

Background: Distinct termination pathways for yeast RNA polymerase II (RNApII) employ proteins (Nrd1 and Rtt103) recognizing different phospho-forms of the RNApII C-terminal domain (CTD).
Results: Alteration of CTD-binding specificity of Nrd1 significantly affects RNApII termination.
Conclusion: Differential interaction between RNApII CTD and termination factors is crucial in choosing a termination pathway.
Significance: CTD-interacting domain of Nrd1 couples termination and RNA processing by the nuclear exosome.
Keywords: mRNA, RNA Polymerase II, RNA Processing, Small Nucleolar RNA (snoRNA), Transcription Termination, C-terminal Domain, Nrd1
Abstract
The RNA polymerase II (RNApII) C-terminal domain (CTD)-interacting domain (CID) proteins are involved in two distinct RNApII termination pathways and recognize different phosphorylated forms of CTD. To investigate the role of differential CTD-CID interactions in the choice of termination pathway, we altered the CTD-binding specificity of Nrd1 by domain swapping. Nrd1 with the CID from Rtt103 (Nrd1(CIDRtt103)) causes read-through transcription at many genes, but can also trigger termination where multiple Nrd1/Nab3-binding sites and the Ser(P)-2 CTD co-exist. Therefore, CTD-CID interactions target specific termination complexes to help choose an RNApII termination pathway. Interactions of Nrd1 with both CTD and nascent transcripts contribute to efficient termination by the Nrd1 complex. Surprisingly, replacing the Nrd1 CID with that from Rtt103 reduces binding to Rrp6/Trf4, and RNA transcripts terminated by Nrd1(CIDRtt103) are predominantly processed by core exosome. Thus, the Nrd1 CID couples Ser(P)-5 CTD not only to termination, but also to RNA processing by the nuclear exosome.
Introduction
Transcription termination is closely linked to RNA 3′-end processing (1) via the C-terminal domain (CTD)5 of RNApII (2, 3). The CTD is differentially phosphorylated at Ser-2 and Ser-5 positions during transcription, with Ser(P)-5 predominating within the first few hundred nucleotides of the promoter and Ser(P)-2 dominant further downstream (4, 5). The distinct CTD phosphorylation patterns act as recognition sites for various RNA processing factors (6–8). Besides these two marks, the recent discoveries of phosphorylation at Ser-7, Thr-4, and Tyr-1 greatly increase the complexity of “CTD code” (9, 10). In mammalian cells, Ser(P)-7 plays a role in snRNA gene expression and processing (11), whereas Thr(P)-4 affects histone mRNA 3′-end processing and transcription elongation (12, 13). Although they are found at all RNApII-transcribed genes, general functions for Ser(P)-7 and Thr(P)-4 remain to be investigated. Tyr(P)-1 increases in coding regions and decreases before polyadenylation (pA) sites, and is proposed to impair recruitment of CTD-interacting domain (CID) proteins Nrd1, Pcf11, and Rtt103 within gene bodies (14). Because the CID proteins function in termination, Tyr(P)-1 may set up a confined window within which each CID protein can trigger RNApII termination.
There are at least two distinct termination pathways for RNApII in Saccharomyces cerevisiae (15). For mRNA genes, cleavage of the nascent RNA transcript at pA sites provides an entry point for the 5′-3′ exoribonuclease Rat1 (Xrn2 in human). Rat1 then degrades the downstream RNA transcripts and may, upon catching up to RNApII, trigger termination by dissociating the elongation complex from the template (16). The Rtt103 CID specifically binds to Ser(P)-2 to facilitate Rat1/Rai1 interactions with the elongating RNApII at the 3′-ends of genes. Similar findings in mammalian cells indicate an evolutionarily conserved role for Rat1 in termination (17).
The second termination pathway occurs independently of RNA cleavage or Rat1, and functions at small nucleolar RNAs (snoRNAs) and cryptic unstable transcripts (15, 18, 19). Several proteins are implicated in this pathway: mutations in either of two RNA-binding proteins (Nrd1 and Nab3) or a helicase (Sen1) cause defective termination at multiple snoRNA genes (15, 20, 21). Sen1 is thought to be a functional homolog of bacterial termination factor Rho. Nrd1 has a CID domain that preferentially binds Ser(P)-5 CTD (22), thus preferentially targeting the snoRNA termination complex to early transcribed regions.
Also associated with Nrd1 are the nuclear exosome and TRAMP (Trf4/Air2/Mtr4 polyadenylation) complexes (23). The core exosome consists of six RNase PH-like proteins (Rrp41, Rrp42, Mtr3, Rrp43, Rrp46, and Rrp45), three S1/KH domain proteins (Rrp4, Csl4, and Rrp40), and the 3′-5′ exoribonucleases (Dis3) (24–27). The nuclear exosome contains the 10-subunit core exosome plus the nuclear-specific 3′-5′ exoribonuclease Rrp6. Together with the TRAMP complex, the nuclear exosome degrades or trims the terminated transcripts in a 3′-5′ direction (28, 29). Thus, the Nrd1 complex couples termination and subsequent RNA 3′-end processing at snoRNAs and cryptic unstable transcripts, but how this coupling is mediated has not been shown.
Because the CID proteins involved in the two termination pathways differ in their CTD-binding specificity, Rtt103 favors Ser(P)-2 CTD, whereas Nrd1 prefers Ser(P)-5, one contributor to the choice between the mRNA and snoRNA pathways may be the CTD phosphorylation state. Indeed, the Ser(P)-5-specific binding of Nrd1 has been suggested as an explanation for why the Nrd1/Nab3/Sen1-dependent termination is preferentially used at short genes (less than 600-nt), where Ser(P)-5 predominates over Ser(P)-2 (22, 30, 31). However, the role of differential CTD-CID interactions in the termination pathway choice has not been explicitly addressed.
We replaced the Nrd1 CID with that from Rtt103 to investigate how altering CTD-CID interactions affects RNApII termination. Nrd1(CIDRtt103) becomes capable of interacting with the Ser(P)-2 CTD and triggering RNApII termination at regions where multiple Nrd1/Nab3-binding sites (NN cluster) and Ser(P)-2 CTD are co-localized. Putting NN clusters alone at the 3′-end of genes was not sufficient to trigger the Nrd1-dependent termination (31), so our results demonstrate that concomitant binding of Nrd1 to both CTD and RNA transcripts promotes efficient RNApII termination by the Nrd1 complex. Therefore, the CTD-CID interaction is a crucial determinant for choosing an RNApII termination pathway.
We also found that the transcripts terminated by the CID-swapped Nrd1 are processed predominantly by the core exosome instead of nuclear exosome. This difference is due to reduced interaction of the Nrd1(CIDRtt103) complex with Rrp6 and Trf4 (TRAMP subunit). Furthermore, Nrd1 CID deletion completely loses the interaction with exosome, resulting in RNA processing defects as previously observed (23, 32). These results demonstrate that it is the Nrd1 CID that connects non-pA termination with RNA processing by the nuclear exosome and TRAMP complex.
EXPERIMENTAL PROCEDURES
Yeast Strains
CID-swapped nrd1 mutants were generated by a delitto perfetto method (33). Briefly, BY4741 (WT) was pre-transformed with pRS316-NRD1. A 4.5-kb PCR product having GAL1-I-SceI, KanMX4, and KlURA3 CORE cassette (from pGSKU) was subsequently inserted into the NRD1 ORF at position +321 (ATG is at +1) by homologous recombination. After galactose induction of SceI, this strain was transformed with a CID switching PCR product to replace the CID of Nrd1 (aa 1–154) with the CID of Rtt103 (aa 1–140). This method was similarly utilized to generate ADH1-TACT1 and CRH1::EGFP strains.
For C-terminal myc tagging, a tandem affinity purification (TAP)-tagged allele for each protein was first switched to 5× myc tag using an epitope switching cassette amplified from pFA6a-Myc-KlURA3 (34). Then a 5× myc-tagging cassette obtained from this strain was transformed into the CID-swapped strains and isogenic WT cells.
Chromatin Immunoprecipitations (ChIPs)
ChIP procedures and quantification were performed as described (7). The sequences of oligonucleotides used in this study are available upon request. ChIPed DNAs were either subjected to conventional PCR in the presence of [α-32P]dATP and PAGE visualization or analyzed in real-time using SYBR Green Supermix and CFX96 cycler (Bio-Rad). Variation between duplicate PCRs is less than 5% (as measured by the internal control PCR product) and variation between experiments was typically less than 20%.
Amplification of ChIP DNA for Microarray Analysis
The α-Rpb3 ChIPed DNAs were amplified and re-amplified with the GenomePlex® Complete Whole Genome Amplification 2 (WGA2) Kit using the Farnham Lab WGA Protocol for ChIP-chip (35). The re-amplification was performed in the presence of 0.4 mm dUTP (Promega U1191) to allow later enzymatic fragmentation.
Tiling Array Hybridization
The enzymatic fragmentation, labeling, hybridization, and array scanning were done according to the manufacturer's instructions (Affymetrix Chromatin Immunoprecipitation Assay Protocol P/N 702238). Enzymatic fragmentation and terminal labeling were performed by application of the GeneChip WT Double-stranded DNA Terminal Labeling Kit (P/N 900812, Affymetrix). About 5.5 μg of fragmented and labeled DNA were hybridized to a high-density custom-made Affymetrix tiling array (36) (PN 520055) at 45 °C for 16 h with constant rotational mixing at 60 rpm in a GeneChip Hybridization Oven 640 (Affymetrix). Washing and staining of the tiling arrays were performed using the FS450_0001 script of the Affymetrix GeneChip Fluidics Station 450. The arrays were scanned using an Affymetrix GeneChip Scanner 3000 7G. The resulting raw data image files (.DAT) were inspected for any impairment. The CEL intensity files were used for bioinformatics analysis.
ChIP-Chip Data Normalization
Normalization and analysis of ChIP-chip data were done as described (37). Briefly, ChIP enrichments for RNApII using α-Rpb3 (Neoclone) were obtained by dividing ChIP intensities by the genomic input intensities. The normalized signal was converted to occupancy values between 0 and 100% by setting the genome-wide 99.8% quantile to 100% occupancy and the 10% quantile to 0% occupancy.
Calculation of Read-through Index (RI)
Genes analyzed to generate the averaged profiles and RI were filtered as described previously (14, 37). Briefly, of the 5,769 non-dubious protein-coding nuclear genes in the Saccharomyces Genome Database, we first selected 4,366 genes (76%) with assigned TSS and pA sites (38). Furthermore, genes were excluded when their TSS was downstream or over 200 nt away from the annotated start codon, when their pA site was upstream or over 200 nt away from the annotated stop codon, or when neighboring genes were less than 200 nt away. Of the resulting 1,786 genes, only 1,140 genes with higher mRNA levels than the median expression level of all yeast genes (39) were further considered for gene-averaged profiles. Among 77 snoRNA genes in the yeast genome, 51 snoRNAs, which are monocistronic and independently transcribed, were selected for analysis.
The normalized ChIP-chip signal (log2 scale) of each 25-mer array probe was used as a representative RNApII signal for the genomic position at the center of the probe. To calculate the RI values for 1,140 mRNA genes and 51 snoRNA genes, RNApII signals from the 3′ upstream region (160 nt upstream of pA site of mRNAs or 80 nt upstream of the 3′-end of mature snoRNAs) and the 3′ downstream region (160 nt downstream of pA site of mRNAs or 160 nt downstream of the 3′-end of mature snoRNAs) were compared. First, at each gene, the median RNApII signal of the upstream region was obtained. Next, all RNApII signals in the upstream and downstream regions were re-scaled by subtracting the median of the upstream region, setting the re-scaled median RNApII signals in the upstream region to zero. The median re-scaled RNApII signals of the downstream regions were calculated and their distributions in wild type and mutant cells were statistically assessed using the paired Student's t test implemented in the R package. Finally, the RI was calculated by subtracting the median signal of the CID-swapped strain (Nrd1(CIDRtt103) Δrtt103) from that of the WT (Nrd1).
Averaged RNApII Occupancy Profiles
The mean ± S.D. of RIs were calculated for mRNA and snoRNA data sets. Three gene groups were defined according to their RI values: “high” (genes with the greater RI values than “mean + S.D.”), “low” (genes with the smaller RI values than “mean − S.D.”), and “medium” (the rest). Averaged RNApII occupancy profiles within these groups were calculated by taking the median over genes at each genomic position.
Northern Blot Analysis
GALpro::RRP41 strains were grown on 2% galactose and 1% raffinose medium at 30 °C to an A600 ≈ 0.8 and shifted to 2% glucose media after washing with sterile water. Cells were then incubated at 30 °C for 12 h, transferred once again to fresh 2% glucose media, and further incubated for 12 h (total 24 h).
Total RNA was isolated using hot phenol extraction and quantified by NanoDrop 2000c (Thermo Scientific). About 45 μg of total RNA was loaded onto a 1.2–1.5% agarose (MOPS-formaldehyde) gel. RNA was transferred by capillary action onto NYTRAN N membrane (Whatman) for 20–24 h, and prehybridization was carried out for 2 h at 65 °C in a solution containing 300 mm sodium phosphate buffer (pH 7.2), 1% BSA (w/v), 7% SDS, 1 mm EDTA. Hybridization was performed in the same conditions with a radiolabeled probe for ∼16 h, and the membranes were washed in 2× SSC, 0.1% SDS and 0.2× SSC, 0.1% SDS at RT or 42 °C. After washing, the membranes were analyzed by a phosphorimager (BAS1500, Fuji). Single-strand probes were generated by unidirectional PCR, as described in Ref. 40. About 5 ng of purified DNA template (150–250 bp) was assembled with 200 μm each of dGTP, dATP, and dTTP, 5 μm oligo (antisense to the RNA being analyzed), 5 μl of [α-32P]dCTP (3,000 Ci/mmol), and Taq DNA polymerase in a total volume of 20 μl. PCR was performed for 40 cycles (94 °C, 30 s; 55 °C, 30 s; 72 °C, 50 s), and unincorporated radionucleotide was removed by ProbeQuant G-50 Micro Column (GE Healthcare). The purified probe was denatured by heating at 95 °C for 5 min, chilled on ice, and added into 15 ml of hybridization solution. SCR1 was used as a loading control.
Reverse Transcription-PCR Analysis
Prior to synthesizing cDNA, 3 μg of total RNA was incubated with 10 μl of a mixture containing 20 mm Tris-HCl (pH 8.4), 50 mm KCl, 2 mm MgCl2, and 1 unit of DNase I (Promega) at RT for 90 min to remove genomic DNA. The reaction was stopped by adding 1 μl of 25 mm EDTA and heating at 65 °C for 15 min. cDNA synthesis was carried out in a 20-μl reaction containing 3 μg of RNA, 10 pmol of antisense primer, 20 mm Tris-HCl (pH 8.4), 50 mm KCl, 5 mm MgCl2, 0.5 mm dNTPs, 10 mm DTT, 16 units of RNasin, and 200 units of M-MLV reverse transcriptase (Macrogen) at 42 °C for 50 min. To stop the reaction, reverse transcriptase was inactivated by heating at 70 °C for 15 min and the RNA of RNA/cDNA hybrids was destroyed by RNase H (New England Biolabs) at 37 °C for 20 min. One-tenth of the reverse transcribed cDNA products were then amplified by PCR for 28 cycles (95 °C, 30 s; 55 °C, 60 s; 72 °C, 60 s). The PCR product was resolved by 2% agarose gel electrophoresis and visualized by ethidium bromide staining. The band intensities of PCR products from the WT-galactose sample were set to 100% and those from the rest of samples were calculated in relative amounts.
Co-immunoprecipitation and Western Blotting Analysis
Cells were grown in SC medium to an A600 ≈ 1.6 and broken by glass beads in lysis buffer (20 mm Tris (pH 7.6), 100 mm NaCl, 5 mm MgCl2, 1 mm EDTA, 10% glycerol, 0.05% Nonidet P-40, 1 mm DTT) supplemented with protease inhibitors and phosphatase inhibitors. About 4 mg of extracts were incubated overnight with IgG-Sepharose beads (GE Healthcare) at 4 °C. Pelleted beads were washed three times with ice-cold lysis buffer and boiled for 5 min with 3× SDS loading buffer. After SDS-PAGE, co-IPed proteins were monitored by specific antibodies: H5 for CTD Ser(P)-2, H14 for CTD Ser(P)-5, 8WG16 for hypophosphorylated CTD, 9E10 for myc-tagged proteins (Covance), anti-Nab3 (Invitrogen), and peroxidase anti-peroxidase for TAP-tagged Nrd1 (Sigma).
RESULTS
Altering CTD Binding Specificity of Nrd1 by CID Swapping
The different CTD-binding specificities of Nrd1 and Rtt103 CIDs prompted us to use domain swapping to directly address the role of CTD-CID interactions in choosing an RNApII termination pathway. The two CIDs are well defined and separated from other domains within each protein, making this approach feasible (22, 41). The Nrd1 CID (1–151 aa) is slightly larger than the Rtt103 CID (1–131 aa), due to extended linkers among 8 α-helices (41). With a few more extra amino acids included, the Nrd1 CID (1–154 aa) was replaced with the CID of Rtt103 (1–140 aa). Using a C-terminal TAP tag, we monitored recruitment of Nrd1(CIDRtt103) on several mRNA genes by ChIP. As predicted by the switch in phospho-CTD binding preference, Nrd1(CIDRtt103) significantly loses 5′-end cross-linking, and instead gets recruited to 3′-ends similarly to Rtt103 at both PMA1 and ADH1 genes (Fig. 1, A–C). Along snoRNA loci, Nrd1(CIDRtt103) is also reduced at the 5′-ends and cross-linking was extended further downstream (up to positions 4 and 5) compared with wild-type Nrd1 (Fig. 1, D and E). In contrast, deletion of the CID (Nrd1ΔCID) does not significantly increase or extend Nrd1 recruitment at 3′-ends (Fig. 1, B–E) but merely results in reduction at 5′-ends as clearly seen at PMA1 and ADH1 (Fig. 1, B and C). Therefore, the 3′-end targeting of Nrd1(CIDRtt103) is due to acquisition of the Rtt103 CID. In contrast, the Nrd1 CID fails to target Rtt103 and Pcf11 to 5′-end of genes (data not shown), suggesting that 5′-end recruitment of Rtt103 and Pcf11 may need additional protein interactions besides the CID recognizing Ser(P)-5 CTD.
FIGURE 1.

CID swapping changes the CTD-binding specificity of Nrd1. A, recruitment of Nrd1(CIDRtt103) on the PMA1 gene was monitored by ChIP assay. Nrd1(CIDRtt103) resembles Rtt103 in the recruitment pattern. Numbers below each lane refer to PCR primers as shown. B, recruitment of Nrd1(CIDRtt103) (either WT or Δrtt103 background) and Nrd1ΔCID was analyzed on the PMA1 genes by ChIP-quantitative PCR. Positions of PCR primers for PMA1 are as in A. C–E, recruitment of Nrd1(CIDRtt103) (either WT or Δctk1 background) and Nrd1ΔCID was analyzed by ChIP-qPCR. Positions of PCR primers are shown below. C, ADH1 gene. D, SNR13 gene. E, SNR33 gene. F, Nrd1(CIDRtt103) gains the ability to bind to the Ser(P)-2 CTD of RNApII as revealed by co-IP/Western blot analyses. All Nrd1 variants analyzed here were C-terminal TAP tagged for immunoprecipitation with IgG beads. Error bars represent S.E. for at least three independent experiments.
Interestingly, at the ADH1 gene the recruitment of wild-type Nrd1 is higher at the 3′-end than 5′-end, but this is unlikely to be due to a small antisense gene (MHF1) immediately downstream of ADH1 (Fig. 1C) because the MHF1 transcription level is as low as the adjacent YOL087C gene (42) and only a near-background Ser(P)-5 signal was detected (43). Instead, it is apparently due to an NN cluster present at the 3′-end of ADH1 (see below), revealing an RNA-dependent recognition mechanism for Nrd1 recruitment. Upon CID swapping, the 3′-end recruitment of Nrd1 increases further (Fig. 1C). These results demonstrate that CTD binding by the CID, along with RNA recognition, is indeed an important recruitment mechanism of Nrd1 to elongating RNApII, and that the Rtt103 CID can target proteins to the 3′-end of genes.
Although one CTD phosphorylation site predominates over others at different stages of transcription, it is likely that multiple phosphorylated states co-exist within a single CTD throughout the transcription cycle. Considering structural studies showing that the CTD binding pocket encompasses approximately two heptapeptide repeats (22, 44, 45), CID proteins may compete with each other for CTD binding, and this competition could be decisive in choosing a termination pathway. In support of this idea, the recruitment of wild-type Nrd1 at the 3′-end of PMA1 slightly increases when Rtt103 is deleted and is augmented even further for Nrd1(CIDRtt103) under the Δrtt103 background (Fig. 1B).
To confirm that Nrd1(CIDRtt103) acquires interaction with Ser(P)-2 CTD, TAP-tagged Nrd1 proteins from either wild-type or CID-swapped cells were precipitated and the CTD phosphorylation state of associated RNApII was analyzed by Western blotting. As previously reported (22), the Nrd1 protein showed little association with Ser(P)-2 CTD, whereas Nrd1(CIDRtt103) preferentially binds to this form (Fig. 1F). Consistent with this result, deletion of the Ser-2 kinase CTK1 results in a significant reduction of Nrd1(CIDRtt103) recruitment at 3′-ends (Fig. 1, C-E). Interestingly, Nrd1(CIDRtt103) remains associated with the Ser(P)-5 CTD (Fig. 1F), implying there might be CID-independent ways to target Nrd1 to Ser-5-phosphorylated RNApII, either directly or indirectly. Most likely, some portion of Ser(P)-5 repeats exist within the co-IPed CTD, even as the majority of CTD repeats are Ser-2 phosphorylated. Although CID swapping does not affect the interaction between Nrd1 and Nab3, the association with Sen1 is decreased (Fig. 1F). A recent report showed that Sen1 associates less with Nrd1/Nab3 when bound to Ser(P)-2 CTD (46). Thus, the reduced amount of Sen1 co-IPed with Nrd1(CIDRtt103) may be because Nrd1(CIDRtt103) is preferentially bound to Ser(P)-2 CTD.
The expression of Nrd1 is autoregulated by early termination of its own gene (47). Consistently, CID swapping increases the protein levels of TAP-tagged Nrd1 as expected from its altered recruitment (Fig. 1F, compare lanes 7 and 9, 8 and 10). Note that the smaller size of the Nrd1-TAP band in Nrd1 Δrtt103 cells (Fig. 1F, lanes 3 and 8) is due to a smaller TAP tag (∼6 kDa smaller than the others) used in this strain. This was done for marker compatibility issues. We observed no differences in immunoprecipitation or Western blotting analyses between the TAP::TRP1 and TAP::HIS3 modules in the same cell background.
RNApII Termination in Nrd1(CIDRtt103) Cells
To investigate how CID swapping of Nrd1 affects RNApII termination, RNApII occupancy was monitored by ChIP assays using α-Rpb3 antibody. At the PMA1 gene, RNApII occupancy at positions 8 and 9, where termination normally occurs in WT cells, was significantly higher in Nrd1(CIDRtt103) (74 and 33% of the promoter RNApII signal, respectively) than WT cells (25 and 7%, respectively). Therefore, the Nrd1(CIDRtt103) mutant leads to a termination defect (Fig. 2A). This effect was aggravated upon deletion of Rtt103, with further increases of the RNApII signal at the 3′-region of PMA1 (112 and 60% at positions 8 and 9, respectively) (Fig. 2A). Presumably, the increased recruitment of Nrd1(CIDRtt103) in the Δrtt103 background might lead to stronger perturbation of RNApII termination at the PMA1 gene. One plausible explanation for this termination defect is that Nrd1(CIDRtt103) effectively competes with mRNA 3′-cleavage/pA factors at the 3′-end of genes, thereby interfering with the pA cleavage-dependent termination. Supporting this model, the recruitment of Rna14, a component of Cleavage Factor IA complex, was significantly reduced at the 3′-end of PMA1 (Fig. 2A).
FIGURE 2.

Distinct RNApII termination profiles in Nrd1(CIDRtt103) cells. A, CID swapping of Nrd1 causes defective RNApII termination in PMA1 as measured by ChIP assay with α-Rpb3 antibody (left panel). Significant decrease of Rna14 recruitment at the 3′-end of PMA1 might result from the strong ectopic recruitment of Nrd1(CIDRtt103) to the same region and competition for CTD binding (right panel). B, CID swapping of Nrd1 does not perturb RNApII termination in ADH1 (left panel), although Rna14 recruitment was severely reduced at the 3′-end (right panel). C and D, RNApII termination begins further downstream, presumably because RNApII needs to transcribe longer where the level of Ser(P)-2 CTD becomes stronger for optimal recruitment of Nrd1(CIDRtt103). C, SNR13 gene. D, SNR33 gene. Positions of PCR primers for each geneare are illustrated. Numbers are average RNApII ChIP signals at each position (the promoter signal is set to 100). Error bars represent S.E. for at least three independent experiments.
The recruitment of Rna14 was also greatly reduced at the 3′-end of ADH1 in Nrd1(CIDRtt103) strains (Fig. 2B). In contrast to PMA1, RNApII occupancy dropped normally at the 3′-end of ADH1 (region between positions 3 and 4) in both Nrd1(CIDRtt103) and WT cells, suggesting that termination of ADH1 was unexpectedly unperturbed (Fig. 2B). Intriguingly, RNApII occupancy is significantly higher at position 3 than position 2 in Nrd1(CIDRtt103) strains, which may reflect extended elongation and/or a pause of RNApII near position 3 triggered by the CID-swapped Nrd1.
At snoRNA genes, the RNApII signal remains high at the 3′-flanking region (position 3 in SNR13 and SNR33) and the reduction reflecting termination occurs further downstream (SNR33) in Nrd1(CIDRtt103) strains (Fig. 2, C and D). This implies that Nrd1(CIDRtt103) affects transcription termination of snoRNAs genes, generating a certain level of read-through transcription (see below), perhaps because RNApII needs to transcribe further downstream where Ser(P)-2 CTD levels increase to promote optimal recruitment of Nrd1(CIDRtt103).
Nrd1(CIDRtt103) Alters RNApII Termination at ADH1 Gene
Distinctive termination phenotypes of Nrd1(CIDRtt103) at the PMA1 and ADH1 genes prompted us to investigate features unique to ADH1 further. First of all, we tested whether the difference comes from the 3′-end sequences by replacing the ADH1 3′-flanking region (183 bp, TADH1) with the ACT1 terminator (TACT1, 273 bp) (Fig. 3A). TACT1 has multiple pA sites, whose usage is greatly affected by mutations in cleavage/pA factors (48, 49). In WT cells, RNApII occupancy at ADH1-TACT1 decreased after TACT1 (77 and 26% of the promoter RNApII signal at positions 4 and 5, respectively), indicating that TACT1 successfully terminates ADH1 (Fig. 3B). However, RNApII occupancy was significantly higher in the Nrd1(CIDRtt103) strain (138 and 48%, respectively), implying that termination is defective at TACT1 in Nrd1(CIDRtt103) cells. A similar degree of termination defect was observed in an Nrd1(CIDRtt103) Δrtt103 mutant (137 and 34% at positions 4 and 5, respectively), when compared with Δrtt103 cells (66 and 16%, respectively) (Fig. 3B).
FIGURE 3.
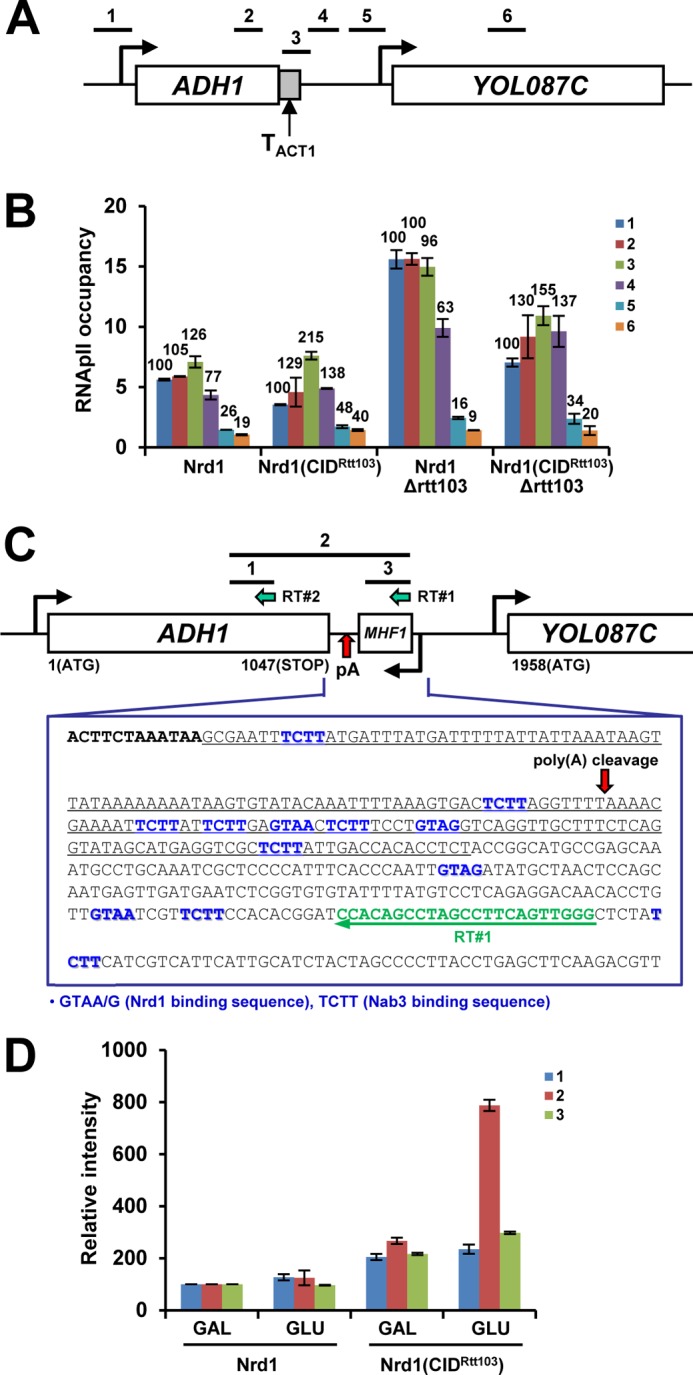
Nrd1(CIDRtt103) alters termination at ADH1. A, schematic diagram of ADH1 with ACT1 terminator. ADH1 3′-flanking region (183 bp as underlined in C, TADH1) was replaced with ACT1 terminator (TACT1, 273 bp). Positions of PCR primers are shown above the diagram. B, termination of ADH1-TACT1 becomes defective in Nrd1(CIDRtt103) strains, indicating that TACT1 may contain cis-acting element(s), responding to Nrd1(CIDRtt103). C, ORF map and sequences downstream of ADH1 are shown. ADH1 has multiple Nrd1/Nab3-binding sites near the pA site: ADH1 ORF in bold, Nrd1/Nab3-binding sites in blue, RT primer (RT#1) in green, and TADH1 (replaced with TACT1 as in A) is underlined. Reverse transcription using RT#1 primer was performed with RNAs from WT (Nrd1) and Nrd1(CIDRtt103) cells grown in either galactose or glucose (24 h) media to deplete Rrp41. Similar analysis was done using RT#2 primer for loading control and band normalization. Bars above the diagram denote PCR products subsequently generated. D, quantification of RT-PCR results. The band intensities of PCR products from the WT-galactose sample were set to 100% and those from the rest of samples were calculated in relative amounts. Results are shown from three repetitions. Error bars represent S.E.
These results suggest that, unlike TPMA1 and TACT1, TACT1 may contain cis-acting elements that respond to Nrd1(CIDRtt103). Interestingly, the ADH1 3′-flanking region has several Nrd1 and Nab3-binding sites (GUA(A/G) and UCUU, respectively) (50), among which five are clustered within 29 nt near the pA site (Fig. 3C). This NN cluster is quite similar to the one found in the SNR13 3′-flanking region (two each of Nrd1 and Nab3-binding sites within 25 nt), a proven binding target of the Nrd1-Nab3 complex (51). Additionally, the ADH1 3′-flanking region contains AU-rich stretches between translation stop codon and pA site (Fig. 3C). These AU-rich regions were shown to be important for ADH1 mRNA 3′-end formation (52) but is also very similar to AU-rich motifs that are recently reported to be associated with nearby Nrd1/Nab3-binding sites as a “supermotif,” triggering Nrd1/Nab3/Sen1-dependent termination (53).
To investigate whether ADH1 3′ end processing is altered in Nrd1(CIDRtt103) strains, we analyzed the ADH1 transcripts in WT and Nrd1(CIDRtt103) strains upon depletion of a core exosome component, Rrp41 (GALpro::RRP41). Total RNAs were isolated and reverse transcribed using a primer (RT#1) complementary to the 3′-downstream region of ADH1. PCR was then carried out with three primer pairs to generate products 1, 2, and 3, respectively (Fig. 3C). The same RNAs were also reverse transcribed with another primer (RT#2) complementary to the coding region, and its subsequent PCR product (product 1) was used for a loading control and band normalization. The band intensities of PCR products from the WT-galactose sample were set to 100 and those from the rest of samples were calculated as relative amounts (Fig. 3D). In WT cells, the amounts of the three PCR products were quite low even upon Rrp41 depletion, implying that nascent ADH1 transcripts that are intact up to RT#1 position are rare and most ADH1 transcripts are already cleaved at the pA site (Fig. 3D). In contrast, Nrd1(CIDRtt103) cells showed significantly higher levels of PCR products 2 and 3 when Rrp41 was depleted. The increase in product 2 spanning the pA site indicates that pA cleavage of the nascent ADH1 transcripts is somehow defective. But because this cleavage defect does not result in a termination defect, ADH1 might terminate, at least in part, by a cleavage-independent mechanism in Nrd1(CIDRtt103) cells.
Nrd1(CIDRtt103) Globally Affects RNApII Termination
To test for genome-wide effects of CID-swapped Nrd1 on RNApII termination, we performed RNApII ChIP-chip experiments with tiling arrays that cover the S. cerevisiae genome at 4-nt resolution. RNApII profiles were compared between wild-type Nrd1 and Nrd1(CIDRtt103) Δrtt103 cells, because the deletion of RTT103 accentuates the 3′-end recruitment and termination defects of Nrd1(CIDRtt103) (Figs. 1B and 2) but has no effect on termination by itself, as revealed by previous ChIP-chip assays (16). Of the 5,769 non-dubious protein-coding nuclear genes, 1,140 were chosen for further analysis (see “Experimental Procedures” for filtering criteria). To quantitate termination at the 3′-end of each gene, we focused on the regions 160 nt upstream and downstream from the pA site. The median RNApII signals from the downstream region were calculated for both WT and Nrd1(CIDRtt103) Δrtt103 cells and their distributions of the medians differ significantly between Nrd1(CIDRtt103) and WT cells (p value < 2.2 × 10−16 in paired t test), showing an overall shift toward the higher median values in the mutant. The averaged RNApII occupancy profile for all 1,140 genes shows higher RNApII signal in the downstream 160-nt region in Nrd1(CIDRtt103) Δrtt103 cells (Fig. 4A, left panel), indicating that read-through transcription occurs at most genes.
FIGURE 4.

Genome-wide ChIP-chip analysis reveals mRNA genes with distinct termination profiles. A, averaged RNApII occupancy profiles of mRNA genes were classified into three groups (high, medium, and low) based on the mean ± S.D. of RI values for the upstream and downstream 160-nt regions from the pA site. B, RNT1 locus and 3′-downstream sequences are shown; RNT1 ORF in bold (left panel). Black bar indicates the probe region used in Northern blotting. Total RNAs isolated from the indicated exosome and/or TRAMP complex mutants were analyzed with a strand-specific radiolabeled probe. The portion of read-through transcripts (marked by red arrowhead) relative to RNT1 mRNAs (set to 100%) upon Rrp41 depletion was quantitated (graph in the middle panel). SCR1 was used to monitor equal loading of RNAs at each lane. Because the same set of total RNAs were analyzed multiple times for different genes, SCR1 internal control is also applicable to C and D. C, internal coding sequences of CRH1 are shown: Nrd1/Nab3-binding sites are shown in blue (left panel). The portion of premature-terminated transcripts (marked by green arrowhead) relative to full-length CRH1 mRNAs (set to 100%) upon Rrp41 depletion is shown in the graph. Note that these short transcripts are not observed in Δrrp6 and/or Δtrf4 mutants. D, internal coding sequences and Northern blots of YVC1 are similarly shown as in C. E, deletion of RRP6 and/or TRF4 does not accumulate shorter transcripts (from CRH1 or YVC1) or pre-snoRNAs (from SNR13 or SNR33, denoted by a blue arrowhead) in Nrd1(CIDRtt103) cells, regardless of carbon sources. SCR1 was used for loading control.
A RI was calculated for each gene as the difference in the re-scaled median ChIP-chip signals of the downstream 160-nt region between Nrd1(CIDRtt103) rtt103 and WT cells (i.e. RI = median signal from Nrd1(IDRtt103) Δrtt103 minus median signal from WT). Among the 1,140 genes analyzed, 725 genes (63.6%) have positive RI values. Based upon the mean value and S.D. of RIs, 1,140 genes were divided into three categories: high (RI > 0.2129, 183 genes, 16.1%), medium (−0.1098 < RI < 0.2129, 796 genes, 69.8%), and low (RI < −0.1098, 161 genes, 14.1%) (Fig. 4A and supplemental Table S1, see “Experimental Procedures” for more details). The averaged RNApII occupancy profiles from each category predict distinctive termination phenotypes, which were put into three categories: strong read-through transcription (high), earlier premature termination (low), and mixed features (medium). We verified the ChIP-chip data for several genes by Northern blotting. To detect various aberrant transcripts, depletion of Rrp41 or deletion of RRP6 (nuclear exosome) and/or TRF4 (TRAMP subunit) were introduced into our strain backgrounds. Indeed, Northern blotting of RNT1 (RI = 0.351, high) showed stronger accumulation of read-through transcripts (marked by red arrowhead) in Nrd1(CIDRtt103) strains than in WT cells when Rrp41 was depleted, but these transcripts were very weakly detected in Δrrp6 and/or Δtrf4 backgrounds in both WT and Nrd1(CIDRtt103) cells (Fig. 4B).
As predicted, at genes with negative RI values (CRH1 (RI = −0.070, medium) and YVC1 (RI = −0.233, low)), RNApII tends to terminate early in Nrd1(CIDRtt103) cells, before reaching the canonical 3′-end termination sites utilized in WT cells, generating multiple shorter transcripts (green arrowheads), which were stabilized upon depletion of Rrp41 but not in Δrrp6 and/or Δtrf4 mutants (Fig. 4, C and D). Because Rrp41 depletion was done by changing carbon sources (from galactose to glucose), whereas the deletion mutants were grown in glucose media all along, we monitored whether the parallel carbon source shift might reveal an effect in Δrrp6 and/or Δtrf4 mutants. However, in either carbon source, we observed no appreciable accumulation of shorter transcripts in Δrrp6 and/or Δtrf4 mutants (Fig. 4E).
Intriguingly, we found that genes producing prematurely terminated transcripts in Nrd1(CIDRtt103) cells often contain internal NN cluster(s) within a short range (typically more than four Nrd1/Nab3-binding sites within ∼60 nt) as shown for the CRH1 and YVC1 genes (Fig. 4, C and D, left panels). Replacing this internal NN cluster with a segment from EGFP that does not contain Nrd1/Nab3-binding sites greatly diminished shorter transcripts from CRH1 (Fig. 5, A and B), indicating that generation of shorter transcripts in Nrd1(CIDRtt103) cells depends upon the NN cluster. Among 195 internal NN clusters from 172 genes (154 genes (89.5%) that belong to the low and medium categories), 136 internal NN clusters (69.7%) are located more than 600 nt away from the transcription start site (TSS): for example, 971 nt (CRH1) and 1227 nt (YVC1) away from TSS. Considering a genome-wide study that the Ser(P)-2 and Ser(P)-5 levels typically crossover at ∼450 nt downstream of TSS (54), the location of these internal NN clusters helps understand why these short transcripts preferentially appear in Nrd1(CIDRtt103) cells. Presumably, these clusters are relatively dormant in WT but become highly recognizable in Nrd1(CIDRtt103) cells, because the CTD of RNApII transcribing up to these internal clusters would have high Ser(P)-2/Ser(P)-5 ratios. The Rtt103 CID consequently allows preferred recruitment of Nrd1(CIDRtt103) over wild-type Nrd1 protein to these downstream NN clusters, resulting in earlier termination. Indeed, at the CRH1 gene, RNApII occupancy was significantly decreased near the internal NN cluster (position 4) in Nrd1(CIDRtt103) Δrtt103 cells, supporting this scenario (Fig. 5, C and D).
FIGURE 5.
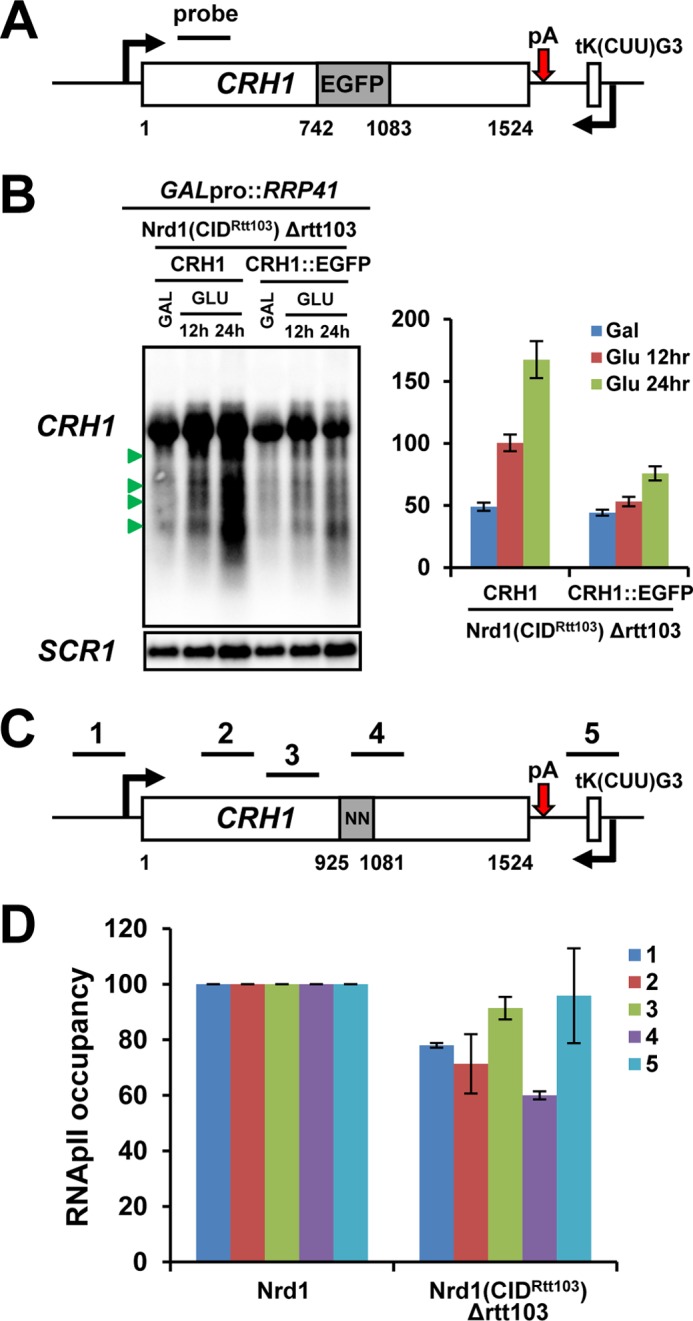
Internal NN cluster mediates premature termination by responding to Nrd1(CIDRtt103) at the CRH1 gene. A, an internal coding region of CRH1 containing the NN cluster was replaced with a same length of DNA fragment from EGFP. B, CRH1 transcripts derived from either wild-type CRH1 or the CRH1::EGFP gene were analyzed by Northern blotting upon depletion of Rrp41. The relative portion of premature-terminated short transcripts (marked by green arrowhead) was quantitated as described in the legend in Fig. 4C. Shorter transcripts were significantly diminished in Nrd1(CIDRtt103) Δrtt103 cells when the NN cluster was removed. C, schematic diagram of CRH1 with internal NN clusters. Positions of ChIP PCR primers are shown above the diagram. D, RNApII occupancy was significantly decreased near the internal NN cluster (position 4) in Nrd1(CIDRtt103) Δrtt103 cells when compared with WT. RNApII occupancy signals at each position from WT were set to 100 and those from Nrd1(CIDRtt103) Δrtt103 cells were calculated in relative amounts after normalization to a non-transcribed telomeric region. Error bars represent S.E. for three independent experiments.
The medium category includes some genes showing phenotypes from read-through to premature termination with varying degrees, but the majority of genes with near zero RI values have no apparent phenotype, implying that termination of these genes is not significantly affected by Nrd1(CIDRtt103). One plausible explanation for near 0 RI would be Nrd1-dependent termination near the normal 3′-end. However, at genes with strong pA signals, termination would still occur by a cleavage-dependent mechanism. Alternatively, there could be a novel termination mechanism not affected by either Nrd1 or cleavage/pA factors.
Nrd1(CIDRtt103) Affects snoRNA Termination
We analyzed monocistronic, independently transcribed snoRNA genes (51 of the 77 annotated snoRNAs in the yeast genome), and the averaged RNApII occupancy profile was calculated for the upstream 80-nt region and downstream 160-nt region from mature 3′-ends. Distribution of the downstream region medians in the CID-swapped strain again differs from that in WT cells (p value = 1.01 × 10−3 in paired t test). As predicted by ChIP assay on the SNR13 and SNR33 genes (Fig. 2, C and D), the averaged RNApII occupancy profile of 51 snoRNA genes indicates read-through transcription at the 3′-ends (Fig. 6A). When RI was measured for the downstream 160-nt region, 35 of 51 (68.6%) snoRNA genes revealed positive RI values in Nrd1(CIDRtt103) Δrtt103 cells (supplemental Table S1). Indeed, read-through transcripts (red arrowhead) were detected on both the SNR13 and SNR33 genes in Nrd1(CIDRtt103) strains when a component of exosome or TRAMP complex was depleted or deleted (Fig. 6B). Surprisingly, in Nrd1(CIDRtt103) cells, the correctly terminated but pre-processed snoRNA transcripts (pre-snoRNA, marked in blue arrowhead) were strongly accumulated when Rrp41 was depleted, but deletion of RRP6 and/or TRF4 has a negligible effect in either glucose or galactose medium condition (Figs. 4E and 6B). This is in contrast to WT cells, where Δrrp6 and/or Δtrf4 mutants have the dominant effect on the accumulation of pre-snoRNAs (Fig. 6B). Similarly, prematurely terminated mRNA transcripts were detected only in Rrp41-depleted cells but not in Δrrp6 or Δtrf4 mutants (Fig. 4, C and D), indicating that the nascent RNA transcripts terminated by Nrd1(CIDRtt103) might be trimmed or degraded independently of nuclear-specific exosome subunits.
FIGURE 6.

Nrd1(CIDRtt103) affects snoRNA termination and processing by the exosome. A, averaged RNApII occupancy profiles of monocistronic independently transcribed snoRNA genes (51 in the yeast genome) for the upstream 80 nt and downstream 160 nt from the mature 3′-end are shown. B, total RNAs were isolated from the indicated strains and analyzed by Northern blotting for SNR13 and SNR33 genes. Read-through transcripts (denoted by red arrowhead) are detected in Nrd1(CIDRtt103) cells. Intriguingly, pre-snoRNA transcripts (denoted by blue arrowhead) are observed upon depletion of the core exosome component, Rrp41, in Nrd1(CIDRtt103) cells. But these transcripts accumulate predominantly in Δrrp6 and Δtrf4 mutants in the WT Nrd1 background. SCR1 loading control shown in Fig. 4B is also applicable here. The portion of read-through transcripts (lower left panel) or pre-snoRNA transcripts (lower right panel) relative to mature snoRNAs (set to 100%) was quantitated. C, co-IP and Western blot analysis using TAP-tagged Nrd1 strains. Interaction with Rrp6, Trf4, and Rrp4 was significantly decreased in Nrd1(CIDRtt103) and Nrd1ΔCID strains. Rtt103 barely interacts with the exosome components tested.
Nrd1(CIDRtt103) Alters snoRNA Processing and Degradation by the Exosome
Because the RNA substrate specificity of exosome was changed in Nrd1(CIDRtt103) cells, we monitored whether the interaction between Nrd1 and exosome/TRAMP components was altered by CID swapping. TAP-tagged Nrd1 proteins were IPed with IgG beads, and co-IPed exosome/TRAMP components (C-terminal 5× myc-tagged) were analyzed by Western blotting. As previously reported (23), all tested exosome and TRAMP components (Rrp6, Trf4, Rrp4, Rrp41, and Dis3) were associated with the wild-type Nrd1 complex, whereas Rtt103 does not significantly interact with exosome components tested (Fig. 6C). Surprisingly, the interaction of Nrd1(CIDRtt103) with Rrp6, Trf4, and Rrp4 was substantially reduced, whereas Dis3 and Rrp41 remained associated. Furthermore, the CID-deleted Nrd1 (Nrd1ΔCID) completely lost the interaction with Rrp6 and Trf4 (Fig. 6C). These results explain, at least in part, why degradation of pre-snoRNAs in Nrd1(CIDRtt103) cells is dependent upon the core exosome subunit Rrp41, instead of nuclear-specific exosome Rrp6 and TRAMP (Fig. 6B). RNA processing and degradation defects previously observed in Nrd1ΔCID mutants (23, 32) could be due to a loss of interaction with Rrp6, Trf4, and Rrp4. Importantly, these data indicate that the Nrd1 CID recruits the nuclear exosome and TRAMP to the Nrd1 complex by interacting with Rrp6, Trf4, and Rrp4 either directly or indirectly. By doing so, the Nrd1 CID would be able to couple NN-mediated RNApII termination and subsequent RNA processing by the nuclear exosome/TRAMP. CID swapping or deletion would obviously break this coupling, resulting in RNA processing by Rrp41-dependent fashion.
DISCUSSION
RNApII has at least two distinct termination pathways in yeast, one for mRNA and one for snoRNA genes. Termination factors involved in the two pathways are recruited to all RNApII-transcribed genes, regardless of gene types, indicating that the termination pathway is not simply determined by differential recruitment of termination factors (15). However, each pathway has a CID protein with distinct CTD-binding specificity (16, 22), and genes affected by sen1 E1597K mutation were much shorter than the average gene length (30). Because the CTD phosphorylation marks are not scaled to gene length (37, 54, 55), these results suggest that differential CTD-CID interactions arising at the 3′-ends of genes of different lengths would be influential in choosing an RNApII termination pathway. Here, we investigated the role of CTD-CID interaction choice by using domain swapping to generate Nrd1 that recognizes Ser(P)-2 instead of Ser(P)-5 CTD. Alteration of the CTD-CID interaction resulted in read-through transcription at many genes, manifesting the importance of correct CTD-CID interaction in RNApII termination. On the other hand, Nrd1(CIDRtt103) was able to terminate mRNA genes either in their coding region or 3′-ends when a substantial level of Ser(P)-2 CTD coincides with NN clusters on the RNA. These results indicate that CID swapping can indeed change the RNApII termination pathway, at least in a subset of gene contexts. Furthermore, this shows that combined cues from CTD and RNA sequences can provide favorable conditions for Nrd1-dependent termination. Finally, we found that trimming and degradation of Nrd1-terminated RNA transcripts becomes independent of the nuclear exosome in Nrd1(CIDRtt103) cells, reflecting a reduced interaction between the Nrd1 complex and Rrp6/Trf4/Rrp4. These results suggest that the Nrd1 CID is likely to mediate the recruitment of nuclear exosome and/or TRAMP complex (Fig. 7).
FIGURE 7.

Model for coupling of termination and RNA processing by the Nrd1 CID. A, the Nrd1 complex binds to Ser(P)-5 CTD once transcription initiates. Soon after it recognizes an NN cluster on the nascent transcript, termination would occur, releasing the Nrd1 CID from CTD. The nuclear exosome (or Rrp6)-TRAMP complex is recruited to the Nrd1 complex via the CID either directly or indirectly, and processes the Nrd1-associated transcripts from the 3′-end. It is currently unknown how the interaction of Nrd1 CID with CTD and/or Rrp6/Trf4/Rrp4 is coordinated in the course of transcription. B, in Nrd1(CIDRtt103) cells, the Nrd1-dependent termination would take place further downstream where Ser(P)-2 predominates over Ser(P)-5. Trimming and degradation of Nrd1-terminated transcripts becomes dependent upon the “core” exosome (containing Rrp41 and Dis3) rather than the nuclear exosome-TRAMP complex, due to reduced interaction between the Nrd1 complex and Rrp6/Trf4/Rrp4.
Multiple mechanisms contribute to recruitment of Nrd1 to RNApII-transcribed genes. These include interaction of the Nrd1 CID with Ser(P)-5 CTD, sequence-specific RNA binding using the Nrd1 RRM (RNA recognition motif), and interaction with Nab3, which contains another RRM. Among these mechanisms, the CID and Nab3-binding domain (amino acid residues 151 to 214) were shown to be required for efficient 5′-end recruitment of Nrd1 (22). Although the deletion of CID resulted in reduced Nrd1 recruitment specifically at the 5′-end of genes, deletion of the Nab3-binding domain caused an overall decrease of Nrd1 recruitment at many genes tested (22). Leaving its RRM and Nab3-binding domain intact, we were able to re-locate Nrd1 to the 3′-end of genes simply by CID swapping (Fig. 1), revealing a critical role of CTD-CID interaction in Nrd1 recruitment.
Genome-wide analysis reveals that a majority of mRNA genes (725 of 1,140 analyzed genes) have positive RI values in a CID-swapped strain, signifying read-through transcription (supplemental Table S1). As seen at the PMA1 gene, this could be due to impaired recruitment of cleavage/pA factors at 3′-ends due to competition with Nrd1(CIDRtt103) (Fig. 2A). However, the RNApII ChIP-chip study also disclosed genes with near 0 or negative RI values, demonstrating that alteration of CTD-binding specificity of Nrd1 affects RNApII termination in diverse ways. Although there is no obvious correlation between gene lengths and RIs, the ADH1 terminator-swapping assay shows that sequences at the 3′-end cause the differences in RI (Fig. 3B). Genes whose termination remains relatively unaffected (ADH1) or occurs prematurely (CRH1, YVC1) in Nrd1(CIDRtt103) cells have NN cluster(s) at the 3′-end or in the coding region, consistent with the NN cluster mediating termination by responding to Nrd1(CIDRtt103), these clusters may be involved in recruiting the nuclear exosome bound to Nrd1 for RNA processing and/or degradation, thereby regulating the transcript level post-transcriptionally. However, these clusters would become eligible to mediate termination in the CID-swapping background, as seen for the CRH1 gene (Fig. 5, B and D). Considering the reduced interaction between Nrd1(CIDRtt103) and Sen1 (Fig. 1F), termination near the NN cluster might be promoted by Nrd1(CIDRtt103) and Nab3, independently of Sen1. However, the low levels of Sen1 associated with Nrd1(CIDRtt103) may still be sufficient for proper termination.
We found that not all NN clusters are functional in mediating Nrd1-dependent termination, suggesting that additional sequence motifs and/or their particular configurations might also be important. In this aspect, the AU-rich motifs recently reported are noteworthy (53), because we often see AU-rich stretches near the Nrd1/Nab3-binding sites.
Genes with an NN cluster near the normal mRNA 3′-end (ADH1) suggest the existence of “switchable” genes. These could become dependent upon the Nrd1-termination pathway upon CID swapping. Supporting this hypothesis, some ribosomal protein (RP) gene paralogs in our analyzed gene set have significantly different RI values and often belong to different categories: for example, RPL42A (RI = 0.270, high) and RPL42B (RI = 0.019, medium). Although there might be some “position effect” from the neighboring genes, given the fact that they have near identical ORFs (probably derived from gene duplication) but their 3′-UTRs are completely diverged over a long period of time, we speculate that eukaryotic termination pathways might have originated from a primordial Nrd1/Nab3/Sen1-like pathway (similar to bacterial Rho-dependent pathway) and later adopted a cleavage-dependent pathway for more efficient and immediate RNA processing and termination during the evolution. In this regard, RPL42B (a switchable gene) may have kept the Nrd1/Nab3 sites, whereas RPL42A lost these “old sequence relics” to become strictly dependent upon the mRNA termination pathway. Considering a previous study that the Nrd1/Nab3 sites near pA signal can mediate “fail-safe” RNApII termination when the pA cleavage pathway does not work (56), the switchable character of these genes would be physiologically relevant for providing a fail-safe mechanism to avoid termination defects even in WT Nrd1 cells.
Nrd1(CIDRtt103) protein has significantly less interaction with Rrp6, Trf4, and Rrp4, but remains bound to appreciable amounts of Rrp41 and Dis3 (Fig. 6C). The associated exosome (containing Rrp41 and Dis3) consequently carries out trimming of pre-snoRNAs (Fig. 6B) and degradation of premature-terminated transcripts (Fig. 4, C and D) independently of Rrp6, Trf4, and Rrp4. Deletion of the Nrd1 CID caused an even more dramatic loss of interaction with Rrp6 and Trf4 (Fig. 6C). Indeed, pre-snoRNAs and cryptic unstable transcripts accumulated similarly in Nrd1ΔCID mutants (22, 32) and Nrd1(CIDRtt103) strains. Thus, swapping or deletion of the Nrd1 CID uncouples the Nrd1 complex from the nuclear exosome-TRAMP complex, causing the associated core exosome to substitute for Rrp6 and Trf4 in RNA trimming and degradation. These results also demonstrate that the Nrd1 CID is responsible for recruitment of Rrp6, Trf4, and Rrp4 to the Nrd1 complex, either directly or indirectly. Given the physical connection between Nrd1(CIDRtt103) and the core exosome subunits (Rrp41 and Dis3), the associated complex is unlikely to be the cytoplasmic exosome, although we cannot rule out completely whether Nrd1(CIDRtt103) shuttles to the cytoplasm.
Rrp6, Trf4, and Rrp4 barely interact with Rtt103 or the Rtt103 CID within the context of Nrd1 (Fig. 6C), indicating that these proteins may specifically recognize the Nrd1 CID. However, it is unclear whether they recognize the CTD binding pocket or a different part of the CID. If the recognition is on the CTD pocket side, this might suggest that CTD and Rrp6/Trf4/Rrp4 could compete for binding to the Nrd1 CID. Thus, reduced interaction between Rrp6/Trf4/Rrp4 and Nrd1(CIDRtt103) could be due to alteration of the CTD binding pocket. Even if Rrp6/Trf4/Rrp4 could similarly recognize the CTD binding pocket of the Rtt103 CID, the ∼20-fold higher affinity of the Rtt103 CID to Ser(P)-2 CTD (Kd = 2.1 ± 0.1 μm) compared with that of the Nrd1 CID to Ser(P)-5 CTD (Kd = 40 ± 3 μm) (22, 41) would greatly limit the available CIDs for binding to Rrp6/Trf4/Rrp4 in Nrd1(CIDRtt103) cells.
Alternatively, Rrp6/Trf4/Rrp4 may recognize a different part of the Nrd1 CID. Although there is no direct competition between CTD and Rrp6/Trf4/Rrp4 in this model, the CTD-CID interaction might allosterically regulate the binding of Rrp6/Trf4/Rrp4. Because RNA processing by Rrp6/Trf4/Rrp4 occurs after transcription is terminated, this allosteric regulation by the CTD would likely be inhibitory to the binding of Rrp6/Trf4/Rrp4. In this model, Nrd1 is recruited to RNApII CTD soon after initiation, but must eventually be released from the CTD and handed over to Rrp6/Trf4/Rrp4 for subsequent RNA processing (Fig. 7). It will be interesting to uncover how the interaction of Nrd1 CID with CTD and/or Rrp6/Trf4/Rrp4 is coordinated during the course of transcription. RNApII termination by the Nrd1 complex, or simply binding to the nascent RNA transcripts using its RRM, might facilitate the dissociation of Nrd1 from the CTD.
Acknowledgments
We thank Francesca Storici and Won-Ki Huh for plasmids and strains.
This work was supported, in whole or in part, by National Institutes of Health Grant GM56663 (to S. B.), Research Center Program Grant EM1302 from the Institute for Basic Science and Basic Science Research Program Grant NRF-2013R1A1A2A10013757 (to M. K.) through the National Research Foundation funded by the Ministry of Education of Korea.

This article contains supplemental Table S1.
Raw ChIP-chip data are available at ArrayExpress under the accession number E-MTAB-1626.
- CTD
- C-terminal domain
- RNApII
- RNA polymerase II
- Ser(P)-5/Ser(P)-2
- phosphorylated serine 5 or 2
- snoRNA
- small nucleolar RNA
- RI
- read-through index
- TSS
- transcription start site
- CID
- CTD-interacting domain
- nt
- nucleotide(s)
- IP
- immunoprecipitation
- aa
- amino acid(s)
- TAP
- tandem affinity purification
- pA
- polyadenylation.
REFERENCES
- 1. Birse C. E., Minvielle-Sebastia L., Lee B. A., Keller W., Proudfoot N. J. (1998) Coupling termination of transcription to messenger RNA maturation in yeast. Science 280, 298–301 [DOI] [PubMed] [Google Scholar]
- 2. Proudfoot N. (2004) New perspectives on connecting messenger RNA 3′ end formation to transcription. Curr. Opin. Cell Biol. 16, 272–278 [DOI] [PubMed] [Google Scholar]
- 3. Buratowski S. (2005) Connections between mRNA 3′ end processing and transcription termination. Curr. Opin. Cell Biol. 17, 257–261 [DOI] [PubMed] [Google Scholar]
- 4. Cho E. J., Kobor M. S., Kim M., Greenblatt J., Buratowski S. (2001) Opposing effects of Ctk1 kinase and Fcp1 phosphatase at Ser-2 of the RNA polymerase II C-terminal domain. Genes Dev. 15, 3319–3329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Komarnitsky P., Cho E. J., Buratowski S. (2000) Different phosphorylated forms of RNA polymerase II and associated mRNA processing factors during transcription. Genes Dev. 14, 2452–2460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cho E. J., Takagi T., Moore C. R., Buratowski S. (1997) mRNA capping enzyme is recruited to the transcription complex by phosphorylation of the RNA polymerase II carboxy-terminal domain. Genes Dev. 11, 3319–3326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kim M., Ahn S. H., Krogan N. J., Greenblatt J. F., Buratowski S. (2004) Transitions in RNA polymerase II elongation complexes at the 3′ ends of genes. EMBO J. 23, 354–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ahn S. H., Kim M., Buratowski S. (2004) Phosphorylation of serine 2 within the RNA polymerase II C-terminal domain couples transcription and 3′ end processing. Mol. Cell 13, 67–76 [DOI] [PubMed] [Google Scholar]
- 9. Buratowski S. (2003) The CTD code. Nat. Struct. Biol. 10, 679–680 [DOI] [PubMed] [Google Scholar]
- 10. Egloff S., Dienstbier M., Murphy S. (2012) Updating the RNA polymerase CTD code. Adding gene-specific layers. Trends Genet. 28, 333–341 [DOI] [PubMed] [Google Scholar]
- 11. Egloff S., O'Reilly D., Chapman R. D., Taylor A., Tanzhaus K., Pitts L., Eick D., Murphy S. (2007) Serine-7 of the RNA polymerase II CTD is specifically required for snRNA gene expression. Science 318, 1777–1779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hsin J. P., Sheth A., Manley J. L. (2011) RNAP II CTD phosphorylated on threonine-4 is required for histone mRNA 3′ end processing. Science 334, 683–686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hintermair C., Heidemann M., Koch F., Descostes N., Gut M., Gut I., Fenouil R., Ferrier P., Flatley A., Kremmer E., Chapman R. D., Andrau J. C., Eick D. (2012) Threonine-4 of mammalian RNA polymerase II CTD is targeted by Polo-like kinase 3 and required for transcriptional elongation. EMBO J. 31, 2784–2797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mayer A., Heidemann M., Lidschreiber M., Schreieck A., Sun M., Hintermair C., Kremmer E., Eick D., Cramer P. (2012) CTD tyrosine phosphorylation impairs termination factor recruitment to RNA polymerase II. Science 336, 1723–1725 [DOI] [PubMed] [Google Scholar]
- 15. Kim M., Vasiljeva L., Rando O. J., Zhelkovsky A., Moore C., Buratowski S. (2006) Distinct pathways for snoRNA and mRNA termination. Mol. Cell 24, 723–734 [DOI] [PubMed] [Google Scholar]
- 16. Kim M., Krogan N. J., Vasiljeva L., Rando O. J., Nedea E., Greenblatt J. F., Buratowski S. (2004) The yeast Rat1 exonuclease promotes transcription termination by RNA polymerase II. Nature 432, 517–522 [DOI] [PubMed] [Google Scholar]
- 17. West S., Gromak N., Proudfoot N. J. (2004) Human 5′ → 3′ exonuclease Xrn2 promotes transcription termination at co-transcriptional cleavage sites. Nature 432, 522–525 [DOI] [PubMed] [Google Scholar]
- 18. Kiss T. (2002) Small nucleolar RNAs. An abundant group of noncoding RNAs with diverse cellular functions. Cell 109, 145–148 [DOI] [PubMed] [Google Scholar]
- 19. Thiebaut M., Kisseleva-Romanova E., Rougemaille M., Boulay J., Libri D. (2006) Transcription termination and nuclear degradation of cryptic unstable transcripts. A role for the nrd1-nab3 pathway in genome surveillance. Mol. Cell 23, 853–864 [DOI] [PubMed] [Google Scholar]
- 20. Steinmetz E. J., Conrad N. K., Brow D. A., Corden J. L. (2001) RNA-binding protein Nrd1 directs poly(A)-independent 3′-end formation of RNA polymerase II transcripts. Nature 413, 327–331 [DOI] [PubMed] [Google Scholar]
- 21. Ursic D., Chinchilla K., Finkel J. S., Culbertson M. R. (2004) Multiple protein/protein and protein/RNA interactions suggest roles for yeast DNA/RNA helicase Sen1p in transcription, transcription-coupled DNA repair and RNA processing. Nucleic Acids Res. 32, 2441–2452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Vasiljeva L., Kim M., Mutschler H., Buratowski S., Meinhart A. (2008) The Nrd1-Nab3-Sen1 termination complex interacts with the Ser-5-phosphorylated RNA polymerase II C-terminal domain. Nat. Struct. Mol. Biol. 15, 795–804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Vasiljeva L., Buratowski S. (2006) Nrd1 interacts with the nuclear exosome for 3′ processing of RNA polymerase II transcripts. Mol. Cell 21, 239–248 [DOI] [PubMed] [Google Scholar]
- 24. Butler J. S. (2002) The yin and yang of the exosome. Trends Cell Biol. 12, 90–96 [DOI] [PubMed] [Google Scholar]
- 25. Liu Q., Greimann J. C., Lima C. D. (2006) Reconstitution, activities, and structure of the eukaryotic RNA exosome. Cell 127, 1223–1237 [DOI] [PubMed] [Google Scholar]
- 26. Lykke-Andersen S., Brodersen D. E., Jensen T. H. (2009) Origins and activities of the eukaryotic exosome. J. Cell Sci. 122, 1487–1494 [DOI] [PubMed] [Google Scholar]
- 27. Houseley J., Tollervey D. (2009) The many pathways of RNA degradation. Cell 136, 763–776 [DOI] [PubMed] [Google Scholar]
- 28. Vanácová S., Wolf J., Martin G., Blank D., Dettwiler S., Friedlein A., Langen H., Keith G., Keller W. (2005) A new yeast poly(A) polymerase complex involved in RNA quality control. PLoS Biol. 3, e189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wyers F., Rougemaille M., Badis G., Rousselle J. C., Dufour M. E., Boulay J., Régnault B., Devaux F., Namane A., Séraphin B., Libri D., Jacquier A. (2005) Cryptic pol II transcripts are degraded by a nuclear quality control pathway involving a new poly(A) polymerase. Cell 121, 725–737 [DOI] [PubMed] [Google Scholar]
- 30. Steinmetz E. J., Warren C. L., Kuehner J. N., Panbehi B., Ansari A. Z., Brow D. A. (2006) Genome-wide distribution of yeast RNA polymerase II and its control by Sen1 helicase. Mol. Cell 24, 735–746 [DOI] [PubMed] [Google Scholar]
- 31. Gudipati R. K., Villa T., Boulay J., Libri D. (2008) Phosphorylation of the RNA polymerase II C-terminal domain dictates transcription termination choice. Nat. Struct. Mol. Biol. 15, 786–794 [DOI] [PubMed] [Google Scholar]
- 32. Kubicek K., Cerna H., Holub P., Pasulka J., Hrossova D., Loehr F., Hofr C., Vanacova S., Stefl R. (2012) Serine phosphorylation and proline isomerization in RNAP II CTD control recruitment of Nrd1. Genes Dev. 26, 1891–1896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Storici F., Resnick M. A. (2006) The delitto perfetto approach to in vivo site-directed mutagenesis and chromosome rearrangements with synthetic oligonucleotides in yeast. Methods Enzymol. 409, 329–345 [DOI] [PubMed] [Google Scholar]
- 34. Sung M. K., Ha C. W., Huh W. K. (2008) A vector system for efficient and economical switching of C-terminal epitope tags in Saccharomyces cerevisiae. Yeast 25, 301–311 [DOI] [PubMed] [Google Scholar]
- 35. O'Geen H., Nicolet C. M., Blahnik K., Green R., Farnham P. J. (2006) Comparison of sample preparation methods for ChIP-chip assays. BioTechniques 41, 577–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. David L., Huber W., Granovskaia M., Toedling J., Palm C. J., Bofkin L., Jones T., Davis R. W., Steinmetz L. M. (2006) A high-resolution map of transcription in the yeast genome. Proc. Natl. Acad. Sci. U.S.A. 103, 5320–5325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mayer A., Lidschreiber M., Siebert M., Leike K., Söding J., Cramer P. (2010) Uniform transitions of the general RNA polymerase II transcription complex. Nat. Struct. Mol. Biol. 17, 1272–1278 [DOI] [PubMed] [Google Scholar]
- 38. Nagalakshmi U., Wang Z., Waern K., Shou C., Raha D., Gerstein M., Snyder M. (2008) The transcriptional landscape of the yeast genome defined by RNA sequencing. Science 320, 1344–1349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dengl S., Mayer A., Sun M., Cramer P. (2009) Structure and in vivo requirement of the yeast Spt6 SH2 domain. J. Mol. Biol. 389, 211–225 [DOI] [PubMed] [Google Scholar]
- 40. Marquardt S., Hazelbaker D. Z., Buratowski S. (2011) Distinct RNA degradation pathways and 3′ extensions of yeast non-coding RNA species. Transcription 2, 145–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lunde B. M., Reichow S. L., Kim M., Suh H., Leeper T. C., Yang F., Mutschler H., Buratowski S., Meinhart A., Varani G. (2010) Cooperative interaction of transcription termination factors with the RNA polymerase II C-terminal domain. Nat. Struct. Mol. Biol. 17, 1195–1201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Xu Z., Wei W., Gagneur J., Perocchi F., Clauder-Münster S., Camblong J., Guffanti E., Stutz F., Huber W., Steinmetz L. M. (2009) Bidirectional promoters generate pervasive transcription in yeast. Nature 457, 1033–1037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kim M., Suh H., Cho E. J., Buratowski S. (2009) Phosphorylation of the yeast Rpb1 C-terminal domain at serines 2, 5, and 7. J. Biol. Chem. 284, 26421–26426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Meinhart A., Cramer P. (2004) Recognition of RNA polymerase II carboxy-terminal domain by 3′-RNA-processing factors. Nature 430, 223–226 [DOI] [PubMed] [Google Scholar]
- 45. Becker R., Loll B., Meinhart A. (2008) Snapshots of the RNA processing factor SCAF8 bound to different phosphorylated forms of the carboxyl-terminal domain of RNA polymerase II. J. Biol. Chem. 283, 22659–22669 [DOI] [PubMed] [Google Scholar]
- 46. Chinchilla K., Rodriguez-Molina J. B., Ursic D., Finkel J. S., Ansari A. Z., Culbertson M. R. (2012) Interactions of Sen1, Nrd1, and Nab3 with multiple phosphorylated forms of the Rpb1 C-terminal domain in Saccharomyces cerevisiae. Eukaryot. Cell 11, 417–429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Arigo J. T., Carroll K. L., Ames J. M., Corden J. L. (2006) Regulation of yeast NRD1 expression by premature transcription termination. Mol. Cell 21, 641–651 [DOI] [PubMed] [Google Scholar]
- 48. Mandart E., Parker R. (1995) Effects of mutations in the Saccharomyces cerevisiae RNA14, RNA15, and PAP1 genes on polyadenylation in vivo. Mol. Cell. Biol. 15, 6979–6986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Vo L. T., Minet M., Schmitter J. M., Lacroute F., Wyers F. (2001) Mpe1, a zinc knuckle protein, is an essential component of yeast cleavage and polyadenylation factor required for the cleavage and polyadenylation of mRNA. Mol. Cell. Biol. 21, 8346–8356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Carroll K. L., Pradhan D. A., Granek J. A., Clarke N. D., Corden J. L. (2004) Identification of cis elements directing termination of yeast nonpolyadenylated snoRNA transcripts. Mol. Cell. Biol. 24, 6241–6252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Carroll K. L., Ghirlando R., Ames J. M., Corden J. L. (2007) Interaction of yeast RNA-binding proteins Nrd1 and Nab3 with RNA polymerase II terminator elements. RNA 13, 361–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Heidmann S., Schindewolf C., Stumpf G., Domdey H. (1994) Flexibility and interchangeability of polyadenylation signals in Saccharomyces cerevisiae. Mol. Cell. Biol. 14, 4633–4642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Porrua O., Hobor F., Boulay J., Kubicek K., D'Aubenton-Carafa Y., Gudipati R. K., Stefl R., Libri D. (2012) In vivo SELEX reveals novel sequence and structural determinants of Nrd1-Nab3-Sen1-dependent transcription termination. EMBO J. 31, 3935–3948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kim H., Erickson B., Luo W., Seward D., Graber J. H., Pollock D. D., Megee P. C., Bentley D. L. (2010) Gene-specific RNA polymerase II phosphorylation and the CTD code. Nat. Struct. Mol. Biol. 17, 1279–1286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bataille A. R., Jeronimo C., Jacques P. É., Laramée L., Fortin M. È., Forest A., Bergeron M., Hanes S. D., Robert F. (2012) A universal RNA polymerase II CTD cycle is orchestrated by complex interplays between kinase, phosphatase, and isomerase enzymes along genes. Mol. Cell. 45, 158–170 [DOI] [PubMed] [Google Scholar]
- 56. Rondón A. G., Mischo H. E., Kawauchi J., Proudfoot N. J. (2009) Fail-safe transcriptional termination for protein-coding genes in S. cerevisiae. Mol. Cell. 36, 88–98 [DOI] [PMC free article] [PubMed] [Google Scholar]