

Background: Kappa opioid receptor (KOR) signaling may produce antinociception through G protein or dysphoria through βarrestin pathways.
Results: Two highly selective, brain penetrant agonist scaffolds bias KOR signaling toward G protein coupling and produce antinociception in mice.
Conclusion: Described are first-in-class small molecule agonists that bias KOR signaling through G proteins.
Significance: Functionally selective KOR agonists can now be used in vivo.
Keywords: Arrestin, Brain, Drug Discovery, ERK, G Protein-coupled Receptors (GPCR), Opiate Opioid, Pain, Dysphoria, Kappa Opioid Receptor
Abstract
The kappa opioid receptor (KOR) is widely expressed in the CNS and can serve as a means to modulate pain perception, stress responses, and affective reward states. Therefore, the KOR has become a prominent drug discovery target toward treating pain, depression, and drug addiction. Agonists at KOR can promote G protein coupling and βarrestin2 recruitment as well as multiple downstream signaling pathways, including ERK1/2 MAPK activation. It has been suggested that the physiological effects of KOR activation result from different signaling cascades, with analgesia being G protein-mediated and dysphoria being mediated through βarrestin2 recruitment. Dysphoria associated with KOR activation limits the therapeutic potential in the use of KOR agonists as analgesics; therefore, it may be beneficial to develop KOR agonists that are biased toward G protein coupling and away from βarrestin2 recruitment. Here, we describe two classes of biased KOR agonists that potently activate G protein coupling but weakly recruit βarrestin2. These potent and functionally selective small molecule compounds may prove to be useful tools for refining the therapeutic potential of KOR-directed signaling in vivo.
Introduction
The kappa opioid receptor (KOR)5 is a seven-transmembrane G protein-coupled receptor (GPCR) (1). The cognate neuropeptides for KOR are endogenous opioids, including the dynorphin peptides. Dynorphins and KOR are widely expressed throughout the central nervous system (2–4). In a canonical sense, KOR activation is defined by agonist-induced coupling of the Gα subunit of the heterotrimeric G proteins in the pertussis toxin-sensitive Gi/o family (5), subsequent inhibition of adenylyl cyclase (6), activation of inward-rectifying potassium channels (7), and blockade of calcium channels (8).
There is considerable evidence that selective KOR agonists produce antinociception in animal models (9–12), and mice lacking KOR expression are no longer responsive to the antinociceptive effects of a selective KOR agonist U50,488 (13). The G protein-mediated signaling events are believed to contribute to the analgesic properties of KOR agonists (14, 15). Unlike MOR agonists, KOR agonists do not cause physical dependence nor do they produce respiratory failure; thus, they are attractive as potent analgesics (16). However, KOR activation has also been implicated in producing an array of undesirable side effects, including dysphoria (17, 18), sedation (10), diuresis (11), hallucination (19), and depression (20). These adverse effects limit the therapeutic potential of KOR agonists as analgesics.
It is becoming widely evident that activation of GPCRs by chemically distinct agonists can promote receptor coupling to distinct pathways in different tissues (21–23). The KOR also has the potential to signal through multiple downstream signaling cascades, and there is increasing evidence that signaling to alternative cascades may promote side effects associated with KOR activation. In addition to G protein coupling, another proximal event following GPCR activation is the agonist-promoted recruitment of βarrestins. As regulatory and scaffolding proteins, βarrestins can lead to desensitization of the receptor by impeding further G protein coupling; in some cases, βarrestins facilitate the assembly of protein scaffolds, thereby acting as signal transducers (24–26). For example, βarrestin2 recruitment has been shown to promote recruitment and activation of MAPKs, including ERK1/2 (27, 28) and p38 (29). Interestingly, studies in AtT-20 cells and mouse striatal neurons suggest that the KOR may signal via a GPCR kinase 3 (GRK3)- and a βarrestin2-dependent mechanism to activate the stress MAPK p38 and that this signaling cascade may mediate the dysphoric effects of KOR agonists (29, 30). Moreover, KOR activation has been shown to lead to ERK1/2 phosphorylation in both neurons and astrocytes through both G protein and βarrestin-dependent mechanisms (28, 29, 31, 32). Endogenously elevated dynorphin levels produced by repeated swim stress tests lead to KOR-dependent ERK1/2 phosphorylation in the mouse brain that was not dependent on GRK3 expression (33). Therefore, ERK activation may be a useful indicator of both βarrestin-dependent and G protein-dependent signaling states downstream of KOR (34).
Because G protein-mediated signaling is implicated in KOR-induced antinociception and βarrestin2-mediated signaling has been implicated in the aversive and dysphoric properties of KOR agonists, the development of G protein-biased agonists may provide a means to optimally tune KOR therapeutics. In two different screening campaigns, we discovered two new classes of KOR agonists, triazole probe 1 and isoquinolinone probe 2 (Fig. 1). The triazole scaffold was identified under the auspices of the Molecular Libraries Probe Production Centers Network via a high throughput screening campaign. In this work, which was done in collaboration with colleagues at Sanford-Burnham Research Institute and Duke University, the National Institutes of Health Small Molecule Repository was screened to identify KOR ligands based on activation of βarrestin2 recruitment (35, 36). The isoquinolinone scaffold arose from a 72-member library prepared by a tandem Diels-Alder acylation reaction that was screened for binding at potential GPCR targets by the NIMH Psychoactive Drug Screening Program (37, 38). The lead isoquinolinone was reported as a KOR agonist showing selectivity and high binding affinity for KOR over MOR, DOR, and other screened GPCRs (37, 38); moreover, the triazole lead also displayed high selectivity for KOR over MOR and DOR (Fig. 1) (35, 36). The isoquinolinone compounds are particularly interesting in that they lack the basic nitrogen center common in small molecule KOR ligands (10, 39–41); the best known ligand lacking this feature is salvinorin A, a natural neoclerdane diterpene found to be a highly selective, potent KOR agonist (19).
FIGURE 1.
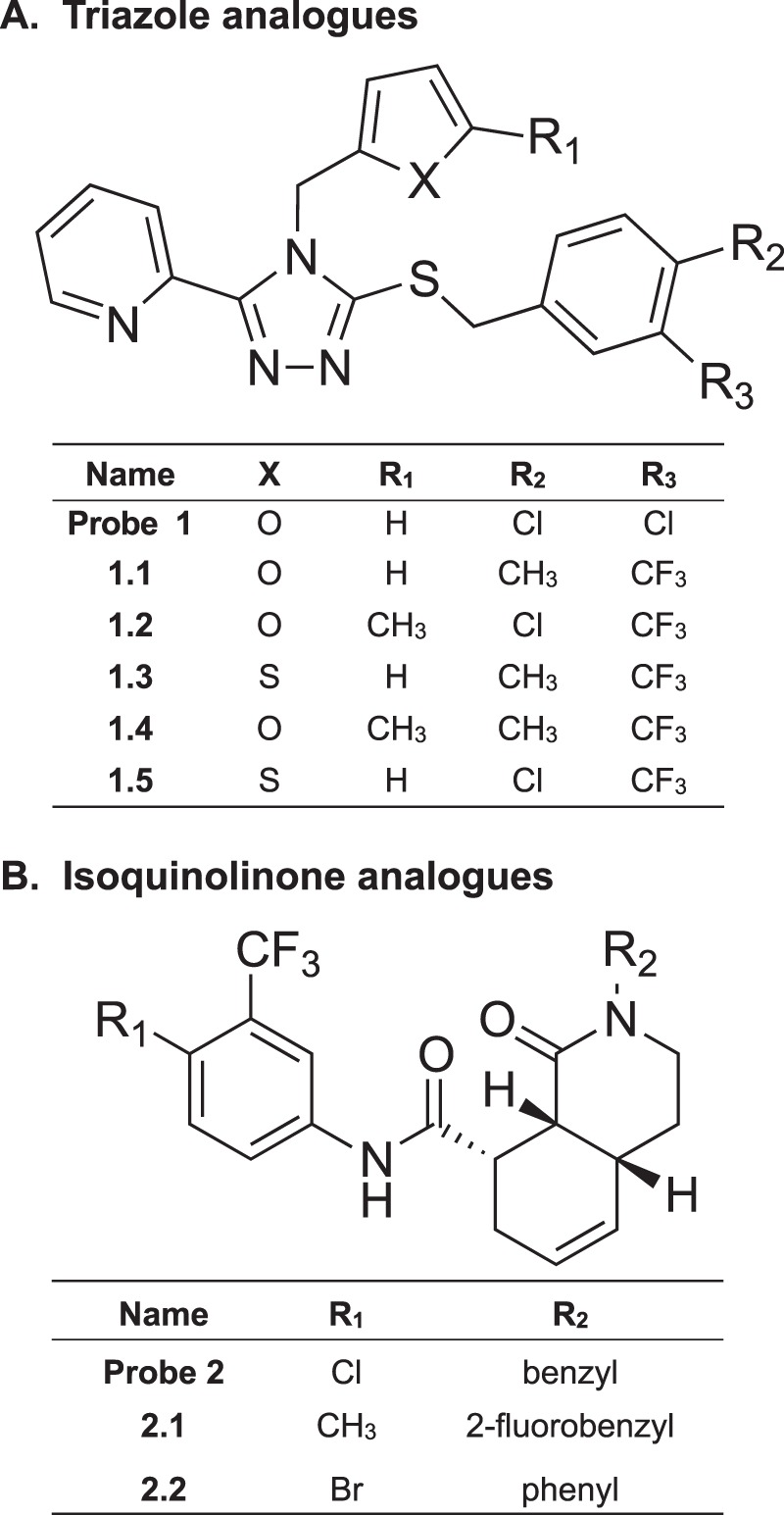
Structure of KOR agonists. A, triazole probes (PubChem compound ID 44601470) were reported to have binding affinity for KOR (Ki = 2.4 nm) over MOR (Ki = 1900 nm) and DOR (Ki = 5351 nm) (36). Analogues of the triazole probe are shown. B, isoquinolinone probe and analogues were reported to have binding affinity to KOR (Ki = 5 nm) over MOR (Ki = 3550 nm) and DOR (Ki > 10 μm) (38).
Following their initial disclosure, these scaffolds were subjected to iterative rounds of medicinal chemistry and structure-activity relationship studies with the goal of developing KOR agonists that are biased toward G protein coupling. Here, we report five triazole analogues and two isoquinolinone analogues (Fig. 1) that activate KOR in a manner that is preferentially biased toward G protein signaling with minimal effects on βarrestin2 recruitment and downstream ERK1/2 activation.
EXPERIMENTAL PROCEDURES
Compounds and Reagents
Reference compound (+)-(5α,7α,8β)-N-methyl-N-(7-(1-pyrrolidinyl)-1-oxaspiro(4.5)dec-8-yl)-benzeneacetamide (U69,593) was purchased from Sigma and was prepared in ethanol as a 10 mm stock. The triazole probe 1 (36) and the isoquinolinone probe 2 (38) were synthesized as described previously. The syntheses of new triazole and isoquinolinone analogues and intermediates are detailed in the supplemental material. Test compounds were prepared as 10 mm stocks in DMSO (Fisher); all reagents were then diluted to working concentrations in vehicle containing equal concentrations of DMSO and ethanol not exceeding 1% of either in any assay. DAMGO, natrindole, trans-(±)-3,4-dichloro-N-methyl-N-(2-(1-pyrrolidinyl)cyclohexyl)benzeneacetamide hydrochloride (U50,488H), and naloxone were obtained from Tocris Bioscience (Ellisville, MO). [35S]GTPγS and radioligands [3H]U69,593, [3H]DAMGO, and [3H]diprenorphine were purchased from PerkinElmer Life Sciences. Antibodies for detecting phospho-ERK1/2 and total ERK1/2 were from Cell Signaling (Beverly, MA). Li-Cor secondary antibodies (anti-rabbit IRDye800CW and anti-mouse IRDye680LT) were purchased from Li-Cor Biosciences. DiscoveRx PathHunterTM enzyme fragment complementation (EFC) assay detection reagent was purchased from DiscoveRx Corp. (Fremont, CA).
Cell Lines and Cell Culture
Chinese hamster ovary (CHO) cells expressing recombinant human kappa opioid receptor (CHO-hKOR cell line) were generated as described previously (42). The CHO-hMOR cell line was a gift from Dr. Richard B. Rothman of the National Institute on Drug Abuse (43). The CHO-hDOR cell line was made by stably transfecting CHO cells with human DOR cDNA (Missouri S&T cDNA Resource Center, Rolla, MO). The CHO cell lines were maintained in DMEM/F-12 media (Invitrogen) supplemented with 10% fetal bovine serum, 1% penicillin/streptomycin, and 500 μg/ml geneticin (the parental CHO-K1 cell line was grown without geneticin). The U2OS cell line stably expressing hKOR and βarrestin2-eGFP (U2OS-hKOR-βarrestin2-GFP) was a gift from Dr. Lawrence Barak, Duke University. These cells were maintained in minimum Eagle's medium with 10% fetal bovine serum, 1% penicillin/streptomycin, 100 μg/ml geneticin, and 50 μg/ml Zeocin. DiscoveRx PathHunterTM U2OS EFC cell line expressing βarrestin2 and hKOR (U2OS-hKOR-βarrestin2-EFC) or hMOR (U2OS-hMOR-βarrestin2-EFC) was purchased from DiscoveRx Corp. (Fremont, CA) and maintained in minimum Eagle's medium with 10% fetal bovine serum, 1% penicillin/streptomycin, 500 μg/ml geneticin, and 250 μg/ml hygromycin B. All cells were grown at 37 °C (5% CO2 and 95% relative humidity).
Animals
C57BL/6J male mice between 5 and 7 months old from The Jackson Laboratory (Bar Harbor, ME) were used in accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals and with approval by The Scripps Research Institute Animal Care; the Scripps vivarium is fully AAALAC-accredited.
Membrane G Protein Signaling
Membranes were prepared according to a modified procedure of Schmid et al. (42). Briefly, CHO-hKOR cells were serum-starved for 1 h in DMEM/F-12 media, collected with 5 mm EDTA, washed with PBS, and stored at −80 °C. For each assay, cell pellets were homogenized via Teflon-on-glass homogenizer in buffer (10 mm Tris-HCl, pH 7.4, 100 mm NaCl, 1 mm EDTA), passed through a 26-gauge needle eight times, centrifuged twice at 20,000 × g for 30 min at 4 °C, and resuspended in assay buffer (50 mm Tris-HCl, pH 7.4, 100 mm NaCl, 5 mm MgCl2, 1 mm EDTA, and 3 μm GDP). For each reaction, 15 μg of membrane protein were incubated in assay buffer containing ∼0.1 nm [35S]GTPγS and increasing concentrations of compounds in a total volume of 200 μl for 1 h at room temperature. The reactions were terminated by rapid filtration over GF/B filters using a 96-well plate harvester (Brandel Inc., Gaithersburg, MD). Filters were dried overnight, and radioactivity was determined with a TopCount NXT high throughput screening microplate scintillation and luminescence counter (PerkinElmer Life Sciences).
Whole Cell G Protein Signaling
CHO-hKOR cells were seeded in a 96-well tissue culture plate (BD Biosciences) at 60,000 cells/well. On the 2nd day, cells were serum-starved for 1 h followed by treatment with saponin (50 μg/ml, Sigma) for 4 min to briefly permeabilize cells (44). Cells were then incubated with [35S]GTPγS and increasing concentrations of compounds in assay buffer in a 200-μl volume for 1 h at room temperature as described for membrane G protein signaling above. The [35S]GTPγS binding was measured using the same methods described above.
Cellular Impedance
CHO-hKOR cells were plated at 60,000 cells per well in CellKeyTM microplates (Molecular Device, Sunnyvale, CA) in complete DMEM/F-12 media. The 2nd day, the cells were equilibrated at room temperature in Hanks' balanced salt solution (Invitrogen) containing 0.1% BSA (fatty acid-free, Sigma) and 20 mm HEPES for 30 min. Changes in impedance due to changes in extracellular current (Ziec) were recorded using CellKeyTM cellular analysis system (Molecular Device) for 35 min after treatment with increasing doses of compounds (45, 46). Maximal changes in impedance were extracted and plotted in dose-response curves.
βArrestin2 Recruitment (EFC)
The βarrestin2 EFC assays were performed according to the manufacturer's (DiscoveRx) protocol with slight modification as described previously (42). Briefly, the U2OS-hKOR-βarrestin2-EFC cells were plated at 5000 cells/well in 20 μl of Opti-MEM media containing 1% FBS for overnight in 384-well white plates. The 2nd day, cells were treated with compounds for 90 min followed by 1-h incubation of detection reagent. Luminescence values generated following the substrate addition were obtained using a Synergy HT luminometer (BioTek, Winooski, VT). All compounds were run in duplicate per assay and normalized to vehicle-treated cells.
βArrestin2 Imaging
For concentration-response studies, U2OS-hKOR-βarrestin2-GFP cells were plated in a 384-well Aurora Black walled, clear bottom plate at a density of 5000 cells per well and incubated overnight. Cells were serum-starved for 30 min prior to treatment (20 min) and then fixed with pre-warmed 4% paraformaldehyde and stained with nuclear dye Hoechst (1:1000) for 30 min. Cells were then washed three times with PBS and stored in PBS until imaged. Images were acquired with a ×20 objective on the CellInsight (Thermo Scientific), and the number of spots per cell was determined using the Cellomics® Spot Detector BioApplication algorithm (version 6.0). For imaging of live cells, an Olympus FluoView IX81 confocal microscope (Olympus, Center Valley, PA) was used. Live cell imaging was assessed in CHO-hKOR cells transfected with βarrestin2-YFP (4 μg) via electroporation plated on collagen-coated glass-bottom dishes (MatTek, Ashland, MA) (42). Single focal plane images were obtained using ×100 objective between 5 and 20 min after drug treatment. Cells that did not respond to compound stimulation were treated with 10 μm U69,593 to validate the cell's potential to respond prior to concluding a negative drug effect (data not shown). Individual images were adjusted for brightness/contrast and size (scale bars are indicated).
Radioligand Binding
Receptor binding assays were performed as described previously (42, 47). Cell pellets (prepared as for G protein coupling) were homogenized in homogenization buffer (10 mm Tris-HCl, pH 7.4, with 1 mm EDTA) and centrifuged twice at 20,000 × g for 30 min at 4 °C. The resulting membrane pellet was homogenized in assay buffer (50 mm Tris-HCl, pH 7.4). For saturation binding studies, 2.5–15 μg of membrane protein was incubated with increasing concentrations of the appropriate radioligand (KOR, [3H]U69,593 (43.6 Ci/mmol); MOR, [3H]DAMGO (51.5 Ci/mmol); DOR, [3H]diprenorphine (50.0 Ci/mmol)) and brought to a final volume of 200 μl. For competition binding studies, membranes were incubated for 1 h at 25 °C in the presence of radioligands (0.4 nm [3H]U69,593, 0.5–1.6 nm [3H]DAMGO, or 0.4–1.2 nm [3H]diprenorphine) and increasing concentrations of the test compounds. Nonspecific binding was determined for each radioligand in the presence of 10 μm U69,593 (KOR), 10 μm DAMGO (MOR), or 10 μm naltrindole (DOR). All binding assays were terminated by filtration through GF/B glass fiber filters (pretreated with 0.1% polyethyleneimine) on a Brandel cell harvester (Brandel Inc., Gaithersburg, MD). Filters were dried and counted with Microscint on a TopCount NXT high throughput screening microplate scintillation and luminescence counter (PerkinElmer Life Sciences).
In-cell Western ERK1/2 Phosphorylation
In-cell Western assays were performed as reported previously by Schmid et al. (42). In brief, CHO-hKOR cells plated on 384-well plates were treated with test compounds for 10 min, fixed with 4% paraformaldehyde, and permeabilized with 0.2% Triton X-100. After a 1-h incubation in blocking buffer (1:1, Li-Cor blocking buffer with PBS, containing 0.05% Tween 20) (Li-Cor), cells were then incubated with primary antibodies detecting phosphorylated ERK1/2 and total-ERK1/2 (Cell Signaling) at 4 °C overnight. After four washes with PBS + 0.1% Tween 20 (PBS-T) and one with Li-Cor blocking buffer + 0.05% Tween 20, cells were incubated with Li-Cor secondary antibodies (anti-rabbit IRDye800CW, 1:500; anti-mouse IRDye680LT, 1:1500) in Li-Cor blocking buffer containing 0.025% Tween 20 for 1 h. Following an additional four washes with PBST, one wash with PBS, 1 wash with water, plates were dried, and fluorescence signal was determined using the Odyssey Infrared Imager (Li-Cor Biosciences, Lincoln, NE) at 700 and 800 nm.
ERK1/2 Western Blot Analysis
CHO-hKOR cells in 6-well plates were serum-starved for 1 h prior to treatment (10 min) as described previously (42). Antibodies to phosphorylated ERK1/2 and total ERK1/2 (Cell Signaling) were used, and the ratio of phosphorylated ERK1/2 to total ERK1/2 was calculated and normalized to vehicle treatment. Percentage of response was calculated according to the maximal responses by 10 μm U69,593 stimulation.
Tissue (Brain) Distribution
Test compounds were formulated as 1 mg/ml solution in 10% DMSO, 10% Tween 80, and sterile distilled H2O and were dosed at 10 mg/kg intraperitoneally in C57Bl-6 mice. Brain and plasma samples were taken at 30 and 60 min. Plasma and brain were mixed with acetonitrile (1:5 v/v or 1:5 w/v, respectively). The brain sample was disrupted with a probe tip sonicator. Samples were centrifuged at 16,000 × g, and the compound concentration in the supernatant was determined using liquid chromatography (Shimadzu, Japan)/tandem mass spectrometry (AB Sciex, Framingham, MA) operated in positive ion mode using multiple reaction monitoring methods. Separate standard curves were prepared in blank plasma and brain matrix. Brain concentration was calculated as compound per mg of tissue and converted to a concentration assuming a density of 1, where 1 g of tissue equals 1 ml.
Pharmacokinetics
Pharmacokinetic parameters were determined in C57Bl-6 mice by intravenous dosing in the tail vein. Mice (n = 3) were dosed with drug at 2 mg/kg, and ∼20 μl of blood was collected at 0, 5, 15, 30, 60, 120, 240, and 480 min post-dose. Plasma was generated by standard centrifugation techniques resulting in ∼10 μl of plasma that was immediately frozen. Drug levels were determined using an ABSciex 5500 mass spectrometer using multiple reaction monitoring and mass transitions of 431.3→81.1 for compound 1.1 and 461.3→286.1 for compound 2.1. Pharmacokinetic parameters were calculated using a noncompartmental model (Phoenix WinNonlin, Pharsight Inc.).
Plasma and Brain Binding
Plasma protein binding and nonspecific brain binding were determined using equilibrium dialysis. All samples were tested in triplicate. A 96-well equilibrium dialysis chamber (HTDialysis, Gales Ferry, CT) with 12,000 molecular weight cutoff dialysis membranes were used. Human plasma and rat plasma were provided by a commercial vendor (Pel-Freez Biologicals, Rogers, AR). Equal volumes of buffer and plasma containing 2.5 μm drug were added to opposite sides of the dialysis membrane. The plate was covered and allowed to shake in a 37 °C incubator for 16 h. Similar methods were used for nonspecific brain binding except a brain homogenate (1 part rat brain, 3 parts buffer) was used in place of the plasma. After 16 h, the concentration of drug in the plasma/brain versus plasma compartments was determined by LC-MS/MS. The fraction bound was calculated as ([plasma] − [buffer])/[plasma].
Determination of logP
Estimation of octanol/water partition coefficient (logP) was determined using a fast gradient reverse phase HPLC method as described by Valko et al. (48) without modification. Briefly, chromatographic retention times for test compounds and 10 standards were determined using a linear acetonitrile gradient on a Luna C18(2) 50 × 4.6 mm 5-mm column. Retention time was used to calculate the chromatographic hydrophobicity indices in acetonitrile (CHIACN). CHIACN and hydrogen bond count were used to calculate the logP according to the formula logP = 0.047 × CHIACN + 0.36 × hydrogen bond count − 1.10. Refer to Valko et al. (48) for details and a full description of the method validation.
Warm Water Tail Immersion
Antinociception was evaluated in the warm water tail-immersion (tail flick) assay (49 °C) as described previously (47, 49, 50). In brief, the tip (∼2 cm) of the mouse's tail was submerged in warm water, and the time until it was withdrawn from the water was recorded. Response latencies were measured prior to (basal) and at the indicated times following drug administration (30 mg/kg, intraperitoneally given as 10 μl/g). U50,488H and the test compounds were prepared in a vehicle containing 10% Tween 80 and 10% DMSO in sterile distilled H2O. A cutoff latency of 30 s was imposed to prevent tissue damage. Vehicle alone had no effect on response latencies (data not shown).
Data Analysis and Statistics
Sigmoidal dose-response curves were generated using three-parameter nonlinear regression analysis in GraphPad Prism 6.01 software (GraphPad, La Jolla, CA). All compounds were run in parallel assays in 2–4 replicates per n. The EC50 values and maximal responses (Emax) of drugs were obtained from the average of each individual experiment following nonlinear regression analyses and are reported as the means ± S.E.
To express the results of each response assay in a format suitable for comparison, each data set was fit, using GraphPad Prism Version 6.01, to the operational model (51) expressed as Equation 1 (52),
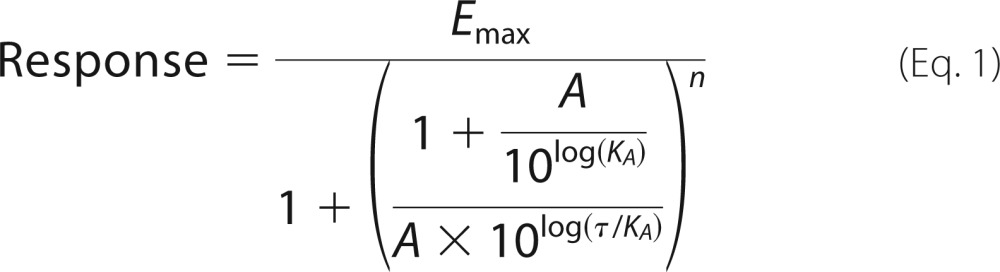 |
where Emax is the maximal response of the system; A is the molar concentration of the drug, and KA is the equilibrium dissociation constant. For each assay, the maximal response (Emax) is constrained to be a shared value. The τ parameter is defined, in the operational model, as the agonist efficacy, and the log(τ/KA) ratio (transduction coefficient) provides a single parameter value that is useful for the comparison of agonist activity (51). For all studies, U69,593 is used as the reference compound and is assayed in parallel with the test compounds. To produce an appropriate estimate of log(τ/KA) for the full reference agonist (U69,593), in each assay, the log KA for the full agonist was set constant at 0 (i.e. KA = 1 m). The resulting log(τ/KA) ratio for the reference agonist is used to produce the “normalized” transduction coefficient (Δlog(τ/KA) ratio) for each of the test compounds shown in Equation 2,
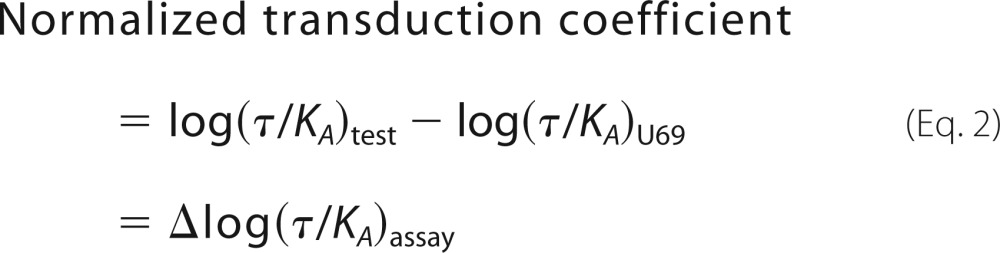 |
For a limited number of test compounds in certain assays, the logKA value in the equation was constrained to be less than 0 (i.e. KA <1 m) to permit convergence to the model. An asterisk in the tables indicates these few individual cases.
Bias factors (52–54) are derived by subtracting the Δlog(τ/KA)test-U69 for each assay (ΔΔlog(τ/KA)assay1 − assay2) and are calculated as shown in Equation 3,
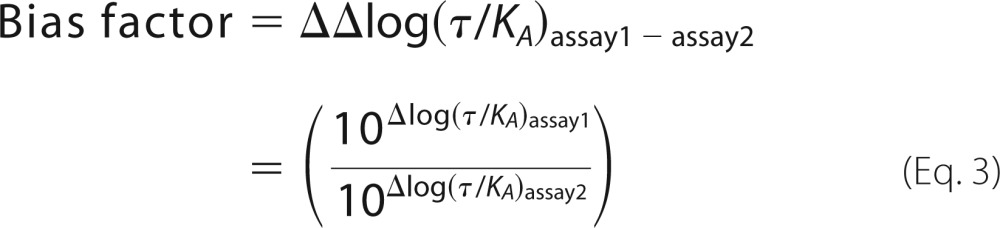 |
The statistical tests used are noted in the figure legends, where Student's t test indicates an unpaired two-tailed analysis. All studies were performed n ≥ 3 independent experiments performed in multiple replicates.
RESULTS
KOR-mediated G protein signaling was evaluated using a [35S]GTPγS binding assay in cell membranes from CHO-K1 cells stably expressing the human KOR (CHO-hKOR) (42). Concentration-response curves were run in parallel with U69,593, a well characterized potent, full agonist at KOR, as a reference agonist for the assay; EC50 values and maximal responses of test compounds were determined by nonlinear regression analysis and are presented in Table 1. Compared with U69,593 (51 nm potency), the triazole and isoquinolinone analogues show similar potencies for stimulating G protein signaling, with EC50 values ranging from ∼30 to 270 nm and with maximal stimulations roughly equivalent to that achieved with U69,593 stimulation (Fig. 2; Table 1). Furthermore, none of these compounds stimulate G protein signaling in CHO cells expressing hMOR when tested at a 10 μm concentration (data not shown) demonstrating selectivity for KOR.
TABLE 1.
Signaling parameters for KOR agonists across functional assay platforms
Emax values are calculated based on U69,593 maximal stimulation. Whereas EC50 values exceeding 3000 nm are generally not considered a good fit, the values are presented here for comparison. EFC refers to the KOR-βarrestin2 enzyme fragment complementation assay; mG refers to membrane G protein signaling assays, and imaging refers to automated high content imaging of βarrestin2-GFP translocation to KOR. NC indicates not converged. n ≥ 3 individual assays; means are presented ± S.E. Maximal stimulation over vehicle treatment with U69,593 was ∼4-fold for [35S]GTPγS binding, ∼8-fold for the EFC assay, and ∼20-fold for the imaging assay.
| Entry | [35S]GTPγS (mG) |
βarrestin2 EFC |
βarrestin2 imaging |
|||
|---|---|---|---|---|---|---|
| EC50 | Emax | EC50 | Emax | EC50 | Emax | |
| nm | % | nm | % | nm | % | |
| U69,593 | 51.5 ± 4.5 | 100 | 131 ± 22.4 | 100 | 205.3 ± 21.4 | 100 |
| 1.1 | 31.0 ± 3.8 | 94 ± 1 | 4129 ± 746 | 97 ± 5 | 3138 ± 400 | 96 ± 10 |
| 1.2 | 101.4 ± 20.5 | 93 ± 4 | 3210 ± 608 | 65 ± 4 | 5991 ± 1055 | 79 ± 26 |
| 1.3 | 66.0 ± 9.5 | 98 ± 1 | 2201 ± 267 | 106 ± 20 | 3154 ± 845 | 92 ± 10 |
| 1.4 | 250.1 ± 53.6 | 92 ± 5 | 6257 ± 2048 | 68 ± 10 | 3993 ± 1115 | 77 ± 29 |
| 1.5 | 32.7 ± 8.1 | 93 ± 1 | 8918 ± 2841 | 72 ± 20 | >10,000 | NC |
| 2.1 | 84.7 ± 12.4 | 89 ± 1 | >10,000 | NC | 3784 ± 797 | 83 ± 2 |
| 2.2 | 264.5 ± 43.4 | 84 ± 2 | > 10,000 | NC | >10,000 | NC |
FIGURE 2.
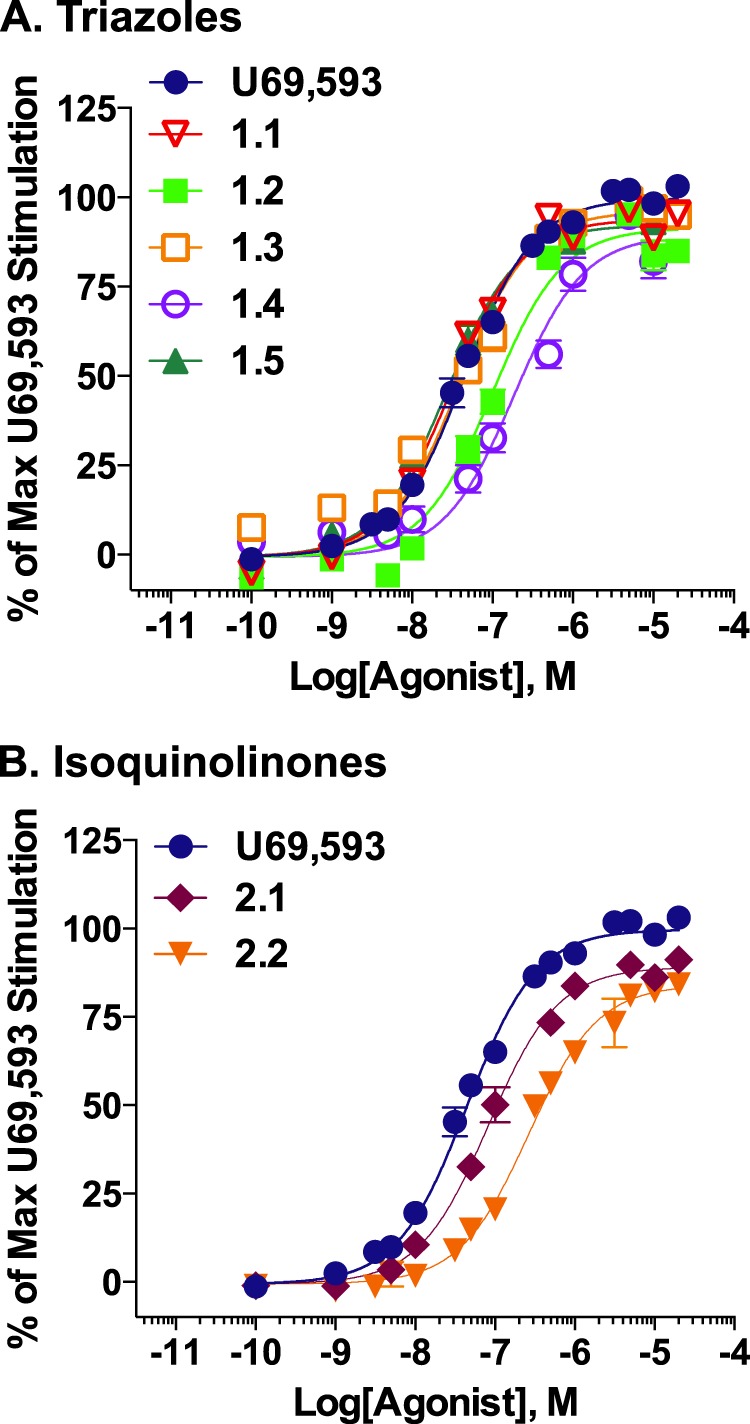
Triazole analogues (A) and isoquinolinone analogues (B) are potent, full agonists in membrane [35S]GTPγS binding assays. CHO-hKOR cell membrane preparations were incubated with increasing concentrations of indicated agonists in the presence of [35S]GTPγS; activity is calculated as percentage of maximal U69,593 stimulation following base-line subtraction. Calculated potencies and efficacies are presented in Table 1. Data are presented as the mean ± S.E. (n ≥ 3).
To determine the relative potencies for recruiting βarrestin2 to the agonist-stimulated KOR, two cell-based assays, as described in the Molecular Libraries Probe Production Centers Network probe report, were used (35). The commercial EFC assay consists of U2OS cells expressing human KOR and βarrestin2 (U2OS-hKOR-βarrestin2-EFC), each tagged with a fragment of β-galactosidase. The degree of βarrestin2 recruitment is measured as increases in luminescence intensities triggered by enzyme complementation occurring upon receptor and βarrestin2 engagement (55). The second assay utilizes U2OS cells expressing human KOR and βarrestin2 tagged with green fluorescent protein (U2OS-hKOR-βarrestin2-GFP) (56, 57). The translocation of βarrestin2-GFP is measured by automated high content imaging analysis that detects a difference in diffuse cellular GFP distribution to the agonist-induced formation of fluorescent punctae (56). In both cases, U69,593 is used as the full agonist reference ligand.
While maintaining potent agonism in the G protein signaling assays, the triazole and isoquinolinone compounds only weakly recruit βarrestin2, with potencies exceeding 2 μm in both assays, whereas the potency of U69,593 is ∼130 nm in the βarrestin2 EFC assay and ∼205 nm in the imaging assay (Fig. 3; Table 1). Examination of the concentration-response curves in both assay platforms (Fig. 3, A and C) reveals that each of the triazole compounds shows minimal βarrestin2 recruitment at lower concentrations (<1 μm). However, stimulation dramatically increases at higher concentrations (>1 μm); this is also true for compound 2.1 of the isoquinolinones (Fig. 3, B and D). This phenomenon was verified for representative compounds using confocal microscopy to visualize βarrestin2-GFP translocation in the U2OS cells (data not shown) that confirmed the results obtained in the high content imaging platform (Fig. 3, C and D).
FIGURE 3.

Compared with U69,593, the triazole and isoquinolinone analogues are weak agonists for βarrestin2 recruitment to KOR. Concentration-response curves of triazole analogues (A) and isoquinolinone analogues (B) in the commercial EFC assay or the high content imaging assay (C, triazoles and D, isoquinolinones) reveal that these compounds lead to βarrestin2 recruitment at low potencies compared with U69,593. Calculated potencies and efficacies are presented in Table 1. Data are presented as the mean ± S.E. E, representative images of CHO-hKOR cells expressing βarrestin2-YFP treated with U69,593, 1.1, or 2.1. U69,593 robustly recruits βarrestin2 at 100 nm and 1 and 10 μm, inducing green fluorescent punctae formation. 1.1 does not induce green fluorescent punctae formation at 100 nm and 1 μm, although βarrestin2 recruitment becomes apparent at 10 μm. 2.1 does not recruit βarrestin2-YFP even at 10 μm dose. Insets: 4× magnifications showing the βarrestin2-YFP punctae (arrows). Scale bars, 20 μm.
G protein signaling and βarrestin2 recruitment were, by design, performed in different cellular backgrounds (CHO-hKOR and U2OS-hKOR-βarrestin2-EFC or U2OS-hKOR-βarrestin2-GFP). The U2OS cell line was used for the commercial enzyme fragment complementation assay because it gave a larger assay window than that observed for the CHO-hKOR-βarrestin2-EFC commercial assay that is also available from the manufacturer. This was preferred to bias the system toward detecting even weak βarrestin2-KOR interactions. Furthermore, even though CHO-hKOR-βarrestin2-EFC cells could be used, it is important to realize that the KOR and βarrestin2 are linked to fragments of β-galactosidase and therefore, although the parental cell line is of the same origin, the stably transfected cell lines are not identical.
To determine whether the cell line (U2OS versus CHO-K1) contributed to the apparent bias, we also evaluated βarrestin2 (tagged with YFP) recruitment in the CHO-hKOR cell line by confocal microscopy for select agonists of each class. In the confocal microscopy studies of CHO-hKOR cells, 1.1 does not promote βarrestin2 recruitment at low concentrations (100 nm and 1 μm) but induces punctae formation at 10 μm (Fig. 3E), consistent with the studies in U2OS cell imaging assays and the EFC βarrestin2 assay. The ability to visualize the agonist-induced βarrestin2 recruitment also suggests that the positive signal at high concentrations is not an artifact of the assay. In contrast to the observations in the other βarrestin2 assay platforms, 2.1 revealed no visible recruitment of βarrestin2-YFP even at 10 μm, in the CHO-hKOR cells (Fig. 3E). The lack of response to 2.1 in the CHO-hKOR cells transfected with βarrestin2-YFP is likely due to a low detection window with the low degree of stimulation falling below detectable thresholds. Finally, when tested in the MOR EFC assay, no stimulation is seen at any dose (1, 10, and 100 μm), which also argues against the effect being an artifact of the assay (data not shown).
The studies presented in Tables 1 and in Figs. 2 and 3 suggest that the triazole and isoquinolinone analogues presented here bias KOR function toward G protein signaling over the recruitment of βarrestin2. To compare the relative differences within and between assays, curves were fit to the operational model (51) to derive transduction coefficients (log(τ/KA)) for the reference ligand (U69,593) and the “test” ligands using GraphPad Prism Version 6.01, as described previously (42, 52, 54). The transduction coefficient represents the relative propensity of the ligand to generate a signal proportional to the agonist's calculated relative affinity for engaging the receptor based on its performance in the assay. Subtracting the log(τ/KA) for U69,593 from the values obtained for the test ligands generates a “normalized” transduction coefficient for the test ligand within the assay, or the Δlog(τ/KA). To determine preference between assays, bias factors were calculated as described under “Experimental Procedures” (Table 2) (54). Although such modeling may be imperfect for extreme cases of bias, we have presented the analysis here for qualitative comparison; each of the compounds displays bias for activation of KOR toward G protein signaling over βarrestin2 recruitment.
TABLE 2.
Analysis of bias comparing G protein signaling and βarrestin2 recruitment in reference to U69,593 activity
Bias factors (ΔΔlog(τ/KA)assay1 − assay2) were calculated as described under “Experimental Procedures.” The asterisk indicates log(τ/KA) values were constrained to <0 (less than 1 m relative affinity) to permit convergence to the operational model. mG indicates membrane G protein signaling; imaging indicates automated imaging analysis. n ≥ 3 individual assays; means are presented ± S.E. βarr2 is βarrestin2.
| Entry | [35S]GTPγS (mG), log(τ/KA) | βArrestin2 EFC, log(τ/KA) | βArrestin2 imaging log(τ/KA) | Bias factors |
|
|---|---|---|---|---|---|
| mG/βarr2 EFC | mG/βarr2 Imaging | ||||
| U69,593 | 7.36 ± 0.01 | 7.09 ± 0.04 | 6.79 ± 0.05 | 1.0 | 1.0 |
| 1.1 | 7.47 ± 0.03 | 5.42 ± 0.10* | 5.61 ± 0.11 | 61.2 | 20.0 |
| 1.2 | 6.95 ± 0.03 | 5.38 ± 0.16* | 5.37 ± 0.15 | 19.9 | 10.4 |
| 1.3 | 7.40 ± 0.04 | 5.66 ± 0.09* | 5.68 ± 0.11 | 30.1 | 14.4 |
| 1.4 | 6.67 ± 0.06 | 4.92 ± 0.13 | 5.47 ± 0.12 | 30.8 | 4.3 |
| 1.5 | 7.52 ± 0.04 | 5.21 ± 0.15 | 5.31 ± 0.65 | 111.7 | 44.3 |
| 2.1 | 7.01 ± 0.05 | 5.25 ± 0.18 | 5.59 ± 0.58 | 31.4 | 7.2 |
| 2.2 | 6.48 ± 0.05 | 4.54 ± 0.81 | 3.91 ± 0.30* | 46.7 | 100.0 |
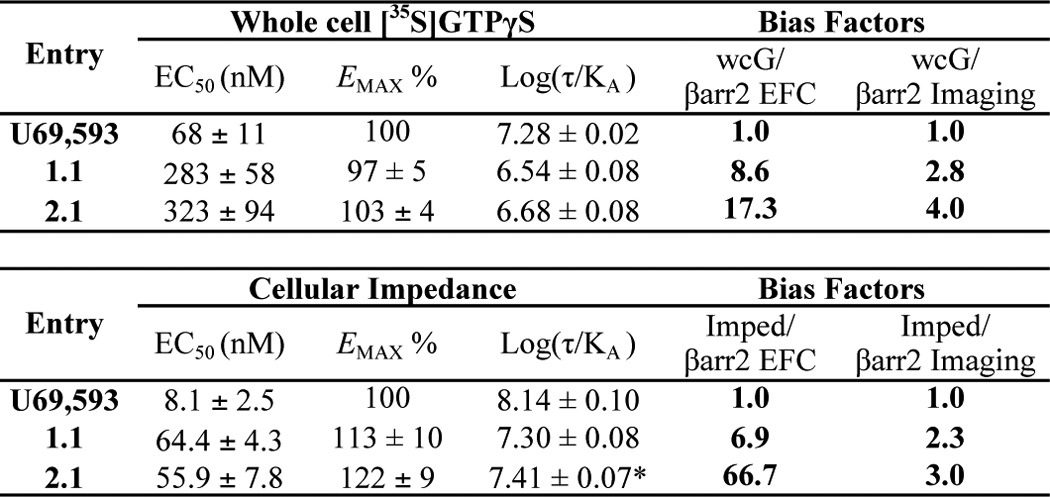
Because the [35S]GTPγS binding assay was performed in membrane preparations whereas the βarrestin2 recruitment studies were performed in whole cells, we considered that these contextual differences could confound our interpretations of bias. Therefore, we selected a representative triazole and isoquinolinone and performed two additional G protein signaling assays in whole cell preparations (Fig. 4). Using a low concentration of saponin to permeabilize the CHO-hKOR cells allowing uptake of the radionucleotide, [35S]GTPγS binding was performed in whole cells plated on 96-well plates (Fig. 4A). U69,593, 1.1, and 2.1 potently stimulate G protein signaling in this whole cell assay. Furthermore, in the absence of the brief permeabilization step, U69,593 does not induce [35S]GTPγS binding due to the lack of available radionucleotide inside the cell.
FIGURE 4.
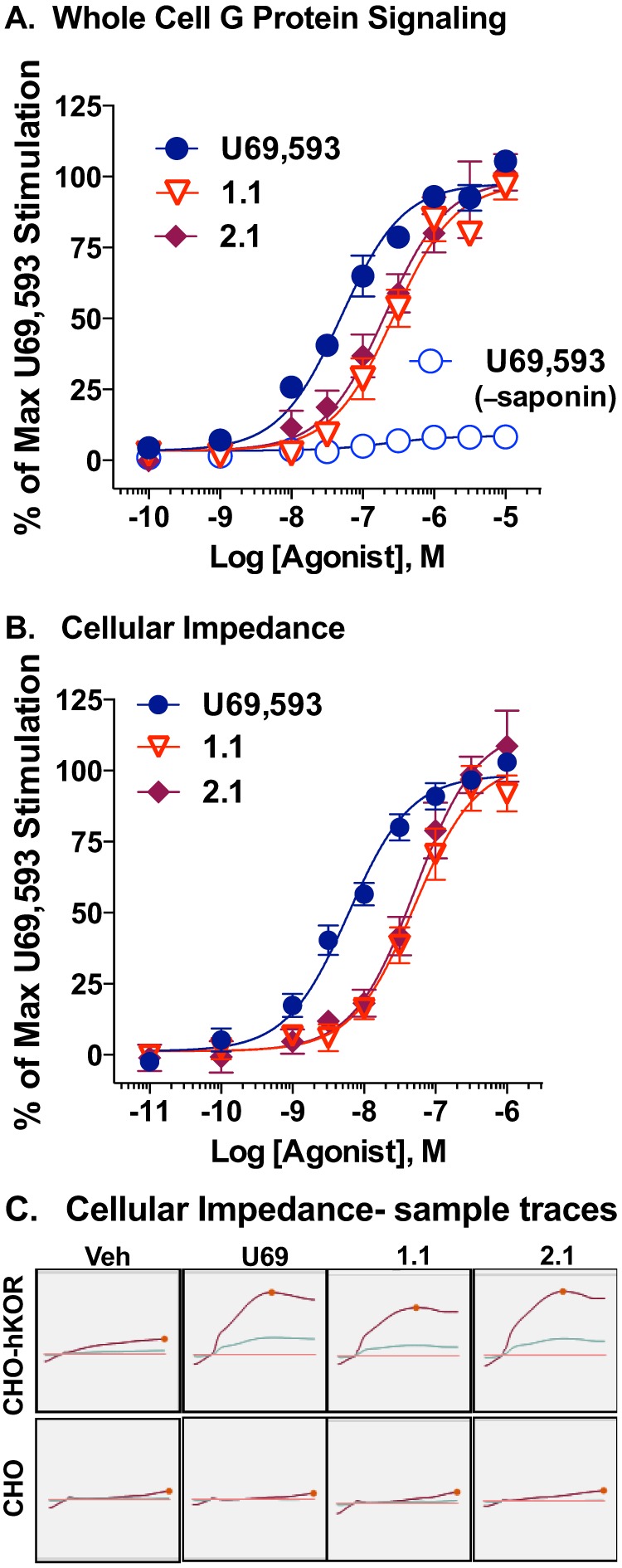
Triazole and isoquinolinone KOR agonists are potent, full agonists in whole cell G protein signaling assays. A, whole cell G coupling assay. CHO-hKOR cells were permeabilized prior to performing agonist-stimulated [35S]GTPγS binding assays directly on plated cells in 96-well plates. Activity is calculated as percentage of maximal U69,593 stimulation following base-line subtraction. U69,593 induces no stimulation in the absence of permeabilization (−saponin). B, cellular impedance assay. Changes in cellular impedance in CHO-hKOR cells were recorded for 35 min after treatment of increasing doses of U69,593, 1.1 and 2.1. Maximal changes in impedance are calculated as percentage of maximal U69,593 (U69) stimulation. C, example traces comparing vehicle (Veh) and the maximum dose of 1 μm of each compound are shown (bottom). Calculated potencies and efficacies for both assays are presented in Table 3. Data are presented as the mean ± S.E. (n ≥ 5).
A label-free, cellular impedance assay was also utilized to assess relative potencies in the living CHO-hKOR cells (Fig. 4B). This assay measures changes in impedance resulting from cytoskeletal re-organization leading to changes in cell shape (45). GPCRs promote actin reassembly patterns reflective of the coupling of particular Gα proteins. In our analysis, the cellular response signature resembles that of a typical Gαi-coupled GPCR (Fig. 4C) (58). Importantly, no response was observed in the parental CHO cell line (data not shown). In this live cell assay format, 1.1 and 2.1 are fully efficacious and nearly as potent as U69,593 in promoting KOR-dependent responses indicative of G protein signaling (Table 3).
TABLE 3.
Representative triazole and isoquinolinone compounds in whole cell G protein signaling assays
EC50 and Emax values were calculated as described in Table 1. Bias factors (ΔΔlog(τ/Ka)assay1 − assay2) were calculated as described under “Experimental Procedures.” βArrestin2 EFC and βarrestin2 imaging transduction coefficients are found in Table 2. The asterisk indicates that log Ka values were constrained to <0 (less than 1 m relative affinity) to permit convergence to the operational model. n ≥ 5 individual assays; means are presented ± S.E. Maximal stimulation over vehicle treatment with U69,593 was ∼2-fold for whole cell [35S]GTPγS binding (wcG) and ∼8-fold for the cellular impedance assay.
Further analysis was undertaken to compare all G protein signaling assays to all βarrestin2 recruitment assays. Data from Tables 2 and 3 were used to determine bias factors that are presented graphically in Fig. 5. In summary, regardless of the assay format and the cell type, the triazole and isoquinolinone maintain bias toward KOR-induced G protein signaling over βarrestin2 recruitment.
FIGURE 5.

Triazole and isoquinolinone agonists bias KOR toward G protein signaling pathways. Transduction efficiencies presented in Tables 2 and 3 for G protein signaling assays and βarrestin2 recruitment assays were used to calculate bias factors. Bias factors are presented in Tables 2 and 3 and are plotted here on a logarithmic scale (base 10). As the reference agonist, the bias of U69,593 conforms to unity in all assays. The pathways represented are as follows: membrane [35S]GTPγS binding (mG protein); βarr2 EFC, βarr2 imaging, cellular impedance, and whole cell [35S]GTPγS binding (wcG protein). Independent of the platform used to assess G protein signaling or βarrestin2 recruitment, both 1.1 and 2.1 display bias for G protein signaling.
To further characterize the compounds at a downstream signaling pathway, ERK1/2 phosphorylation was investigated. GPCRs can utilize both G protein-dependent and βarrestin-mediated signaling pathways to activate ERK1/2 MAPKs (34). Interestingly, both classes of compounds activate ERK with potencies between 300 and 6000 nm as compared with ∼5 nm for U69,593. Although this appears to correlate with their relative potency for recruiting βarrestin2 (Fig. 6, A and B, and Table 4), further experiments must be undertaken to further define these pathways.
FIGURE 6.

Ligands induced ERK1/2 phosphorylation in CHO-hKOR cells. A, CHO-hKOR cells were treated with increasing doses of triazole (A) and isoquinolinone analogues (B) for 10 min, and phosphorylated (p-ERK) and total ERK (t-ERK) were detected by fluorescence intensity in 96-well plate “In-cell Western” assays. The ratio of p-ERK1/2 to total ERK1/2 was calculated and reported normalized to the percentage of maximal response induced by U69,593. C and D, Western blot analysis confirms that the p42 and p44 bands increase with drug treatment when examined with the p-ERK1/2 antibody. The high degree of ERK1/2 phosphorylation in CHO-hKOR cells induced by the triazole (C) or isoquinolinone (D) stimulation exceeds that observed for U69,593 (U69) (10 μm, 10 min), confirming the observations made in the In-cell Western format. Ratios of p-ERK1/2 over total ERK1/2 were normalized to the maximal U69,593 stimulation observed (n ≥ 3; *, p < 0.05; **, p < 0.01, Student's t test); molecular masses are indicated in kDa. Veh, vehicle.
TABLE 4.
Signaling parameters for KOR agonists in ERK1/2 phosphorylation studies
Emax values are calculated based on U69,593 maximal stimulation. n ≥ 3 individual assays; means are presented ± S.E. Maximal stimulation over vehicle treatment with U69,593 was ∼4 fold.
| Entry | ERK1/2 phosphorylation |
|
|---|---|---|
| EC50 | Imax | |
| nm | % | |
| U69,593 | 5.4 ± 0.78 | 100 |
| 1.1 | 329 ± 84 | 109 ± 7 |
| 1.2 | 2282 ± 426 | 139 ± 11 |
| 1.3 | 432 ± 152 | 167 ± 14 |
| 1.4 | 2778 ± 605 | 173 ± 9 |
| 1.5 | 5642 ± 2215 | 241 ± 49 |
| 2.1 | 2812 ± 1116 | 149 ± 20 |
| 2.2 | 553 ± 238 | 93 ± 10 |
It is noteworthy that although these agonists are not very potent in this assay, their efficacy for activating ERK exceeds that of the reference compound U69,593. Because the assay utilized here involves an immunohistochemistry approach, it is possible that the increase in fluorescence could be due to the induction of other kinases that may be recognized by the phospho-specific ERK1/2 antibodies used. Therefore, Western blot analysis was performed to determine whether the increase in phosphorylation detected could be attributed to the 42- and 44-kDa bands, p-ERK1 and p-ERK2, respectively. Western blot analysis confirmed that the intensity of 42- and 44-kDa bands are elevated over that observed for U69,593-induced phosphorylation suggesting that this effect is not an artifact of the 384-well plate immunohistochemistry approach (Fig. 6C). Furthermore, none of the compounds activate ERK1/2 in the CHO-K1 parental cell line when tested at 1, 10, or 20 μm suggesting that these effects are KOR-dependent (data not shown). Because the efficacy of the test compounds exceeded that of the reference compound in this assay, U69,593 was insufficient to define the maximum potential of the system, and therefore, these data were not fit to the operational model. Additional studies, investigating differences in ERK1/2 populations (cytosol and nuclear) as well as the temporal regulation of ERK1/2 phosphorylation, will be needed to make accurate comparisons between the agonists.
It is possible that, given the different receptor expression levels and cellular context, the ligands may not have comparable opportunities to interact with the receptor. To address this question, radioligand competition binding assays were used to determine whether the test ligands maintained comparable affinity and ability to displace [3H]U69,593 binding to membranes from the CHO-hKOR and U2OS-hKOR-βarrestin2-EFC cell lines. Saturation binding of [3H]U69,593 was performed initially to determine equilibrium dissociation constants (KD values) and receptor numbers (Bmax values) in each cell line (with unlabeled U69,593 at 10 μm defining the nonspecific binding). The CHO-hKOR cell line expresses 2.07 ± 0.48 pmol/mg membrane protein with a [3H]U69,593 binding affinity of 0.27 ± 0.11 nm (Table 5). The commercial U2OS-hKOR-βarrestin2-EFC cell line expresses nearly 6-fold more receptors (11.45 ± 2.26 pmol/mg of membrane protein), and the affinity for [3H]U69,593 binding is 0.36 ± 0.10 nm. When the test compounds were compared in each of the cell lines, the triazole and isoquinolinone compounds all fully displace [3H]U69,593 binding and all do so with high affinity (0.25–1.79 nm), confirming comparable opportunity to bind to the KOR (Table 5). Further studies were performed to determine selectivity profiles of binding to MOR and DOR expressed in CHO-K1 cells. The triazole and isoquinolinone derivatives, like the initial probes, display high selectivity for KOR binding over mu and delta opioid receptors (Table 5) (35, 36, 38).
TABLE 5.
Binding affinities for KOR agonists in the U2OS-hKOR-βarrestin2-EFC, CHO-hKOR, CHO-hDOR, and CHO-hMOR cell lines
Binding parameters were determined for KOR using [3H]U69,593 and 10 μm unlabeled U69,593 to determine the nonspecific binding. Saturation binding assays revealed 11.45 ± 2.26 pmol/mg receptors in the U2OS-hKOR-βarrestin2-EFC cell line and 2.07 ± 0.48 pmol/mg receptor in the CHO-hKOR cell line. All of the compounds fully displaced [3H]U69,593 binding in a manner that did not significantly differ from displacement achieved with U69,593 (extra sum of squares F test, p > 0.05). To determine selectivity, [3H]diprenorphine was used for DOR with 10 μm unlabeled naltrindole to determine nonspecific binding; [3H]DAMGO was used for MOR binding with 10 μm naloxone used to determine nonspecific binding. Saturation binding assays revealed an affinity of 0.342 ± 0.07 nm and 5.51 ± 1.0 pmol/mg receptors in the CHO-hDOR cell line and an affinity of 0.443 ± 0.02 nm and 5.36 ± 0.76 pmol/mg receptor in the CHO-hMOR cell line. None of the compounds fully displaced radioligand binding to DOR or MOR at the highest concentrations tested (10 μm); Ki values were calculated for curves that displaced more than 50% of radioligand binding; those that did not displace more than 50% are listed as >10,000 nm. n ≥ 3 individual assays; means are presented ± S.E. Ki ratios were calculated based on the Ki values indicated (10,000 is used where >10,000 is indicated to define the upper limit of affinity).
| Entry | EFC-KOR, Ki or KD | CHO-hKOR, Ki or KD | CHO-hDOR |
CHO-hMOR |
Ki ratios in CHO lines |
|||
|---|---|---|---|---|---|---|---|---|
| Ki | % at 10 μm | Ki | % at 10 μm | DOR/KOR | MOR/KOR | |||
| nm | nm | nm | nm | |||||
| U69 | 0.36 ± 0.10 | 0.27 ± 0.11 | >10,000 | 48 ± 12 | 1638 ± 548 | 62 ± 12 | >36,946 | 6052 |
| 1.1 | 0.33 ± 0.05 | 0.25 ± 0.02 | 2225 ± 571 | 58 ± 10 | >10,000 | 31 ± 11 | 8905 | >40,011 |
| 1.2 | 0.55 ± 0.05 | 0.54 ± 0.09 | >10,000 | 48 ± 8 | 712 ± 482 | 77 ± 13 | >18,703 | 1332 |
| 1.3 | 0.47 ± 0.07 | 0.25 ± 0.06 | 2681 ± 192 | 60 ± 4 | 1687 ± 251 | 68 ± 10 | 10299 | 6636 |
| 1.4 | 1.79 ± 0.18 | 1.16 ± 0.14 | 2061 ± 181 | 67 ± 8 | 2720 ± 714 | 60 ± 15 | 1779 | 2348 |
| 1.5 | 0.67 ± 0.13 | 0.47 ± 0.11 | >10,000 | 31 ± 16 | >10,000 | 33 ± 11 | >21,216 | >21,216 |
| 2.1 | 0.28 ± 0.05 | 0.35 ± 0.13 | >10,000 | 33 ± 10 | 368 ± 136 | 80 ± 8 | >28,542 | 1051 |
| 2.2 | 1.45 ± 0.10 | 1.31 ± 0.33 | >10,000 | 9 ± 11 | >10,000 | 20 ± 11 | >7,649 | >7,649 |
Ultimately, these compounds, of which there is a growing collection, may become important tools for ascertaining the relative contributions of G protein-dependent, βarrestin2-independent signaling at KOR in vivo. As an early step in characterization, the pharmacokinetic properties of 1.1 and 2.1 were assessed in mice. Brain and plasma compound contents were determined following a single systemic, intraperitoneal administration (10 mg/kg, intraperitoneal), and both compounds were detected at low micromolar concentrations in brain at 30 and 60 min (Table 6). Pharmacokinetic distribution and clearance rates were determined following a 2 mg/kg (i.v.) tail vein injection and timed blood collections. Plasma protein binding and nonspecific brain binding were also determined for these compounds. These parameters, along with the determined “logP” values are presented in Table 6 and indicate good brain exposure and reasonable pharmacokinetic properties. Together these findings suggest that these scaffolds may represent favorable leads for development of biased KOR agonists with CNS penetration.
TABLE 6.
Triazole (1.1) and isoquinolinone (2.1) brain and blood distribution, pharmacokinetics, and logP values
The abbreviations used are as follows: t½ (h), half-life of compound; tmax (h), time point of maximal compound concentration detected; AUClast (μm·h), area under the plasma concentration − time curve; Cl_obs (ml/min/kg), plasma clearance rate; Vss_obs (liter/kg), steady-state volume of distribution.
| Assay | 1.1 | 2.1 |
|---|---|---|
| [Brain] (μm) | ||
| 30 min | 1.91 ± 0.83 | 1.09 ± 0.04 |
| 60 min | 1.31 ± 0.15 | 1.21 ± 0.11 |
| [Plasma] (μm) | ||
| 30 min | 0.43 ± 0.15 | 0.79 ± 0.06 |
| 60 min | 0.22 ± 0.02 | 0.82 ± 0.02 |
| Brain (% bound) | ||
| Rat | >99.5 | 99.20 |
| Plasma (% bound) | ||
| Human | >99.5 | >99.5 |
| Mouse | >99.5 | 96.90 |
| Rat | >99.5 | 99.20 |
| Pharmacokinetics | ||
| t½ | 2.80 h | 2.03 h |
| tmax | 0.25 h | 0.14 h |
| Cmax | 4.94 μm | 4.16 μm |
| AUClast | 4.62 μm·h | 6.83 μm·h |
| Cl_obs | 31.53 ml/min/kg | 26.82 ml/min/kg |
| Vss_obs | 4.20 liter/kg | 3.43 liter/kg |
| Determined logP | 3.68 | 4.16 |
Because G protein-mediated KOR signaling has been proposed as the mechanism underlying KOR-mediated antinociception, the antinociceptive effects of 1.1 and 2.1 were then assessed using the warm water tail-flick assay (Fig. 6) (10, 59). Mice were injected with 30 mg/kg, intraperitoneal doses of either 1.1, 2.1, or the selective KOR agonist U50,488H, as this dose of U50,488H has been shown to induce antinociception in mice in this assay (60). The triazole and isoquinolinone compounds significantly increased the latency to tail withdrawal similar to that seen with U50,488H, with the effects peaking at 20 min post drug treatment (Fig. 7) demonstrating that these agonists are capable of inducing antinociception in mice.
FIGURE 7.
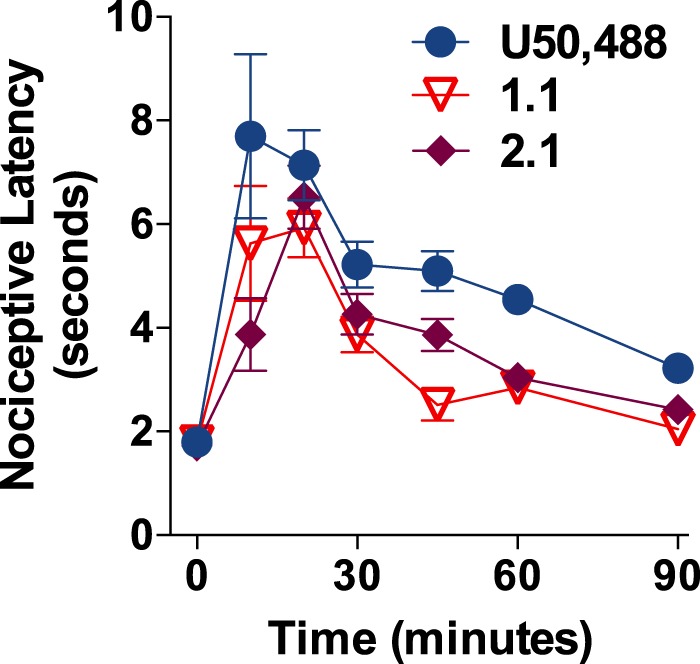
Compounds 1.1 and 2.1 induce antinociceptive effects in mice. C57BL/6J male mice were treated with U50,488H, 1.1, or 2.1 at 30 mg/kg, and intraperitoneal and tail withdrawal response latencies were recorded in response to exposure of the tail to warm water (49 °C) at 10, 20, 30, 45, 60, and 90 min post-treatment. Data are presented as the mean ± S.E. U50,488H and both test compounds induced similar time-dependent antinociceptive responses (one-way ANOVA, for U50,488H, F(6,49) = 5.58, p < 0.0001; for 1.1, F(6,49) = 10.55, p < 0.0001; for 2.1, F(6,49) = 13.42, p < 0.0001; Bonferroni's post test, basal versus treatment time within each drug treatment: U50,488H (10, 20 min, p < 0.001; 30, 45 min, p < 0.01; 60 min, p < 0.05) and 1.1 (10, 20 min, p < 0.001; 30, 45 min, p < 0.05), 2.1 (20, 30 min, p < 0.001, 10; 45 min, p < 0.01) n = 8–10 mice per drug treatment).
DISCUSSION
The triazole and isoquinolinone analogues described herein are highly selective KOR agonists that induce receptor signaling biased toward G protein signaling over βarrestin2 recruitment. When tested in vivo, 1.1 and 2.1, a triazole and an isoquinolinone, respectively, prove to be brain-penetrant. Subsequently, 1.1 and 2.1 produce antinociception in the mouse tail flick test, which correlates with the ability of these compounds to potently activate G protein signaling in cell-based assays.
Prior studies suggest that KOR agonists that do not engage βarrestin2-mediated signaling pathways may prove to have less aversive properties such as dysphoria (30, 61, 62) (Fig. 8). It is hopeful that the generation of compounds, such as those in the series described here, will provide pharmacological tools that will aid in the elucidation of KOR-mediated signaling cascades in cellular systems and, importantly, in vivo. The fact that these series possess very high affinity and selectivity for KOR over other opioid receptors, are brain penetrant, and have favorable pharmacokinetic parameters will certainly aid in these ventures.
FIGURE 8.
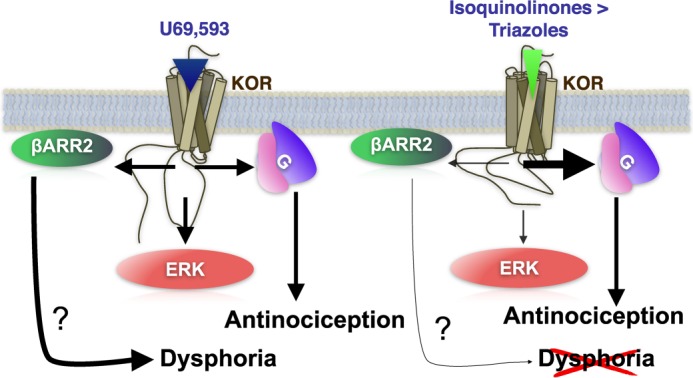
Schematic of isoquinolinone and triazole compound signaling at KOR compared with U69,593. Activation at KOR mediates multiple signaling cascades leading to G protein coupling, βarrestin recruitment, and ERK kinase phosphorylation. The isoquinolinones and triazoles act as biased agonists, preferentially inducing G protein coupling over βarrestin2 recruitment. This lack of preference for βarrestin2 recruitment appears to correlate with a loss in potency for ERK1/2 activation. Because KOR-mediated antinociception has been attributed to G protein signaling mechanisms and because KOR-induced interactions with βarrestin2 are proposed to induce dysphoria, biasing KOR activation toward G protein signaling and away from βarrestin2 pathways may be the key to induce antinociception and avoid dysphoria (30, 57, 58).
Recently, Rives et al. (62) found the naltrindole-derived ligand 6′-GNTI to be a potent partial agonist for KOR-stimulated G protein signaling yet an antagonist for recruiting βarrestins. From our laboratory, Schmid et al. (42) recently confirmed the delineation of this bias in vitro and further explored it in cultured striatal neurons. Although 6′-GNTI potently activates ERK1/2 in CHO-hKOR cells, in neurons 6′-GNTI does not activate ERK1/2 although U69,593 does so in a βarrestin2-dependent manner, suggesting that KOR utilizes βarrestin2 to activate ERK in the endogenous setting (42). The triazole and isoquinolinone compounds investigated herein differ from 6′-GNTI in that they are full agonists in G protein signaling and they are able to activate βarrestin2 recruitment in the cell-based assays, although at a greatly diminished potency. Furthermore, the triazoles and isoquinolinones do not potently activate ERK1/2, further distinguishing them from 6′-GNTI. The increasing number of compounds identified with diverse signaling profiles downstream of KOR activation may be useful for docking studies to the continually refined crystal structure of these GPCRs (63), which may provide insight into the chemical signatures driving specific receptor confirmations and signaling events.
The triazoles and isoquinolinones consistently activate ERK1/2 with less potency than that observed for U69,593. Therefore, it is attractive to speculate that the triazoles and isoquinolinones are biased toward G protein signaling over ERK1/2 phosphorylation. We hesitate, however, to make this claim based on two confounding variables. In this particular pathway, U69,593 does not adequately serve as a reference compound. The second issue, and perhaps the most important, is that ERK1/2 activation is a downstream effector that represents the consolidation of multiple upstream cascades (potentially originating from affecting different G proteins, βarrestins, calcium influx, etc.). Although our negative controls demonstrate that the activation of ERK by the test compounds is due to KOR (no response in parental CHO-K1 cells), it is difficult to interpret what component of cellular signaling is participating more (overshoot in efficacy) or less (rightward shift in potency) in generating the response profile elicited by these compounds downstream of KOR. Regardless, the compounds consistently reveal more potency in the G protein signaling assay than in the ERK1/2 and βarrestin assays. Overall, these results are intriguing and represent an area for further explorations into the compartmental (nuclear versus cytoplasmic (64)) and temporal (transient or sustained (28, 64)) aspects of GPCR-stimulated ERK activation in respect to βarrestin and G protein-dependent mechanisms.
It will be of great interest to utilize these compounds in additional behavioral and physiological assays to determine the contribution of βarrestin2 to KOR signaling in vivo. However, it will first be important to determine that the signaling bias observed across the various cell-based assay platforms is maintained in vivo and whether these pathways are associated with behaviors. If indeed these newly discovered KOR-biased agonists, which are brain penetrant following systemic injections, prove to maintain signaling bias in the endogenous setting, these compounds may serve as important tools for investigating the contributions of ERK activation and/or βarrestin2 recruitment to KOR-mediated effects in vivo, and it is hopeful that these new pharmacological tools will serve the community for exploring relevant KOR biology in an endogenous setting.
Acknowledgments
We thank Ben Neuenswander and Patrick Porubsky (Kansas University) for HPLC-MS analysis and Larry Barak (Duke University) for the U2OS-hKOR-βarrestin2-GFP cell line. We are grateful to Arthur Christopoulos (Monash University, Australia) for helpful discussions on determining bias factors.
This work was supported, in whole or in part, by National Institutes of Health Grant R01 DA031927 (to L. M. B. and J. A.).

This article contains supplemental text.
- KOR
- kappa opioid receptor
- GPCR
- G protein-coupled receptor
- h
- human
- EFC
- enzyme fragment complementation
- GTPγS
- guanosine 5′-3-O-(thio)triphosphate
- p-ERK
- phosphorylated ERK
- MOR
- mu opioid receptor
- DOR
- delta opioid receptor
- DAMGO
- [d-Ala2,N-Me-Phe4,Gly-ol5]-enkephalin.
REFERENCES
- 1. Cox B. M. (2013) Recent developments in the study of opioid receptors. Mol. Pharmacol. 83, 723–728 [DOI] [PubMed] [Google Scholar]
- 2. Chavkin C., James I. F., Goldstein A. (1982) Dynorphin is a specific endogenous ligand of the kappa opioid receptor. Science 215, 413–415 [DOI] [PubMed] [Google Scholar]
- 3. Mansour A., Fox C. A., Burke S., Meng F., Thompson R. C., Akil H., Watson S. J. (1994) Mu, delta, and kappa opioid receptor mRNA expression in the rat CNS: an in situ hybridization study. J. Comp. Neurol. 350, 412–438 [DOI] [PubMed] [Google Scholar]
- 4. Minami M., Satoh M. (1995) Molecular biology of the opioid receptors: structures, functions and distributions. Neurosci. Res. 23, 121–145 [DOI] [PubMed] [Google Scholar]
- 5. Prather P. L., McGinn T. M., Claude P. A., Liu-Chen L. Y., Loh H. H., Law P. Y. (1995) Properties of a kappa-opioid receptor expressed in CHO cells: interaction with multiple G-proteins is not specific for any individual Gα subunit and is similar to that of other opioid receptors. Brain Res. Mol. Brain Res. 29, 336–346 [DOI] [PubMed] [Google Scholar]
- 6. Lawrence D. M., Bidlack J. M. (1993) The kappa opioid receptor expressed on the mouse R1.1 thymoma cell line is coupled to adenylyl cyclase through a pertussis toxin-sensitive guanine nucleotide-binding regulatory protein. J. Pharmacol. Exp. Ther. 266, 1678–1683 [PubMed] [Google Scholar]
- 7. Ma G. H., Miller R. J., Kuznetsov A., Philipson L. H. (1995) kappa-Opioid receptor activates an inwardly rectifying K+ channel by a G protein-linked mechanism: coexpression in Xenopus oocytes. Mol. Pharmacol. 47, 1035–1040 [PubMed] [Google Scholar]
- 8. Macdonald R. L., Werz M. A. (1986) Dynorphin A decreases voltage-dependent calcium conductance of mouse dorsal root ganglion neurones. J. Physiol. 377, 237–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pasternak G. W. (1980) Multiple opiate receptors: [3H]ethylketocyclazocine receptor binding and ketocyclazocine analgesia. Proc. Natl. Acad. Sci. U.S.A. 77, 3691–3694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vonvoigtlander P. F., Lahti R. A., Ludens J. H. (1983) U-50,488: a selective and structurally novel non-Mu (kappa) opioid agonist. J. Pharmacol. Exp. Ther. 224, 7–12 [PubMed] [Google Scholar]
- 11. Dykstra L. A., Gmerek D. E., Winger G., Woods J. H. (1987) Kappa opioids in rhesus monkeys. I. Diuresis, sedation, analgesia and discriminative stimulus effects. J. Pharmacol. Exp. Ther. 242, 413–420 [PubMed] [Google Scholar]
- 12. Millan M. J., Czl̸onkowski A., Lipkowski A., Herz A. (1989) Kappa-opioid receptor-mediated antinociception in the rat. II. Supraspinal in addition to spinal sites of action. J. Pharmacol. Exp. Ther. 251, 342–350 [PubMed] [Google Scholar]
- 13. Simonin F., Valverde O., Smadja C., Slowe S., Kitchen I., Dierich A., Le Meur M., Roques B. P., Maldonado R., Kieffer B. L. (1998) Disruption of the kappa-opioid receptor gene in mice enhances sensitivity to chemical visceral pain, impairs pharmacological actions of the selective kappa-agonist U-50,488H and attenuates morphine withdrawal. EMBO J. 17, 886–897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Goicoechea C., Ormazábal M. J., Abalo R., Alfaro M. J., Martín M. I. (1999) Calcitonin reverts pertussis toxin blockade of the opioid analgesia in mice. Neurosci. Lett. 273, 175–178 [DOI] [PubMed] [Google Scholar]
- 15. Gullapalli S., Ramarao P. (2002) Role of L-type Ca2+ channels in pertussis toxin induced antagonism of U50,488H analgesia and hypothermia. Brain Res. 946, 191–197 [DOI] [PubMed] [Google Scholar]
- 16. Raehal K. M., Bohn L. M. (2005) Mu opioid receptor regulation and opiate responsiveness. AAPS J. 7, E587–E591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pfeiffer A., Brantl V., Herz A., Emrich H. M. (1986) Psychotomimesis mediated by kappa opiate receptors. Science 233, 774–776 [DOI] [PubMed] [Google Scholar]
- 18. Land B. B., Bruchas M. R., Lemos J. C., Xu M., Melief E. J., Chavkin C. (2008) The dysphoric component of stress is encoded by activation of the dynorphin kappa-opioid system. J. Neurosci. 28, 407–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Roth B. L., Baner K., Westkaemper R., Siebert D., Rice K. C., Steinberg S., Ernsberger P., Rothman R. B. (2002) Salvinorin A: a potent naturally occurring nonnitrogenous kappa opioid selective agonist. Proc. Natl. Acad. Sci. U.S.A. 99, 11934–11939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Knoll A. T., Carlezon W. A., Jr. (2010) Dynorphin, stress, and depression. Brain Res. 1314, 56–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bohn L. M. (2009) in Functional Selectivity of G Protein-coupled Receptor Ligands (Neve K. A., ed) 1st Ed., pp. 71–85, Humana Press Inc., Totowa, NJ [Google Scholar]
- 22. Urban J. D., Clarke W. P., von Zastrow M., Nichols D. E., Kobilka B., Weinstein H., Javitch J. A., Roth B. L., Christopoulos A., Sexton P. M., Miller K. J., Spedding M., Mailman R. B. (2007) Functional selectivity and classical concepts of quantitative pharmacology. J. Pharmacol. Exp. Ther. 320, 1–13 [DOI] [PubMed] [Google Scholar]
- 23. Reiter E., Ahn S., Shukla A. K., Lefkowitz R. J. (2012) Molecular mechanism of β-arrestin-biased agonism at seven-transmembrane receptors. Annu. Rev. Pharmacol. Toxicol. 52, 179–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Luttrell D. K., Luttrell L. M. (2003) Signaling in time and space: G protein-coupled receptors and mitogen-activated protein kinases. Assay Drug Dev. Technol. 1, 327–338 [DOI] [PubMed] [Google Scholar]
- 25. Schmid C. L., Bohn L. M. (2009) Physiological and pharmacological implications of β-arrestin regulation. Pharmacol. Ther. 121, 285–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shenoy S. K., Lefkowitz R. J. (2011) β-Arrestin-mediated receptor trafficking and signal transduction. Trends Pharmacol. Sci. 32, 521–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lefkowitz R. J., Shenoy S. K. (2005) Transduction of receptor signals by β-arrestins. Science 308, 512–517 [DOI] [PubMed] [Google Scholar]
- 28. McLennan G. P., Kiss A., Miyatake M., Belcheva M. M., Chambers K. T., Pozek J. J., Mohabbat Y., Moyer R. A., Bohn L. M., Coscia C. J. (2008) Kappa opioids promote the proliferation of astrocytes via Gβγ and β-arrestin 2-dependent MAPK-mediated pathways. J. Neurochem. 107, 1753–1765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bruchas M. R., Macey T. A., Lowe J. D., Chavkin C. (2006) Kappa opioid receptor activation of p38 MAPK is GRK3- and arrestin-dependent in neurons and astrocytes. J. Biol. Chem. 281, 18081–18089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bruchas M. R., Land B. B., Aita M., Xu M., Barot S. K., Li S., Chavkin C. (2007) Stress-induced p38 mitogen-activated protein kinase activation mediates kappa-opioid-dependent dysphoria. J. Neurosci. 27, 11614–11623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bohn L. M., Belcheva M. M., Coscia C. J. (2000) Mitogenic signaling via endogenous kappa-opioid receptors in C6 glioma cells: evidence for the involvement of protein kinase C and the mitogen-activated protein kinase signaling cascade. J. Neurochem. 74, 564–573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Belcheva M. M., Clark A. L., Haas P. D., Serna J. S., Hahn J. W., Kiss A., Coscia C. J. (2005) Mu and kappa opioid receptors activate ERK/MAPK via different protein kinase C isoforms and secondary messengers in astrocytes. J. Biol. Chem. 280, 27662–27669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bruchas M. R., Xu M., Chavkin C. (2008) Repeated swim stress induces kappa opioid-mediated activation of extracellular signal-regulated kinase 1/2. Neuroreport 19, 1417–1422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bruchas M. R., Chavkin C. (2010) Kinase cascades and ligand-directed signaling at the kappa opioid receptor. Psychopharmacology 210, 137–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hedrick M. P., Gosalia P., Frankowski K., Shi S., Prisinzano T. E., Schoenen F., Aube J., Su Y., Vasile S., Sergienko E., Gray W., Hariharan S., Ghosh P., Milan L., Heynen-Genel S., Chung T. D. Y., Dad S., Caron M., Bohn L. M., Barak L. S. (2010) Probe Reports from the NIH Molecular Libraries Program, Bethesda, MD [Google Scholar]
- 36. Frankowski K. J., Hedrick M. P., Gosalia P., Li K., Shi S., Whipple D., Ghosh P., Prisinzano T. E., Schoenen F. J., Su Y., Vasile S., Sergienko E., Gray W., Hariharan S., Milan L., Heynen-Genel S., Mangravita-Novo A., Vicchiarelli M., Smith L. H., Streicher J. M., Caron M. G., Barak L. S., Bohn L. M., Chung T. D., Aubé J. (2012) Discovery of small molecule kappa opioid receptor agonist and antagonist chemotypes through a HTS and hit refinement strategy. ACS Chem. Neurosci. 3, 221–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Frankowski K. J., Hirt E. E., Zeng Y., Neuenswander B., Fowler D., Schoenen F., Aubé J. (2007) Synthesis of N-alkyl-octahydroisoquinolin-1-one-8-carboxamide libraries using a tandem Diels-Alder/acylation sequence. J. Comb. Chem. 9, 1188–1192 [DOI] [PubMed] [Google Scholar]
- 38. Frankowski K. J., Ghosh P., Setola V., Tran T. B., Roth B. L., Aubé J. (2010) N-Alkyl-octahydroisoquinolin-1-one-8-carboxamides: a novel class of selective, nonbasic, nitrogen-containing kappa-opioid receptor ligands. ACS Med. Chem. Lett. 1, 189–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lahti R. A., Mickelson M. M., McCall J. M., Von Voigtlander P. F. (1985) [3H]U-69593 a highly selective ligand for the opioid kappa receptor. Eur. J. Pharmacol. 109, 281–284 [DOI] [PubMed] [Google Scholar]
- 40. Jones R. M., Portoghese P. S. (2000) 5′-Guanidinonaltrindole, a highly selective and potent kappa-opioid receptor antagonist. Eur. J. Pharmacol. 396, 49–52 [DOI] [PubMed] [Google Scholar]
- 41. Thomas J. B., Atkinson R. N., Rothman R. B., Fix S. E., Mascarella S. W., Vinson N. A., Xu H., Dersch C. M., Lu Y., Cantrell B. E., Zimmerman D. M., Carroll F. I. (2001) Identification of the first trans-(3R,4R)-dimethyl-4-(3-hydroxyphenyl)piperidine derivative to possess highly potent and selective opioid kappa receptor antagonist activity. J. Med. Chem. 44, 2687–2690 [DOI] [PubMed] [Google Scholar]
- 42. Schmid C. L., Streicher J. M., Groer C. E., Munro T. A., Zhou L., Bohn L. M. (2013) Functional selectivity of 6′-guanidinonaltrindole (6′-GNTI) at kappa opioid receptors in striatal neurons. J. Biol. Chem. 288, 22387–22398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ananthan S., Saini S. K., Dersch C. M., Xu H., McGlinchey N., Giuvelis D., Bilsky E. J., Rothman R. B. (2012) 14-Alkoxy- and 14-acyloxypyridomorphinans: mu agonist/delta antagonist opioid analgesics with diminished tolerance and dependence side effects. J. Med. Chem. 55, 8350–8363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Breivogel C. S., Walker J. M., Huang S. M., Roy M. B., Childers S. R. (2004) Cannabinoid signaling in rat cerebellar granule cells: G-protein activation, inhibition of glutamate release and endogenous cannabinoids. Neuropharmacology 47, 81–91 [DOI] [PubMed] [Google Scholar]
- 45. McGuinness R. (2007) Impedance-based cellular assay technologies: recent advances, future promise. Curr. Opin. Pharmacol. 7, 535–540 [DOI] [PubMed] [Google Scholar]
- 46. Robinson J., Smith A., Sturchler E., Tabrizifard S., Kamenecka T., McDonald P. (2013) Development of a high-throughput screening-compatible cell-based functional assay to identify small molecule probes of the galanin 3 receptor (GalR3). Assay Drug Dev. Technol. 10.1089/adt.2013.526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bohn L. M., Gainetdinov R. R., Lin F. T., Lefkowitz R. J., Caron M. G. (2000) Mu-opioid receptor desensitization by β-arrestin-2 determines morphine tolerance but not dependence. Nature 408, 720–723 [DOI] [PubMed] [Google Scholar]
- 48. Valko K., My Du C., Bevan C., Reynolds D. P., Abraham M. H. (2001) Rapid method for the estimation of octanol/water partition coefficient (log P(oct)) from gradient RP-HPLC retention and a hydrogen bond acidity term (ζα(2)(H)). Curr. Med. Chem. 8, 1137–1146 [DOI] [PubMed] [Google Scholar]
- 49. Bohn L. M., Lefkowitz R. J., Gainetdinov R. R., Peppel K., Caron M. G., Lin F. T. (1999) Enhanced morphine analgesia in mice lacking β-arrestin 2. Science 286, 2495–2498 [DOI] [PubMed] [Google Scholar]
- 50. Raehal K. M., Schmid C. L., Medvedev I. O., Gainetdinov R. R., Premont R. T., Bohn L. M. (2009) Morphine-induced physiological and behavioral responses in mice lacking G protein-coupled receptor kinase 6. Drug Alcohol Depend. 104, 187–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Black J. W., Leff P. (1983) Operational models of pharmacological agonism. Proc. R. Soc. Lond B Biol. Sci. 220, 141–162 [DOI] [PubMed] [Google Scholar]
- 52. Kenakin T., Christopoulos A. (2013) Signalling bias in new drug discovery: detection, quantification and therapeutic impact. Nat. Rev. Drug Discov. 12, 205–216 [DOI] [PubMed] [Google Scholar]
- 53. Evans B. A., Broxton N., Merlin J., Sato M., Hutchinson D. S., Christopoulos A., Summers R. J. (2011) Quantification of functional selectivity at the human α(1A)-adrenoceptor. Mol. Pharmacol. 79, 298–307 [DOI] [PubMed] [Google Scholar]
- 54. Kenakin T., Watson C., Muniz-Medina V., Christopoulos A., Novick S. (2012) A simple method for quantifying functional selectivity and agonist bias. ACS Chem. Neurosci. 3, 193–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bohn L. M., McDonald P. H. (2010) Seeking ligand bias: Assessing GPCR coupling to beta-arrestins for drug discovery. Drug Discov. Today Technol. 7, e37–e42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Barak L. S., Ferguson S. S., Zhang J., Caron M. G. (1997) A β-arrestin/green fluorescent protein biosensor for detecting G protein-coupled receptor activation. J. Biol. Chem. 272, 27497–27500 [DOI] [PubMed] [Google Scholar]
- 57. Barak L. S., Warabi K., Feng X., Caron M. G., Kwatra M. M. (1999) Real-time visualization of the cellular redistribution of G protein-coupled receptor kinase 2 and β-arrestin 2 during homologous desensitization of the substance P receptor. J. Biol. Chem. 274, 7565–7569 [DOI] [PubMed] [Google Scholar]
- 58. Peters M. F., Scott C. W. (2009) Evaluating cellular impedance assays for detection of GPCR pleiotropic signaling and functional selectivity. J. Biomol. Screen. 14, 246–255 [DOI] [PubMed] [Google Scholar]
- 59. McLaughlin J. P., Myers L. C., Zarek P. E., Caron M. G., Lefkowitz R. J., Czyzyk T. A., Pintar J. E., Chavkin C. (2004) Prolonged kappa opioid receptor phosphorylation mediated by G-protein receptor kinase underlies sustained analgesic tolerance. J. Biol. Chem. 279, 1810–1818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Mogil J. S., Wilson S. G., Chesler E. J., Rankin A. L., Nemmani K. V., Lariviere W. R., Groce M. K., Wallace M. R., Kaplan L., Staud R., Ness T. J., Glover T. L., Stankova M., Mayorov A., Hruby V. J., Grisel J. E., Fillingim R. B. (2003) The melanocortin-1 receptor gene mediates female-specific mechanisms of analgesia in mice and humans. Proc. Natl. Acad. Sci. U.S.A. 100, 4867–4872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Chavkin C. (2011) The therapeutic potential of kappa-opioids for treatment of pain and addiction. Neuropsychopharmacology 36, 369–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Rives M. L., Rossillo M., Liu-Chen L. Y., Javitch J. A. (2012) 6′-Guanidinonaltrindole (6′-GNTI) is a G protein-biased kappa-opioid receptor agonist that inhibits arrestin recruitment. J. Biol. Chem. 287, 27050–27054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wu H., Wacker D., Mileni M., Katritch V., Han G. W., Vardy E., Liu W., Thompson A. A., Huang X. P., Carroll F. I., Mascarella S. W., Westkaemper R. B., Mosier P. D., Roth B. L., Cherezov V., Stevens R. C. (2012) Structure of the human kappa-opioid receptor in complex with JDTic. Nature 485, 327–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ahn S., Shenoy S. K., Wei H., Lefkowitz R. J. (2004) Differential kinetic and spatial patterns of β-arrestin and G protein-mediated ERK activation by the angiotensin II receptor. J. Biol. Chem. 279, 35518–35525 [DOI] [PubMed] [Google Scholar]