

Background: The contribution of caveolae to physiological interaction between two major ion channels, large conductance Ca2+-activated K+ (BKCa) and Ca2+ (Cav1.2) channels, is unknown in vascular myocytes.
Results: The loss of caveola by caveolin-1 deficiency reduced BKCa-Cav1.2 coupling, Cav1.2 clustering, and membrane excitability regulation.
Conclusion: Caveolin-1 provides platform for BKCa-Cav1.2 molecular complex.
Significance: Caveolin-1-BKCa-Cav1.2 in caveola forms a novel Ca2+ signal domain for arterial tonus regulation.
Keywords: Calcium Signaling, Caveolin, Fluorescence Resonance Energy Transfer (FRET), Imaging, Single Molecule Biophysics, Vascular Smooth Muscle Cells, L-type Voltage-dependent Calcium Channel, Large Conductance Calcium-activated Potassium Channel, Total Internal Reflection Fluorescent Microscopy
Abstract
L-type voltage-dependent Ca2+ channels (LVDCC) and large conductance Ca2+-activated K+ channels (BKCa) are the major factors defining membrane excitability in vascular smooth muscle cells (VSMCs). The Ca2+ release from sarcoplasmic reticulum through ryanodine receptor significantly contributes to BKCa activation in VSMCs. In this study direct coupling between LVDCC (Cav1.2) and BKCa and the role of caveoline-1 on their interaction in mouse mesenteric artery SMCs were examined. The direct activation of BKCa by Ca2+ influx through coupling LVDCC was demonstrated by patch clamp recordings in freshly isolated VSMCs. Using total internal reflection fluorescence microscopy, it was found that a large part of yellow fluorescent protein-tagged BKCa co-localized with the cyan fluorescent protein-tagged Cav1.2 expressed in the plasma membrane of primary cultured mouse VSMCs and that the two molecules often exhibited FRET. It is notable that each BKα subunit of a tetramer in BKCa can directly interact with Cav1.2 and promotes Cav1.2 cluster in the molecular complex. Furthermore, caveolin-1 deficiency in knock-out (KO) mice significantly reduced not only the direct coupling between BKCa and Cav1.2 but also the functional coupling between BKCa and ryanodine receptor in VSMCs. The measurement of single cell shortening by 40 mm K+ revealed enhanced contractility in VSMCs from KO mice than wild type. Taken together, caveolin-1 facilitates the accumulation/clustering of BKCa-LVDCC complex in caveolae, which effectively regulates spatiotemporal Ca2+ dynamics including the negative feedback, to control the arterial excitability and contractility.
Introduction
The calcium ion (Ca2+) is a major second messenger responsible for variety of physiological responses, including neurotransmitter/hormone release, muscular contraction, cell proliferation, apoptosis, etc. It has been demonstrated that voltage-dependent Ca2+ channel (VDCC)2 and Ca2+ effectors often accumulate spatially and form Ca2+ microdomains (1, 2) to increase the local Ca2+ level and activate specific signal transduction events.
In smooth muscle cells (SMCs), it has been established that the L-type VDCC (LVDCC), ryanodine receptor (RyR), and large conductance Ca2+-activated K+ channel (BKCa, KCa1.1) constitute the Ca2+ microdomains. Two types of local Ca2+ events, Ca2+ hotspots (3) arising from depolarization-evoked Ca2+-induced Ca2+ release (CICR) (4) and spontaneous Ca2+ release (Ca2+ sparks) (5), play a crucial role in the regulation of SMC contraction and relaxation. These two Ca2+ events occur in the same distinct local areas in SMC so as to couple with BKCa activity and, respectively, contribute to the action potential repolarization phase and spontaneous transient outward current (STOC) (6). Thus, BKCa is thought to get activated mainly by Ca2+ release from the sarcoplasmic reticulum through RyRs, which loosely couple with LVDCC on the plasma membrane in excitation-contraction coupling (7). In this study the relationship between LVDCC and BKCa via RyR activation is termed as “loose coupling.”
Caveolae are composed of an omega-shaped structure on plasma membrane. Caveolin-1 is an essential factor for the properly formed caveolae structure and accumulates many types of signaling molecules (8, 9). The BKCa protein contains a binding motif to caveolin-1 and is often co-localized in caveolae (10, 11). Several reports indicate that caveolae are involved in Ca2+ hotspot and spark generation (12, 13). Caveolin-1 knock-out (KO) mice congenitally lack caveolae and exhibit many types of cardiovascular abnormalities (9, 14, 15).
The interaction of BKCa with several functional molecules in addition to caveolin-1 has been identified (16). In some neurons it has been reported that BKCa is directly activated by Ca2+ influx through the tightly coupled VDCC (17–20). On the other hand, in tsA-201 cells, which express LVDCC and BKCa, the Ca2+ entry through single LVDCCs rarely evokes BKCa opening (21). These authors suggested that the BKCa selectively interacts with N-type VDCC rather than L-type VDCC. To our knowledge, the direct molecular and/or functional interaction between BKCa and VDCC has not been reported in muscles, including skeletal, cardiac, and smooth muscles, even though BKCa co-localizes with Cav1.1 in the distrophin molecular complex (22) in skeletal muscle and can interact with caveolin-3 (23). In this study this direct coupling between LVDCC and BKCa on the same plasma membrane is known as “tight coupling.” The exact molecular mechanisms underlying direct physical interaction between BKCa and Cav1.2 have not yet been clarified. Subtypes of VDCCs acting as molecular partners for BKCa depend upon the local tissues. The LVDCC encoded by Cav1.2 (α1C) is highly expressed and serves as a predominant Ca2+ entry pathway in SMCs (24). Because tissue-specific splice variants with differential functions have been found in both BKCa (25) and Cav1.2 (26), it remains to be totally resolved whether BKCa physically couples with Cav1.2 and the coupling is functionally significant in SMCs.
In the previous study we first demonstrated that BKα forms tetramer and directly interacts with caveolin-1 in living aortic SMCs using total internal reflection fluorescence (TIRF) microscopy. In the TIRF system, an evanescent wave excites fluorescent molecules within a 200-nm depth of a glass bottom. This enables visualization of the fluorescent-labeled molecules localized on the plasma membrane in living cells (27). The present study was undertaken to examine the possible BKCa-Cav1.2 complex and the potential roles of caveolin-1 for this complex formation in vascular SMCs (VSMCs) by use of TIRF microscopy imaging methods. To demonstrate functional coupling between LVDCC and BKCa, whole cell patch clamp recording using two different Ca2+ chelators, EGTA and 1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid (BAPTA) was performed (18, 28). Results suggest that the BKCa-Cav1.2 complex is mostly co-localized in the caveolae of VSMCs. Furthermore, data from KO mice denote the importance of caveolae as a platform of the ion channel complex formation that effectively regulates properly excitability and contractility of VSMCs.
EXPERIMENTAL PROCEDURES
Animals, Cell Isolation, and Culture
Caveolin-1 knock-out (KO) mice on the C57BL/6 background were obtained from The Jackson Laboratory (stock number 004585) (Bar Harbor, ME). Wild-type (WT) control mice (C57BL/6) were purchased (Japan SLC, Hamamatsu, Japan). All experiments were approved by the Ethics Committee of Nagoya City University and were conducted in accordance with the Guide for the Care and Use of Laboratory Animals of the Japanese Pharmacological Society. The superior mesenteric arteries were removed from male mice (8–14 weeks) and cleansed of connective tissue in Ca2+-Mg2+-free Krebs solution containing 112 mm NaCl, 4.7 mm KCl, 1.2 mm KH2PO4, 25 mm NaHCO3, 14 mm glucose. From the third to the fifth branch were removed and incubated in Ca2+-Mg2+-free Krebs solution containing 0.4% collagenase (Amano Enzyme, Nagoya, Japan) and 0.1% papain (Sigma) for 45 min at 37 °C. After incubation, these tissues were washed in Ca2+-Mg2+-free Krebs solution and dispersed mechanically. For electrophysiological experiments, a few drops of the cell suspension were placed in a recording chamber. For culture, single cells were suspended in DMEM supplemented with 10% heat-inactivated FBS, 20 units/ml penicillin, and 20 μg/ml streptomycin (Sigma). After settling on glass-bottom dishes, cells were transiently transfected with fluorescent-labeled cDNA and cultured for 24–48 h.
Electrophysiological Recording
Electrophysiological studies were performed using a whole cell voltage clamp technique with a CEZ-2400 amplifier (Nihon Kohden, Tokyo, Japan), an analog-digital converter (DIGIDATA 1320A; Molecular Devices, Sunnyvale, CA), and a pCLAMP software (Version 8.2; Molecular Devices) in vascular cells as described previously (29). To examine whether the functional coupling between BKCa and Cav1.2 was tight or loose, we used two types of pipette solution containing 1) 140 mm KCl, 4 mm MgCl2, 10 mm HEPES, 2 mm Na2ATP, and 5 mm EGTA or 2) 140 mm KCl, 4 mm MgCl2, 10 mm HEPES, 2 mm Na2ATP, and 10 mm BAPTA (pH 7.2 with KOH). Here 10 mm BAPTA was used to inhibit both tight and loose coupling, and 5 mm EGTA was used to inhibit loose coupling. We defined the EGTA-resistant, BAPTA-sensitive current component as a BKCa current directly activated by Ca2+ influx through tightly coupled Cav1.2 channels. For simultaneous recording of STOC and Ca2+ sparks, the pipette solution contained 140 mm KCl, 4 mm MgCl2, 10 mm HEPES, 2 mm Na2ATP, and 0.1 mm fluo-4 (Invitrogen) (pH 7.2 with KOH). The extracellular solution (normal HEPES-buffered solution) had an ionic composition of 137 mm NaCl, 5.9 mm KCl, 2.2 mm CaCl2, 1.2 mm MgCl2, 14 mm glucose, and 10 mm HEPES. The pH was adjusted to 7.4 with NaOH. For VDCC current measurements, the pipette solution contained 120 mm CsCl, 20 mm tetraethylammonium chloride (Tokyo Chemical Industry, Tokyo, Japan), 1 mm MgCl2, 10 mm HEPES, 2 mm Na2ATP, and 20 mm BAPTA (pH 7.2 with CsOH). The bath solution contained 92 mm NaCl, 5.9 mm KCl, 30 mm BaCl2, 1.2 mm MgCl2, 14 mm glucose, and 10 mm HEPES. The pH was adjusted to 7.4 with NaOH. Whole cell BKCa currents and VDCC currents were activated from a holding potential of −60 mV by applying 150-ms voltage steps to voltages between −50 and +40 mV in increments of 10 mV. To detect tight-coupling between BKCa and Cav1.2, depolarizing stimuli from a holding potential of −60 to 0 or +10 mV for 150 ms were applied. STOCs were measured at a steady membrane potential of −40 mV.
Plasmid Constructs and Transfection
The full-length of cDNA encoding the rat KCNMA1 (BKα subunit, GenBankTM Accession numbers U93052 and AY330293.2) was subcloned into pEYFP-N1 (BKα-YFP), pEYFP-C1 (YFP-BKα), pECFP-N1 (BKα-CFP), and pAcGFP1-N1 (BKα-GFP) (Clontech Laboratories, Mountain View, CA). Human CACNA1C (NM_000719), SCN5A (NM_198056.2), and CAV1 (NM_001753) were also subcloned into pECFP-N1 (Cav1.2-CFP), pEYFP-N1 (Cav1.2-YFP), pAcGFP1-N1 (Cav1.2-GFP), and pECFP-C1 (CFP-caveolin1 and CFP-Nav1.5). All constructs were confirmed by DNA sequencing. Functions of each channel were examined by whole cell patch clamp recording. Primary-cultured myocytes and HEK293 cells were transiently transfected with fluorescent-labeled cDNA (each 2 μg for co-expression) using Lipofectamine 2000 (Invitrogen). Experiments were performed 24–48 h after transfection. Under these conditions, BKCa current density measured in aortic myocytes expressing BKα-YFP was comparable with that measured in myocytes expressing yellow fluorescent proteins (YFP) alone. This meant that in our system artifacts by overexpression of fluorescent protein-labeled ion channels were minimized due to their relatively low expression levels (30).
Single-molecule Imaging
Single-molecule imaging was performed using a TIRF imaging system with an objective lens (CFI Plan Apo TIRF 60×/1.45 or CFI Apo TIRF 100×/1.49, oil immersion; Nikon, Tokyo, Japan) as described previously (30). Data were collected with an EM-CCD camera and analyzed by AQUACOSMOS software (Hamamatsu Photonics, Hamamatsu, Japan). Cyan fluorescent proteins (CFP)-fused proteins were excited with a 405-nm blue diode, and YFP- and green fluorescent protein (GFP)-fused proteins were excited with a 488-nm argon laser (Coherent, Santa Clara, CA), respectively, and reflected off dichroic mirrors (427–441/490–510 nm; Omega Optical, Brattleboro, VT). CFP/YFP emissions were collected through dichroic mirrors and dual band-pass filters (454–479/523–567 nm; Omega Optical). A resolution of images was 71 per pixel (x-y) and less than 200 nm (z). TIRF images were collected at 465-ms exposure times and scanned every 1651 ms. Normal HEPES-buffered solution was used during recording. All experiments were carried out at room temperature (25 °C).
Fluorescence Resonance Energy Transfer (FRET) Analysis
The efficiency of FRET (EFRET) was evaluated based on the acceptor photobleaching method, in which the emission of the donor fluorophore is compared before and after the photobleaching of the acceptor (31). The fluorescence of YFP was photobleached using a mercury lamp (100 W, C-SHG1; Nikon) and a G-2A filter cube (Ex510–560/DM575/BA590; Nikon) for 2.5 min. FRET efficiency (EFRET) was calculated as the percentage increase in CFP emission after YFP photobleaching, as described previously (30).
Single-molecule GFP Bleaching
We counted subunits of BKCa and Cav1.2 in membranes of HEK293 cells and arterial myocytes by observing bleaching steps of GFP fused to BKα and VDCCα1C in a single particle, as described previously (30, 32, 33). HEK293 cells or primary-cultured myocytes were transiently transfected with 2 μg of cDNA encoding BKα-GFP or Cav1.2-GFP using Lipofectamine 2000. At 24 h after transfection, cells were fixed for 10 min with 4% paraformaldehyde in PBS, rinsed, and placed in PBS solution before the experiment. GFP was excited with a 488-nm laser and imaged with a B-2A filter cube (DM505/BA520; Nikon) and an objective (100×/1.49). Images (256 × 256 pixels; 1 pixel = 107 nm) were acquired at 10 Hz for 100–120 s. Fluorescence intensity in a region of interest (3 × 3 pixels) was calculated by subtracting the background in 16 pixels around the region of interest. The number of bleaching steps was determined by eye from the fluorescence signal trace. The steps within each single trace were similar in amplitude but varied between different traces. This is a consequence of the Gaussian profile of the laser and complex topology of the plasma membrane, which results in different local illumination intensities of the evanescent field. We used the following criteria for discarding signals: (a) a signal exhibits an elliptical shape, (b) a signal is very close to other signals (<4 pixels), (c) a signal that fluorescent intensity fluctuates too much to be accurately determined bleaching steps, (d) a signal does not show complete bleaching.
Fluorescent Labeling of Freshly Isolated Myocytes
BKCa and LVDCC in freshly isolated mesenteric arterial myocytes were labeled with polyclonal anti-BKα antibody (APC-107, Alomone Laboratories, Jerusalem, Israel) and DM-BODIPY (−)-dihydropyridine (Invitrogen). Cells were immunostained as described previously (34). Dissociated cells settled on glass-bottom dishes (Matsunami Glass Industry, Osaka, Japan) were labeled with 1:100 diluted antibodies for 12 h at 4 °C after fixation and permeabilization. Then cells were washed and incubated with Alexa 405-conjugated anti-rabbit IgG goat antiserum (Invitrogen) for 1 h at room temperature. After washing, cells were loaded with 100 nm DM-BODIPY(−)-dihydropyridine for 5 min and subsequently washed in PBS. Fluorescently labeled cells were observed using a TIRF imaging system and AQUACOSMOS software (mentioned above). Alexa405 and BODIPY were excited with the blue diode and the argon laser, respectively. The emissions of Alexa405 and BODIPY were collected using CFP-HQ (DM450/BA460–510; Nikon) and YFP-HQ (DM510/BA520–560; Nikon) filter cubes, respectively.
Measurement of the Fluo-4 Signal
For simultaneous measurements of Ca2+ sparks and STOCs in VSMCs, the Ca2+ images were obtained using a TIRF imaging system and AQUACOSMOS software (mentioned above) under the whole cell-clamp mode. A myocyte was loaded with 100 μm fluo-4 by diffusion from the recording pipette. An argon laser (488 nm) and a B-2A filter were used for excitation light and emission collection, respectively. Images were collected every 14 ms for 12 s. Resolution of images was 142 or 214 nm/pixel. Fluorescence intensity (F) in the region of interest was measured as an average from the pixels included in the area of a 2.14 × 2.14 μm square. The data are shown as ΔF/F0 (%), where F0 is the basal F value obtained as the average intensity of the regions of interest acquired during the measurements, and ΔF is the difference between F and F0.
To measure the averaged Ca2+ concentration from whole cell area, myocytes were loaded with 10 μm fluo-4 AM (Invitrogen), and fluorescent images were acquired using AQUACOSMOS software. The minimum fluorescence intensity (Fmin) and maximum fluorescence intensity (Fmax) were obtained by applying Ca2+-free HEPES-buffered solution (CaCl2 was removed from and 5 mm EGTA was added to normal HEPES-buffered solution) and 10 μm ionomycin in normal HEPES-buffered solution containing 2 mm Ca2+, respectively. To induce a contraction in a myocyte, 40 mm KCl HEPES-buffered solution (40 KCl), which contained 102.9 mm NaCl, 40 mm KCl, 2.2 mm CaCl2, 1.2 mm MgCl2, 14 mm glucose, and 10 mm HEPES (pH 7.4 with NaOH), was applied. Ca2+ elevation from base line was normalized to the maximum Ca2+ change (i.e. the difference between Fmin and Fmax). Images were collected every 2 s. A resolution of images was 267 nm/pixel.
Single Cell Shortening Measurements
Transmitted light images were acquired using the above-mentioned microscope and an objective lens (20×, dry, Nikon). 40 KCl and/or 1 μm paxilline (Pax) (Tocris Bioscience, Bristol, UK) were applied to myocytes. Cellular contraction was estimated as the decrease in cell area from the control condition. Images were collected every 1 s for 20 min. A resolution of images was 533 nm/pixel.
Statistics
Pooled data are shown as the mean ± S.E. Statistical significance between two groups was determined by Student's t test. Statistical significance among groups was evaluated by Tukey's test. Significant difference is expressed in the figures as p < 0.05 (*) or p < 0.01 (**).
RESULTS
BKCa Activation by Ca2+ Influx through Tightly Coupled LVDCC in VSMCs
At first, membrane currents elicited by depolarization were measured in mesenteric artery SMCs from WT or KO mice using low Ca2+ buffering pipette solution (50 μm EGTA). A large part of the outward current upon depolarization was blocked by the addition of 1 μm Pax (Fig. 1A) or 50 μm Cd2+, indicating that BKCa activation as one of the major outward currents is largely due to Ca2+ influx through VDCC. In highly excitable SMCs such as those of bladder and vas deferens, the Ca2+ source for BKCa activation upon depolarization may also include Ca2+ from the sarcoplasmic reticulum via Ca2+-induced Ca2+ release (CICR) (4, 35). This is also the case in mesenteric SMCs, and the outward currents upon depolarization showed irregular shapes and occurred concomitantly with local subcellular Ca2+ transients (Fig. 1B). To prevent the component of BKCa current activation due to CICR during depolarization, the following experiments were performed under much stronger Ca2+ buffering conditions (36).
FIGURE 1.
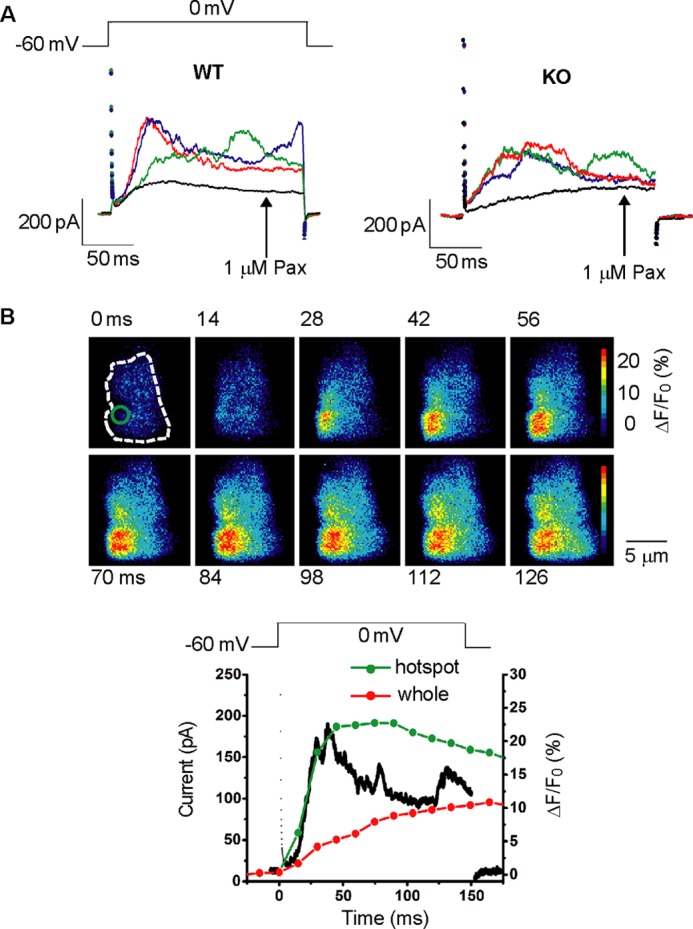
The Ca2+ images and membrane currents during depolarization in mesenteric artery SMCs under whole cell voltage clamp using the low Ca2+ buffer pipette solution. A, the membrane currents upon depolarization from −60 to 0 mV were recorded 3 times with the interval of 15 s from the same cell in the control conditions (left, myocyte from WT mouse; right, myocyte from KO mouse). The black lines indicate the currents after the addition of 1 μm paxiline (Pax). B, single mesenteric artery SMC from WT mouse was depolarized from a holding potential of −60 to 0 mV for 150 ms under the whole cell voltage clamp. The Ca2+ images were simultaneously obtained using the TIRF microscopy system. Images obtained every 14 ms are shown with recording times after the start of depolarization. The Ca2+-indicator fluo-4 was applied from the recording pipette. The dotted line indicates the SMC area attached to the bottom of the recording chamber. Changes in relative fluorescent intensity (ΔF/F0) in the hotspot area indicated by a circle in the image at zero time were plotted against time with the green line. Changes in the average ΔF/F0 from whole cell area are also plotted with the red line. The black line indicates the membrane current.
Accordingly, we used two different Ca2+ chelators, EGTA and BAPTA, to examine the possible tight-coupling between BKCa and Cav1.2 in VSMCs (18). These Ca2+ buffers have similar binding affinities for Ca2+, but the binding rate constant of BAPTA is ∼150 times faster than EGTA (18, 28). Thus, BAPTA is much more effective in preventing the diffusion of free Ca2+ away from the entrance site in LVDCC on the plasma membrane. Based on these distinct characteristics, tight-coupling is effectively interfered by 10 mm BAPTA but not by 5 mm EGTA, whereas loose coupling is equally sensitive to 10 mm BAPTA and 5 mm EGTA (18).
In the presence of 5 mm EGTA in the pipette solution, a fast-activated and inactivated outward current remained in SMCs of the WT but not those of KO (Fig. 2A). When the pipette solution contained 10 mm BAPTA, no such transient outward current was observed in either the WT or KO myocytes (Fig. 2A). Thus, the fast inactivation recorded by the pipette solution containing 5 mm EGTA was presumably due to Ca2+ removal by the slow Ca2+ chelation by EGTA. The slope of the rising phase of the outward current was compared and is shown in Fig. 2B (in WT: 0.80 ± 0.27 (pA/pF)/ms with EGTA (n = 5) and 0.08 ± 0.02 (pA/pF)/ms with BAPTA (n = 5); in KO: 0.16 ± 0.04 (pA/pF)/ms with EGTA (n = 6) and 0.10 ± 0.02 (pA/pF)/ms with BAPTA (n = 8); p < 0.01 versus WT with EGTA). The time to the peak of the EGTA-resistant currents was 35.8 ± 11.5 ms (n = 5) in WT and 55.2 ± 10.8 ms (n = 6) in KO (p > 0.05). The peak amplitude of the EGTA-resistant outward currents was 243.3 ± 38.4 pA (n = 5) in the WT and 111.7 ± 19.4 pA (n = 6) in the KO myocytes (p < 0.05). Thus, the EGTA-resistant transient outward current was presumably activated by Ca2+ influx via tight-coupled Cav1.2. This EGTA-resistant current was significantly larger and activated faster in the WT than KO myocytes.
FIGURE 2.

Functional coupling of BKCa and LVDCC in mesenteric arterial SMCs. A, myocytes were depolarized from −60 to 0 mV to activate LVDCC and BKCa. To estimate the direct contribution of Ca2+ influx through Cav1.2 to BKCa activation, pipette solutions containing 5 mm EGTA or 10 mm BAPTA were used to avoid the contamination of BKCa current component activated by Ca2+ released from sarcoplasmic reticulum. Currents in the presence of 1 μm Pax in WT (left) and KO (right) are superimposed with gray lines. B, the slope of the rising phase of the outward current at 0 mV in WT (EGTA n = 5, BAPTA n = 5) and KO (EGTA n = 6, BAPTA n = 8) is summarized. **, p < 0.01 versus WT with EGTA. C, I-V relationships of Pax-sensitive BKCa currents were measured using a pipette solution containing EGTA or BAPTA in WT (Ca) and KO (Cb). The BKCa current resistant to 5 mm EGTA but inhibited by 10 mm BAPTA in WT (EGTA n = 5, BAPTA n = 5) appears to be larger than that in KO (EGTA n = 6, BAPTA n = 8) at potentials positive to 0 mV. D, the Pax-sensitive current density of the EGTA- or BAPTA-resistant component in WT (EGTA n = 5, BAPTA n = 5) and KO (EGTA n = 6, BAPTA n = 8) is summarized. *, p < 0.05; **, p < 0.01 versus EGTA-resistant component in WT.
Fig. 2C illustrates the I-V relationships of BKCa currents detected as a component sensitive to 1 μm Pax in WT (Fig. 2Ca) and KO (Fig. 2Cb). In WT, the BKCa current density resistant to 5 mm EGTA in the pipette solution was higher than that of the 10 mm BAPTA-resistant component at potentials positive to 0 mV. The summarized data at +10 mV in WT show that the EGTA-resistant and BAPTA-resistant BKCa currents were 1.7 ± 0.3 pA/pF (n = 5) and 0.3 ± 0.3 pA/pF (n = 5), respectively (p < 0.01, Fig. 2D). Furthermore, the components in KO were 0.8 ± 0.2 pA/pF (n = 6) and 0.3 ± 0.2 pA/pF (n = 8), respectively (p > 0.05) (Fig. 2D). It is notable that the amplitude of the EGTA-resistant component in the WT was significantly larger than that in the KO myocytes (p < 0.05).
In the next series of experiments, the functional expression of LVDCC and BKCa in mesenteric arterial SMCs was compared between WT and KO. In mesenteric arterial SMCs, Cav1.2 was expressed so abundantly that substantial inward currents through LVDCC were detected at positive potentials to −20 mV and peaked at +10 mV under the conditions, where outward currents were blocked. The LVDCC currents were detected as the component blocked by the addition of 50 μm Cd2+. The peak LVDCC currents were recorded at +10 or +20 mV, and neither cell capacitance nor the peak LVDCC currents density at +10 mV were changed by the deletion of caveolin-1 (WT: 25.0 ± 1.0 pF, 2.9 ± 0.5 pA/pF, n = 5; KO: 26.8 ± 2.5 pF, 3.0 ± 0.6 pA/pF, n = 7; p > 0.05). The BKCa current density at +40 mV was not changed by caveolin-1 deficiency (WT: 27.1 ± 1.7 pF, 1.8 ± 0.3 pA/pF, n = 5; KO: 27.4 ± 1.8 pF, 1.7 ± 0.2 pA/pF, n = 8; p > 0.05). These results indicate that the functional expression levels of LVDCC and BKCa in KO are comparable with those in WT.
Similar experiments were performed in aortic myocytes. The current density of BKCa (WT: 1.4 ± 0.2 pA/pF, n = 7; KO: 1.2 ± 0.2 pA/pF, n = 8; at +40 mV; p > 0.05) and Cav1.2 (WT: 0.34 ± 0.06 pA/pF, n = 3; KO: 0.36 ± 0.10 pA/pF, n = 6; at +10 mV; p > 0.05) in the aortic myocytes from KO mice was similar to that from WT mice, respectively. EGTA-resistant current was detected in the WT but not KO in aortic myocytes (in WT, 1.3 ± 0.3 pA/pF with EGTA (n = 7), 0.3 ± 0.1 pA/pF with BAPTA (n = 7), p < 0.05; in KO, 0.3 ± 0.1 pA/pF with EGTA (n = 8), 0.4 ± 0.2 pA/pF with BAPTA (n = 8), p > 0.05). Taken together, it can be suggested that caveolin-1 deficiency diminished the tight-coupling between BKCa and Cav1.2 in freshly isolated arterial SMCs regardless of the vessel diameter.
BKCa and Cav1.2 Form Molecular Complex in Caveolae of VSMCs
To visualize BKCa-Cav1.2 molecular complex in VSMCs and investigate the contribution of caveolin-1 to the complex formation, we performed TIRF microscopy molecular imaging (30).
First, BKCa and LVDCC in freshly isolated mesenteric SMCs were stained with an anti-BKα antibody and DM-BODIPY (−)-dihydropyridine, respectively. In TIRF microscope images, fluorescent particles were detected by the Alexa405 and BODIPY dyes binding to these channels in the plasma membrane (Fig. 3A). A part of the particles from the BKCa were co-localized with those from LVDCC in both WT and KO myocytes, but the ratio of co-localized particles against total BKCa particles was significantly lower in KO than WT myocytes (WT: 45.8 ± 1.9%, n = 8; KO: 24.1 ± 8.0%, n = 7; p < 0.05; Fig. 3B).
FIGURE 3.

Co-localization of BKCa and Cav1.2 in caveolae in mesenteric arterial SMCs. A, fluorescent labeling of BKα and Cav1.2 in freshly isolated mesenteric SMCs from WT (upper) and KO (lower) mice were visualized using TIRF microscope. The area surrounded by a white line in the transmitted light images (left) are shown as TIRF images. BKα and Cav1.2 were labeled with anti-BKα antibody and DM-BODIPY (−)-dihydropyridine, respectively. Fluorescent signals from particles corresponding to BKα, Cav1.2, and co-localization were shown in green, red, and yellow (indicated by arrowheads), respectively. B, the ratio of BKα and Cav1.2 co-localization particle number to total BKα particle number in WT (n = 8) and KO (n = 7) myocytes (*, p < 0.05). C, typical TIRF images of mesenteric SMCs expressing YFP- or CFP-tagged channels from WT (left) and KO (right) mice were shown (see also supplemental Movies 1 and 2). The green, red, and yellow particles indicate BKα-YFP alone, Cav1.2-CFP alone, and BKα-YFP and Cav1.2-CFP co-localization, respectively. Trajectory of the channel for 60 s is also shown in right of each image. D, the ratio of BKα-YFP and Cav1.2-CFP co-localization to BKα-YFP alone in WT (n = 10) and KO (n = 9) myocytes (*, p < 0.05).
Next, BKα and VDCCα1C, which were labeled with YFP and CFP at the C termini (BKα-YFP and Cav1.2-CFP, respectively), were transiently co-expressed in SMCs isolated from mesenteric artery (Fig. 3, 4) and aorta, which were then primary-cultured. The transient expression of the labeled molecules was carefully performed by regulating the amount of cDNA to minimize the artifacts due to overexpression (see “Experimental Procedures”). Thus, the influence of transient expression of BKα-YFP on the functional expression of BKCa was examined in aortic myocytes. BKα-YFP or YFP alone were transiently expressed in aortic myocytes from WT mice, and the functional expression of BKCa was measured by the whole cell patch clamp recording. The pipette filling the solution contained 10 mm BAPTA to minimize the influence by changes in intracellular Ca2+ concentration by Ca2+ influx and release. The density of Pax-sensitive currents was 1.4 ± 0.3 pA/pF at +40 mV (n = 4) in the control myocytes (i.e. expressing YFP alone; mean cell capacitance of 15.3 ± 0.3 pF) and 1.1 ± 0.2 pA/pF (n = 6, p > 0.05 versus control) in BKα-YFP-expressing myocytes (mean cell capacitance of 17.3 ± 1.1 pF). The density of BKCa current in aortic myocytes was not significantly changed by the expression of BKα-YFP.
FIGURE 4.
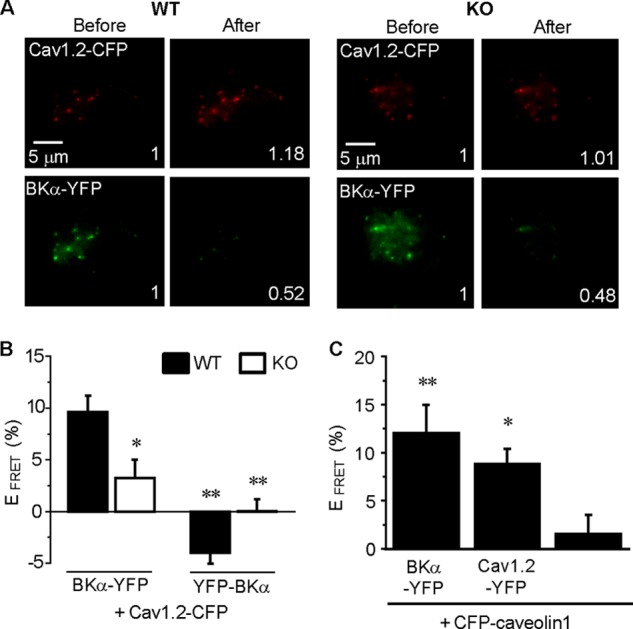
Caveolin-1 in caveolae interacts with BKCas and Cav1.2 channels and markedly facilitates the direct molecular coupling between BKCa and Cav1.2 in mesenteric arterial SMCs. A, FRET analysis was performed in mesenteric arterial myocytes co-expressing BKα-YFP and Cav1.2-CFP from WT (left panels) and KO (right panels) mice. Panels show Cav1.2-CFP and BKα-YFP emissions before and after selective photobleaching of YFP. Values in each image indicate the fluorescence intensity relative to pre-bleaching. B, FRET efficiency (EFRET) is summarized for the WT and KO. EFRET was calculated as the percent increase in CFP emission after YFP photobleaching (n = 4 ∼ 36; *, p < 0.05; **, p < 0.01 versus BKα-YFP + Cav1.2-CFP in WT). C, EFRET between BKα-YFP and CFP-caveolin-1 and between Cav1.2-YFP and CFP-caveolin-1 in mesenteric myocytes from WT mice was calculated. Cells expressing only CFP-caveolin-1 were used as a negative control (n = 9 ∼ 13; *, p < 0.05; **, p < 0.01 versus CFP-caveolin-1).
The detection of single molecule or the cluster of BKα-YFP alone, Cav1.2-CFP alone, and BKα-YFP/Cav1.2-CFP co-localization was performed as shown by the green, red, and yellow dots in the TIRF images, respectively (Fig. 3C and supplemental Movies 1 and 2). The ratio of the co-localization against BKα-YFP alone was compared between mesenteric arterial SMCs from KO and WT (Fig. 3D). The ratio of co-localization was significantly smaller in KO than WT (WT: 4.61 ± 0.91, n = 10; KO: 2.09 ± 0.7, n = 9; p < 0.05).
To detect protein-protein interactions between BKCa and Cav1.2, FRET analyses were performed using the acceptor photobleaching method (31) (Fig. 4). The validity of the system was confirmed with HEK293 cells co-expressing BKα-YFP and BKα-CFP as a positive control. We also co-expressed BKα-YFP and CFP-labeled Nav1.5 (CFP-Nav1.5) or expressed Cav1.2-CFP alone as a negative control (BKα-YFP+BKα-CFP: 18.2 ± 1.8%, n = 5; BKα-YFP+CFP-Nav1.5: 2.5 ± 2.7%, n = 8; Cav1.2-CFP: −0.7 ± 2.2%, n = 14).
In WT myocytes expressing BKα-YFP and Cav1.2-CFP, the fluorescence intensity of Cav1.2-CFP was increased after the bleaching of BKα-YFP. The increase in Cav1.2-CFP fluorescence intensity was attenuated in KO myocytes (Fig. 4A). The EFRET values shown in Fig. 4A were 15.3% in the WT and 0% in the KO. As summarized in Fig. 4B, EFRET was significantly smaller in the KO than WT myocytes (WT: 9.6 ± 1.5%, n = 34; KO: 3.3 ± 1.8%, n = 26; p < 0.05). Similar data were obtained in aortic myocytes (WT: 8.3 ± 1.2%, n = 23; KO: 0.0 ± 2.0%, n = 28; p < 0.01). In addition, BKα, which was labeled with YFP at the extracellular N terminus (YFP-BKα), was also used for FRET analyses. No FRET interaction was detected between YFP-BKα and Cav1.2-CFP (WT: −4.0 ± 1.0%, n = 10; KO: 0.1 ± 1.1%, n = 4). These data indicate that BKCa directly interacts with Cav1.2 in VSMCs, and caveolin-1 facilitates the formation of the complex.
The direct couplings between caveolin-1 and BKCa, and caveolin-1 and LVDCC in mesenteric SMCs were also examined by FRET analyses (Fig. 4C). Myocytes expressing only CFP-caveolin-1 were used as a negative control (1.6 ± 2.0%, n = 11). FRET interaction was observed in cells expressing CFP-caveolin-1+BKα-YFP (12.0 ± 3.0%, n = 9, p < 0.01) and CFP-caveolin-1+Cav1.2-YFP (8.8 ± 1.6%, n = 13, p < 0.05).
Molecular Assembly of Cav1.2 and BKCa Complex and the Contribution of Caveolin-1
Furthermore, we applied single-molecule fluorescence bleaching analyses (30, 32, 37) to clarify the number of GFP-tagged BKα and VDCCα1C in the single fluorescent particles. Again, to minimize the artifacts of overexpression, the expression of GFP labeled BKCa and/or Cav1.2 was kept relatively low.
At first the usefulness and accuracy of the system for single-molecule fluorescence bleaching analyses were verified using HEK293 cells expressing Cav1.2-GFP. Single Cav1.2 as an α1C-subunit can form a functional channel on its own without other Ca2+ channel subunits (37) (Fig. 5A). The majority of fluorescent particles displayed a single bleaching step (1 step, 79.3 ± 5.3%; 2 steps, 18.4 ± 5.2%; 3 steps, 2.4 ± 2.4%; 4 steps, 0%, analyzed from 46 spots from 6 cells; Fig. 5B). A small part of spots bleached in two or three steps, and these probably arose from the rare co-localization of channels within a diffraction-limited area (37).
FIGURE 5.

Counting of BKα-GFP or Cav1.2-GFP subunits within single fluorescent particles. BKα-GFP or Cav1.2-GFP was transiently expressed in HEK293 cells and mesenteric myocytes. Based on the single-molecule photobleaching method, the bleaching steps of the GFP signals in a TIRF region were counted to determine the number of BKα-GFP subunits or Cav1.2-GFP within a single fluorescent particle. A, change in fluorescence intensity of Cav1.2-GFP expressed in HEK293 cells after the photobleaching stimulation. The arrows and solid lines indicate the bleaching step(s). The dotted line indicates complete bleaching (basal) level. B, the number of bleaching steps in HEK293 cells expressing Cav1.2-GFP (n = 6) is shown as a histogram. C and D, time course of fluorescence intensity of BKα-GFP and Cav1.2-GFP expressed in mesenteric myocytes after the photobleaching stimulation. E, the observed number of bleaching steps in mesenteric myocytes expressing BKα-GFP (n = 5 and n = 4 in WT and KO myocytes, respectively; top) and Cav1.2-GFP (n = 7 and n = 9 in WT and KO myocytes, respectively; bottom) is summarized as histograms (*, p < 0.05 versus WT).
In the WT and KO myocytes, the fluorescent particles of BKα-GFP exhibited mainly 1–4 bleaching steps (1 step, 35.7 ± 8.8 and 41.6 ± 9.2%; 2 steps, 23.8 ± 5.1 and 26.2 ± 8.6%; 3 steps, 25.3 ± 9.4 and 19.3 ± 6.4%; 4 steps, 15.2 ± 4.4 and 12.9 ± 8.4%, analyzed from 39 and 22 spots in 5 and 4 myocytes from WT and KO mice, respectively) (Fig. 5, C–E). These results confirmed our previous observation that only a part of BKCas contains BKα-GFP as components of the tetramer with 1–3 native BKα molecules in mesenteric arterial myocytes under the experimental conditions in this study (30).
A single Cav1.2 molecule is thought to form the LVDCC channel pore as the α1C subunit as shown in Fig. 5A. Thus, it was a rather unexpected result that many of the fluorescent particles of Cav1.2-GFP exhibited more than 1 bleaching step (1 step, 32.0 ± 7.5 and 50.4 ± 6.9%; 2 steps, 27.9 ± 8.5 and 30.9 ± 4.7%; 3 steps, 20.5 ± 5.3 and 12.4 ± 3.3%; 4 steps, 19.4 ± 3.8 and 6.3 ± 2.9%, p < 0.05), analyzed from 57 and 77 spots in 7 and 9 myocytes from WT and KO mice, respectively) (Fig. 5, C–E). Notably, the proportion of the particles exhibiting four steps of Cav1.2-GFP bleaching in KO myocytes was significantly smaller than that in WT myocytes. Overall the number of bleaching steps in KO myocytes tended to be smaller than WT myocytes. These data may suggest that Cav1.2 preferentially formed homo-clusters. However, based on the finding that BKCa forms a complex with Cav1.2 in caveolae, it is more likely that each BKα molecule of the tetramer formation in a BKCa interacts with a single Cav1.2 to form hetero-clusters as a consequence.
Smaller STOC Frequency in KO Than in WT Myocytes
To examine the contribution of caveolae to the loose coupling between BKCa and RyR, we performed simultaneous recordings of Ca2+ sparks and STOCs at −40 mV in mesenteric arterial SMCs freshly isolated from WT and KO mice. Typical TIRF images are shown in supplemental Movies 3 and 4. Changes in the membrane currents and cytosolic Ca2+ levels at the sites indicated by circles in the images were shown in Fig. 6A. The STOC frequency in the KO myocytes (1.1 ± 0.2 Hz, n = 10) was significantly smaller than the WT (2.4 ± 0.4 Hz, n = 5; p < 0.01) (Fig. 6B). The averaged STOC amplitude in the WT myocytes (25.1 ± 1.7 pA, n = 5) was similar to that in KO (29.7 ± 3.5 pA, n = 10; p > 0.05) (Fig. 6B). On the other hand, the Ca2+ spark frequency (1.22 ± 0.06 Hz, n = 5) and amplitude (1.3 ± 0.2%, n = 5) in WT myocytes were comparable with those in KO (0.99 ± 0.10 Hz, n = 10; 1.8 ± 0.3%, n = 10, respectively; p > 0.05, Fig. 6C). These results suggest that the lack of caveolae attenuates the coupling between BKCa and RyR and thereby reduces the efficacy to translate Ca2+ spark signals to electrical STOC signals.
FIGURE 6.
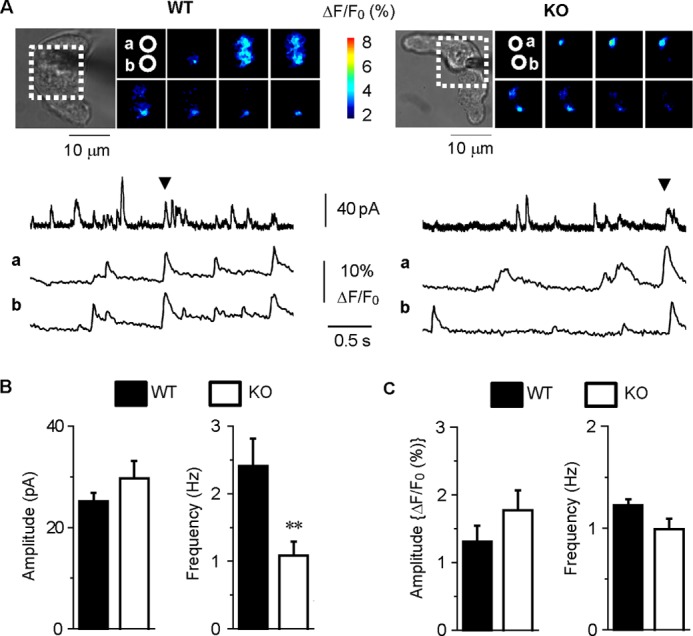
Simultaneous recordings of STOCs and Ca2+ sparks in WT and KO myocytes. A, fluorescent images surrounded by a dotted line in the transmitted light images (upper) were collected at the time points indicated by arrowheads in the membrane current recordings (middle). The fluorescent images were also shown in supplemental Movies 3 and 4. Changes in cytosolic Ca2+ levels at the two sites (a and b) circled in the fluorescent images are correspondingly shown (lower). B, the mean STOC amplitude and frequency in WT (n = 5) and KO (n = 10) cells are summarized (**, p < 0.01, versus WT). C, the mean Ca2+ spark amplitude and frequency in WT (n = 5) and KO (n = 10) cells are summarized (p > 0.05).
Attenuated Negative Feedback of Ca2+ Regulation via BKCa Activation in KO Myocytes
Next, we examined the contribution of caveorin-1 to the regulation of SMC contraction via BKCa activity. When 40 KCl was applied to freshly isolated mesenteric arterial SMCs, the contraction in the KO myocytes (−10.3 ± 1.6%, n = 19) was larger in WT myocytes (−5.0 ± 0.9%, n = 21; p < 0.01) (Fig. 7, A and B). Ca2+ imaging showed that this difference was attributable to the enhanced Ca2+ rise in KO myocytes (the increase in fluorescent intensity in WT, 6.2 ± 1.4% n = 8; KO, 12.8 ± 2.2%, n = 9; p < 0.05) (Fig. 7, C and D). The application of 1 μm Pax induced a smaller contraction in the KO myocytes (−2.6 ± 0.3%, n = 20) than in the WT (−4.6 ± 0.7%, n = 12; p < 0.01) (Fig. 7E). An additional application of 40 KCl generated a comparable contraction in the WT myocytes (−11.5 ± 2.3%, n = 12) and KO (−11.1 ± 2.0%, n = 20; p > 0.05) (Fig. 7F). These results suggest that depolarization-induced cell contraction in KO myocytes is larger than that in WT myocytes, because the negative feedback mechanism by BKCa activation to membrane excitation was attenuated in KO myocytes.
FIGURE 7.
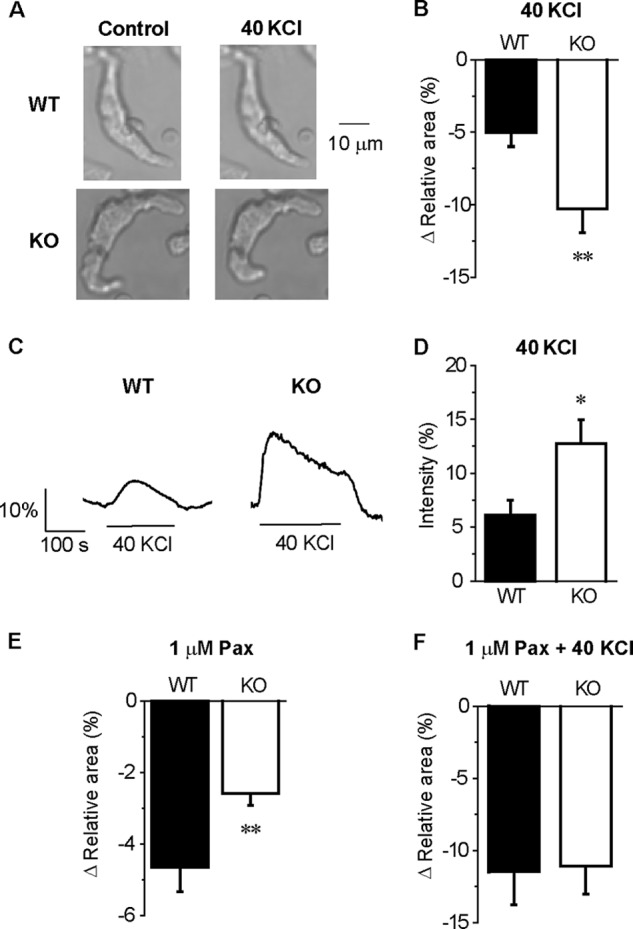
BKCa activity is lower in KO than in WT myocytes. A, images of single cell contraction caused by 40 KCl. B, summarized data of the 40 KCl-induced decrease in cell area in the images relative to that before 40 KCl application in WT (n = 21) and KO (n = 19) myocytes, respectively. C, change in the intracellular Ca2+ level caused by 40 KCl. D, summarized data of 40 KCl induced Ca2+ elevation in the WT (n = 8) and KO (n = 9) myocytes. E and F, summarized data of a 1 μm Pax-induced (E) and 1 μm Pax + 40 KCl-induced (F) decrease in cell area in WT (n = 12) and KO (n = 20) myocytes. *, p < 0.05; **, p < 0.01 versus WT.
DISCUSSION
This report is, to our knowledge, the first to demonstrate that caveolin-1 facilitates molecular interaction between Cav1.2 and BKCa and their accumulation as a molecular complex in caveola in living VSMCs. It is suggested that each of tetrameric BKα subunits, which form a functional BKCa, can directly interact with Cav1.2 molecule to promote Cav1.2 clustering in the molecular complex in caveolae.
Several technologies, including analyses using patch clamp, biochemical procedures, mass spectrometry, and morphological analyses, have been applied to determine the molecular components and their molecular and/or functional couplings in Ca2+ microdomains (1, 2, 20). To date, molecular interaction between two molecules among BKCa, Cav1.2, and caveolin-1 has been reported in several types of cells including expression systems. We also showed that caveolin-1 directly interacts with BKCa or Cav1.2 with FRET analysis. The caveolin-1 N terminus contains a caveolin-1 scaffolding domain (9) that interacts with various types of signaling molecules containing the caveolin-1 binding motif (10, 38). Caveolin-1 binding motifs are characterized as ΦXΦXXXXΦ and ΦXXXXΦXXΦ, where Φ is an aromatic amino acid, and X is any amino acid (38). It has been reported that BKCa has a caveolin-1 binding motif (1042YNMLCFGIY1050) within its C terminus and presumably accumulates in caveolae (10, 11). Caveolin-3, which is mainly expressed in skeletal muscle, also directly interacts with BKCa at the same binding site and presumably contributes to form the dystrophin microdomain (23).
VDCCα1C also contains the motif (1479FDYLTRDW1486) at the C terminus, and its association with caveolin-1 and caveolin-3 has been reported in several different tissues (39, 40). Although the binding sites remain to be determined, the functional tight-coupling between BKCa and Cav1.2 and their co-immunoprecipitation have been demonstrated in neurons, oocytes, and CHO cells (18). In this study the direct interaction between BKCa and Cav1.2 was first visualized by FRET analyses in living cells. In addition, their functional coupling was demonstrated in mesenteric and aortic SMCs using two different Ca2+ chelators, EGTA and BAPTA, in combination with the whole cell patch clamp recordings as has been shown in neurons (1, 18, 20). Because caveolin-1 deficiency resulted in caveolae disruption, a significant decrease in EGTA-resistant BKCa current density, co-localization ratio, and FRET efficiency between BKα-YFP and Cav1.2-CFP, it is clear that caveolin-1 facilitates the BKCa-Cav1.2 interaction in VSMCs. It was, however, also suggested that caveolin-1 is not an essential factor for BKCa-Cav1.2 tight coupling, because a small but nonetheless significant amount of coupling was observed in caveolin-1 KO. Caveolin-1 may indirectly promote BKCa-Cav1.2 complex formation by offering a platform for both BKCa and Cav1.2 to be accumulated in caveolae and prove higher probability to BKCa-Cav1.2 complex formation in caveolae than in other cell membrane areas.
Single-molecule bleaching analyses revealed that single fluorescent spots derived from BKα-GFP exhibited 1–4 bleaching steps in VSMCs. These findings confirm the previous observation that a tetrameric BKα assembly included 1–4 fluorescent protein-labeled BKαs in VSMCs (30). On the other hand, a portion of the fluorescent particles of Cav1.2-GFP also exhibited more than one bleaching step in VSMCs, although only one VDCCα1C subunit can act as a functional Ca2+ channel in HEK293 cells. The possibility that the BKCa-Cav1.2 complex may consist of tetrameric BKα assembly and four Cav1.2 channels has been speculated from the consequence of two-dimensional gel electrophoresis (18) and in a review (20). The result reported here that single fluorescent spots from Cav1.2-GFP exhibited multistep bleaching in mesenteric arterial myocytes provides the first direct evidence supporting this hypothesis (Fig. 8A). It has been also demonstrated that Cav1.2 can interact with nearby Cav1.2 channels via their C termini, and this interaction enables coupled gating of these channels in arterial myocytes (41). In the present study, however, the number of Cav1.2-GFP within single fluorescent particles analyzed by the step-bleaching in mesenteric arterial myocytes of KO appeared to be apparently smaller than WT. There are two limitations in this approach (37). (i) When a single fluorescent spot consists of a substantial number of multimers (>10), the detection of discrete steps becomes more difficult. In this case, the number of multimers can be estimated from the size of a single bleaching step and the total fluorescence, but the accuracy of this estimation may be limited. (ii) The other is that the GFP fluorescence emission occurs at the probability of ∼80% in distinct fusion proteins when being excited continuously. This may result in the underestimation of the number.
FIGURE 8.
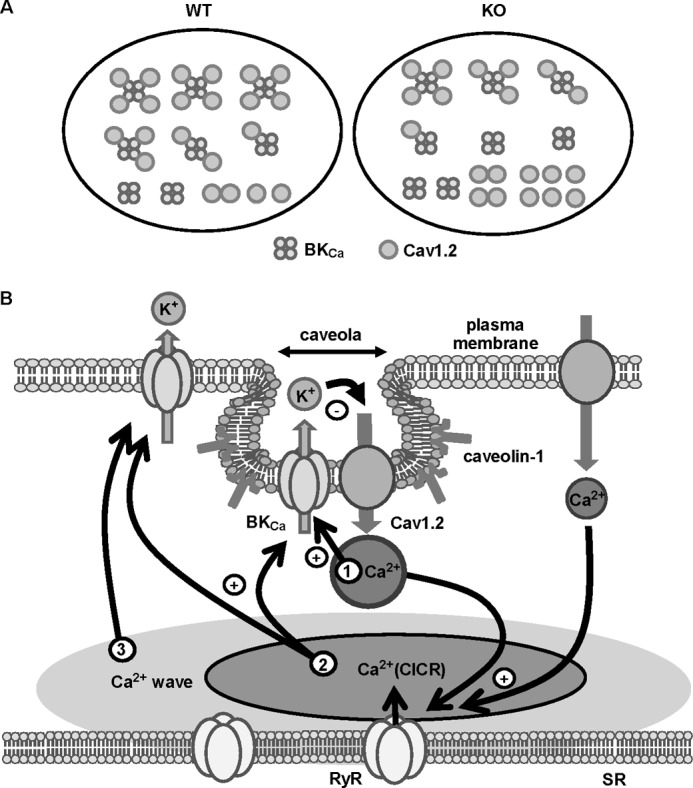
BKCa-Cav1.2 complex in VSMCs. A, BKCa-Cav1.2 complex can consist of one BKCa (a tetrameric BKα assembly) and four Cav1.2 channels. In caveolin-1-deficient (KO) myocytes, the interaction between BKCa and Cav1.2 is attenuated by following reasons. The BKCa-Cav1.2 complex, which is facilitated by the accumulation of these channels in caveolae by the interaction with caveolin-1, is reduced in KO myocytes. The number of Cav1.2 within the BKCa-Cav1.2 complex is also reduced in KO myocytes. B, BKCa can be activated by three steps of Ca2+ increase. The first is Ca2+ influx from tightly coupled Cav1.2. The second is Ca2+ release from loosely coupled RyR, i.e. CICR. The third step is global Ca2+ level elevation evoked by the Ca2+ wave. We propose that large portions of the BKCa-Cav1.2 complexes are localized in caveolae and that the complex of 1 BKCa with 4 Cav1.2 cluster may contribute to initiate Ca2+ local events in caveolae as the key membrane structure in Ca2+ microdomain, which, however, includes the effective negative feedback mechanism for cytosolic Ca2+ regulation.
In addition, SMCs from animal tissues, which expressed endogenous channels, were used in this study. In the preparation, fluorescent spots contained native subunits without GFP labeling that led to the underestimation of the subunit number within a single spot. This meant that the distribution histogram shown in Fig. 5E may be shifted to the left by the included native subunit (i.e. fluorescent spots contain more number of subunits than that estimated by bleaching steps counting). Taken together, it can be strongly suggested that caveolin-1 localizes/accumulates Cav1.2 and BKCa in caveolae and promotes effective coupling between these channels and their clustering, particularly that of Cav1.2 (Fig. 8B).
The tight-coupling between Cav1.2 and BKCa and their accumulation in the caveolae of VSMCs may be much more significant in terms of physiological impact in mesenteric arterial SMCs than aortic SMCs. The LVDCC density in mesenteric arterial myocytes is sufficiently high as to elicit action potentials due to sympathetic nerve stimulation (42). The fast outward current due to BKCa activation via BKCa-Cav1.2 tight coupling (EGTA-resistant BKCa current) upon depolarization may contribute to early repolarization (20). It is notable that this fast outward current was not observed in KO myocytes, where BKCa-Cav1.2 FRET efficiency was significantly reduced. The ensuing robust outward current due to CICR upon depolarization may contribute to the late repolarization and/or after hyperpolarization of an action potential (4), as has been also suggested in neurons (19). Alternatively, it is possible that the accumulation of efflux K+ in caveolae may induce a further activation rather than a decrease in Cav1.2 activity. This assumption seems, however, unlikely, as the membrane excitability and contraction in the mesenteric myocytes from KO mice were apparently increased more than in the WT. In fact, an application of 40 KCl to KO myocytes evoked a larger intracellular Ca2+ elevation and contraction than in the WT. Moreover, in the presence of Pax, the muscular contraction in the WT was comparable with that in the KO myocytes. It is thus strongly suggested that the enhancement of BKCa-Cav1.2 tight-coupling by caveolin-1 significantly contributes to the negative feedback regulation of membrane excitability and contraction in mesenteric arterial SMCs. RhoA and Rho kinase cause Ca2+-induced Ca2+ sensitization in response to depolarization (43). Because caveolin-1 attenuates RhoA activity (44, 45), caveolin-1 deficiency may activate RhoA and induce synergistic enhancement of tonic contraction. Thus, the augmentation of two pathways, (i) CICR and (ii) Ca2+-induced Ca2+-sensitization in KO cells, may enhance smooth muscle cell contraction.
So far the information about the relation between caveolae and STOC generation is controversial; both an increase (36) and decrease (14) in STOC frequency have been demonstrated upon caveolae disruption. In the present study data from a simultaneous recording of Ca2+ sparks and STOCs revealed that the characteristics of the Ca2+ sparks were unchanged, but the frequency of STOCs was decreased. In single myocytes, Pax induced a smaller contraction in KO than WT myocytes. This finding strongly suggests that cell excitability and contractility in the resting state are higher in KO than WT myocytes. It can be speculated that caveola deficiency makes BKCa more inaccessible to the RyR (13), and this leads to the lower frequency of detectable STOCs. Thus, caveolae and caveolin-1 play obligatory roles in both tight and loose coupling in the Ca2+ microdomain of SMCs. In addition to Ca2+ sparks, Ca2+ sparklets have been known as a quantal factor in Ca2+ elevation by single LVDCC opening at rest (46, 47). In cells where BKCa-Cav1.2 tight coupling is present, STOCs due to Ca2+ sparklets may concomitantly occur.
Guia et al. (48) have revealed that in coronary myocytes, Ca2+ influx through LVDCC directly activated BKCa at −30 mV in the presence of BayK8644, ryanodine, and cyclopiazonic acid using the cell-attached patch clamp technique. This means that the Ca2+ sparklet may activate the tight-coupled BKCa around the resting membrane potential and induce STOCs. However, these experiments were performed under limited conditions, where LVDCCs were well activated by BayK8644. Therefore, it is unclear whether Ca2+ sparklets evoke or potentiate STOCs under physiological conditions. The major contributor to the STOCs generation in the resting state may be Ca2+ sparks rather than Ca2+ sparklets, because the amplitude of the BKCa current due to a sparklet may be too small to explain the STOC amplitude in arterial myocytes.
In urinary bladder SMCs (49), Ca2+ sparklets often trigger CICR and subsequent BKCa current activation mainly via loose coupling, but a smaller component that is due to direct BKCa activation is also included.3 In the present study, however, the simultaneous measurement of Ca2+ sparklet and STOCs was not systematically performed, and this remains to be determined.
Caveolin-1 acts as an endothelial nitric-oxide synthase inhibitor; accordingly, caveolin-1 ablation induces an elevation of the NO level (14). On the other hand, caveolin-1 deficiency causes remodeling of resistant vessels (50) and increases the responsiveness to adrenergic stimulation by an elevation in protein kinase C activity (44). Furthermore the production of endothelium-derived hyperpolarization factor is impaired in the KO artery because the activities of connexin 43 and transient receptor potential vanilloid 4 (TRPV4) in the endothelium are attenuated (51). It has been also reported that the number of caveolae is reduced in endothelial cells of hypertensive rat aortae (52) and that the ratio of caveolin-1 dimer is decreased in mesenteric arterial myocytes from spontaneously hypertensive rats (53), i.e. hypertension disassembles caveolae. Our data from single isolated myocytes suggest that the lack of caveolae reduced the BKCa-Cav1.2 complex and BKCa activation by Ca2+ influx via Cav1.2. The negative feedback mechanism for Cav1.2 activity regulation may, therefore, be reduced in KO myocytes. It is apparent that the effects of caveolin-1 deficiency on systemic arterial pressure are much more complex.
In conclusion, BKCa activation upon depolarization takes place by three distinct steps of Ca2+ increase (Fig. 8B). The first step is Ca2+ influx through tightly coupled Cav1.2. The second step is local CICR via loosely coupled RyR in discrete sarcoplasmic reticula. The third step is global Ca2+ level elevation due to CICR conduction in whole myocytes. It was demonstrated that large portions of the BKCa-Cav1.2 complexes are accumulated in caveolae by the interaction with caveolin-1 serving as a microdomain that plays an obligatory role in the control of Ca2+ signaling, excitability, and contractility in VSMCs.
This work was supported by Grant-in-aid for Scientific Research on Priority Areas 20056027 and 23136512 (to Y. I.) from the Ministry of Education, Culture, Sports, Science, and Technology, Grant-in-aid for Scientific Research (B) 23390020 (to Y. I.), and Grant-in-aid for Japan Society for the Promotion of Science Fellows 226641 (to Y. S.) from the Japan Society for the Promotion of Science.

This article contains supplemental Movies 1–4.
H. Yamamura and Y. Suzuki, unpublished data.
- VDCC
- voltage-dependent Ca2+ channel
- VSMC
- vascular smooth muscle cell
- BAPTA
- 1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid
- BKCa
- large conductance Ca2+-activated K+ channel
- CFP
- cyan fluorescent protein
- YFP
- yellow fluorescent protein
- CICR
- Ca2+-induced Ca2+ release
- LVDCC
- L-type voltage-dependent Ca2+ channel
- Pax
- paxilline
- RyR
- ryanodine receptor
- SMC
- smooth muscle cell
- STOC
- spontaneous transient outward current
- TIRF
- total internal reflection fluorescence
- 40 KCl
- 40 mm KCl HEPES-buffered solution
- pF
- picofarads.
REFERENCES
- 1. Augustine G. J., Santamaria F., Tanaka K. (2003) Local calcium signaling in neurons. Neuron 40, 331–346 [DOI] [PubMed] [Google Scholar]
- 2. Parekh A. B. (2008) Ca2+ microdomains near plasma membrane Ca2+ channels. Impact on cell function. J. Physiol. 586, 3043–3054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Imaizumi Y., Torii Y., Ohi Y., Nagano N., Atsuki K., Yamamura H., Muraki K., Watanabe M., Bolton T. B. (1998) Ca2+ images and K+ current during depolarization in smooth muscle cells of the guinea pig vas deferens and urinary bladder. J. Physiol. 510, 705–719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Morimura K., Ohi Y., Yamamura H., Ohya S., Muraki K., Imaizumi Y. (2006) Two-step Ca2+ intracellular release underlies excitation-contraction coupling in mouse urinary bladder myocytes. Am. J. Physiol. Cell Physiol. 290, C388–C403 [DOI] [PubMed] [Google Scholar]
- 5. Nelson M. T., Cheng H., Rubart M., Santana L. F., Bonev A. D., Knot H. J., Lederer W. J. (1995) Relaxation of arterial smooth muscle by calcium sparks. Science 270, 633–637 [DOI] [PubMed] [Google Scholar]
- 6. Ohi Y., Yamamura H., Nagano N., Ohya S., Muraki K., Watanabe M., Imaizumi Y. (2001) Local Ca2+ transients and distribution of BK channels and ryanodine receptors in smooth muscle cells of guinea-pig vas deferens and urinary bladder. J. Physiol. 534, 313–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Collier M. L., Ji G., Wang Y., Kotlikoff M. I. (2000) Calcium-induced calcium release in smooth muscle. Loose coupling between the action potential and calcium release. J. Gen. Physiol. 115, 653–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pani B., Singh B. B. (2009) Lipid rafts/caveolae as microdomains of calcium signaling. Cell Calcium 45, 625–633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cohen A. W., Hnasko R., Schubert W., Lisanti M. P. (2004) Role of caveolae and caveolins in health and disease. Physiol. Rev. 84, 1341–1379 [DOI] [PubMed] [Google Scholar]
- 10. Alioua A., Lu R., Kumar Y., Eghbali M., Kundu P., Toro L., Stefani E. (2008) Slo1 caveolin-binding motif, a mechanism of caveolin-1-Slo1 interaction regulating Slo1 surface expression. J. Biol. Chem. 283, 4808–4817 [DOI] [PubMed] [Google Scholar]
- 11. Lu T., Zhang D. M., Wang X. L., He T., Wang R. X., Chai Q., Katusic Z. S., Lee H. C. (2010) Regulation of coronary arterial BK channels by caveolae-mediated angiotensin II signaling in diabetes mellitus. Circ. Res. 106, 1164–1173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hotta S., Yamamura H., Ohya S., Imaizumi Y. (2007) Methyl-β-cyclodextrin prevents Ca2+-induced Ca2+ release in smooth muscle cells of mouse urinary bladder. J. Pharmacol. Sci. 103, 121–126 [DOI] [PubMed] [Google Scholar]
- 13. Löhn M., Fürstenau M., Sagach V., Elger M., Schulze W., Luft F. C., Haller H., Gollasch M. (2000) Ignition of calcium sparks in arterial and cardiac muscle through caveolae. Circ. Res. 87, 1034–1039 [DOI] [PubMed] [Google Scholar]
- 14. Drab M., Verkade P., Elger M., Kasper M., Lohn M., Lauterbach B., Menne J., Lindschau C., Mende F., Luft F. C., Schedl A., Haller H., Kurzchalia T. V. (2001) Loss of caveolae, vascular dysfunction, and pulmonary defects in caveolin-1 gene-disrupted mice. Science 293, 2449–2452 [DOI] [PubMed] [Google Scholar]
- 15. Hnasko R., Lisanti M. P. (2003) The biology of caveolae. Lessons from caveolin knockout mice and implications for human disease. Mol. Interv. 3, 445–464 [DOI] [PubMed] [Google Scholar]
- 16. Lu R., Alioua A., Kumar Y., Eghbali M., Stefani E., Toro L. (2006) MaxiK channel partners. Physiological impact. J. Physiol. 570, 65–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Marrion N. V., Tavalin S. J. (1998) Selective activation of Ca2+-activated K+ channels by co-localized Ca2+ channels in hippocampal neurons. Nature 395, 900–905 [DOI] [PubMed] [Google Scholar]
- 18. Berkefeld H., Sailer C. A., Bildl W., Rohde V., Thumfart J. O., Eble S., Klugbauer N., Reisinger E., Bischofberger J., Oliver D., Knaus H. G., Schulte U., Fakler B. (2006) BKCa-Cav channel complexes mediate rapid and localized Ca2+-activated K+ signaling. Science 314, 615–620 [DOI] [PubMed] [Google Scholar]
- 19. Fakler B., Adelman J. P. (2008) Control of KCa channels by calcium nano/microdomains. Neuron 59, 873–881 [DOI] [PubMed] [Google Scholar]
- 20. Berkefeld H., Fakler B., Schulte U. (2010) Ca2+-activated K+ channels. From protein complexes to function. Physiol. Rev. 90, 1437–1459 [DOI] [PubMed] [Google Scholar]
- 21. Loane D. J., Lima P. A., Marrion N. V. (2007) Co-assembly of N-type Ca2+ and BK channels underlies functional coupling in rat brain. J. Cell Sci. 120, 985–995 [DOI] [PubMed] [Google Scholar]
- 22. Kim H., Pierce-Shimomura J. T., Oh H. J., Johnson B. E., Goodman M. B., McIntire S. L. (2009) The dystrophin complex controls BK channel localization and muscle activity in Caenorhabditis elegans. PLoS Genet 5, e1000780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Suzuki Y., Yamamura H., Ohya S., Imaizumi Y. (2013) Direct molecular interaction of caveolin-3 with KCa1.1 channel in living HEK293 cell expression system. Biochem. Biophys. Res. Commun. 430, 1169–1174 [DOI] [PubMed] [Google Scholar]
- 24. Navedo M. F., Amberg G. C., Westenbroek R. E., Sinnegger-Brauns M. J., Catterall W. A., Striessnig J., Santana L. F. (2007) Cav1.3 channels produce persistent calcium sparklets, but Cav1.2 channels are responsible for sparklets in mouse arterial smooth muscle. Am. J. Physiol. Heart Circ. Physiol. 293, H1359–1370 [DOI] [PubMed] [Google Scholar]
- 25. Dinardo M. M., Camerino G., Mele A., Latorre R., Conte Camerino D., Tricarico D. (2012) Splicing of the rSlo gene affects the molecular composition and drug response of Ca2+-activated K+ channels in skeletal muscle. PLoS ONE 7, e40235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liao P., Yu D., Li G., Yong T. F., Soon J. L., Chua Y. L., Soong T. W. (2007) A smooth muscle Cav1.2 calcium channel splice variant underlies hyperpolarized window current and enhanced state-dependent inhibition by nifedipine. J. Biol. Chem. 282, 35133–35142 [DOI] [PubMed] [Google Scholar]
- 27. Axelrod D. (1981) Cell-substrate contacts illuminated by total internal reflection fluorescence. J. Cell Biol. 89, 141–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Naraghi M., Neher E. (1997) Linearized buffered Ca2+ diffusion in microdomains and its implications for calculation of [Ca2+] at the mouth of a calcium channel. J. Neurosci. 17, 6961–6973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Imaizumi Y., Muraki K., Watanabe M. (1989) Ionic currents in single smooth muscle cells from the ureter of the guinea-pig. J. Physiol. 411, 131–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yamamura H., Ikeda C., Suzuki Y., Ohya S., Imaizumi Y. (2012) Molecular assembly and dynamics of fluorescent protein-tagged single KCa1.1 channel in expression system and vascular smooth muscle cells. Am. J. Physiol. Cell Physiol. 302, C1257–C1268 [DOI] [PubMed] [Google Scholar]
- 31. Miyawaki A., Tsien R. Y. (2000) Monitoring protein conformations and interactions by fluorescence resonance energy transfer between mutants of green fluorescent protein. Methods Enzymol. 327, 472–500 [DOI] [PubMed] [Google Scholar]
- 32. Leake M. C., Chandler J. H., Wadhams G. H., Bai F., Berry R. M., Armitage J. P. (2006) Stoichiometry and turnover in single, functioning membrane protein complexes. Nature 443, 355–358 [DOI] [PubMed] [Google Scholar]
- 33. Nakajo K., Ulbrich M. H., Kubo Y., Isacoff E. Y. (2010) Stoichiometry of the KCNQ1-KCNE1 ion channel complex. Proc. Natl. Acad. Sci. U.S.A. 107, 18862–18867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ohba T., Sawada E., Suzuki Y., Yamamura H., Ohya S., Tsuda H., Imaizumi Y. (2013) Enhancement of Ca2+ influx and ciliary beating by membrane hyperpolarization due to ATP-sensitive K+ channel opening in mouse airway epithelial cells. J. Pharmacol. Exp. Ther. 347, 145–153 [DOI] [PubMed] [Google Scholar]
- 35. Ohya S., Yamamura H., Muraki K., Watanabe M., Imaizumi Y. (2001) Comparative study of the molecular and functional expression of L-type Ca2+ channels and large conductance, Ca2+-activated K+ channels in rabbit aorta, and vas deferens smooth muscle. Pflugers Arch. 441, 611–620 [DOI] [PubMed] [Google Scholar]
- 36. Cheng X., Jaggar J. H. (2006) Genetic ablation of caveolin-1 modifies Ca2+ spark coupling in murine arterial smooth muscle cells. Am. J. Physiol. Heart Circ. Physiol. 290, H2309–H2319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ulbrich M. H., Isacoff E. Y. (2007) Subunit counting in membrane-bound proteins. Nat. Methods 4, 319–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Couet J., Li S., Okamoto T., Ikezu T., Lisanti M. P. (1997) Identification of peptide and protein ligands for the caveolin-scaffolding domain. Implications for the interaction of caveolin with caveolae-associated proteins. J. Biol. Chem. 272, 6525–6533 [DOI] [PubMed] [Google Scholar]
- 39. Daniel E. E., El-Yazbi A., Cho W. J. (2006) Caveolae and calcium handling, a review and a hypothesis. J. Cell. Mol. Med. 10, 529–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Balijepalli R. C., Foell J. D., Hall D. D., Hell J. W., Kamp T. J. (2006) Localization of cardiac L-type Ca2+ channels to a caveolar macromolecular signaling complex is required for β2-adrenergic regulation. Proc. Natl. Acad. Sci. U.S.A. 103, 7500–7505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Navedo M. F., Cheng E. P., Yuan C., Votaw S., Molkentin J. D., Scott J. D., Santana L. F. (2010) Increased coupled gating of L-type Ca2+ channels during hypertension and Timothy syndrome. Circ. Res. 106, 748–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Itoh T., Kuriyama H., Suzuki H. (1983) Differences and similarities in the noradrenaline- and caffeine-induced mechanical responses in the rabbit mesenteric artery. J. Physiol. 337, 609–629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sakurada S., Takuwa N., Sugimoto N., Wang Y., Seto M., Sasaki Y., Takuwa Y. (2003) Ca2+-dependent activation of Rho and Rho kinase in membrane depolarization-induced and receptor stimulation-induced vascular smooth muscle contraction. Circ. Res. 93, 548–556 [DOI] [PubMed] [Google Scholar]
- 44. Shakirova Y., Bonnevier J., Albinsson S., Adner M., Rippe B., Broman J., Arner A., Swärd K. (2006) Increased Rho activation and PKC-mediated smooth muscle contractility in the absence of caveolin-1. Am. J. Physiol. Cell Physiol. 291, C1326–C1335 [DOI] [PubMed] [Google Scholar]
- 45. Nuno D. W., England S. K., Lamping K. G. (2012) RhoA localization with caveolin-1 regulates vascular contractions to serotonin. Am. J. Physiol. Regul. Integr Comp. Physiol. 303, R959–R967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Santana L. F., Navedo M. F. (2009) Molecular and biophysical mechanisms of Ca2+ sparklets in smooth muscle. J. Mol. Cell Cardiol. 47, 436–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Navedo M. F., Amberg G. C., Nieves M., Molkentin J. D., Santana L. F. (2006) Mechanisms underlying heterogeneous Ca2+ sparklet activity in arterial smooth muscle. J. Gen. Physiol. 127, 611–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Guia A., Wan X., Courtemanche M., Leblanc N. (1999) Local Ca2+ entry through L-type Ca2+ channels activates Ca2+-dependent K+ channels in rabbit coronary myocytes. Circ. Res. 84, 1032–1042 [DOI] [PubMed] [Google Scholar]
- 49. Yamamura H., Imaizumi Y. (2012) Total internal reflection fluorescence imaging of Ca2+-induced Ca2+ release in mouse urinary bladder smooth muscle cells. Biochem. Biophys. Res. Commun. 427, 54–59 [DOI] [PubMed] [Google Scholar]
- 50. Albinsson S., Shakirova Y., Rippe A., Baumgarten M., Rosengren B. I., Rippe C., Hallmann R., Hellstrand P., Rippe B., Swärd K. (2007) Arterial remodeling and plasma volume expansion in caveolin-1-deficient mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 293, R1222–R1231 [DOI] [PubMed] [Google Scholar]
- 51. Saliez J., Bouzin C., Rath G., Ghisdal P., Desjardins F., Rezzani R., Rodella L. F., Vriens J., Nilius B., Feron O., Balligand J. L., Dessy C. (2008) Role of caveolar compartmentation in endothelium-derived hyperpolarizing factor-mediated relaxation. Ca2+ signals and gap junction function are regulated by caveolin in endothelial cells. Circulation 117, 1065–1074 [DOI] [PubMed] [Google Scholar]
- 52. Rodrigues G. J., Restini C. B., Lunardi C. N., Neto Mdos A., Moreira J. E., Bendhack L. M. (2010) Decreased number of caveolae in endothelial cells impairs the relaxation induced by acetylcholine in hypertensive rat aortas. Eur. J. Pharmacol. 627, 251–257 [DOI] [PubMed] [Google Scholar]
- 53. Weston A. H., Porter E. L., Harno E., Edwards G. (2010) Impairment of endothelial SKCa channels and of downstream hyperpolarizing pathways in mesenteric arteries from spontaneously hypertensive rats. Br. J. Pharmacol. 160, 836–843 [DOI] [PMC free article] [PubMed] [Google Scholar]