

Background: Serine acetyltransferase (SAT) catalyzes the limiting step in cysteine biosynthesis.
Results: Analysis of soybean SAT provides insight into catalysis and protein-protein interactions.
Conclusion: Key structural features are required for catalysis and formation of a stable macromolecular complex.
Significance: A new role for protein complex formation in plant cysteine biosynthesis is proposed.
Keywords: Biosynthesis, Crystal Structure, Enzyme Structure, Plant Biochemistry, Plant Molecular Biology
Abstract
Serine acetyltransferase (SAT) catalyzes the limiting reaction in plant and microbial biosynthesis of cysteine. In addition to its enzymatic function, SAT forms a macromolecular complex with O-acetylserine sulfhydrylase. Formation of the cysteine regulatory complex (CRC) is a critical biochemical control feature in plant sulfur metabolism. Here we present the 1.75–3.0 Å resolution x-ray crystal structures of soybean (Glycine max) SAT (GmSAT) in apoenzyme, serine-bound, and CoA-bound forms. The GmSAT-serine and GmSAT-CoA structures provide new details on substrate interactions in the active site. The crystal structures and analysis of site-directed mutants suggest that His169 and Asp154 form a catalytic dyad for general base catalysis and that His189 may stabilize the oxyanion reaction intermediate. Glu177 helps to position Arg203 and His204 and the β1c-β2c loop for serine binding. A similar role for ionic interactions formed by Lys230 is required for CoA binding. The GmSAT structures also identify Arg253 as important for the enhanced catalytic efficiency of SAT in the CRC and suggest that movement of the residue may stabilize CoA binding in the macromolecular complex. Differences in the effect of cold on GmSAT activity in the isolated enzyme versus the enzyme in the CRC were also observed. A role for CRC formation as a molecular chaperone to maintain SAT activity in response to an environmental stress is proposed for this multienzyme complex in plants.
Introduction
As a critical nutrient for plants, sulfur is required for a range of biological functions and is assimilated from the surrounding environment (1, 2). In plant sulfur metabolism, cysteine synthesis plays a central role in fixing inorganic sulfur from the environment into the metabolic precursor for cellular thiol-containing compounds (3–6). As the first step in cysteine biosynthesis (Fig. 1), serine acetyltransferase (SAT)3 catalyzes the formation of O-acetylserine from acetyl-CoA and serine (7–10). Multiple studies suggest that SAT is the limiting step in cysteine biosynthesis (7, 10–12). For example, in many plants, SAT activity is much lower than that of the second step in the pathway, and overexpression of SAT in plants leads to large increases in cysteine content (5, 7, 10). In the second step of the pathway, O-acetylserine sulfhydrylase (OASS), which is a member of the β-substituted alanine synthase enzyme family, catalyzes formation of cysteine from O-acetylserine and sulfide using pyridoxal-5′-phosphate as a cofactor (13–16).
FIGURE 1.

Overview of plant cysteine biosynthesis. The overall reactions catalyzed by SAT and OASS are shown.
A key biochemical control feature of this pathway in plants is the association of SAT and OASS to form the cysteine regulatory complex (CRC) (17–25). Under high sulfur conditions, formation of the CRC enhances the catalytic efficiency of SAT for O-acetylserine production. Although assembly of the CRC inhibits OASS activity in the complex, a cellular excess of OASS maintains the formation of cysteine. In a nutrient-poor state, depletion of sulfur leads to an accumulation of O-acetylserine, which disassociates the CRC to attenuate SAT activity. Physiologically, this multienzyme assembly acts as a sensor to coordinate sulfur assimilation and modulate cysteine production in plants.
Plants typically encode multiple SAT isoforms sharing ∼60–80% amino acid sequence homology with specific cellular localization patterns (i.e. cytosolic, chloroplastic, and mitochondrial forms). For example, Arabidopsis thaliana (thale cress) and Glycine max (soybean) contain SAT gene families with five and eight isoforms, respectively (5, 7, 9, 26–32). In soybean, two SAT isoforms (Glyma16g03080/GmSAT1;1 and Glyma18g08910/GmSAT2;1) have been functionally characterized with regard to their biochemical properties and regulation of activity (31, 32). GmSAT1;1 is cytosolic, and GmSAT2;1 is targeted to the cytosol and plastid. Both soybean SAT isoforms are sensitive to feedback inhibition by cysteine but to varying degrees. Only the role of GmSAT1;1 in the CRC has been examined (23). Clones of SAT from Citrullus vulgaris (watermelon), Spinacea oleracea (spinach), and Allium cepa (onion) have also been reported (8, 33, 34).
Comparative genomics and metabolic studies indicate that the SAT gene family is essential for plant growth and that O-acetylserine plays a role as a plant growth regulatory signal (35–37). Differential expression of SAT family members is linked to responses to cadmium exposure in A. thaliana and to nickel hyperaccumulation in Thlaspi goesingense (38–41). Studies in soybean and A. thaliana also suggest that the cysteine biosynthetic enzymes interact with signaling systems through phosphorylation-dependent pathways as a means of coordinating sulfur, nitrogen, and carbon metabolism (32, 42). Moreover, efforts aimed at improving resistance to oxidative stress and enhancing nutritional content by increasing the levels of sulfur-containing amino acids in rice, wheat, potato, and soybean have focused on engineering cysteine biosynthesis (43–47).
At the molecular level, the bacterial SAT are more thoroughly studied than the plant forms of the enzyme. Biochemical studies of the plant SAT are largely limited to analysis of steady-state kinetic parameters (8–9, 26–34). Early studies of the SAT from spinach and Salmonella typhimurium suggested a ping pong bi bi kinetic mechanism involving a possible acetyl-enzyme intermediate (48–49); however, more recent analyses of the SAT from Escherichia coli and Haemophilus influenzae indicate either a random or an ordered mechanism, respectively (50, 51). Examination of the chemical mechanism in the H. influenzae SAT implicates two histidine residues as important for a general base catalysis in which a proton is abstracted from serine for nucleophilic attack on acetyl-CoA (52–53). Crystal structures of the SAT from various bacteria and the protozoan parasite Entamoeba histolytica reveal that these enzymes form a dimer of trimers with each monomer defined by an N-terminal α-helical domain and a C-terminal left-handed β-helix domain, which forms the active site of the enzyme at the interface of two neighboring SAT monomers (54–58). The plant SAT appear to function as either trimeric or hexameric proteins with oligomerization depending upon protein concentration (23, 24). Although various plant SAT exhibit similar kinetic properties (8, 9, 26–34), differences in feedback inhibition by cysteine, phosphorylation state, and formation of the CRC have been linked to the C-terminal 10–15 residues of the enzyme (9, 23, 24, 32, 59). To date, no structural information has been reported for the plant SAT.
Here we present the crystal structures of soybean SAT (GmSAT) in the apoenzyme, serine-bound, and CoA-bound forms. Structure-guided mutagenesis identifies critical residues required for catalysis and substrate interaction in the SAT reaction mechanism and a conserved arginine residue that may change position to interact with CoA during CRC formation to enhance SAT activity. The effect of CRC formation on SAT activity under low temperature conditions may allow OASS to act as a molecular chaperone of SAT in plants when the demand for sulfur assimilation and cysteine production increases in response to low temperatures.
EXPERIMENTAL PROCEDURES
Expression Constructs and Site-directed Mutagenesis
Generation of the pET-28a-GmSAT and the pET-28a-GmSATΔC10 expression constructs was previously described (13, 23). Site-directed mutants of GmSAT were generated using the QuikChange PCR method (Agilent Technologies).
Protein Expression and Purification
GmSAT was expressed in E. coli BL21(DE3) grown at 37 °C in Terrific broth containing 50 μg ml−1 kanamycin until A600 nm ∼0.8. After induction with 1 mm isopropyl 1-thio-β-d-galactopyranoside, the cultures were grown overnight at 18 °C. Cells were pelleted by centrifugation (10,000 × g; 10 min) and suspended in 50 mm Tris (pH 8.0), 500 mm NaCl, 20 mm imidazole, 1 mm β-mercaptoethanol, 10% (v/v) glycerol, and 1% (v/v) Tween 20. All protein purification steps were performed at room temperature. After sonication and centrifugation (30,000 × g; 30 min), the supernatant was passed over a Ni2+-NTA column equilibrated in suspension buffer. The column was then sequentially washed with buffer minus Tween 20 and with 40 mm imidazole (wash buffer) and then with wash buffer containing 20 mm O-acetylserine to remove bound E. coli OASS from GmSAT. Wash buffer with 250 mm imidazole was used to elute His-tagged protein from the column. Size exclusion chromatography of the eluted protein was performed using a Superdex-200 26/60 HiLoad column equilibrated in 10 mm Tris (pH 8.0), 50 mm NaCl, and 5 mm β-mercaptoethanol. Fractions containing GmSAT were concentrated to 10–20 mg ml−1 with protein quantified by a Bradford assay using bovine serum albumin as a standard. Protein was stored at room temperature without significant loss of activity for up to 30 days. Expression and purification of GmOASS and GmSATΔC10 and formation of the CRC by GmSAT and GmOASS were as described previously (23).
Protein Crystallization and Structure Determination
Crystals of the apoenzyme form of GmSAT were obtained by the vapor diffusion method in 10-μl hanging drops of a 1:1 mixture of protein and crystallization buffer (1.8 m ammonium phosphate, 100 mm imidazole, pH 8) at 20 °C over a 0.5-ml reservoir. Crystals of the GmSAT-serine complex were obtained in the same condition with the addition of 100 mm serine. Crystals of the GmSAT-CoA complex grew in 12% ethanol, 100 mm Tris (pH 7.8), 200 mm MgCl2, 1 mm Tris(2-carboxyethyl)phosphine hydrochloride, and 10 mm CoA using sitting drops. All crystals were stabilized in cryoprotectant (crystallization solution and 20% glycerol) before flash freezing in liquid nitrogen. Diffraction data (100 K) were collected at beamline 19BM of the Argonne National Laboratory Advanced Photon Source with indexing, integration, and scaling performed with HKL3000 (60). The structure of the GmSAT apoenzyme form was solved by molecular replacement implemented in PHASER (61) using the structure of E. coli SAT without ligands or water molecules (55) (Protein Data Bank entry 1TD3) as a search model. Model building of the GmSAT structure was performed in COOT (62), and all refinements were performed with PHENIX (63). The final model of the GmSAT apoenzyme includes residues 14–255 in chain A and residues 12–255 in chain B. The crystal structure of the GmSAT-serine complex was determined by the difference Fourier method using the refined wild-type structure as a starting model. In the GmSAT-serine complex, residues 13–255 are modeled in both chains of the asymmetric unit. To determine the structure of the GmSAT-CoA complex, molecular replacement implemented in PHASER used the GmSAT apoenzyme structure as a search model to find six molecules in the asymmetric unit. The final model includes residues 17–103, 107–196, and 200–255 of chain A; residues 17–197 and 202–255 of chain B; residues 15–105, 108–196, and 203–255 of chain C; residues 17–103, 107–196, and 201–255 of chain D; residues 17–196 and 199–255 of chain E; and residues 17–103, 108, 196, and 200–255 of chain F. Crystal parameters, data collection statistics, and refinement statistics for these three structures are summarized in Table 1. Coordinates and structure factors for the GmSAT apoenzyme (4N69), the GmSAT-serine complex (4N6A), and the GmSAT-CoA complex (4N6B) have been deposited in the RCSB Protein Data Bank.
TABLE 1.
Crystallographic statistics
| GmSAT | GmSAT-serine | GmSAT-CoA | |
|---|---|---|---|
| Crystal | |||
| Space group | R3 | R3 | C2 |
| Cell dimensions | a = b = 110.87 Å, c = 144.01 Å | a = b = 111.37 Å, c = 143.46 Å | a = 207.08 Å, b = 99.18 Å, c = 120.26 Å; β = 117.2° |
| Data collection | |||
| Wavelength (Å) | 0.979 | 0.979 | 0.979 |
| Resolution range (Å) (highest shell resolution) | 25.0–1.75 (1.80–1.75) | 32.1–1.80 (1.85–1.80) | 38.0–3.0 (3.05–3.0) |
| Reflections (total/unique) | 202,566 / 65,219 | 231,622 / 59,901 | 156,040/42,487 |
| Completeness (highest shell) | 98.8% (100%) | 97.4% (99.4%) | 99.4% (100%) |
| I/σ (highest shell) | 38.7 (3.0) | 33.7 (2.8) | 11.4 (1.9) |
| Rsyma (highest shell) | 4.8% (47.5%) | 5.3% (48.0%) | 7.5% (56.0%) |
| Model and refinement | |||
| Rcrystb/Rfreec | 18.0% /20.6% | 16.7%/19.6% | 22.2%/26.4% |
| No. of protein atoms | 3,704 | 3,694 | 10,525 |
| No. of water molecules | 411 | 465 | 0 |
| No. of ligand atoms | 5 | 24 | 240 |
| Root mean square deviation, bond lengths (Å) | 0.007 | 0.006 | 0.009 |
| Root mean square deviation, bond angles (degrees) | 1.022 | 0.987 | 1.48 |
| Average B-factor (Å2): protein, water, ligand | 39.3, 52.4, 74.9 | 35.2, 48.2, 73.3 | 67.2, 104.0 |
| Stereochemistry: most favored, allowed, outliers | 98.4, 1.6, 0% | 98.4, 1.6, 0% | 93.1, 6.8, 0.1% |
a Rsym = Σ|Ih − 〈Ih〉|/ΣIh, where 〈Ih〉 is the average intensity over symmetry.
b Rcryst = Σ|Fo − 〈Fc〉|/ΣFo, where summation is over the data used for refinement.
c Rfree is defined the same as Rcryst but was calculated using 5% of data excluded from refinement.
Enzyme Assays
SAT assays coupled the appearance of CoA to production of 5-thio-2-nitrobenzoate (TNB; ϵ412 nm = 14,150 m−1 cm−1) by a disulfide exchange reaction with 5,5′-dithiobis(2-nitrobenzoate) (53, 64). Initial velocity rates were calculated from reactions (0.5 ml) performed at room temperature with 100 mm HEPES (pH 7.3), 0.45 mm 5,5′-dithiobis(2-nitrobenzoate), variable amounts of acetyl-CoA and serine, and appropriate amounts of either wild-type or mutant GmSAT or CRC. Steady-state kinetic parameters were determined by non-linear fitting of initial velocity versus substrate concentration to the Michaelis-Menten equation in SigmaPlot.
RESULTS
Overall Three-dimensional Structure of GmSAT
To examine the three-dimensional structure of a plant SAT, the 1.75 Å resolution x-ray crystal structure of the apoenzyme form GmSAT was solved by molecular replacement (Table 1). There were two monomers (chains A and B) per asymmetric unit. The GmSAT monomer is divided into an N-terminal domain (residues Val12–Phe151) consisting of eight α-helices and the C-terminal left-handed β-helix domain (Ala152–Val255) formed by multiple β-strand triplets (Fig. 2A). The overall fold of the GmSAT monomer is similar to the bacterial and Entamoeba homologs with a root mean square deviation of 1.0–2.8 Å for Cα atoms over ∼190–230 residues (54–58). Additional structural homology with a variety of acyltransferases, including QdtC required for synthesis of dTDP-3-acetamido-3,6-dideoxy-α-d-glucose, perosamine N-acetyltransferase, N-acetyl-glucosamine-1-phosphate uridyltransferase, and PglD from the UDP-N,N′-diacetylbacillosamine synthesis pathway (65–68), was detected using the DALI server and displayed 1.1–7.2 Å root mean square deviation of Cα atoms over ∼90–130 residues, largely in the left-handed β-helix domain. Crystallographic symmetry of GmSAT generates a trimeric structure from each chain that adopts the dimer of trimers packing observed for the bacterial SAT structures (Fig. 2B). The 3-fold axis runs the length of the trimer and allows for extensive interaction between monomers (Fig. 2, C and E). The packing interface between trimers is formed by α1–α3 of each monomer and is a mix of charged and uncharged surface regions (Fig. 2D). Although the overall structure of GmSAT shares a common fold with the bacterial and Entamoeba enzymes, structures of GmSAT in complex with either serine or CoA provide new information about substrate interaction in these enzymes.
FIGURE 2.

Overall structure of GmSAT. A, monomer structure of GmSAT. The N-terminal α-helical domain and the C-terminal left-handed β-helix domain are colored rose and blue, respectively. Secondary structure features are labeled, as are the N and C termini. B, dimer of trimer arrangement of GmSAT. In the GmSAT apoenzyme and serine complex, crystallographic symmetry of chain A (red) and chain B (rose) complete the trimers shown on the left and right, respectively. C, view down the N-terminal 3-fold symmetry axis indicated by a triangle. D, surface electrostatic view of the N-terminal trimer, as in C. E, view (rotated 180° from that of C) down the C-terminal 3-fold symmetry axis indicated by a triangle. The β1c-β2c loop that forms part of the serine binding site is indicated.
Structures of the GmSAT-Serine and GmSAT-CoA Complexes
Crystal structures of GmSAT with serine and CoA bound were determined to 1.8 and 3.0 Å resolution, respectively (Table 1). Both structures define the active site at the C-terminal interfaces between each monomer (Fig. 3A). Clear electron density for the ligands in the GmSAT-serine and GmSAT-CoA complexes were observed (Fig. 3, B and C).
FIGURE 3.

Active site location at the interface of monomers in GmSAT. A, binding of CoA (space-filling model) is shown to highlight the location of the GmSAT active site at the interfaces between monomers. The C-terminal view is shown with each monomer colored differently. B, electron density of serine in the GmSAT-serine complex. A 2Fo − Fc omit map (1.0 σ) is shown. C, electron density of CoA in the GmSAT-CoA complex. A 2Fo − Fc omit map (1.0 σ) is shown.
In GmSAT, the serine binding site is located at the N-terminal side of the left-handed β-helix domain between the second and third β-strand repeats of two adjacent monomers (Fig. 4). One side of the binding site is delineated by the position of Asp168 and His169 of one monomer and the other by the β1c-β2c loop (residues 193–205) of an adjacent monomer (Fig. 4B). This loop is mobile, because it is disordered in the GmSAT-CoA complex structure. Movement of the β1c-β2c loop positions Arg203 and His204 for charge-charge and hydrogen bonding interactions, respectively, with the substrate. Interaction between Glu177 and Arg203 may help stabilize the conformation of the β1c-β2c loop for serine binding. Within the serine binding site, Asp154 hydrogen-bonds with the Nδ of His169, which orients the Nϵ toward the substrate hydroxyl group. The position of this residue implicates it as critical for catalysis. The substrate amine interacts with a carboxylate oxygen of Asp168. The side chains of Arg203 and His204 form additional contacts with the serine carboxylate group.
FIGURE 4.
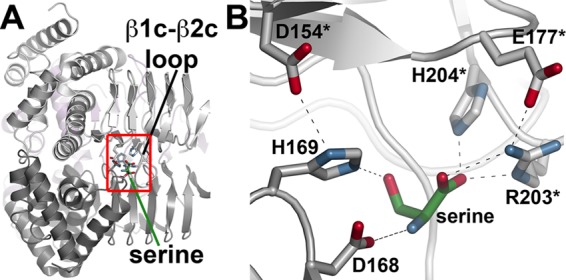
Structure of the serine binding site. A, view of the location of serine binding between two GmSAT monomers. The flexible β1c-β2c loop and serine are indicated. The red box shows the area highlighted in B. B, the serine binding site of GmSAT. Residues contributed by each monomer are colored gray and white, respectively, with an asterisk to indicate the second monomer.
Binding of CoA occurs along the interface of the left-handed β-helix domains to position the thioester group near the serine binding site and catalytic residues (Figs. 3A and 5A). In contrast to the GmSAT·serine complex structure, the β1c-β2c loop is disordered in the GmSAT·CoA complex. Overall, the CoA binding site is a long groove at the interface of two adjacent monomers, which provides extensive van der Waals contacts between the protein and ligand (Fig. 5A). Specific bonding interactions are contributed largely by residues from one monomer (Fig. 5B). Lys230 and Thr246 interact with the pyrophosphate portion of CoA. Ala215 and Ala233, along with Gly195 of the adjacent monomer, hydrogen-bond with the pantothenate arm of CoA. An additional basic residue (Arg253) is positioned within 5–7 Å of the CoA phosphate group (Fig. 5, B and C). These interactions position the thioester group of CoA in proximity to the serine binding site and two histidines (His169 and His189) that may serve catalytic roles in the SAT reaction mechanism. Of these two residues, His169 is closer (3.6 Å) to the CoA. As modeled in the GmSAT·CoA complex, His189 is 6.4 Å away from the CoA thiol, but rotation of the side chain into the active site can place the imidiazole ring ∼3.5 Å from the thiol.
FIGURE 5.

Structure of the GmSAT CoA binding site. A, stereoview of the CoA binding site along the left-handed β-helix domain. Surfaces corresponding to monomers are colored differently. The position of serine from the GmSAT-serine complex is shown for reference. The β1c-β2c loop is disordered in the GmSAT-CoA complex. B, GmSAT active site view. Residues interacting with CoA are shown, with an asterisk indicating residues from the adjacent monomer. The position of serine from the GmSAT-serine complex is shown for reference. C, C-terminal view of GmSAT showing the location of Arg253 (blue surface) in each monomer. CoA molecules are shown as space-filling models.
Sequence comparison of the SAT from soybean (GmSAT), A. thaliana (AtSAT), E. coli (EcSAT), H. influenzae (HiSAT), and E. histoyltica (EhSAT) reveals that residues in the serine binding site and the catalytic site are highly conserved across these organisms (Fig. 6). Amino acids in the CoA binding site are also conserved, although some sequence variation is observed between homologs.
FIGURE 6.
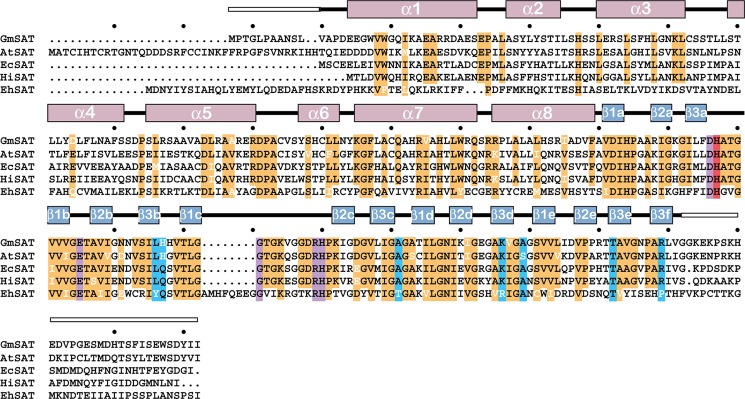
Sequence comparison of SAT from different organisms. The amino acid sequences of the SAT from soybean (GmSAT; NP_001235640.1), A. thaliana (AtSAT; AAC37474.1), E. coli (EcSAT; NP_290190.1), H. influenzae (HiSAT; NP_438764.1), and E. histoyltica (EhSAT; BAA82868.1) are shown. The secondary structure of GmSAT is shown above the alignment with α-helices (rose) and β-strands (blue) indicated. White boxes indicate disordered regions of the GmSAT structure. Highly conserved residues (orange), residues in the serine binding site (purple), residues in the CoA binding site (blue), and the catalytic histidine (red) in the SAT are highlighted.
Steady-state Kinetic Analysis of GmSAT Active Site Mutants
The structures of GmSAT in complex with serine and CoA suggest possible roles for key residues in catalysis and substrate binding (Figs. 4 and 5). To probe the contributions of these residues to GmSAT function, a series of site-directed mutations (D154A, D154N, D168A, D168N, H169A, H169N, E177Q, H189A, H189N, R203A, R203K, H204A, H204N, A215V, K230M, A233V, T426A, and R253A) were introduced into GmSAT. With the exception of the D154A and D154N mutants, which yielded insoluble protein, each GmSAT mutant behaved like wild type during expression and purification.
Mutations of Asp168, His169, and His189 showed the largest effects on the steady-state kinetic parameters of GmSAT (Table 2). Substitution of either His169 or His189 with an alanine led to inactive enzyme; however, the H169N and H189N mutants retained activity. The H169N mutant resulted in a 1,400-fold reduction in turnover rate compared with wild-type enzyme. In comparison, the H189N mutant displayed a 30-fold slower turnover rate and increased Km values for both substrates. Mutations of Asp168 to alanine and asparagine decreased catalytic efficiency ∼1,000- and 10-fold, respectively. The three-dimensional structures of GmSAT and these results suggest potential functional roles for these residues
TABLE 2.
Steady-state kinetic parameters of wild-type and mutant GmSAT
Reactions were performed as described under “Experimental Procedures.” All parameters are expressed as mean ± S.E. for n = 3. ND, activity was not detected.
| Protein | V/Et | Kmacetyl-CoA | kcat/Kmacetyl-CoA | Kmserine | kcat/Kmserine |
|---|---|---|---|---|---|
| s−1 | μm | m−1 s−1 | μm | m−1 s−1 | |
| Wild type | 70 ± 10 | 321 ± 17 | 218,069 | 617 ± 102 | 113,452 |
| Catalytic site | |||||
| D168A | 0.15 ± 0.05 | 716 ± 182 | 209 | 1,810 ± 292 | 83 |
| D168N | 5.5 ± 0.4 | 244 ± 41 | 22,541 | 1,203 ± 205 | 4,572 |
| H169A | ND | ||||
| H169N | 0.05 ± 0.01 | 330 ± 72 | 152 | 404 ± 54 | 124 |
| H189A | ND | ||||
| H189N | 2.3 ± 0.2 | 2,890 ± 553 | 796 | 3,100 ± 761 | 742 |
| Serine site | |||||
| E177Q | 16 ± 5 | 370 ± 70 | 43,243 | 1,767 ± 387 | 9,055 |
| R203A | ND | ||||
| R203K | 7.8 ± 1.0 | 590 ± 83 | 13,220 | 36,022 ± 5,680 | 217 |
| H204A | 7.9 ± 1.9 | 506 ± 86 | 15,613 | 1,850 ± 358 | 4,270 |
| H204N | 57 ± 8 | 438 ± 41 | 130,137 | 845 ± 279 | 67,456 |
| CoA site | |||||
| A215V | 93 ± 8 | 598 ± 77 | 155,519 | 908 ± 79 | 102,423 |
| K230M | 1.9 ± 0.2 | 5,052 ± 1,030 | 376 | 702 ± 114 | 2,707 |
| A233V | 62 ± 15 | 561 ± 138 | 110,517 | 635 ± 125 | 97,638 |
| T246A | 41 ± 18 | 1,834 ± 656 | 22,356 | 935 ± 237 | 43,850 |
| R253A | 74 ± 9 | 567 ± 146 | 130,511 | 625 ± 76 | 118,400 |
Altering residues in the serine and CoA binding sites also affected the steady-state kinetic parameters of GmSAT to varying degrees (Table 2). Within the serine binding site, alteration of Arg203 resulted in either a loss of activity (R203A) or up to a 500-fold decrease in kcat/Km (R203K) compared with wild-type enzyme. Mutation of His204, which also forms hydrogen bond interactions with the substrate, to either alanine or asparagine showed that a complete loss of the imidazole ring led to decreased turnover rates and increased Km values for serine. Substitution of a glutamine for Glu177, which interacts with Arg203 to potentially stabilize the position of the β1c-β2c loop over the serine binding site, also reduced catalytic efficiency 13-fold with serine.
The A215V, K230M, A233V, and T246A mutants targeted residues in the CoA binding site (Table 2). Of these mutants, substitution of a methionine for Lys230 resulted in a 580-fold decrease in kcat/Km for acetyl-CoA, which combined a 35-fold decrease in turnover rate and a 16-fold increase in Km for the substrate. A lesser effect was observed with the T246A mutant, which had a 6-fold increase in the Km for acetyl-CoA. The A215V and A233V mutants had modest effects on steady-state kinetic parameters. The R253A mutant was not significantly different from wild-type GmSAT with regard to steady-state kinetic parameters. Overall, the mutagenesis results suggest that critical ionic protein-ligand interactions are important for binding of both substrates in the GmSAT active site.
Role of Arg253 in the CRC
Although the GmSAT R253A mutant displayed steady-state kinetic parameters comparable with those of wild-type enzyme, the arginine residue is conserved in the SAT from soybean, Arabidopsis, and bacteria that interact with OASS to form the CRC (Fig. 6). In the E. histolytica SAT, which does not bind with OASS, the corresponding residue is a proline. Because the disordered C-terminal tail region of SAT is essential for protein-protein interaction with OASS (13, 17, 21–25), the position of Arg253 on the surface of the C-terminal of SAT (Fig. 5C) led us to examine the effect of mutation on SAT activity in the CRC.
Previous work analyzing macromolecular assembly of the CRC showed that association of GmSAT and GmOASS improved the catalytic efficiency of the SAT reaction (23). Formation of the CRC with either wild-type or R253A GmSAT was confirmed by size-exclusion chromatography. As observed previously, the elution profile suggests interaction of three GmOASS dimers with a trimeric form of either GmSAT form (23). The catalytic efficiency of the CRC containing the GmSAT R253A mutant was 4-fold higher than that of the GmSAT R253A mutant alone (Table 3); however, this was significantly less than the 20-fold improvement of SAT activity observed with formation of a CRC containing wild-type SAT (Table 3). Overall, formation of the CRC with the GmSAT R253A mutant led to an 8-fold reduction in catalytic efficiency compared with that of the wild-type CRC. This result suggests that Arg253 may change position to interact with CoA during CRC formation to enhance SAT activity.
TABLE 3.
Comparison of steady-state kinetic parameters for wild-type and R253A SAT as isolated protein and in the CRC
Reactions were performed as described under “Experimental Procedures.” All parameters are expressed as mean ± S.E. for n = 3. Values for GmSAT and GmSAT R253A are from Table 2 and are provided here for comparison.
| V/Et | Kmacetyl-CoA | kcat/Kmacetyl-CoA | Kmserine | kcat/Kmserine | |
|---|---|---|---|---|---|
| s−1 | μm | m−1 s−1 | μm | m−1 s−1 | |
| GmSAT | 70 ± 10 | 321 ± 17 | 218,069 | 617 ± 102 | 113,452 |
| GmSAT R253A | 74 ± 9 | 567 ± 146 | 130,511 | 625 ± 76 | 118,400 |
| GmSAT−GmOASS (CRC) | 203 ± 38 | 45 ± 6 | 4,511,111 | 830 ± 73 | 244,578 |
| GmSAT R253A−GmOASS (CRC) | 125 ± 28 | 231 ± 57 | 541,125 | 731 ± 170 | 171,000 |
Effect of CRC Formation on Protein Stability
Association of SAT and OASS to form the CRC provides a mechanism for altering enzymatic activity of both cysteine biosynthesis enzymes (17–25). During protein expression and purification of GmSAT, we observed that temperature was a factor in the stability of GmSAT because the protein was inactivated by cold (0–4 °C). To examine the effects of temperature on SAT activity, full-length GmSAT, GmSAT lacking the C-terminal tail (GmSATΔC10), and the CRC were incubated at 4 and 25 °C for 60 min. The enzymatic activities of the full-length and truncated forms of GmSAT were significantly reduced following cold treatment (Table 4). The activity loss was reversible with recovery of ∼90% of original activity upon return to 25 °C. In comparison, association of GmSAT with GmOASS as part of the CRC protected SAT activity. Similar experiments with GmOASS showed no effect of incubation temperature on enzyme function.
TABLE 4.
Comparison of the effect of temperature on SAT activity
Reactions were performed as described under “Experimental Procedures” following a 60-min incubation at the indicated temperatures.
| Relative SAT activity |
||
|---|---|---|
| 25 °C | 4 °C | |
| % | % | |
| GmSAT | 100 | 19.4 |
| GmSATΔC10 | 100 | 17.1 |
| GmSAT-GmOASS (CRC) | 100 | 92.4 |
DISCUSSION
SAT catalyzes the first step in the cysteine biosynthesis pathway by formation of O-acetylserine from acetyl-CoA and serine (Fig. 1) (7–12). Subsequent reaction with metabolic sulfide leads to generation of cysteine, which is an essential compound for the synthesis of multiple sulfur-containing molecules, such as methionine, glutathione, phytochelatin heavy metal-binding peptides, iron-sulfur clusters, vitamin cofactors, and an array of specialized natural products (3, 6). The crystal structures of the SAT from soybean in complex with either serine or CoA and the biochemical analysis of GmSAT site-directed mutants provide new insights into the molecular mechanism of cysteine biosynthesis in plants.
GmSAT shares a common three-dimensional fold with its homologs from bacteria and the protozoa Entamoeba and maintains a highly conserved set of active site residues for catalysis and substrate recognition (Figs. 2 and 6). The overall structure of the hexapeptide repeat that defines the left-handed β-helical fold of SAT and other O-acyltransferases provides a highly adaptable scaffold for tailoring of substrate specificity (65–69). Across the O-acyltransferase family of enzymes, oligomerization of monomers into trimeric assemblies with the length of the left-handed β-helical fold running along the 3-fold axis provides for substrate binding at the interfaces between monomers (65–69) (Fig. 3). In addition to their overall structural organization, the x-ray crystal structures of various O-acyltransferases show that a loop corresponding to the β1c-β2c loop of GmSAT extends from the β-helix of one monomer to interact with a neighboring subunit to act as a cover of the active site (65–69). Moreover, the active sites of these enzymes share a common catalytic mechanism that relies on a histidine as the general base (65–69).
The reaction mechanism of the bacterial SAT has been well studied, with pH dependence and solvent deuterium isotope effect experiments suggesting that a single proton is involved in the rate-limiting step and that the acetylation of serine relies on general base catalysis (52). The structures of GmSAT (Figs. 4 and 5 and Table 2) and the steady-state kinetic analysis of site-directed mutants suggest a similar reaction mechanism (Fig. 7). Based on the GmSAT structure, Asp154 and His169 are positioned to interact as a dyad that enhances the basicity of the imidazole ring. Substitution of His169 either eliminates SAT activity (H169A) or leads to a 1,400-fold slower rate of reaction (H169N), which is consistent with the loss of a general base in the reaction. Although mutants of Asp154 were insoluble and could not be assayed, interaction between the carboxylate side chain and His169 would facilitate catalysis. The effects of the His169 mutants are similar to those described for the H. influenzae SAT (53). In the proposed SAT mechanism (Fig. 7), His169 acts as a general base to abstract a proton from the serine hydroxyl group. The activated serine side chain undergoes nucleophilic attack on the carbonyl of acetyl-CoA, which leads to formation of a tetrahedral reaction intermediate. Within the GmSAT active site, His189 may serve to stabilize the oxyanion of the intermediate. Although His189 is oriented away from CoA in the GmSAT structure, rotation of the side chain into the active site may provide an oxyanion hole, as suggested by the kinetic data obtained with the H189Q and H189A mutants. Subsequent collapse of the intermediate leads to formation of O-acetylserine and release of CoA with the histidine serving as a general acid to donate a proton to the CoA sulfur.
FIGURE 7.

Proposed reaction mechanism for GmSAT.
The combined crystallographic and mutagenesis study of GmSAT also identifies other critical active site features necessary for substrate interaction with the enzyme that have not been previously described. Comparison of the structures of GmSAT in complex with either serine or CoA suggests flexibility of the β1c-β2c loop helps stabilize positioning of the active site lid (Fig. 4B and Table 2). Indeed, mutagenesis of Glu177 reduces catalytic efficiency with serine by potentially altering the interaction with Arg203 for substrate binding. Likewise, mutation of Arg203 to either lysine or alanine compromises GmSAT activity by altering the kinetic parameters for serine. The position of the serine in the GmSAT structure is similar to the binding of cysteine, an inhibitor, in the bacterial SAT structures (55, 56). The GmSAT-serine structure confirms the mode of cysteine inhibition as a competitor for the amino acid substrate (55–56); however, it also explains the observed effects on acetyl-CoA by the inhibitor because binding at the serine site would lock the β1c-β2c loop over the cleft between adjacent monomers to alter acetyl-CoA access to the active site.
Kinetic analysis of point mutants of residues in the CoA binding site (A215V, K230M, A233V, and T246A) implicates the interaction made between Lys230 and the backbone phosphates of the ligand as a critical determinant for substrate binding (Table 2). In general, the SAT active site relies on key ionic protein-ligand interactions for association with both substrates.
The GmSAT-CoA complex structure also led to the discovery of Arg253 as a mediator of CoA interaction following formation of the CRC (Table 3). Arg253, which is conserved in the SAT that associate with OASS to form the CRC (Fig. 6), is positioned near the CoA phosphate group (Fig. 5C) but does directly interact with the ligand in the crystal structure. Because multiple studies demonstrate the essential role of the C-terminal region of SAT for interaction with the second enzyme from the cysteine biosynthesis pathway (i.e. OASS) in the CRC (17–25), the location of Arg253 on the C-terminal surface of the left-handed β-helix region. Mutation of Arg253 did not significantly alter the steady-state kinetic properties of GmSAT as an isolated protein. In contrast, as part of the CRC, the R253A mutant displayed an attenuated enhancement of catalytic efficiency for the SAT reaction compared with that of wild-type SAT in the CRC (Table 3). Based on the position of Arg253 in the GmSAT structure and kinetic analysis of wild-type and mutant SAT and CRC, we suggest that binding of OASS at the C-terminal tail of SAT shifts the arginine toward the CoA binding site to enhance interaction with the substrate and improve catalytic efficiency.
In our efforts to obtain protein for structural and functional studies of GmSAT, we observed that temperature is a critical factor in the stability of GmSAT because the protein is inactivated by cold (4 °C) temperature. Interestingly, SAT activity in the CRC is largely maintained following incubation at the same temperature. Removal of the C-terminal region of GmSAT, which prevents association with GmOASS (23), does not alter cold inactivation (Table 4). Similar experiments with GmOASS showed no effect of incubation temperature on activity. Cold inactivation and/or denaturation occurs when a protein loses its folded structure as a result of temperature decreases that produce a favorable change in free energy between water and nonpolar groups that weaken hydrophobic interactions to disrupt protein structure (70–74). These can be either global or localized structural changes. Formation of the soybean CRC alters activity of SAT and OASS in the complex, and experimental studies suggest that association with OASS may reorganize the C-terminal region of SAT (22, 23). In addition to effects on reaction kinetics and inhibition, these changes may allow the CRC to function as a molecular chaperone to prevent inactivation of SAT under stress conditions, in particular cold temperature. As shown here, SAT activity is sensitive to cold (4 °C), but formation of the CRC largely prevents the loss of activity. This is intriguing because multiple studies suggest that expression and activity of enzymes in plant thiol metabolism increase under cold stress. A similar protective role of CRC formation was demonstrated against feedback inhibition by cysteine (24).
Multiple metabolic and cellular changes occur at the molecular level in response to low temperature stress, including the generation of reactive oxygen species that cause cellular damage (75). Plants subjected to low temperature accumulate hydrogen peroxide and the activities of reactive oxygen species detoxification enzymes require enhanced glutathione synthesis, which places increased demands on the sulfur assimilation and cysteine synthesis pathways (75–81). Thus, CRC formation may allow OASS to act as a molecular chaperone of SAT activity in plants when the demand for sulfur assimilation and cysteine production increases in response to low temperatures.
Cold adaptation of plant proteins is not well explored, but it could be a possible mechanism for sessile organisms to tolerate this environmental stress. In general, understanding cold adaptation of enzymes has a number of potential biotechnology applications for the use of cold-active biocatalysts in food processing, cold temperature detergents, and the biosynthesis of volatile intermediates (72). A possible role for CRC formation as a molecular chaperone to maintain SAT activity, which is the rate-limiting step of cysteine synthesis, in response to an environmental stress is a new role for this multienzyme complex in plants. This is relevant for plant biotechnology because understanding the role of the CRC in maintaining SAT activity may be useful for determining which SAT and/or CRC to use for improving cold tolerance in early season crops at risk for cold damage.
Acknowowledgment
Portions of this research were carried out at the Argonne National Laboratory Structural Biology Center of the Advanced Photon Source, a national user facility operated by the University of Chicago for the Department of Energy Office of Biological and Environmental Research (DE-AC02-06CH11357).
This work was supported by United States Department of Agriculture Grant NRI-2005-02518 (to J. M. J.).
The atomic coordinates and structure factors (codes 4N69, 4N6A, and 4N6B) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- SAT
- serine O-acetyltransferase (EC 2.3.1.30)
- CRC
- cysteine regulatory complex
- OASS
- O-acetylserine sulfhydrylase (also O-acetylserine(thiol)lyase; EC 2.5.1.47)
- GmSAT and GmOASS
- G. max SAT and OASS, respectively
- TNB
- 5-thio-2-nitrobenzoate.
REFERENCES
- 1. Amtmann A., Armengaud P. (2009) Effects of N, P, K, and S on metabolism. New knowledge gained from multi-level analysis. Curr. Opin. Plant Biol. 12, 275–283 [DOI] [PubMed] [Google Scholar]
- 2. Schachtman D. P., Shin R. (2007) Nutrient sensing and signaling. NPKS. Annu. Rev. Plant Biol. 58, 47–69 [DOI] [PubMed] [Google Scholar]
- 3. Takahashi H., Kopriva S., Giordano M., Saito K., Hell R. (2011) Sulfur assimilation in photosynthetic organisms. Molecular functions and regulations of transporters and assimilatory enzymes. Annu. Rev. Plant Biol. 62, 157–184 [DOI] [PubMed] [Google Scholar]
- 4. Yi H., Galant A., Ravilious G. E., Preuss M. L., Jez J. M. (2010) Sensing sulfur conditions. Simple to complex biochemical regulatory mechanisms in plant thiol metabolism. Mol. Plant 3, 269–279 [DOI] [PubMed] [Google Scholar]
- 5. Yi H., Ravilious G. E., Galant A., Krishnan H. B., Jez J. M. (2010) Thiol metabolism in soybean. Sulfur to homoglutathione. Amino Acids 39, 963–978 [DOI] [PubMed] [Google Scholar]
- 6. Ravilious G. E., Jez J. M. (2012) Structural biology of plant sulfur metabolism. From assimilation to biosynthesis. Nat. Prod. Rep. 29, 1138–1152 [DOI] [PubMed] [Google Scholar]
- 7. Ruffet M. L., Lebrun M., Droux M., Douce R. (1995) Subcellular distribution of serine acetyltransferase from Pisum sativum and characterization of an Arabidopsis thaliana putative cytosolic isoform. Eur. J. Biochem. 227, 500–509 [DOI] [PubMed] [Google Scholar]
- 8. Saito K., Yokoyama H., Noji M., Murakoshi I. (1995) Molecular cloning and characterization of a plant serine acetyltransferase playing a regulatory role in cysteine biosynthesis from watermelon. J. Biol. Chem. 270, 16321–16326 [DOI] [PubMed] [Google Scholar]
- 9. Noji M., Inoue K., Kimura N., Gouda A., Saito K. (1998) Isoform-dependent differences in feedback regulation and subcellular localization of serine acetyltransferase involved in cysteine biosynthesis from Arabidopsis thaliana. J. Biol. Chem. 273, 32739–32745 [DOI] [PubMed] [Google Scholar]
- 10. Droux M. (2003) Plant serine acetyltransferase. New insights for regulation of sulphur metabolism in plant cells. Plant Physiol. Biochem. 41, 619–627 [Google Scholar]
- 11. Haas F. H., Heeg C., Queiroz R., Bauer A., Wirtz M., Hell R. (2008) Mitochondrial serine acetyltransferase functions as a pacemaker of cysteine synthesis in plant cells. Plant Physiol. 148, 1055–1067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wirtz M., Beard K. F., Lee C. P., Boltz A., Schwarzländer M., Fuchs C., Meyer A. J., Heeg C., Sweetlove L. J., Ratcliffe R. G., Hell R. (2012) Mitochondrial cysteine synthase complex regulates O-acetylserine biosynthesis in plants. J. Biol. Chem. 287, 27941–27947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bonner E. R., Cahoon R. E., Knapke S. M., Jez J. M. (2005) Molecular basis of plant cysteine biosynthesis. Structural and functional analysis of O-acetylserine sulfhydrylase from Arabidopsis thaliana. J. Biol. Chem. 280, 38803–38813 [DOI] [PubMed] [Google Scholar]
- 14. Watanabe M., Kusano M., Oikawa A., Fukushima A., Noji M., Saito K. (2008) Physiological roles of the β-substituted alanine synthase gene family in Arabidopsis. Plant Physiol. 146, 310–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yi H., Jez J. M. (2012) Assessing functional diversity in the soybean β-substituted alanine synthase enzyme family. Phytochemistry 83, 15–24 [DOI] [PubMed] [Google Scholar]
- 16. Yi H., Juergens M., Jez J. M. (2012) Structure of soybean β-cyanoalanine synthase and the molecular basis for cyanide detoxification in plants. Plant Cell 24, 2696–2706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jez J. M., Dey S. (2013) The cysteine regulatory complex from plants and microbes. What was old is new again. Curr. Opin. Struct. Biol. 23, 302–310 [DOI] [PubMed] [Google Scholar]
- 18. Bogdanova N., Hell R. (1997) Cysteine synthesis in plants. Protein-protein interactions of serine acetyltransferase from Arabidopsis thaliana. Plant J. 11, 251–262 [DOI] [PubMed] [Google Scholar]
- 19. Berkowitz O., Wirtz M., Wolf A., Kuhlmann J., Hell R. (2002) Use of biomolecular interaction analysis to elucidate the regulatory mechanism of the cysteine synthase complex from Arabidopsis thaliana. J. Biol. Chem. 277, 30629–20634 [DOI] [PubMed] [Google Scholar]
- 20. Droux M., Ruffet M. L., Douce R., Job D. (1998) Interactions between serine acetyltransferase and O-acetylserine (thiol) lyase in higher plants. Structural and kinetic properties of the free and bound enzymes. Eur. J. Biochem. 255, 235–245 [DOI] [PubMed] [Google Scholar]
- 21. Francois J. A., Kumaran S., Jez J. M. (2006) Structural basis for interaction of O-acetylserine sulfhydrylase and serine acetyltransferase in the Arabidopsis cysteine synthase complex. Plant Cell 18, 3647–3655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kumaran S., Jez J. M. (2007) Thermodynamics of the interaction between O-acetylserine sulfhydrylase and the C-terminus of serine acetyltransferase. Biochemistry 46, 5586–5594 [DOI] [PubMed] [Google Scholar]
- 23. Kumaran S., Yi H., Krishnan H. B., Jez J. M. (2009) Assembly of the cysteine synthase complex and the regulatory role of protein-protein interactions. J. Biol. Chem. 284, 10268–10275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wirtz M., Birke H., Heeg C., Müller C., Hosp F., Throm C., König S., Feldman-Salit A., Rippe K., Petersen G., Wade R. C., Rybin V., Scheffzek K., Hell R. (2010) Structure and function of the hetero-oligomeric cysteine synthase complex in plants. J. Biol. Chem. 285, 32810–32817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Feldman-Salit A., Wirtz M., Hell R., Wade R. C. (2009) A mechanistic model of the cysteine synthase complex. J. Mol. Biol. 386, 37–59 [DOI] [PubMed] [Google Scholar]
- 26. Murillo M., Foglia R., Diller A., Lee S., Leustek T. (1995) Serine acetyltransferase from Arabidopsis thaliana can functional complement the cysteine requirement of a cysE mutant in cysteine biosynthesis of Escherichia coli. Cell Mol. Biol. Res. 41, 425–433 [PubMed] [Google Scholar]
- 27. Bogdanova N., Bork C., Hell R. (1995) Cysteine biosynthesis in plants. Isolation and functional identification of a cDNA encoding a serine acetyltransferase from Arabidopsis thaliana. FEBS Lett. 358, 43–47 [DOI] [PubMed] [Google Scholar]
- 28. Roberts M. A., Wray J. L. (1996) Cloning and characterization of an Arabidopsis cDNA clone encoding an organellar isoform of serine acetyltransferase. Plant Mol. Biol. 30, 1041–1049 [DOI] [PubMed] [Google Scholar]
- 29. Howarth J. R., Roberts M. A., Wray J. L. (1997) Cysteine biosynthesis in higher plants. New members of the Arabidopsis thaliana serine acetyltransferase small gene family obtained by functional complementation of an Escherichia coli cysteine auxotroph. Biochim. Biophys. Acta 1350, 123–127 [DOI] [PubMed] [Google Scholar]
- 30. Kawashima C. G., Berkowitz O., Hell R., Noji M., Saito K. (2005) Characterization and expression analysis of a serine acetyltransferase gene family involved in a key step of the sulfur assimilation pathway in Arabidopsis. Plant Physiol. 137, 220–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chronis D., Krishnan H. B. (2004) Sulfur assimilation in soybean (Glycine max [L.] Merr.). Molecular cloning and characterization of a cytosolic isoform of serine acetyltransferase. Planta 218, 417–426 [DOI] [PubMed] [Google Scholar]
- 32. Liu F., Yoo B. C., Lee J. Y., Pan W., Harmon A. C. (2006) Calcium-regulated phosphorylation of soybean serine acetyltransferase in response to oxidative stress. J. Biol. Chem. 281, 27405–27415 [DOI] [PubMed] [Google Scholar]
- 33. Noji M., Takagi Y., Kimura N., Inoue K., Saito M., Horikoshi M., Saito F., Takahashi H., Saito K. (2001) Serine acetyltransferase involved in cysteine biosynthesis from spinach. Molecular cloning, characterization, and expression analysis of a cDNA encoding a plastidic isoform. Plant Cell Physiol. 42, 627–634 [DOI] [PubMed] [Google Scholar]
- 34. McManus M. T., Leung S., Lambert A., Scott R. W., Pither-Joyce M., Chen B., McCallum J. (2005) Molecular and biochemical characterisation of a serine acetyltransferase of onion, Allium cepa (L.). Phytochemistry 66, 1407–1416 [DOI] [PubMed] [Google Scholar]
- 35. Watanabe M., Mochida K., Kato T., Tabata S., Yoshimoto N., Noji M., Saito K. (2008) Comparative genomics and reverse genetics analysis reveal indispensable functions of the serine acetyltransferase gene family in Arabidopsis. Plant Cell 20, 2484–2496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Krueger S., Niehl A., Lopez Martin M. C., Steinhauser D., Donath A., Hildebrandt T., Romero L. C., Hoefgen R., Gotor C., Hesse H. (2009) Analysis of cytosolic and plastidic serine acetyltransferase mutants and subcellular metabolite distributions suggests interplay of the cellular compartments for cysteine biosynthesis in Arabidopsis. Plant Cell Environ. 32, 349–367 [DOI] [PubMed] [Google Scholar]
- 37. Hubberten H. M., Klie S., Caldana C., Degenkolbe T., Willmitzer L., Hoefgen R. (2012) Additional role of O-acetylserine as a sulfur status-independent regulator during plant growth. Plant J. 70, 666–677 [DOI] [PubMed] [Google Scholar]
- 38. Howarth J. R., Domínguez-Solís J. R., Gutiérrez-Alcalá G., Wray J. L., Romero L. C., Gotor C. (2003) The serine acetyltransferase gene family in Arabidopsis thaliana and the regulation of its expression by cadmium. Plant Mol. Biol. 51, 589–598 [DOI] [PubMed] [Google Scholar]
- 39. Freeman J. L., Persans M. W., Nieman K., Salt D. E. (2005) Nickel and cobalt resistance engineered in Escherichia coli by overexpression of serine acetyltransferase from the nickel hyperaccumulator Thlaspi goesingense. Appl. Environ. Microbiol. 71, 8627–8633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Freeman J. L., Salt D. E. (2007) The metal tolerance profile of Thlaspi goesingense is mimicked in Arabidopsis thaliana heterologously expressing serine acetyltransferase. BMC Plant Biol. 7, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Na G., Salt D. E. (2011) Differential regulation of serine acetyltransferase is involved in nickel hyperaccumulation in Thlaspi goesingense. J. Biol. Chem. 286, 40423–40432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Shin R., Jez J. M., Basra A., Zhang B., Schachtman D. P. (2011) 14-3-3 Proteins fine-tune plant nutrient metabolism. FEBS Lett. 585, 143–147 [DOI] [PubMed] [Google Scholar]
- 43. Blaszczyk A., Brodzik R., Sirko A. (1999) Increased resistance to oxidative stress in transgenic tobacco plants overexpressing bacterial serine acetyltransferase. Plant J. 20, 237–243 [DOI] [PubMed] [Google Scholar]
- 44. Harms K., von Ballmoos P., Brunold C., Höfgen R., Hesse H. (2000) Expression of a bacterial serine acetyltransferase in transgenic potato plants leads to increased levels of cysteine and glutathione. Plant J. 22, 335–343 [DOI] [PubMed] [Google Scholar]
- 45. Tabe L., Wirtz M., Molvig L., Droux M., Hell R. (2010) Overexpression of serine acetyltransferase produced large increases in O-acetylserine and free cysteine in developing seeds of a grain legume. J. Exp. Bot. 61, 721–733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kim W. S., Chronis D., Juergens M., Schroeder A. C., Hyun S. W., Jez J. M., Krishnan H. B. (2012) Transgenic soybean plants overexpressing O-acetylserine sulfhydrylase accumulate enhanced levels of cysteine and Bowman-Birk protease inhibitor in seeds. Planta 253, 13–23 [DOI] [PubMed] [Google Scholar]
- 47. Nguyen H. C., Hoefgen R., Hesse H. (2012) Improving the nutritive value of rice seeds. Elevation of cysteine and methionine contents in rice plants by ectopic expression of a bacterial serine acetyltransferase. J. Exp. Bot. 63, 5991–6001 [DOI] [PubMed] [Google Scholar]
- 48. Ruffet M. L., Droux M., Douce R. (1994) Purification and kinetic properties of serine acetyltransferase free of O-acetylserine(thiol)lyase from spinach chloroplasts. Plant Physiol. 104, 597–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Leu L. S., Cook P. F. (1994) Kinetic mechanism of serine acetyltransferase from Salmonella typhimurium. Biochemistry 33, 2667–2671 [DOI] [PubMed] [Google Scholar]
- 50. Hindson V. J., Shaw W. V. (2003) Random-order ternary complex reaction mechanism of serine acetyltransferase from Escherichia coli. Biochemistry 42, 3113–3119 [DOI] [PubMed] [Google Scholar]
- 51. Johnson C. M., Huang B., Roderick S. L., Cook P. F. (2004) Kinetic mechanism of the serine acetyltransferase from Haemophilus influenzae. Arch. Biochem. Biophys. 429, 115–122 [DOI] [PubMed] [Google Scholar]
- 52. Johnson C. M., Huang B., Roderick S. L., Cook P. F. (2004) Chemical mechanism of the serine acetyltransferase from Haemophilus influenzae. Biochemistry 43, 15534–15539 [DOI] [PubMed] [Google Scholar]
- 53. Guan R., Roderick S. L., Huang B., Cook P. F. (2008) Roles of histidines 154 and 189 and aspartate 139 in the active site of serine acetyltransferase from Haemophilus influenzae. Biochemistry 47, 6322–6328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hindson V. J., Moody P. C., Rowe A. J., Shaw W. V. (2000) Serine acetyltransferase from Eschericia coli is a dimer of trimers. J. Biol. Chem. 275, 461–466 [DOI] [PubMed] [Google Scholar]
- 55. Pye V. E., Tingey A. P., Robson R. L., Moody P. C. (2004) Structure and mechanism of serine acetyltransferase from Escherichia coli. J. Biol. Chem. 279, 40729–40736 [DOI] [PubMed] [Google Scholar]
- 56. Olsen L. R., Huang B., Vetting M. W., Roderick S. L. (2004) Structure of serine acetyltransferase in complex with CoA and its cysteine feedback inhibitor. Biochemistry 43, 6013–6019 [DOI] [PubMed] [Google Scholar]
- 57. Gorman J., Shapiro L. (2004) Structure of serine acetyltransferase from Haemophilus influenzae Rd. Acta Crystallogr. D 60, 1600–1605 [DOI] [PubMed] [Google Scholar]
- 58. Kumar S., Raj I., Nagpal I., Subbarao N., Gourinath S. (2011) Structural and biochemical studies of serine acetyltransferase reveal why the parasite Entamoeba histoyltica cannot form a cysteine synthase complex. J. Biol. Chem. 286, 12533–12541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Inoue K., Noji M., Saito K. (1999) Determination of the sites required for the allosteric inhibition of serine acetyltransferase by l-cysteine in plants. Eur. J. Biochem. 266, 220–227 [DOI] [PubMed] [Google Scholar]
- 60. Minor W., Cymborowski M., Otwinowski Z., Chruszcz M. (2006) HKL-3000. The integration of data reduction and structure solution from diffraction images to initial model in minutes. Acta Crystallogr. D 62, 859–866 [DOI] [PubMed] [Google Scholar]
- 61. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Emsley P., Cowtan K. (2004) Coot. Model-building tools for molecular graphics. Acta Crystallogr. D 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 63. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) Phenix. A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Cook P. F., Wedding R. T. (1978) Cysteine synthase from Salmonella typhimurium LT-2. Aggregation, kinetic behavior, and effect of modifiers. J. Biol. Chem. 253, 7874–7879 [PubMed] [Google Scholar]
- 65. Thoden J. B., Cook P. D., Schäffer C., Messner P., Holden H. M. (2009) Structural and functional studies of QdtC. An N-acetyltransferase required for the biosynthesis of dTDP-3-acetamido-3,6-dideoxy-α-d-glucose. Biochemistry 48, 2699–2709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Thoden J. B., Reinhardt L. A., Cook P. D., Menden P., Cleland W. W., Holden H. M. (2012) Catalytic mechanism of perosamine N-acetyltransferase revealed by high-resolution x-ray crystallographic studies and kinetic analyses. Biochemistry 51, 3433–3444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Olivier N. B., Imperiali B. (2008) Crystal structure and catalytic mechanism of PglD from Campylobacter jejuni. J. Biol. Chem. 283, 27937–27946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Sulzenbacher G., Gal L., Peneff C., Fassy F., Bourne Y. (2001) Crystal structure of Streptococcus pneumoniae N-acetylglucosamine-1-phosphate uridyltransferase bound to acetyl-coenzyme A reveals a novel active site architecture. J. Biol. Chem. 276, 11844–11851 [DOI] [PubMed] [Google Scholar]
- 69. Raetz C. R., Roderick S. L. (1995) A left-handed parallel β helix in the structure of UDP-N-acetylglucosamine acyltransferase. Science 270, 997–1000 [DOI] [PubMed] [Google Scholar]
- 70. Privalov P. L. (1990) Cold denaturation of proteins. Crit. Rev. Biochem. Mol. Biol. 25, 281–305 [DOI] [PubMed] [Google Scholar]
- 71. Marshall C. J. (1997) Cold-adapted enzymes. Trends Biotechnol. 15, 359–364 [DOI] [PubMed] [Google Scholar]
- 72. Georlette D., Blaise V., Collins T., D'Amico S., Gratia E., Hoyoux A., Marx J. C., Sonan G., Feller G., Gerday C. (2004) Some like it cold. Biocatalysis at low temperatures. FEMS Microbiol. Rev. 28, 25–42 [DOI] [PubMed] [Google Scholar]
- 73. Almog O., Kogan A., de Leeuw M., Gdalevsky G. Y., Cohen-Luria R., Parola A. H. (2008) Structural insights on cold inactivation of tryptophanase and cold adaptation of subtilisin S41. Biopolymers 89, 354–359 [DOI] [PubMed] [Google Scholar]
- 74. Taguchi S., Komada S., Momose H. (2000) The complete amino acid substitutions at position 131 that are positively involved in cold adaptation of subtilisin BPN′. Appl. Environ. Microbiol. 66, 1410–1415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Iba K. (2002) Acclimative response to temperature stress in higher plants. Approaches of gene engineering for temperature tolerance. Annu. Rev. Plant Biol. 53, 225–245 [DOI] [PubMed] [Google Scholar]
- 76. Kocsy G., Szalai G., Vágújfalvi A., Stéhli L., Orosz G., Galiba G. (2000) Genetic study of glutathione accumulation during cold hardening in wheat. Planta 210, 295–301 [DOI] [PubMed] [Google Scholar]
- 77. Kocsy G., von Ballmoos P., Suter M., Rüegsegger A., Galli U., Szalai G., Galiba G., Brunold C. (2000) Inhibition of glutathione synthesis reduces chilling tolerance in maize. Planta 211, 528–536 [DOI] [PubMed] [Google Scholar]
- 78. Kocsy G., Galiba G., Brunold C. (2001) Role of glutathione in adaptation and signalling during chilling and cold acclimation in plants. Physiol. Plant 113, 158–164 [DOI] [PubMed] [Google Scholar]
- 79. Gómez L. D., Vanacker H., Buchner P., Noctor G., Foyer C. H. (2004) Intercellular distribution of glutathione synthesis in maize leaves and its response to short-term chilling. Plant Physiol. 134, 1662–1671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Phartiyal P., Kim W. S., Cahoon R. E., Jez J. M., Krishnan H. B. (2006) Soybean ATP sulfurylase, a homodimeric enzyme involved in sulfur assimilation, is abundantly expressed in roots and induced by cold treatment. Arch. Biochem. Biophys. 450, 20–29 [DOI] [PubMed] [Google Scholar]
- 81. Phartiyal P., Kim W. S., Cahoon R. E., Jez J. M., Krishnan H. B. (2008) The role of 5-adenylylsulfate reductase in the sulfur assimilation pathway of soybean. Molecular cloning, gene expression, and kinetic characterization. Phytochemistry 69, 356–364 [DOI] [PubMed] [Google Scholar]