

Background: Nox1, an oxidant source in colon carcinoma and vascular disease, is activated by NOXA1.
Results: A NOXA1 peptide blocked NOXA1-Nox1 binding and inhibited colon carcinoma and endothelial oxidants and migration.
Conclusion: The findings identify a NOXA1-activating domain and an isoform-specific Nox1 inhibitor.
Significance: The data provide insight into Nox1 regulation and present a potential therapy for suppressing oxidative stress-related disease.
Keywords: Enzyme Inhibitors, Enzyme Mechanisms, NADPH Oxidase, Peptides, Reactive Oxygen Species (ROS), Nox1
Abstract
Excessive vascular and colon epithelial reactive oxygen species production by NADPH oxidase isoform 1 (Nox1) has been implicated in a number of disease states, including hypertension, atherosclerosis, and neoplasia. A peptide that mimics a putative activation domain of the Nox1 activator subunit NOXA1 (NOXA1 docking sequence, also known as NoxA1ds) potently inhibited Nox1-derived superoxide anion (O2⨪) production in a reconstituted Nox1 cell-free system, with no effect on Nox2-, Nox4-, Nox5-, or xanthine oxidase-derived reactive oxygen species production as measured by cytochrome c reduction, Amplex Red fluorescence, and electron paramagnetic resonance. The ability of NoxA1ds to cross the plasma membrane was tested by confocal microscopy in a human colon cancer cell line exclusively expressing Nox1 (HT-29) using FITC-labeled NoxA1ds. NoxA1ds significantly inhibited whole HT-29 carcinoma cell-derived O2⨪ generation. ELISA and fluorescence recovery after photobleaching experiments indicate that NoxA1ds, but not its scrambled control, binds Nox1. FRET experiments conducted using Nox1-YFP and NOXA1-CFP illustrate that NoxA1ds disrupts the binding interaction between Nox1 and NOXA1, whereas a control peptide did not. Moreover, hypoxia-induced human pulmonary artery endothelial cell O2⨪ production was completely inhibited by NoxA1ds. Human pulmonary artery endothelial cell migration under hypoxic conditions was also reduced by pretreatment with NoxA1ds. Our data indicate that a peptide recapitulating a putative activation subdomain of NOXA1 (NoxA1ds) is a highly efficacious and selective inhibitor of Nox1 activity and establishes a critical interaction site for Nox1-NOXA1 binding required for enzyme activation.
Introduction
NADPH oxidases (Noxes)2 represent a diverse family of transmembrane proteins with the primary catalytic function of generating reactive oxygen species (ROS), including superoxide (O2⨪) and hydrogen peroxide (H2O2) (1). The effects of ROS signaling in the cell are diverse and key to cell functions; thus, it remains critical to precisely define the regulation of ROS sources and the species produced. The diverse roles of individual types of ROS are illustrated by the ability of H2O2 to oxidize enzymatic cofactors such as the tetrahydrobiopterin utilized by nitric oxide synthase to produce NO, thus uncoupling the enzyme (2); independently, however, O2⨪ reacts with NO neutralizing its bioactivity and forming the tyrosine-nitrating compound peroxynitrite (ONOO−) (3, 4). Additionally, O2⨪ and its dismuted metabolite H2O2 oxidize thiols, causing functional alterations in protein structure/function and enhancing growth factor signaling by inactivation of protein phosphatases that subserve normal cell signaling (5–7). To complicate matters, most mammalian cells abundantly express superoxide dismutase (SOD), which rapidly converts O2⨪ to the relatively stable H2O2 and facilitates the generation of downstream-oxidizing ROS (8). Excessive stimulation of Noxes often leads to hyperproliferative and migratory phenotypes as well as oxidative stress, resulting in end-organ damage (9). Increasingly, the roles of individual Nox isoforms are being appreciated as playing highly localized, specific, and temporal roles in disease progression. Indeed, Nox1's localization to smooth muscle caveolae and endosomes could limit Nox1's effect to a specific signaling pathway. Thus, specific inhibitors of Nox1 are likely to facilitate the identification of these links while also serving as a basis for therapy.
The production of ROS by Nox enzymes is achieved through a conserved catalytic mechanism contained within the enzyme's transmembrane domain that causes the transfer of electrons from NADPH to molecular oxygen to form O2⨪ and/or H2O2 (10). Although this catalytic core is conserved among Nox isoforms, it alone is insufficient for activity in a subclass of Noxes, including Nox1. Indeed, key interactions between Nox1 and its co-activating cytosolic subunits play a major role in activation of this enzyme domain by forming a heterodimeric complex essential for ROS generation (1, 11). Canonical Nox2 oxidase, the closest homologue of Nox1 and for which much more is known, requires the GTPase Rac, organizer subunit p47phox, and activator subunit p67phox for activation (12–14), whereas canonical Nox1 requires interaction with the homologous organizer NOXO1 and activator NOXA1 (15). Nox1 also requires the small GTPase Rac and the transmembrane protein p22phox (1). The assembled complex of subunits becomes the functional Nox1 oxidase. The biochemical structure and function of NOXA1 and NOXO1 are largely unknown due to their more recent discovery. That said, p67phox contains an “activation domain” spanning amino acids 190–210 that participates in the catalytic reduction of FAD (16). This domain shares 50 and 80% homology with corresponding residues 191–211 and 198–208 of NOXA1, respectively (17). Herein, we provide evidence that residues 195–205 of NOXA1 serve a function similar to that of the activation domain of p67phox and, in turn, hypothesize that this peptide could serve as a viable inhibitor of NOXA1-Nox1 binding and activation. With the knowledge that mutagenesis of a tyrosine for an alanine at residue 199 of NOXA1 reduces Nox1-derived O2⨪ production by >95% (18), we postulated that the same substitution would render this truncated peptide as a highly effective inhibitor of Nox1-derived O2⨪ production. In addition, by including amino acids that are homologous (residues 200 to 205) and nonhomologous between NOXA1 and p67phox (residues 195–198), we surmised that the peptide would be rendered specific for the Nox1 oxidase. The peptide representing amino acids 195–205 of NOXA1 with a Y199A substitution is heretofore referred to as NOXA1 docking sequence (NoxA1ds) and was tested for its efficacy as a Nox1 inhibitor (Scheme 1).
SCHEME 1.
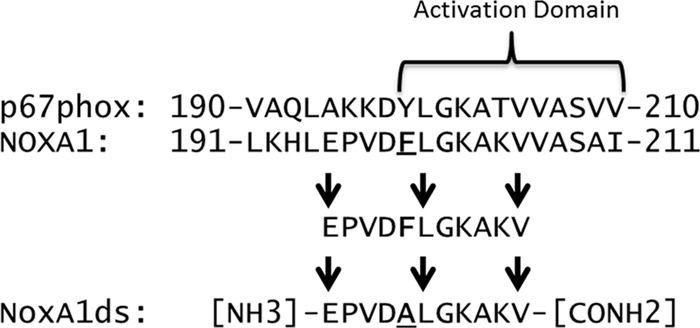
Design of NoxA1ds. Alignment of p67phox and NOXA1 indicates a highly homologous region between the two sequences within the activation domain. Phe-199 (F199) in NOXA1 was previously shown to be critical for enzymatic activity of Nox1. We selected an 11-mer sequence that contained critical portions of the activation domain (i.e. Phe-199), substituted Phe-199 to Ala, and used the resulting peptide as NoxA1ds.
Tissue distribution of the Noxes is wide and variable with Nox1 being uniquely expressed in the colon epithelium and prevalent in blood vessels (19, 20). In the colonic epithelium, Nox1 acts to potentiate phosphotyrosine signaling and to decrease bacterial virulence (21) as well as participate in carcinogenesis. Nox1 has also been broadly identified as a contributor to tumor growth by enhancing angiogenesis and a range of cardiovascular diseases, including ischemia reperfusion injury, neointimal cell hyperplasia, and hypertension-induced hypertrophy (22–29).
Targeted and specific inhibition of Nox1 has long been desired, and yet there remains a dearth of Nox1-specific inhibitors with a validated target and established mechanism. Here, we report that a peptide derived from the activation domain of NOXA1 is an isoform-specific inhibitor of Nox1 and that this peptide not only binds Nox1 but also disrupts its interaction with NOXA1. The current findings elucidate a key domain of NOXA1 as a binding partner for Nox1, shed light on the mechanism of Nox1 oxidase assembly, and provide a useful tool for investigating the role of Nox1 in a variety of cellular processes and pathologies.
EXPERIMENTAL PROCEDURES
Cytochrome c, superoxide dismutase (SOD), lithium dodecyl sulfate, catalase, horseradish peroxidase (HRP), hypoxanthine, PMA, and rhodamine B were purchased from Sigma. Xanthine oxidase was obtained from Calbiochem. Amplex Red was purchased from Invitrogen. Protease inhibitor mixture was purchased from Roche Diagnostics. NoxA1ds, Nox2dsTAT, and scrambled NoxA1ds were synthesized by the Tufts University Core Facility (Boston). The sequence of human NoxA1ds is as follows: NH3-EPVDALGKAKV-CONH2. The scrambled NoxA1ds sequence (SCRMB) is as follows: NH3-LVKGPDAEKVA-CONH2. In both cases, the NH3 group represents the amino end and CONH2 represents the amide of the C terminus, a consequence of the synthetic procedure. Each peptide was prepared in several batches, with no batch having a purity of less than 90%. FITC-labeled NoxA1ds was also synthesized by Tufts University and is identical in sequence to NoxA1ds with FITC linked to the N terminus of NoxA1ds via an α-hydroxyl acid. Rhodamine-labeled NoxA1ds was synthesized by Fisher and is identical in sequence to NoxA1ds with rhodamine B linked to the N terminus of NoxA1ds via an α-hydroxyl acid.
Cell Lines
COS-22 (COS-7 cells stably expressing human p22phox) and COS-Nox2 (also known as COS-phox) cells (COS-7 cells stably expressing human p22phox, Nox2, p47phox, and p67phox) were kindly provided by Dr. Mary C. Dinauer (Indiana University School of Medicine). COS-22 cells were maintained in Dulbecco's modified Eagle's medium (Cellgro) with 4.5 g/liter glucose, l-glutamine, and sodium pyruvate containing 10% heat-inactivated fetal bovine serum (FBS, Invitrogen), 100 units/ml penicillin, and 100 μg/ml streptomycin (Invitrogen) supplemented with 1.8 mg/ml G418 (Calbiochem). COS-Nox2 cells were maintained in otherwise identical media supplemented with 1 μg/ml puromycin (Sigma) and 0.2 mg/ml hygromycin B (Invitrogen). HEK-Nox5 (HEK-293 cells stably expressing human Nox5) were kindly provided by Dr. David Fulton (Georgia Health Sciences University) and were maintained in DMEM with 4.5 g/liter glucose, l-glutamine, and sodium pyruvate containing 10% FBS, 100 units/ml penicillin, and 100 μg/ml streptomycin. HT-29 cells were purchased from ATCC and cultured in McCoy's 5a modified medium (Manassas, VA). Human pulmonary artery endothelial cells (HPAEC) and their growth medium (EBM-2) were purchased from Lonza (Basel, Switzerland). COS, HEK, and HT-29 cells were used within 7–10 passages for all experiments, and HPAEC were used within 5–8 passages. Murine peritoneal macrophages were isolated as described by Zhang et al. (30).
Plasmid Preparation, Amplification, and Purification
Plasmids encoding full-length human cDNAs for Nox1 (pcDNA3.1-hNox1), NOXO1 (pcDNA3.1-hNOXO1), NOXA1 (pCMVsport 6-hNOXA1), and Nox4 (pcDNA3-hNox4) were kindly provided by Dr. David Lambeth (Emory University, Atlanta, GA) (31, 32). Plasmids encoding full-length human cDNAs for Nox1-YFP and NoxA1-CFP were custom subclones purchased from OriGene (Rockville, MD). Nox1-YFP was subcloned into the pCMV6-AC-mYFP plasmid, and NOXA1-CFP was subcloned into the pCMV6-AC-mCFP plasmid. Both plasmids placed the fluorophore at the C terminus of the Nox protein sequence. A membrane-targeted CFP with extensive N-terminal myristoylation (CFPm) was kindly provided by Dr. Jean-Pierre Vilardaga (Pittsburgh, PA) (33). Plasmids encoding Nox1, NOXO1, NOXA1, Nox1-YFP, NOXA1-CFP, or CFPm were transformed and amplified into Escherichia coli strain TOP10 (Invitrogen). Plasmids were purified using a QIAfilter plasmid purification kit (Qiagen Inc., Valencia, CA.).
Transfection
Cell transfection was carried out using Lipofectamine LTX and Plus reagent (Invitrogen), according to the manufacturer's instructions. COS-22 cells were singly or in combination transiently transfected with the above Nox plasmids. Cells were used for experiments 24 h after transfection.
Detection of Nox1/2/5-derived O2⨪ Production
Nox1- and Nox2-derived O2⨪ production was measured as described by Csanyi et al. (34) with minor modifications. Briefly, separate populations of COS-22 cells were transfected with either pcDNA3.1-hNox1 alone (COS-Nox1) or a combination of pcDNA3.1-hNOXO1 and pCMVsport 6-hNOXA1 (COS-NOXO1/A1). Adherent cells were harvested by incubating with 0.05% trypsin/EDTA for 5 min at 37 °C. Following addition of DMEM, 10% FBS to neutralize the trypsin, the cells were pelleted by centrifugation at 1000 × g for 5 min at 4 °C and subsequently resuspended at 5 × 107 cells/ml in lysis buffer (8 mm potassium, sodium phosphate buffer, pH 7.0, 131 mm NaCl, 340 mm sucrose, 2 mm NaN3, 5 mm MgCl2, 1 mm EGTA, 1 mm EDTA, 1 mm DTT, and protease inhibitor mixture) (35). The cells were lysed by freeze/thaw cycles (five cycles) and passed through a 30-gauge needle five times to further lyse the cells and then centrifuged at 1000 × g for 10 min at 4 °C to remove unbroken cells. The supernatant was then centrifuged at 160,000 × g for 60 min at 4 °C to yield a membrane-enriched pellet (membrane fraction). The membrane fraction from COS-Nox1 was resuspended in lysis buffer and retained, whereas the COS-NOXO1/A1 cytosolic fraction was retained. O2⨪ generation was measured in oxidase assay buffer (65 mm sodium phosphate buffer, pH 7.0, 1 mm EGTA, 10 μm FAD, 1 mm MgCl2, 2 mm NaN3, and 0.2 mm cytochrome c with added 1000 units/ml catalase to prevent H2O2-mediated oxidation of cytochrome c (35)). The components of the cell-free system were added in the following order: oxidase assay buffer, COS-Nox1 cell membrane fraction (5 × 105 cell eq/well, ∼5 μg of protein/well), and NoxA1ds/SCRMB peptides followed by 10 min incubation on ice, after which COS-NOXO1/A1-containing cytosolic fractions were added (5 × 105 cell eq/well). Plates were placed on an orbital shaker for 5 min at 120 movements/min at room temperature before addition of 180 μm NADPH to initiate O2⨪ production. The production of O2⨪ was calculated from the initial linear rate (over 15 min) of SOD-inhibitable cytochrome c reduction quantified at 550 nm using an extinction coefficient of 21.1 mm−1 cm−1 (Biotek Synergy 4 Hybrid Multi-Mode Microplate Reader). The concentration of NoxA1ds peptide that caused 50% inhibition of O2⨪ production (IC50) in COS-Nox1 cell lysates was calculated by Prism 5 (GraphPad Software, Inc., La Jolla, CA).
To measure Nox2-derived O2⨪ production, COS-Nox2 cells were separated into membrane and cytosolic fractions as described above. The production of O2⨪ was measured using identical methods with the addition of 130 μm lithium dodecyl sulfate after incubation with NoxA1ds or SCRMB. Measurement of HEK-Nox5 O2⨪ production was determined using methods exactly as described previously with NoxA1ds and SCRMB peptides being added to HEK-Nox5 membrane lysates prior to the addition of calcium (final concentration 20 μm) (13). Throughout these procedures, extreme care was taken to maintain the lysate at a temperature close to 4 °C.
O2⨪ production by peritoneal macrophages was determined after re-plating cells into 96-well white micro-plates (Greiner-Bio One GmbH, Germany) at a density of 5 × 104 cells/well. Cells were preincubated with 10 μm NoxA1ds for 1 h in reduced serum media at 37 °C before addition of 1 μm PMA and 400 μm luminol derivative L-012 for 30 min. Luminescence was quantified over time using a Biotek Synergy 4 Hybrid Multi-Mode Microplate Reader.
Nox4-derived H2O2 Production
H2O2 production was quantified in COS-Nox4 cell lysates as described previously (36). It is important to note that COS-Nox4 cells do not produce O2⨪ (34). COS-Nox4 and COS-22 cells were suspended to a concentration of 5 × 107 cells/ml in ice-cold disruption buffer (PBS containing 0.1 mm EDTA, 10% glycerol, protease inhibitor mixture, and 0.1 mm PMSF). The cells were lysed by five freeze/thaw cycles and passed through a 30-gauge needle five times to further lyse the cells. Incubation of COS-Nox4 cell lysate (10 μg/100 μl) with NoxA1ds was performed in assay buffer (25 mm HEPES, pH 7.4, containing 120 mm NaCl, 3 mm KCl, 1 mm MgCl2, 25 μm FAD, 0.1 mm Amplex Red, and 0.32 units/ml of HRP) for 15 min at room temperature on an orbital shaker (120 movements/min), before the addition of 36 μm NADPH, to initiate H2O2 production. This relatively low concentration of NADPH was used because it was found that higher concentrations interfered with Amplex Red fluorescence. Fluorescence measurements were made using a Biotek Synergy 4 Hybrid Multi-Mode Microplate Reader (excitation wavelength, 560 nm; emission wavelength, 590 nm). A standard curve of known H2O2 concentrations was developed using the Amplex Red assay (as per the manufacturer's instructions) and was used to quantify H2O2 production in the COS-Nox4 cell-free system. The reaction was monitored at room temperature for 60 min. The emission increase was linear during this interval. The effect of NoxA1ds on Nox4-derived H2O2 production was expressed as percent inhibition of Nox4, which was calculated by designating the amount H2O2 production by control mixtures in the absence of peptide as 100%.
Xanthine Oxidase-derived O2⨪ Production
The nitroxide spin probe 1-hydroxy-3-methoxycarbonyl-2,2,5,5-tetramethylpyrrolidine hydrochloride (CPH; Alexis Corp., San Diego) was used to examine O2⨪ production using a Bruker eScan Table-Top EPR spectrometer (Bruker Biospin). O2⨪ production in semi-purified preparations of xanthine oxidase initiated by the addition of 100 μm xanthine was measured in Krebs/HEPES buffer (100 mm NaCl, 5 mm KCl, 2.5 mm CaCl2, 1.2 mm MgSO4, 25 mm NaHCO3, 1.0 mm KH2PO4, 5.6 mm d-glucose, 20 mm Na-HEPES) supplemented with 50 μm CPH. Purified xanthine oxidase was incubated with NoxA1ds or SCRMB for 5 min at room temperature. Analyses of the CPH up-field spectra peak height were used to quantify the amount of O2⨪ produced by the lysates and were compared with buffer-only control spectra or spectra in the presence of NoxA1ds, SCRMB, or 200 units/ml SOD. To minimize the deleterious effects of contaminating metals, the buffers were treated with Chelex resin and contained 25 μm deferoxamine (Noxygen Science Transfer, Germany). The EPR instrument settings were as follows: field sweep, 50 G; microwave frequency, 9.78 GHz; microwave power 20 milliwatts; modulation amplitude, 2 G; conversion time, 327 ms; time constant, 655 ms; receiver gain, 1 × 105, DETC (Noxygen Science Transfer, Germany).
Detection of O2⨪ Production by Whole Cells Treated with NoxA1ds
HT-29 cells at 80% confluence were serum-starved in media containing 0.5% FBS for 12 h, and NoxA1ds (final concentrations of 0.1, 0.3, 1, 3, or 5 μm) was added directly to the growth media for 1 h prior to cell lysis and membrane preparation for cytochrome c assay. Alternatively, cells were suspended in Krebs/HEPES buffer with NoxA1ds for 1 h before addition of CPH to detect O2⨪ production via EPR.
To measure HPAEC O2⨪ production, cells were grown to 80% confluence prior to 12 h of serum starvation (0.2% FBS). Cells were placed in normoxic (20% O2) or hypoxic (1% O2) conditions for 23 h and then treated with NoxA1ds (10 μm) or SCRMB (10 μm) for 1 h.
For both HT-29 and HPAEC, cells were lysed in lysis buffer by five freeze/thaw cycles and five passages through a 30-gauge needle. Membranes fractions were collected by centrifugation (28,000 × g, 20 min). Membranes were suspended in oxidase assay buffer, and NADPH-dependent O2⨪ production was determined using cytochrome c reduction.
Enzyme-linked Immunosorbent Assay (ELISA)
ELISA was used to determine whether NoxA1ds binds Nox1. Neutravidin-coated plates (Thermo Scientific, Rockford, IL) were incubated with 10 μm biotinylated NoxA1ds or 10 μm biotinylated SCRMB (Tufts University Core Facility, Boston) for 2 h at room temperature. The plates were washed three times with wash buffer (25 mm Tris, 150 mm NaCl, and 0.05% Tween 20, pH 7.2). After 1 h of incubation at room temperature with 50 μg of membrane fractions prepared from COS-22, COS-Nox1, or COS-Nox2 cells were added to plates in phosphate-buffered saline, pH 7.2, at room temperature and allowed to incubate for 1 h. Rabbit polyclonal Nox1 antibody (1:500; Santa Cruz Biotechnology, Santa Cruz, CA) was added to detect Nox1 bound to NoxA1ds or SCRMB, respectively. After 1 h of incubation and extensive washing, bound primary antibodies were detected by the addition of FITC-labeled goat anti-rabbit IgG antibody (1:1000; 30 min Sigma). The fluorescence of each well was measured using a Biotek Synergy 4 Hybrid Multi-Mode Microplate Reader (excitation, 488 nm and emission, 518 nm; BioTek, Winooski, VT).
Fluorescence Recovery after Photobleaching (FRAP)
FRAP was performed using an Olympus FV1000 confocal microscope with minor modifications from studies by Wheeler et al. (37). Briefly, circular regions of interest (1.0 μm2) were selected and bleached with a 400-ms pulse from a 488-nm (YFP) and a 559-nm (rhodamine) laser line using the SIM scanner; recovery data were acquired using the instrument's main scanner and the 488- and 559-nm lines of an argon gas laser. This short pulse was selected to ensure a Gaussian bleaching spot. To maximize reproducibility of the experimental conditions, all data were acquired in the photon-counting mode of the instrument. Thirty five images were then collected at intervals of ∼7 s. Average fluorescence intensities of the bleached and control regions in other cells or the far-removed regions of the same cell were obtained and used to account for bleaching resulting from image acquisition.
FRET
COS-22 or HPAEC cells seeded at 20% density on glass-bottomed tissue culture plates were co-transfected with Nox1-YFP and NOXA1-CFP or Nox1-YFP and CFPm. After 24 h, cells were washed with PBS and fixed in 2% paraformaldehyde. The interaction between Nox1 or CFPm and NOXA1 was then detected using a combination laser scanning microscope system (Nikon A1 confocal) and quantified by acceptor photobleaching. To achieve excitation, the 458 nm line of an argon ion laser was focused through the Nikon ×60 oil differential interference contrast objective. Emissions of YFP (the FRET acceptor) and CFP were collected through 530–575- and 475–500-nm barrier filters, respectively. Photobleaching was performed with 50 iterations and 100% intensity of a 514-nm laser. Using methods described previously, average fluorescence intensities/pixel were calculated following background subtraction (38). To determine the effect of NoxA1ds on Nox1-NOXA1 association, cells transfected with Nox1-YFP and NOXA1-CFP were incubated with 10 μm NoxA1ds or SCRMB 24 h post-transfection for 1 h before fixing the cells. To determine the effect of VEGF on Nox1-NOXA1 association, HPAEC transfected with Nox1-YFP and NOXA1-CFP were treated with 20 nm VEGF for 1 h, 24 h post-transfection. FRET efficiency was calculated as follows: EFRET = (CFPpost/CFPpre −1)·100. The efficiency of the fluorescence resonance energy transfer directly reflects the distance separating the donor and the acceptor.
Cell Migration Assay
Scratch assay was performed as described by Liang et al. (39) with minor modification. Briefly, HPAEC were grown to confluence in 35-mm dishes before growth media were replaced with starvation media (1:10 dilution of growth media). Each dish was marked on its bottom with ink bisecting the dish into two halves. HPAEC were starved for 16 h before the cell monolayer was disrupted with a P1000 pipette tip in one stroke that passed through both halves of the dish. The scratch was photographed at positions immediately above and below the halfway mark. Cells were treated with ±20 nm VEGF, ±10 μm NoxA1ds, ±10 μm SCRMB, and ±10 μm Nox2dsTAT and placed in either 1.0% O2 or 20% O2 environment at 37 °C for 24 h. VEGF was added only at time 0, whereas peptides were added at time 0 and every 4 h thereafter. After 24 h, the cells were again photographed immediately above and below the halfway mark on the dish. ImageJ software was used to calculate the change in distance between the cell fronts as compared with the time 0 photograph.
Statistical Analysis
All results are expressed as means ± S.E. Student's t test was used for simple comparisons between two data sets. One-way ANOVA with replication followed by a Bonferroni post-test was used to discern point differences in data sets with more than two groups. Statistical analyses and IC50 determinations were performed using GraphPad Prism 5 software. A p value of <0.05 was considered statistically significant.
RESULTS
NoxA1ds Is a Potent Inhibitor of Nox1-derived O2⨪ in Cell-free Preparations
To investigate the ability of NoxA1ds (NH3-EPVDALGKAKV-CONH2) to inhibit Nox1, O2⨪ was assessed in a reconstituted canonical COS-Nox1 oxidase system expressing Nox1, p22phox, NOXA1, and NOXO1. Previous reports suggested that a related activation domain in NOXA1 homologue p67phox interacts with FAD sites on Nox2's C-terminal tail (40). To maximize the potential for NoxA1ds and Nox1 to interact in this manner, membrane-integrated fractions from COS-Nox1 cells containing holoprotein Nox1 with its C-terminal tail were incubated with cumulative concentrations of NoxA1ds (10−12–10−5 m) before adding cytosolic fractions containing NOXA1 and NOXO1. NoxA1ds concentration-dependently inhibited O2⨪ production with an IC50 of 20 nm (Fig. 1A). Maximal inhibition of Nox1 was achieved at 1.0 μm NoxA1ds. In concert with these experiments, we designed a scrambled control peptide consisting of an identical amino acid composition in randomized order (SCRMB, NH3-LVKGPDAEKVA-CONH2).
FIGURE 1.

NoxA1ds inhibits nox1-derived O2⨪ but not related enzymes. Production of O2⨪ was calculated by monitoring the reduction of cytochrome c for 15 min post-NADPH addition and subtracting base-line cytochrome c reduction occurring in the presence of SOD. A, production of O2⨪ as measured by the reduction of cytochrome c by cell lysates from COS cells transiently transfected with the Nox1 oxidase. Increasing concentrations (from 0.1 to 10,000 nm) of NoxA1ds caused a dose-dependent inhibition of O2⨪ production with an IC50 of 19 nm. Maximal inhibition of 88% of total O2⨪ production was achieved at a dose of 1.0 μm NoxA1ds. B, increasing concentrations of SCRMB did not inhibit Nox1-derived O2⨪. C, production of O2⨪ as measured by the reduction of cytochrome c by cell lysates from COS cells transiently transfected with the Nox2 oxidase. Lysates were treated with increasing concentrations of NoxA1ds (from 0.1 to 10,000 nm) prior to stimulation of enzyme assembly with 130 μm lithium dodecyl sulfate and enzyme activation with 180 μm NADPH. D, production of H2O2 as measured by Amplex Red fluorescence 15 min post-NADPH addition by membrane fractions prepared from COS cells transiently transfected with the Nox4 oxidase. Lysates were treated with increasing concentrations of NoxA1ds (from 100 to 10,000 nm). E, production of O2⨪ as measured by the reduction of cytochrome c by cell lysates from HEK293 cells stably transfected with the Nox5 oxidase stimulated with Ca2+ as described by Banfi et al. (13) and treated with increasing concentrations of NoxA1ds (from 0.1 to 10,000 nm). F, production of O2⨪ as measured by electron paramagnetic resonance from pure xanthine oxidase enzyme preparations, stimulated with hypoxanthine and pretreated with increasing concentrations of NoxA1ds (from 0.1 to 10,000 nm). In all panels, enzyme activity was evaluated 15 min post-enzyme activation with substrate (20 min post-NoxA1ds treatment), and no inhibition of the signal was observed in B–F. n = 9–12, three to four separate experiments.
Multiple variations of SCRMB with different amino acid sequences were initially considered as control peptides. This sequence was chosen as the control peptide as it did not preserve the order of charged residues nor the central key amino acid sequence of the activation domain (VDAL). SCRMB did not inhibit Nox1 (Fig. 1B).
NoxA1ds Is an Isoform-specific Inhibitor of Nox1
Isoform specificity was explored by testing the ability of NoxA1ds to inhibit Nox2, Nox4, and Nox5. Cell lysates were prepared from COS-Nox2, COS-Nox4, or HEK-Nox5 cells, and O2⨪ (Nox2/Nox5) or H2O2 (Nox4) production was measured using the reduction of cytochrome c or Amplex Red fluorescence, respectively. NoxA1ds was applied to prepared membrane fractions before addition of fractions containing cytosolic subunits (absent in Nox4 and Nox5 preparations) to maximize its ability to inhibit the oxidase. NoxA1ds did not inhibit Nox2-derived O2⨪ production, Nox4-derived H2O2 production, or Nox5-derived O2⨪ production (Fig. 1, C–E). Additionally, the ability of NoxA1ds to inhibit O2⨪ production by xanthine oxidase (XO), a molybdenum-dependent enzyme that produces O2⨪, was investigated. XO produces O2⨪ as a result of purine catabolism where electrons from the substrate (hypoxanthine) are transferred to oxygen as the enzyme produces xanthine and uric acid (41, 42). The hydroxylamine EPR spin probe CPH was used to detect O2⨪ produced by XO in the presence or absence of O2⨪ scavenger SOD or the XO-specific inhibitor allopurinol (measuring CP• signal intensity). Concentrations of NoxA1ds up to 10 μm did not inhibit XO-derived O2⨪ as compared with SOD and allopurinol controls, which abolished the signal (Fig. 1F). Each Nox and XO was also tested for the effect of SCRMB control peptide; no effect on ROS production by any of these oxidases was observed (data not shown). These data indicate that NoxA1ds does not inhibit Nox2-, Nox4-, Nox5-, or XO-derived ROS. In addition, the data indicate that NoxA1ds does not scavenge either O2⨪ or H2O2.
NoxA1ds Inhibits O2⨪ in Whole Cells
We used HT-29 cells to explore whether NoxA1ds could be used as a viable pharmacological inhibitor in intact cells. HT-29 are a unique population of colon carcinoma cells that express Nox1 without expression of other Nox isoforms (43). To test for permeability, HT-29 cells were treated with a FITC-labeled NoxA1ds variant for 1 h before imaging. Confocal microscopy revealed that NoxA1ds permeated the cell membrane of HT-29 cells and localized to the cytoplasm (Fig. 2, A–D). To test whether NoxA1ds inhibits Nox1-derived O2⨪ production in intact cells containing Nox1, HT-29 cells were incubated with increasing NoxA1ds concentrations, and O2⨪ was measured in membrane fractions using cytochrome c. Fig. 2E illustrates concentration-dependent inhibition of HT-29 O2⨪ that was absent in cells treated with SCRMB control peptide. In whole HT-29 cells, the IC50 was 100 nm with maximal inhibition at 5.0 μm NoxA1ds. Nox1 inhibition in HT-29 cells by NoxA1ds was matched by Nox1 siRNA control (Fig. 2, F and G). As an additional control, we investigated whether NoxA1ds inhibited O2⨪ production in cells that do not express Nox1. Peritoneal macrophages isolated from Nox1-null mice were pretreated with NoxA1ds for 1 h before addition of PMA to stimulate O2⨪ production. No inhibition of O2⨪ production was observed using L-012 luminescence (Fig. 2H).
FIGURE 2.

NoxA1ds is cell-permeant and effective in whole colon adenocarcinoma cells yet ineffective in Nox1−/y cells. FITC-labeled NoxA1ds and native NoxA1ds were incubated with HT-29 colon adenocarcinoma cells. HT-29 cells bear the distinction of having abundant Nox1 expression while not expressing Nox2, Nox4, or Nox5. A–D, FITC-labeled NoxA1ds was incubated with HT-29 colon adenocarcinoma cells for 1 h prior to imaging. Confocal microscopy of these cells indicates that FITC-NoxA1ds penetrated the extracellular membrane and that its distribution is cytosolic and perinuclear. DIC, differential interference contrast. E, as measured by SOD inhibitable reduction of cytochrome c, increasing concentrations of NoxA1ds caused concentration-dependent inhibition of O2⨪ production by HT-29 cells. SCRMB peptide did not inhibit O2⨪ production, ***, p < 0.05 for 0.5, 1, and 5 μm NoxA1ds versus 0 μm NoxA1ds by one-way ANOVA. F, as measured by EPR, 10 μm NoxA1ds and Nox1 siRNA both significantly inhibited O2⨪ production by HT29 cells, and p values were determined by one-way ANOVA. G, Western blot analysis of Nox1/β-actin protein from Nox1 siRNA or control (Ctrl)-treated HT29 cells. H, peritoneal macrophages were isolated from Nox1 null mice, and O2⨪ production was measured by L-012 chemiluminescence after 2 h of incubation with NoxA1ds and subsequent PMA stimulation. No difference was observed between NoxA1ds-treated and -untreated samples. *, p < 0.05 for control versus siRNA, t test; AU is arbitrary units. All values are expressed as n = 9, except for H, which was quantified via three separate experiments; n = 10–12 cells from four individual mice.
NoxA1ds Binds to Nox1
An ELISA-based assay was used to test whether NoxA1ds binds to Nox1. Biotin-tagged NoxA1ds (B-NoxA1ds) applied to neutravidin-coated 96-well plates was added to membrane fractions from Nox1-transfected or nontransfected COS-22 cells followed by treatment with primary antibodies and then fluorescent secondary antibodies. After repeated washings, a 30% increase in fluorescence was observed in wells treated with membrane fractions of Nox1-transfected cells versus untransfected cells. This increase was not observed in wells treated with Nox1-transfected cell membranes added to wells containing biotin-tagged SCRMB control peptide (Fig. 3A). A similar ELISA-based assay indicated that Nox2 from COS-Nox2 membrane fractions did not bind NoxA1ds (Fig. 3B).
FIGURE 3.

NoxA1ds binds to Nox1 but does not detectably bind Nox2. A, neutravidin-coated plates were incubated with biotin-tagged NoxA1ds (Biotin-NoxA1ds) or biotin-tagged SCRMB (Biotin-SCRMB) before addition of cell membranes prepared from cells transfected with Nox1 (Nox1 membrane) or transfected with an empty vector (COS22 membrane). Bound Nox1 was detected through a FITC-conjugated secondary antibody bound to the Nox1 primary antibody. FITC fluorescence was expressed as binding as % COS22 membranes on each experimental day. There was no difference in binding between COS22 membranes and Nox1 membranes incubated with Biotin-SCRMB, whereas membranes incubated with Biotin-NoxA1ds showed a significant increase in binding when transfected with Nox1. n = 10–12, three separate experiments, *, p < 0.05, two-way ANOVA with Bonferroni post-test. B, neutravidin-coated plates were incubated with biotin-tagged NoxA1ds (Biotin-NoxA1ds) or biotin-tagged SCRMB (Biotin-SCRMB) before addition of cell membranes prepared from cells transfected with Nox2 (Nox2 membrane) or transfected with an empty vector (COS22 membrane). Bound Nox2 was detected through an Alexa 488-conjugated secondary antibody bound to the Nox1 primary antibody. Fluorescence was expressed as binding as % COS22 membranes on each experimental day. There was no difference in binding between COS22 membranes and Nox2 membranes incubated with Biotin-SCRMB or Biotin-NoxA1ds, C, FRAP of COS22 cells treated with 70 nm rhodamine B-labeled NoxA1ds (R-NoxA1ds) in the absence and presence of Nox1YFP transfection and FRAP of Nox1YFP alone. Panel to the right represents mobile fraction of these groups after 250 s. *, p < 0.05, one-way ANOVA with Bonferroni post test.
To corroborate these results, FRAP of a rhodamine-labeled NoxA1ds (R-NoxA1ds) in untransfected cells and those transfected with Nox1-YFP was quantified. As seen in Fig. 3B, R-NoxA1ds completely recovered after 250 s in untransfected cells, although R-NoxA1ds recovery was significantly slower and markedly attenuated in cells transfected with Nox1-YFP, i.e. by 250 s, and R-NoxA1ds had recovered less than 50% in Nox1-YFP-transfected cells. The significant decrease in the mobile fraction of R-NoxA1ds- and Nox1-YFP-transfected versus nontransfected cells corroborates the ELISA data, indicating binding between Nox1 and NoxA1ds (Fig. 3C).
NoxA1ds Interrupts Nox1-NOXA1 Association
Upon observation of NoxA1ds-Nox1 binding, we proposed that NoxA1ds would interrupt association of Nox1 and NOXA1. To test this, we employed a FRET-based assay in which COS-22 cells were transfected with Nox1-YFP and NOXA1-CFP. Cells were analyzed by fixing followed by irreversible photobleaching using FRET. FRET is observed indicating protein interaction between two partners when the donor emission (CFP) signal increases after a nearby acceptor fluorophore (YFP) is inactivated by irreversible photobleaching. In these cells, photobleaching of the YFP label resulted in a concomitant increase in CFP fluorescence, together with a decrease in YFP signal intensity, indicating a direct interaction between Nox1-YFP and NOXA1-CFP (Fig. 4A). Hence, photobleaching effectively inactivated the acceptor (YFP-Nox1) that was associated with increased donor (CFP) fluorescence. We used a myristoylated CFP (CFPm), which is exclusively targeted to the cell membrane as a negative control (33). In cells expressing CFPm and Nox1-YFP, photobleaching of YFP did not result in an increase in CFP fluorescence, indicating FRET was not a result of coincidental membrane abundance and co-localization of these proteins (data not shown).
FIGURE 4.

NoxA1ds disrupts Nox1-NOXA1 interaction. FRET between Nox1-YFP- and NoxA1-CFP-transfected COS22 cells in the presence or absence of 10 μm NoxA1ds or SCRMB was evaluated. Relative fluorescence of CFP is green, and YFP is red. Traces below the images indicate fluorescent intensities of CFP and YFP below the arrow overlaid on each cell. A, transfected COS22 cells were treated with vehicle for 1 h prior to imaging cells; photobleaching of Nox1-YFP was complete and resulted in a concomitant increase in CFP fluorescence. B, transfected COS22 cells were treated with 10 μm SCRMB peptide for 1 h prior to imaging cells; photobleaching of Nox1-YFP was complete and also resulted in a concomitant increase in CFP fluorescence. C, transfected COS22 cells were treated with 10 μm NoxA1ds peptide for 1 h prior to imaging cells; photobleaching of Nox1-YFP was complete but did not result in a concomitant increase in CFP fluorescence. D, quantification of FRET efficiency from images A–C. Values expressed as n = 8, three separate experiments; ***, p < 0.001 by one-way ANOVA and Bonferroni post-test.
COS-22 cells transfected with Nox1-YFP and NOXA1-CFP were then incubated with 10 μm SCRMB NoxA1ds or NoxA1ds for 1 h before imaging. The SCRMB control peptide had no effect on CFP/YFP FRET coupling (Fig. 4B). Conversely, NoxA1ds significantly reduced FRET efficiency (Fig. 4C). These results indicate that Nox1 and NOXA1 directly interact and that this interaction is disrupted by NoxA1ds but not by control peptide (Fig. 4D).
Nox1-derived O2⨪ Mediates Endothelial Cell Migration
To test the role of this key Nox1-NOXA1 interaction in a pathophysiologically relevant vascular cell phenotype, we investigated whether Nox1-NoxA1ds binding was effective in HPAEC as these cells express abundant Nox1 with Nox4 being the most highly expressed (Fig. 5B). ELISA data indicated that NoxA1ds binds to Nox1 but not Nox2 or -4 in HPAEC (Fig. 5A). HPAEC were exposed to 1.0% oxygen for 24 h and treated with 10 μm NoxA1ds or SCRMB for 1 h before measuring O2⨪ production. Hypoxia caused a 3-fold increase in O2⨪ production that was completely inhibited by preincubation with NoxA1ds but not with SCRMB (Fig. 5, C and D). The effect of NoxA1ds was partially replicated using Nox1 siRNA, with detectable yet statistically insignificant inhibition of O2⨪ production. The incomplete inhibition of O2⨪ production was likely due to incomplete knockdown of Nox1 by the siRNA (Fig. 5E). To determine the effect of NoxA1ds on migration, we performed a wound assay on VEGF-stimulated HPAEC migration under hypoxic conditions (1.0% O2). NoxA1ds caused a significant reversion in endothelial cell migration to vehicle control levels, an effect that was not observed in cells treated with SCRMB (Fig. 6, A and B). Additionally, HPAEC transfected with Nox1-YFP and NOXA1-CFP showed an increase in FRET when treated with 20 nm VEGF that was inhibitable by NoxA1ds, indicating that VEGF promotes Nox1-NOXA1 association (Fig. 7).
FIGURE 5.

NoxA1ds attenuates hypoxia-induced O2⨪ production. A, NoxA1ds binds to Nox1 but not Nox2 or Nox4 in HPAEC. Neutravidin-coated plates were incubated with biotin-tagged NoxA1ds (B-NoxA1ds) or biotin-tagged SCRMB (B-SCRMB) before addition of HPAEC cell membranes. Captured Noxes were detected through a Alexa 488-conjugated secondary antibody bound to the Nox1, -2, or -4 primary antibody. Fluorescence was expressed with background lysis buffer fluorescence subtracted. When fluorescence was detected via Nox1 primary antibody, there was a significant increase in binding as compared with B-SCRMB. n = 4, *, p < 0.05, unpaired t test. B, relative Nox expression in HPAEC quantified by quantitative PCR. C, in hypoxia and normoxia, SCRMB and NoxA1ds peptides were added to cells at 10 μm for 1 h prior to cell lysis and quantification of enzyme activity. Cells were transfected with Nox1 siRNA 24 h prior to 24-h normoxic/hypoxic treatment followed by cell lysis and quantification of enzyme activity. SCRMB, NoxA1ds, and Nox1 siRNA had a negligible effect on O2⨪ production under normoxic conditions. Hypoxia (1.0% O2, 24 h) treatment resulted in a 3-fold increase in O2⨪ production that was unaffected by SCRMB. Upon treatment with NoxA1ds, O2⨪ production by cells subjected to hypoxia returned to the amount observed under normoxia. D, representative experimental trace for enzyme activity in B shown as the SOD-inhibitable reduction of cytochrome c over time. n = 9, three separate experiments. E, Western blot analysis of Nox1/β-actin protein from Nox1 siRNA or control-treated HPAEC. Nox1 knockdown was incomplete and approximates the degree of knockdown observed as O2⨪ production. F, hypoxia (1.0% O2, 24 h) treatment resulted in a 2-fold increase in HPAEC O2⨪ production that was unaffected by Nox2dsTAT or its control (SCRMB). n = 9, three separate experiments. *, p < 0.05; **, p < 0.01, one-way ANOVA with Bonferroni post-test.
FIGURE 6.

NoxA1ds inhibits VEGF-stimulated wound healing. Confluent HPAEC were scratched with a P1000 pipette tip, photographed, treated with 20 nm VEGF ± 10 μm NoxA1ds, and photographed again after 24 h. A, representative images of HPAEC immediately after scratch wounding and 24 h post-scratch wound. B, quantification of HPAEC wound closure VEGF ± 10 μm NoxA1ds. C, quantification of HPAEC wound closure treated with VEGF ± 10 μm Nox2dsTAT. Values represent n = 6–8, three to four separate experiments, *, p < 0.05 one-way ANOVA with Bonferroni post-test as compared with vehicle treatment.
FIGURE 7.

NoxA1ds disrupts VEGF-stimulated Nox1-NOXA1 interaction. FRET between Nox1-YFP and NoxA1-CFP transfected HPAEC in the presence or absence of 20 nm VEGF and/or 10 μm NoxA1ds. Relative fluorescence of CFP is green, and YFP is red. Traces below the images indicate fluorescent intensities of CFP and YFP below the arrow overlaid on each cell. A, transfected HPAEC were treated with vehicle for 1 h prior to imaging cells; photobleaching of Nox1-YFP was complete and resulted in a concomitant increase in CFP fluorescence. B, transfected HPAEC were treated with 20 nm VEGF for 1 h prior to imaging cells; photobleaching of Nox1-YFP was complete and resulted in an increase in CFP fluorescence of greater magnitude than without VEGF. C, transfected HPAEC were treated with 10 μm NoxA1ds peptide for 1 h prior 20 nm VEGF for 1 h. Cells were then imaged after VEGF treatment. Photobleaching of Nox1-YFP was complete but did not result in a concomitant increase in CFP fluorescence. D, quantification of FRET efficiency from images A–C. Values expressed as n = 6, two separate experiments; *, p < 0.05 versus vehicle; ***, p < 0.001 versus VEGF, one-way ANOVA and Bonferroni post-test.
Nox2 inhibition by Nox2ds-tat in HPAEC had a negligible effect on O2⨪ production and wound healing (Figs. 5F and 6C, respectively). Nox4 inhibition by siRNA significantly decreased cell viability, further corroborating its importance in HPAEC signaling, yet preventing quantitative ROS detection and wound healing measurements.
DISCUSSION
The lack of specific Nox inhibitors has stymied progress into the role of these enzymes in physiology and disease. In this study, we provide evidence for a potent and isoform-specific inhibitor of Nox1-based oxidase, NoxA1ds. To date, existing Nox1 inhibitors include DPI, ML171, and GKT137831 and RNA silencing technology (44–47). DPI has long been considered a potent Nox inhibitor, yet it is burdened by its lack of specificity and inhibition of other flavoproteins (44, 45). The compound ML171 was recently reported as a specific Nox1 inhibitor with no reported inhibition of Nox2 or Nox4 activity; however, no mechanism or target validation data have been presented (46). It also remains unknown whether ML171 acts on Nox5 or XO. The compound GKT137831 is a potent Nox1/4 inhibitor and bears the distinction of being the first Nox inhibitor to have reached phase I clinical trials (47). DPI and GKT137831 are limited by their lack of specificity against a single Nox isoform, and reports on ML171 and GKT137831 lack mechanistic insight and target validation. On a different note, RNA silencing, although often specific and effective in vitro, is generally not considered useful as therapeutic or pervasive in in vivo settings. Moreover, it requires days for effective protein knockdown and is limited by the requirement for cell transfection reagents and the unknown consequences of these agents on cell physiology.
We report here on the design, mechanism, and target validation of a specific Nox1 inhibitor (NoxA1ds) that is cell-permeant and highly potent. NoxA1ds was derived from a peptide whose structure is based on a short sequence of an essential Nox subunit; in this case, a putative activation domain of NOXA1. We demonstrate here that NoxA1ds binds directly to NOX1 and displaces NOXA1 to inhibit enzymatic activity and biological function.
NoxA1ds recapitulates a putative activation subdomain of NOXA1 with a Y199A substitution in the sequence of NOXA1 stretching from residues 195 to 205 (195EPVD(Y→A)LGKAKV205). This modification mimics a mutation first created in the holoprotein by Maehara et al. (18) that caused a 75% loss in activity in the reconstituted canonical Nox1 oxidase system. Although the function of this domain in NOXA1 is unclear, a related domain exists in p67phox, the activator of Nox2 (40). The activation domain of p67phox is known to be critical for catalytic Nox2 oxidase activity. In designing NoxA1ds as a potential competitive and specific inhibitor of Nox1-NOXA1 binding, we selected an activation subdomain in NOXA1 containing residues that are both conserved and nonconserved between NOXA1 and p67phox.
The defined p67phox activation domain (amino acids 190–210) and corresponding region in NOXA1 share 50% homology. NoxA1ds was intentionally derived from a corresponding portion of this region in which 46% of the amino acids are dissimilar between p67phox and NOXA1, i.e. NoxA1ds is derived from amino acids 195 to 205, in which the first four amino acids are EPVD as opposed to AKKD in p67phox. We postulated that this difference confers isoform specificity of NoxA1ds for Nox1. The lack of observed inhibition in Nox4 or Nox5 systems is likely a result of these Nox systems having no cytosolic activator subunit requirement. Finally, the lack of inhibition of XO by NoxA1ds is further confirmation that its inhibitory effect is not germane to all oxidases and is not a consequence of ROS scavenging.
NoxA1ds could potentially mimic functional sites in other proteins and thus interfere with their function. We used BLAST to compare the sequence of NoxA1ds to the National Institutes of Health translated nucleotide database to determine potential nonspecific protein interactions with NoxA1ds. We were surprised to observe that fewer than five mammalian proteins outside of the NADPH oxidase family shared significant homology with NoxA1ds. These hits were hypothetical proteins based on isolated DNA sequences. As such, we believe nonspecific actions of NoxA1ds should be negligible.
When NoxA1ds was added to cell-free preparations of the canonical Nox1 oxidase, composed of catalytic subunit Nox1, activating subunit NOXA1 and organizing subunit NOXO1 along with Rac, NoxA1ds inhibited Nox1-derived O2⨪ production with an IC50 of 19 nm achieving maximum inhibition of 90% at 1.0 μm. In addition, we showed that NoxA1ds does not scavenge either O2⨪ or H2O2 and does not inhibit Nox2, Nox4, Nox5, or XO activity. These results support NoxA1ds as an isoform-specific inhibitor of Nox1.
We extended our studies to determine the in vitro efficacy of NoxA1ds and observed that fluorescent analogues of NoxA1ds are capable of crossing the cellular membrane and localizing to the cytosol. Many short peptides require the small HIV TAT peptide moiety to penetrate cell membranes (48, 49). In contrast, our findings herein demonstrated that TAT is unnecessary for NoxA1ds to cross cell membranes, and we attribute this in part to the positively charged lysines in the C terminus of NoxA1ds and the alternating hydrophobic/hydrophilic amino acid structure of NoxA1ds. Indeed, other short peptides with similar hydropathy patterns have proven highly cell permeant and thus support our findings (50).
Upon proving its permeability in whole cells, we tested whether NoxA1ds was an effective inhibitor of O2⨪ production in a primary cancer cell line (HT-29) that exclusively expresses Nox1 oxidase and in HPAEC, which also express Nox2 and Nox4 (43, 51). We observed that NoxA1ds inhibited O2⨪ production in HT-29 and HPAEC cell lines. In conjunction with our data from COS cell lysates, we demonstrated the ability of NoxA1ds to inhibit Nox1-derived O2⨪ production in two distinct human cell populations as well as a heterologous COS system.
ELISA and FRAP data revealed that NoxA1ds binds to the catalytic Nox1 subunit, and FRET demonstrated that NoxA1ds disrupted the critical association of Nox1 with NOXA1 and that this binding is specific to Nox1 in HPAEC. Our FRET data indicate that not only is this putative activation domain critical for enzyme activity in Nox1 but that this domain is also a key region for the formation of Nox1-NOXA1 complexes (18). Maehara et al. (18) reported that the NOXA1 Y199A mutation interferes with FAD reduction in the enzyme complex and thus may prevent catalytic O2⨪ production. Notably, mutations in this region of p67phox are found in chronic granulomatous disease, an immunodeficiency disorder associated with inefficient Nox-derived ROS production (52). Nevertheless, our data are more in line with NoxA1ds preventing Nox1-NOXA1 binding and inhibiting O2⨪ production. Thus, we propose that this domain is crucial for the binding and activation of Nox1. To determine the relevance of our findings to physiological processes, we sought to investigate the contribution of Nox1-derived O2⨪ in primary cells. Hypoxia and ischemia/reperfusion injury have long been recognized as conditions that promote excessive ROS production in vitro and in vivo, and recently, we demonstrated that Nox1 can function as a key element in reducing vascular flow (27).
Considering the known effect of Nox1 on EC migration and our observation that hypoxia stimulates ROS (53, 54), we investigated the ability of NoxA1ds to inhibit hypoxia-induced endothelial cell ROS production and inhibit migration of endothelial cells. At 1.0% O2, we observed that O2⨪ production increases significantly, an effect that is normalized to normoxic levels after treatment with NoxA1ds. Finally, HPAEC migration stimulated by VEGF under hypoxia was significantly reduced as a result of treatment with NoxA1ds. Furthermore, we demonstrated that VEGF stimulates Nox1-NOXA1 interaction and that NoxA1ds disrupts this assembly supporting our findings with endothelial cell migration (Figs. 6 and 7). These data confirm the efficacy of NoxA1ds in a physiological process known to be Nox1-dependent. Taken together, our data indicate that the peptide NoxA1ds is a specific inhibitor of Nox1 and that this inhibition occurs via binding to the catalytic Nox1 subunit and blockade of Nox1-NOXA1 binding (Fig. 8).
FIGURE 8.
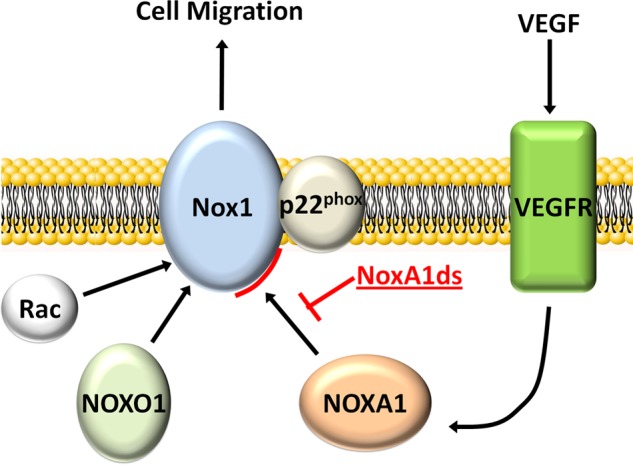
NoxA1ds mechanism of action schematic. The data presented here indicate that NoxA1ds binds to Nox1 and disrupts/prevents its VEGF-stimulated interaction with NOXA1, inhibiting endothelial cell migration.
Notwithstanding the broadly demonstrated effectiveness of related peptides by parenteral, peritoneal, subcutaneous, and direct application to blood vessels using gene therapy (46, 50), the limitations in the use of peptides as “druggable” therapeutics are obvious. These include a very limited oral bioavailability due to peptide degradation in the gut. As mentioned earlier, however, these issues are being circumvented by novel technologies, including the use of nanotechnologies (55) as well as the aerosolization of NoxA1ds directly into lungs to target pulmonary diseases with unpublished studies by our group3 indicating that this treatment protocol markedly reduces right ventricular hypertrophy in pulmonary hypertension. Beyond targeted peptide delivery, there are a number of peptide modifications that can support bioavailability by reducing protease degradation. These include hydrocarbon stapling and partial substitution of d-amino acids (56, 57).
The data we present here identify a novel and specific Nox1 inhibitor called NoxA1ds. NoxA1ds selectively inhibits Nox1-derived O2⨪ production by binding Nox1 and preventing the association of Nox1 with NOXA1. Additionally, as NoxA1ds was designed with modification from the corresponding activation domain of its homologue p67phox (residues 195–211), it has shed important new light on the identity and mechanism of an activation domain in the Nox1 oxidase system. Moreover, our findings characterize this region as not only critically important in catalytic activity and cell phenotypic changes but as a key binding interaction as well. By applying NoxA1ds to myriad biological systems in vitro and in vivo, it will now be possible to pharmacologically determine a role for Nox1. We also expect that NoxA1ds will become a platform for other Nox1 therapeutics and may serve as a plausible treatment in its own right.
This work was supported, in whole or in part, by National Institutes of Health Grant HL079207; Grants HL096376, HL097376, and HL098174 (to R. K. M.); and Grant HL116472 (to B. B. C.). This work was also supported by a merit review award from the United States Department of Veterans Affairs. This work is the subject of a pending United States patent application No. 61/560,075 titled “Novel Inhibitors of Nox1,” on which D. J. R. and P. J. P. are listed as inventors.
D. J. Ranayhossaini, I. Al Ghouleh, S. P. Tofovic, and P. J. Pagano, unpublished results.
- Nox
- NADPH oxidase
- COS-22
- COS-7 cells transfected with p22phox
- COS-NOX1
- COS-22 cells transfected with Nox1, NOXO1, and NOXA1
- COS-NOX2
- COS-22 cells transfected with, Nox2, p47phox, and p67phox
- COS-NOX4
- COS-22 cells transfected with Nox4
- HPAEC
- human pulmonary artery endothelial cells
- NOXA1
- Nox activator subunit 1
- NOXO1
- Nox Organizer subunit 1
- O2⨪
- superoxide anion
- ROS
- reactive oxygen species
- SOD
- superoxide dismutase
- XO
- xanthine oxidase
- X
- xanthine
- FRAP
- fluorescence recovery after photobleaching
- FRET
- fluorescence resonance energy transfer
- ANOVA
- analysis of variance
- PMA
- phorbol myristate acetate
- DPI
- diphenyleneiodonium chloride
- CFP
- cyan fluorescent protein
- YFP
- yellow fluorescent protein
- CPH
- 1-hydroxy-3-methoxycarbonyl-2,2,5,5-tetramethylpyrrolidine hydrochloride.
REFERENCES
- 1. Al Ghouleh I., Khoo N. K., Knaus U. G., Griendling K. K., Touyz R. M., Thannickal V. J., Barchowsky A., Nauseef W. M., Kelley E. E., Bauer P. M., Darley-Usmar V., Shiva S., Cifuentes-Pagano E., Freeman B. A., Gladwin M. T., Pagano P. J. (2011) Oxidases and peroxidases in cardiovascular and lung disease: new concepts in reactive oxygen species signaling. Free Radic. Biol. Med. 51, 1271–1288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kuzkaya N., Weissmann N., Harrison D. G., Dikalov S. (2003) Interactions of peroxynitrite, tetrahydrobiopterin, ascorbic acid, and thiols: implications for uncoupling endothelial nitric-oxide synthase. J. Biol. Chem. 278, 22546–22554 [DOI] [PubMed] [Google Scholar]
- 3. White C. R., Brock T. A., Chang L. Y., Crapo J., Briscoe P., Ku D., Bradley W. A., Gianturco S. H., Gore J., Freeman B. A., et al. (1994) Superoxide and peroxynitrite in atherosclerosis. Proc. Natl. Acad. Sci. U.S.A. 91, 1044–1048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Beckman J. S., Beckman T. W., Chen J., Marshall P. A., Freeman B. A. (1990) Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc. Natl. Acad. Sci. U.S.A. 87, 1620–1624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Juarez J. C., Manuia M., Burnett M. E., Betancourt O., Boivin B., Shaw D. E., Tonks N. K., Mazar A. P., Doñate F. (2008) Superoxide dismutase 1 (SOD1) is essential for H2O2-mediated oxidation and inactivation of phosphatases in growth factor signaling. Proc. Natl. Acad. Sci. U.S.A. 105, 7147–7152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cardey B., Foley S., Enescu M. (2007) Mechanism of thiol oxidation by the superoxide radical. J. Phys. Chem. A 111, 13046–13052 [DOI] [PubMed] [Google Scholar]
- 7. Winterbourn C. C., Metodiewa D. (1999) Reactivity of biologically important thiol compounds with superoxide and hydrogen peroxide. Free Radic. Biol. Med. 27, 322–328 [DOI] [PubMed] [Google Scholar]
- 8. McCord J. M., Fridovich I. (1969) Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). J. Biol. Chem. 244, 6049–6055 [PubMed] [Google Scholar]
- 9. Sedeek M., Hébert R. L., Kennedy C. R., Burns K. D., Touyz R. M. (2009) Molecular mechanisms of hypertension: role of Nox family NADPH oxidases. Curr. Opin. Nephrol. Hypertens. 18, 122–127 [DOI] [PubMed] [Google Scholar]
- 10. Lassègue B., Clempus R. E. (2003) Vascular NAD(P)H oxidases: specific features, expression, and regulation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 285, R277–R297 [DOI] [PubMed] [Google Scholar]
- 11. Ambasta R. K., Kumar P., Griendling K. K., Schmidt H. H., Busse R., Brandes R. P. (2004) Direct interaction of the novel Nox proteins with p22phox is required for the formation of a functionally active NADPH oxidase. J. Biol. Chem. 279, 45935–45941 [DOI] [PubMed] [Google Scholar]
- 12. Leto T. L., Garrett M. C., Fujii H., Nunoi H. (1991) Characterization of neutrophil NADPH oxidase factors p47-phox and p67-phox from recombinant baculoviruses. J. Biol. Chem. 266, 19812–19818 [PubMed] [Google Scholar]
- 13. Bánfi B., Tirone F., Durussel I., Knisz J., Moskwa P., Molnár G. Z., Krause K. H., Cox J. A. (2004) Mechanism of Ca2+ activation of the NADPH oxidase 5 (NOX5). J. Biol. Chem. 279, 18583–18591 [DOI] [PubMed] [Google Scholar]
- 14. Lyle A. N., Deshpande N. N., Taniyama Y., Seidel-Rogol B., Pounkova L., Du P., Papaharalambus C., Lassègue B., Griendling K. K. (2009) Poldip2, a novel regulator of Nox4 and cytoskeletal integrity in vascular smooth muscle cells. Circ. Res. 105, 249–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bánfi B., Clark R. A., Steger K., Krause K. H. (2003) Two novel proteins activate superoxide generation by the NADPH oxidase NOX1. J. Biol. Chem. 278, 3510–3513 [DOI] [PubMed] [Google Scholar]
- 16. Han C. H., Freeman J. L., Lee T., Motalebi S. A., Lambeth J. D. (1998) Regulation of the neutrophil respiratory burst oxidase. Identification of an activation domain in p67(phox). J. Biol. Chem. 273, 16663–16668 [DOI] [PubMed] [Google Scholar]
- 17. Ambasta R. K., Schreiber J. G., Janiszewski M., Busse R., Brandes R. P. (2006) Noxa1 is a central component of the smooth muscle NADPH oxidase in mice. Free Radic. Biol. Med. 41, 193–201 [DOI] [PubMed] [Google Scholar]
- 18. Maehara Y., Miyano K., Yuzawa S., Akimoto R., Takeya R., Sumimoto H. (2010) A conserved region between the TPR and activation domains of p67phox participates in activation of the phagocyte NADPH oxidase. J. Biol. Chem. 285, 31435–31445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Suh Y. A., Arnold R. S., Lassegue B., Shi J., Xu X., Sorescu D., Chung A. B., Griendling K. K., Lambeth J. D. (1999) Cell transformation by the superoxide-generating oxidase Mox1. Nature 401, 79–82 [DOI] [PubMed] [Google Scholar]
- 20. Csányi G., Taylor W. R., Pagano P. J. (2009) NOX and inflammation in the vascular adventitia. Free Radic. Biol. Med. 47, 1254–1266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Corcionivoschi N., Alvarez L. A., Sharp T. H., Strengert M., Alemka A., Mantell J., Verkade P., Knaus U. G., Bourke B. (2012) Mucosal reactive oxygen species decrease virulence by disrupting Campylobacter jejuni phosphotyrosine signaling. Cell Host Microbe 12, 47–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Arbiser J. L., Petros J., Klafter R., Govindajaran B., McLaughlin E. R., Brown L. F., Cohen C., Moses M., Kilroy S., Arnold R. S., Lambeth J. D. (2002) Reactive oxygen generated by Nox1 triggers the angiogenic switch. Proc. Natl. Acad. Sci. U.S.A. 99, 715–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ushio-Fukai M., Nakamura Y. (2008) Reactive oxygen species and angiogenesis: NADPH oxidase as target for cancer therapy. Cancer Lett. 266, 37–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Garrido-Urbani S., Jemelin S., Deffert C., Carnesecchi S., Basset O., Szyndralewiez C., Heitz F., Page P., Montet X., Michalik L., Arbiser J., Rüegg C., Krause K. H., Imhof B. A., Imhof B. (2011) Targeting vascular NADPH oxidase 1 blocks tumor angiogenesis through a PPARα mediated mechanism. PLoS One 6, e14665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ardanaz N., Pagano P. J. (2006) Hydrogen peroxide as a paracrine vascular mediator: regulation and signaling leading to dysfunction. Exp. Biol. Med. 231, 237–251 [DOI] [PubMed] [Google Scholar]
- 26. Cascino T., Csanyi G., Al Ghouleh I., Montezano A. C., Touyz R. M., Haurani M. J., Pagano P. J. (2011) Adventitia-derived hydrogen peroxide impairs relaxation of the rat carotid artery via smooth muscle cell p38 mitogen-activated protein kinase. Antioxid. Redox Signal. 15, 1507–1515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Csányi G., Yao M., Rodríguez A. I., Al Ghouleh I., Sharifi-Sanjani M., Frazziano G., Huang X., Kelley E. E., Isenberg J. S., Pagano P. J. (2012) Thrombospondin-1 regulates blood flow via CD47 receptor-mediated activation of NADPH oxidase 1. Arterioscler. Thromb. Vasc. Biol. 32, 2966–2973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jacobson G. M., Dourron H. M., Liu J., Carretero O. A., Reddy D. J., Andrzejewski T., Pagano P. J. (2003) Novel NAD(P)H oxidase inhibitor suppresses angioplasty-induced superoxide and neointimal hyperplasia of rat carotid artery. Circ. Res. 92, 637–643 [DOI] [PubMed] [Google Scholar]
- 29. Dikalova A., Clempus R., Lassègue B., Cheng G., McCoy J., Dikalov S., San Martin A., Lyle A., Weber D. S., Weiss D., Taylor W. R., Schmidt H. H., Owens G. K., Lambeth J. D., Griendling K. K. (2005) Nox1 overexpression potentiates angiotensin II-induced hypertension and vascular smooth muscle hypertrophy in transgenic mice. Circulation 112, 2668–2676 [DOI] [PubMed] [Google Scholar]
- 30. Zhang X., Goncalves R., Mosser D. M. (2008) Curr. Protoc. Immunol. Chapter 14, Unit 14.11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cheng G., Cao Z., Xu X., van Meir E. G., Lambeth J. D. (2001) Homologs of gp91phox: cloning and tissue expression of Nox3, Nox4, and Nox5. Gene 269, 131–140 [DOI] [PubMed] [Google Scholar]
- 32. Cheng G., Lambeth J. D. (2004) NOXO1, regulation of lipid binding, localization, and activation of Nox1 by the Phox homology (PX) domain. J. Biol. Chem. 279, 4737–4742 [DOI] [PubMed] [Google Scholar]
- 33. Vilardaga J. P., Nikolaev V. O., Lorenz K., Ferrandon S., Zhuang Z., Lohse M. J. (2008) Conformational cross-talk between α2A-adrenergic and μ-opioid receptors controls cell signaling. Nat. Chem. Biol. 4, 126–131 [DOI] [PubMed] [Google Scholar]
- 34. Csányi G., Cifuentes-Pagano E., Al Ghouleh I., Ranayhossaini D. J., Egaña L., Lopes L. R., Jackson H. M., Kelley E. E., Pagano P. J. (2011) Nox2 B-loop peptide, Nox2ds, specifically inhibits the NADPH oxidase Nox2. Free Radic. Biol. Med. 51, 1116–1125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Molshanski-Mor S., Mizrahi A., Ugolev Y., Dahan I., Berdichevsky Y., Pick E. (2007) Cell-free assays: the reductionist approach to the study of NADPH oxidase assembly, or “all you wanted to know about cell-free assays but did not dare to ask”. Methods Mol. Biol. 412, 385–428 [DOI] [PubMed] [Google Scholar]
- 36. Nisimoto Y., Jackson H. M., Ogawa H., Kawahara T., Lambeth J. D. (2010) Constitutive NADPH-dependent electron transferase activity of the Nox4 dehydrogenase domain. Biochemistry 49, 2433–2442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wheeler D., Sneddon W. B., Wang B., Friedman P. A., Romero G. (2007) NHERF-1 and the cytoskeleton regulate the traffic and membrane dynamics of G protein-coupled receptors. J. Biol. Chem. 282, 25076–25087 [DOI] [PubMed] [Google Scholar]
- 38. Chen B. B., Mallampalli R. K. (2009) Masking of a nuclear signal motif by monoubiquitination leads to mislocalization and degradation of the regulatory enzyme cytidylyltransferase. Mol. Cell. Biol. 29, 3062–3075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Liang C.-C., Park A. Y., Guan J.-L. (2007) In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nat. Protoc. 2, 329–333 [DOI] [PubMed] [Google Scholar]
- 40. Nisimoto Y., Motalebi S., Han C. H., Lambeth J. D. (1999) The p67(phox) activation domain regulates electron flow from NADPH to flavin in flavocytochrome b558. J. Biol. Chem. 274, 22999–23005 [DOI] [PubMed] [Google Scholar]
- 41. Kellogg E. W., 3rd, Fridovich I. (1975) Superoxide, hydrogen peroxide, and singlet oxygen in lipid peroxidation by a xanthine oxidase system. J. Biol. Chem. 250, 8812–8817 [PubMed] [Google Scholar]
- 42. Harrison R. (2002) Structure and function of xanthine oxidoreductase: where are we now? Free Radic. Biol. Med. 33, 774–797 [DOI] [PubMed] [Google Scholar]
- 43. Gianni D., Bohl B., Courtneidge S. A., Bokoch G. M. (2008) The involvement of the tyrosine kinase c-Src in the regulation of reactive oxygen species generation mediated by NADPH oxidase-1. Mol. Biol. Cell 19, 2984–2994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. McGuire J. J., Anderson D. J., Bennett B. M. (1994) Inhibition of the biotransformation and pharmacological actions of glyceryl trinitrate by the flavoprotein inhibitor, diphenyleneiodonium sulfate. J. Pharmacol. Exp. Ther. 271, 708–714 [PubMed] [Google Scholar]
- 45. Hancock J. T., Jones O. T. (1987) The inhibition by diphenyleneiodonium and its analogues of superoxide generation by macrophages. Biochem. J. 242, 103–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gianni D., Taulet N., Zhang H., DerMardirossian C., Kister J., Martinez L., Roush W. R., Brown S. J., Bokoch G. M., Rosen H. (2010) A novel and specific NADPH oxidase-1 (Nox1) small-molecule inhibitor blocks the formation of functional invadopodia in human colon cancer cells. ACS Chem. Biol. 5, 981–993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gaggini F., Laleu B., Orchard M., Fioraso-Cartier L., Cagnon L., Houngninou-Molango S., Gradia A., Duboux G., Merlot C., Heitz F., Szyndralewiez C., Page P. (2011) Design, synthesis and biological activity of original pyrazolo-pyrido-diazepine, -pyrazine and -oxazine dione derivatives as novel dual Nox4/Nox1 inhibitors. Bioorg. Med. Chem. 19, 6989–6999 [DOI] [PubMed] [Google Scholar]
- 48. Mishra A., Lai G. H., Schmidt N. W., Sun V. Z., Rodriguez A. R., Tong R., Tang L., Cheng J., Deming T. J., Kamei D. T., Wong G. C. (2011) Translocation of HIV TAT peptide and analogues induced by multiplexed membrane and cytoskeletal interactions. Proc. Natl. Acad. Sci. U.S.A. 108, 16883–16888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Rey F. E., Cifuentes M. E., Kiarash A., Quinn M. T., Pagano P. J. (2001) Novel competitive inhibitor of NAD(P)H oxidase assembly attenuates vascular O(2)(−) and systolic blood pressure in mice. Circ. Res. 89, 408–414 [DOI] [PubMed] [Google Scholar]
- 50. Poillot C., Bichraoui H., Tisseyre C., Bahemberae E., Andreotti N., Sabatier J. M., Ronjat M., De Waard M. (2012) Small efficient cell-penetrating peptides derived from scorpion toxin maurocalcine. J. Biol. Chem. 287, 17331–17342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Dennis K. E., Aschner J. L., Milatovic D., Schmidt J. W., Aschner M., Kaplowitz M. R., Zhang Y., Fike C. D. (2009) NADPH oxidases and reactive oxygen species at different stages of chronic hypoxia-induced pulmonary hypertension in newborn piglets. Am. J. Physiol. Lung Cell. Mol. Physiol. 297, L596–L607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Roos D., Kuhns D. B., Maddalena A., Bustamante J., Kannengiesser C., de Boer M., van Leeuwen K., Köker M. Y., Wolach B., Roesler J., Malech H. L., Holland S. M., Gallin J. I., Stasia M. J. (2010) Hematologically important mutations: the autosomal recessive forms of chronic granulomatous disease (second update). Blood Cells Mol. Dis. 44, 291–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mittal M., Roth M., König P., Hofmann S., Dony E., Goyal P., Selbitz A. C., Schermuly R. T., Ghofrani H. A., Kwapiszewska G., Kummer W., Klepetko W., Hoda M. A., Fink L., Hänze J., Seeger W., Grimminger F., Schmidt H. H., Weissmann N. (2007) Hypoxia-dependent regulation of nonphagocytic NADPH oxidase subunit NOX4 in the pulmonary vasculature. Circ. Res. 101, 258–267 [DOI] [PubMed] [Google Scholar]
- 54. Schröder K., Kohnen A., Aicher A., Liehn E. A., Büchse T., Stein S., Weber C., Dimmeler S., Brandes R. P. (2009) NADPH oxidase Nox2 is required for hypoxia-induced mobilization of endothelial progenitor cells. Circ. Res. 105, 537–544 [DOI] [PubMed] [Google Scholar]
- 55. Kinoshita M., Hynynen K. (2005) Intracellular delivery of Bak BH3 peptide by microbubble-enhanced ultrasound. Pharm. Res. 22, 716–720 [DOI] [PubMed] [Google Scholar]
- 56. Walensky L. D., Kung A. L., Escher I., Malia T. J., Barbuto S., Wright R. D., Wagner G., Verdine G. L., Korsmeyer S. J. (2004) Activation of apoptosis in vivo by a hydrocarbon-stapled BH3 helix. Science 305, 1466–1470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Tugyi R., Uray K., Iván D., Fellinger E., Perkins A., Hudecz F. (2005) Partial d-amino acid substitution: Improved enzymatic stability and preserved Ab recognition of a MUC2 epitope peptide. Proc. Natl. Acad. Sci. U.S.A. 102, 413–418 [DOI] [PMC free article] [PubMed] [Google Scholar]