTPase complex with the RNA distal end (Fig. 9C).
CONCLUSION
The large scale rearrangement of the Ffh-FtsY GTPase complex to the SRP RNA distal end enables the activation of this GTPase complex to be spatially segregated from the site of its initial assembly. This provides an attractive mechanism to mediate coordinated unloading and handover of the cargo from the SRP-SR complex to the SecYEG translocation channel in the membrane and to precisely coordinate the timing of GTPase activation with this cargo unloading event (17, 28). Collectively, our results here show that despite the absence of highly sequence-specific interactions, the fidelity of this long range GTPase movement is ensured by using two distinct sites to provide a bidentate interaction for the GTPase complex at the RNA distal end. The correct positioning of these two sites with respect to one another and to the catalytic base, mediated by an evolutionarily conserved intervening loop, ensures that the GTPase complex is specifically recruited to and activated at the RNA distal end during the protein targeting reaction.
This work was supported, in whole or in part, by National Institutes of Health Grants GM078024 (to S.-o. S.) and GM048123 (to J. F.) and a National Institutes of Health instrument supplement to Grant GM45162 (to D. C. Rees). This work was also supported by Caltech Matching Fund 350270 for the single molecule instruments, the David and Lucile Packard Fellowship in Science and Engineering (to S.-o. S.), and a Henry Dreyfus teacher-scholar award (to S.-o. S.).

This article contains supplemental Tables S1 and S2.
- SRP
- signal recognition particle
- GMPPNP
- guanyl 5′-(β,γ-imino)triphosphate
- HMM
- hidden Markov modeling
- HSQC
- heteronuclear single quantum coherence
- RDC
- residual dipolar coupling
- RNC
- ribosome-nascent chain complex
- SR
- SRP receptor.
REFERENCES
- 1. Akopian D., Shen K., Zhang X., Shan S. (2013) Signal recognition particle: an essential protein-targeting machine. Annu. Rev. Biochem. 82, 693–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Keenan R. J., Freymann D. M., Stroud R. M., Walter P. (2001) The signal recognition particle. Annu. Rev. Biochem. 70, 755–775 [DOI] [PubMed] [Google Scholar]
- 3. Walter P., Johnson A. E. (1994) Signal sequence recognition and protein targeting to the endoplasmic reticulum membrane. Annu. Rev. Cell Biol. 10, 87–119 [DOI] [PubMed] [Google Scholar]
- 4. Halic M., Becker T., Pool M. R., Spahn C. M., Grassucci R. A., Frank J., Beckmann R. (2004) Structure of the signal recognition particle interacting with the elongation-arrested ribosome. Nature 427, 808–814 [DOI] [PubMed] [Google Scholar]
- 5. Halic M., Blau M., Becker T., Mielke T., Pool M. R., Wild K., Sinning I., Beckmann R. (2006) Following the signal sequence from ribosomal tunnel exit to signal recognition particle. Nature 444, 507–511 [DOI] [PubMed] [Google Scholar]
- 6. Janda C. Y., Li J., Oubridge C., Hernández H., Robinson C. V., Nagai K. (2010) Recognition of a signal peptide by the signal recognition particle. Nature 465, 507–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pool M. R., Stumm J., Fulga T. A., Sinning I., Dobberstein B. (2002) Distinct modes of signal recognition particle interaction with the ribosome. Science 297, 1345–1348 [DOI] [PubMed] [Google Scholar]
- 8. Schaffitzel C., Oswald M., Berger I., Ishikawa T., Abrahams J. P., Koerten H. K., Koning R. I., Ban N. (2006) Structure of the E. coli signal recognition particle bound to a translating ribosome. Nature 444, 503–506 [DOI] [PubMed] [Google Scholar]
- 9. Egea P. F., Shan S. O., Napetschnig J., Savage D. F., Walter P., Stroud R. M. (2004) Substrate twinning activates the signal recognition particle and its receptor. Nature 427, 215–221 [DOI] [PubMed] [Google Scholar]
- 10. Focia P. J., Shepotinovskaya I. V., Seidler J. A., Freymann D. M. (2004) Heterodimeric GTPase core of the SRP targeting complex. Science 303, 373–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gilmore R., Blobel G., Walter P. (1982) Protein translocation across the endoplasmic reticulum. I. Detection in the microsomal membrane of a receptor for the signal recognition particle. J. Cell Biol. 95, 463–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gilmore R., Walter P., Blobel G. (1982) Protein translocation across the endoplasmic reticulum. II. Isolation and characterization of the signal recognition particle receptor. J. Cell Biol. 95, 470–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Driessen A. J., Nouwen N. (2008) Protein translocation across the bacterial cytoplasmic membrane. Annu. Rev. Biochem. 77, 643–667 [DOI] [PubMed] [Google Scholar]
- 14. Rapoport T. A. (2007) Protein translocation across the eukaryotic endoplasmic reticulum and bacterial plasma membranes. Nature 450, 663–669 [DOI] [PubMed] [Google Scholar]
- 15. Peluso P., Shan S. O., Nock S., Herschlag D., Walter P. (2001) Role of SRP RNA in the GTPase cycles of Ffh and FtsY. Biochemistry 40, 15224–15233 [DOI] [PubMed] [Google Scholar]
- 16. Wilson C., Connolly T., Morrison T., Gilmore R. (1988) Integration of membrane proteins into the endoplasmic reticulum requires GTP. J. Cell Biol. 107, 69–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Akopian D., Dalai K., Shen K., Duong F., Shan S. (2013) SecYEG activates GTPases to drive the completion of cotranslational protein targeting. J. Cell Biol. 200, 397–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lam V. Q., Akopian D., Rome M., Shen Y., Henningsen D., Shan S. O. (2010) Lipid activation of the signal recognition particle receptor provides spatial coordination of protein targeting. J. Cell Biol. 190, 623–635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shan S. O., Stroud R., Walter P. (2004) Mechanism of association and reciprocal activation of two GTPases. PloS Biol. 2, e320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang X., Kung S., Shan S. (2008) Demonstration of a multistep mechanism for assembly of the SRP × SRP receptor complex: implications for the catalytic role of SRP RNA. J. Mol. Biol. 381, 581–593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang X., Schaffitzel C., Ban N., Shan S. (2009) Multiple conformational switches in a GTPase complex control co-translational protein targeting. Proc. Natl. Acad. Sci. U.S.A. 106, 1754–1759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Batey R. T., Rambo R. P., Lucast L., Rha B., Doudna J. A. (2000) Crystal structure of the ribonucleoprotein core of the signal recognition particle. Science 287, 1232–1239 [DOI] [PubMed] [Google Scholar]
- 23. Batey R. T., Sagar M. B., Doudna J. A. (2001) Structural and energetic analysis of RNA recognition by a universally conserved protein from the signal recognition particle. J. Mol. Biol. 307, 229–246 [DOI] [PubMed] [Google Scholar]
- 24. Shen K., Shan S. (2010) Transient tether between the SRP RNA and SRP receptor ensures efficient cargo delivery during cotranslational protein targeting. Proc. Natl. Acad. Sci. U.S.A. 107, 7698–7703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Peluso P., Herschlag D., Nock S., Freymann D. M., Johnson A. E., Walter P. (2000) Role of 4.5S RNA in assembly of the bacterial signal recognition particle with its receptor. Science 288, 1640–1643 [DOI] [PubMed] [Google Scholar]
- 26. Shen K., Zhang X., Shan S. (2011) Synergistic actions between the SRP RNA and translating ribosome allow efficient delivery of the correct cargos during cotranslational protein targeting. RNA 17, 892–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ataide S. F., Schmitz N., Shen K., Ke A., Shan S. O., Doudna J. A., Ban N. (2011) The crystal structure of the signal recognition particle in complex with its receptor. Science 331, 881–886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shen K., Arslan S., Akopian D., Ha T., Shan S. O. (2012) Activated GTPase movement on an RNA scaffold drives co-translational protein targeting. Nature 492, 271–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Becker T., Bhushan S., Jarasch A., Armache J. P., Funes S., Jossinet F., Gumbart J., Mielke T., Berninghausen O., Schulten K., Westhof E., Gilmore R., Mandon E. C., Beckmann R. (2009) Structure of monomeric yeast and mammalian Sec61 complexes interacting with the translating ribosome. Science 326, 1369–1373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mitra K., Schaffitzel C., Shaikh T., Tama F., Jenni S., Brooks C. L., 3rd, Ban N., Frank J. (2005) Structure of the E. coli protein-conducting channel bound to a translating ribosome. Nature 438, 318–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Joo C., Balci H., Ishitsuka Y., Buranachai C., Ha T. (2008) Advances in single-molecule fluorescence methods for molecular biology. Annu. Rev. Biochem. 77, 51–76 [DOI] [PubMed] [Google Scholar]
- 32. McKinney S. A., Joo C., Ha T. (2006) Analysis of single-molecule FRET trajectories using hidden Markov modeling. Biophys. J. 91, 1941–1951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Guillerez J., Lopez P. J., Proux F., Launay H., Dreyfus M. (2005) A mutation in T7 RNA polymerase that facilitates promoter clearance. Proc. Natl. Acad. Sci. U.S.A. 102, 5958–5963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dieckmann T., Feigon J. (1997) Assignment methodology for larger RNA oligonucleotides: application to an ATP-binding RNA aptamer. J. Biomol. NMR 9, 259–272 [DOI] [PubMed] [Google Scholar]
- 35. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 36. Flinders J., Dieckmann T. (2006) NMR spectroscopy of ribonucleic acids. Prog. Nucl. Magn. Reson. Spectrosc. 48, 137–159 [Google Scholar]
- 37. Meissner A., Sørensen O. W. (1999) The role of coherence transfer efficiency in design of TROSY-type multidimensional NMR experiments. J. Magn. Reson. 139, 439–442 [DOI] [PubMed] [Google Scholar]
- 38. Zhang Q., Stelzer A. C., Fisher C. K., Al-Hashimi H. M. (2007) Visualizing spatially correlated dynamics that directs RNA conformational transitions. Nature 450, 1263–1267 [DOI] [PubMed] [Google Scholar]
- 39. Prestegard J. H., al-Hashimi H. M., Tolman J. R. (2000) NMR structures of biomolecules using field oriented media and residual dipolar couplings. Q. Rev. Biophys. 33, 371–424 [DOI] [PubMed] [Google Scholar]
- 40. Hansen A. L., Al-Hashimi H. M. (2006) Insight into the CSA tensors of nucleobase carbons in RNA polynucleotides from solution measurements of residual CSA: towards new long-range orientational constraints. J. Magn. Reson. 179, 299–307 [DOI] [PubMed] [Google Scholar]
- 41. Musselman C., Pitt S. W., Gulati K., Foster L. L., Andricioaei I., Al-Hashimi H. M. (2006) Impact of static and dynamic A-form heterogeneity on the determination of RNA global structural dynamics using NMR residual dipolar couplings. J. Biomol. NMR 36, 235–249 [DOI] [PubMed] [Google Scholar]
- 42. Bailor M. H., Musselman C., Hansen A. L., Gulati K., Patel D. J., Al-Hashimi H. M. (2007) Characterizing the relative orientation and dynamics of RNA A-form helices using NMR residual dipolar couplings. Nat. Protoc. 2, 1536–1546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lavery R., Sklenar H. (1989) Defining the structure of irregular nucleic acids: conventions and principles. J. Biomol. Struct. Dyn. 6, 655–667 [DOI] [PubMed] [Google Scholar]
- 44. Stelzer A. C., Kratz J. D., Zhang Q., Al-Hashimi H. M. (2010) RNA dynamics by design: biasing ensembles towards the ligand-bound state. Angew. Chem. Int. Ed. Engl. 49, 5731–5733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhang Q., Sun X., Watt E. D., Al-Hashimi H. M. (2006) Resolving the motional modes that code for RNA adaptation. Science 311, 653–656 [DOI] [PubMed] [Google Scholar]
- 46. Ryter J. M., Schultz S. C. (1998) Molecular basis of double-stranded RNA-protein interactions: structure of a dsRNA-binding domain complexed with dsRNA. EMBO J. 17, 7505–7513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lentzen G., Moine H., Ehresmann C., Ehresmann B., Wintermeyer W. (1996) Structure of 4.5S RNA in the signal recognition particle of Escherichia coli as studied by enzymatic and chemical probing. RNA 2, 244–253 [PMC free article] [PubMed] [Google Scholar]
- 48. Voigts-Hoffmann F., Schmitz N., Shen K., Shan S. O., Ataide S. F., Ban N. (2013) The structural basis of FtsY recruitment and GTPase activation by SRP RNA. Mol. Cell, 10.1016/j.molcel.2013.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]