

Abstract
Relaxin is a potent vasodilator of small resistance arteries and modifies arterial compliance in some systemic vascular beds, yet receptors for relaxin, such as RXFP1, have only been localized to vascular smooth muscle. This study first aimed to localize RXFP1 in rat arteries and veins from different organ beds and determine whether receptors are present in endothelial cells. We then tested the hypothesis that region-specific vascular effects of relaxin may be influenced by the cellular localization of RXFP1 within different blood vessels. The aorta, vena cava, mesenteric artery, and vein had significantly higher (P<0.05) RXFP1 immunostaining in endothelial cells compared with vascular smooth muscle, whereas the femoral artery and vein and small pulmonary arteries had higher (P<0.01) RXFP1 immunostaining in the vascular smooth muscle. Male rats were treated subcutaneously with recombinant human relaxin-2 (serelaxin; 4 μg/h) for 5 d; vasodilation and compliance in mesenteric and femoral arteries and veins were compared with placebo controls. Serelaxin significantly (P=0.04) reduced wall stiffness and increased volume compliance in mesenteric arteries but not in the other vessels examined. This was associated with changes in geometrical properties, and not compositional changes in the extracellular matrix. Serelaxin treatment had no effect on acetylcholine-mediated relaxation but significantly (P<0.001) enhanced bradykinin (BK)-mediated relaxation in mesenteric arteries, involving enhanced nitric oxide but not endothelium-derived hyperpolarization or vasodilatory prostanoids. In conclusion, there is differential distribution of RXFP1 on endothelial and smooth muscle across the vasculature. In rats, mesenteric arteries exhibit the greatest functional response to chronic serelaxin treatment.—Jelinic, M., Leo, C-H., Post Uiterweer, E. P., Sandow, S. L., Gooi, J. H., Wlodek, M. E., Conrad, K. P., Parkington, H., Tare, M., Parry, L. J. Localization of relaxin receptors in arteries and veins, and region-specific increases in compliance and bradykinin-mediated relaxation after in vivo serelaxin treatment.
Keywords: RXFP1, vasodilation
It is well established that the peptide hormone relaxin plays an important role in the profound maternal vascular adaptations that occur in pregnancy, specifically targeting the renal and uterine vasculature (1–3). Beneficial cardiovascular effects of relaxin, which are attributed to alterations in the renal and systemic vasculature, have also been demonstrated in congestive and acute heart failure patients in preclinical studies and phase II/III clinical trials (4–6). In conscious normotensive and hypertensive male and female rats, acute intravenous and chronic subcutaneous administration of relaxin increases cardiac output and global arterial compliance and reduces systemic vascular resistance, without affecting mean arterial pressure (7, 8). Relaxin has also been shown to reduce mean arterial pressure in rat models of hypertension (9, 10) and increase coronary flow in rat and guinea pig hearts (11).
These beneficial cardiovascular effects of relaxin in normotensive rats occur in parallel with increases in renal plasma flow and glomerular filtration rate (12), and are underpinned by a reduction in myogenic constriction of the small renal arteries (13). Vasodilatory responses to in vivo relaxin treatment have also been demonstrated in rat mesenteric arteries (13–15), whereas ex vivo relaxin treatment induces vasodilation in isolated rodent small renal and mesenteric arteries (16–18) and human small gluteal and subcutaneous arteries but not pulmonary resistance arteries (18, 19). Only one study to date has assessed veins; incubation of mesenteric veins with relaxin causes relaxation but does not affect myogenic reactivity (17). In small resistance arteries, vascular tone is modulated by endothelium-derived factors, including nitric oxide (NO), prostacyclin (PGI2), and non-NO/PGI2 endothelium-derived hyperpolarization (EDH) (20). Relaxin appears to act through an endothelium-dependent NO-mediated vasodilator pathway (13–15). In small renal arteries, rapid relaxin-induced vasodilation involves phosphatidylinositol-3-kinase (PI3K; ref. 18). Relaxin also directly stimulates NO production from cultured human coronary artery and aortic endothelial cells, with endothelial NO synthase (eNOS) phosphorylation via PI3K activation and phosphorylation of protein kinase B (Akt; Ser473) (18). Currently, it is not known whether PGI2 and EDH contribute to the vasodilator effects of relaxin.
The vascular actions of relaxin extend to modification of passive wall compliance. Chronic subcutaneous relaxin infusion in rats and mice increases arterial compliance in small renal (7, 21), mesenteric (17), uterine (22), brain parenchymal (23, 24) and carotid arteries (25). In mice administered relaxin, the relative increase in small renal artery compliance is mediated by both geometric and compositional (decreased collagen) remodeling (21). Specifically, there is an increase in unpressurized wall area and wall-thickness-to-lumen area ratio, increased smooth muscle cell density and a decrease in collagen-to-total-protein. Relaxin infusion in nonpregnant rats also increases wall thickness and inner diameter of brain parenchymal arterioles (23). The effects of relaxin on arterial compliance appear to be region specific, as relaxin treatment has no effect on the external iliac (21) and middle cerebral arteries (23), or mesenteric veins (17).
One explanation for the region-specific vascular effects of relaxin might be differential expression of relaxin/insulin-like family peptide receptor 1 (RXFP1) in arteries and veins and/or between endothelial and smooth muscle cells. RXFP1 gene and protein are expressed in the thoracic aorta and small renal and mesenteric arteries, but not in the cerebral vasculature, of male and female rodents (14, 24, 26, 27) but these studies did not localize RXFP1 within the artery wall. We recently colocalized RXFP1 with α-smooth muscle actin (α-SMA) muscle-positive cells in the media of uterine arteries (3), demonstrating expression of relaxin receptors in the vascular smooth muscle. Localization of RXFP1 in endothelial cells is less convincing, despite evidence of a direct action of relaxin on aortic and coronary artery endothelial cells to evoke NO release (18, 28). In summary, the specific localization of RXFP1 in vascular smooth muscle cells is consistent with the hypothesis that relaxin acts directly on blood vessels to influence arterial remodeling and compliance, but RXFP1 is yet to be localized to endothelial cells in intact arteries or veins.
Therefore, in this study we tested the hypothesis that region-specific vascular effects of relaxin may be influenced by the distribution of RXFP1 within different blood vessels in males. The main objectives of this study were to localize RXFP1 in a variety of arteries and veins from different organ beds and determine whether receptors are present in smooth muscle or endothelial cells; and compare the effects of chronic subcutaneous in vivo relaxin treatment on specific vasodilator pathways and passive mechanical wall properties in mesenteric and femoral arteries and veins.
MATERIALS AND METHODS
Animal preparation
All experimental protocols were approved by The University of Melbourne Faculty of Science Animal Experimentation Ethics Committee. Male Wistar rats aged 12–14 wk (∼300 g body weight) were purchased from Monash Animal Services (Clayton, VIC, Australia). Male rats were used in all experiments because nonpregnant females produce estrogen from the ovaries, which could confound the experimental outcomes. Rats were housed in the Animal Facility (Department of Zoology, The University of Melbourne, VIC, Australia) on a 12/12-h light-dark cycle and had access to standard rat chow (Specialty Feed, Glen Forrest, WA, Australia) and water ad libitum.
Experimental protocols
Localization of RXFP1 in arteries and veins
Rats were anesthetized with isofluorane and euthanized by cervical dislocation. Thoracic and abdominal aortae, pulmonary, mesenteric, femoral arteries, and mesenteric and femoral veins were dissected from each animal (n=3–4). Blood vessels were then placed in ice-cold 0.1 M phosphate buffered saline (PBS), and excess fat and loose connective tissue were removed before fixing in 10% neutral buffered formalin (NBF). The kidneys were dissected, sectioned bilaterally, and fixed in PLP fixative (8% paraformaldehyde, 0.2 M lysine, 0.01 M periodate in 0.1 M PBS) for 24 h, and then transferred to 0.1 M PBS.
Human mesenteric artery segments were also obtained from 3 male patients, ages 58, 60, and 64. None were smokers, had diabetes, or were on medications that influenced vascular function; all had normal blood pressure and were undergoing cancer-related surgery. Immediately after surgery, the mesenteric arcade was excised from noncancerous gut and cleaned of fat and loose connective tissue. The arteries were washed in ice-cold physiological saline solution (PSS; 120 mM NaCl, 5 mM KCl, 25 mM NaHCO3, 1 mM KH2PO4, 1.2 mM MgSO4, 11 mM glucose, and 2.5 mM calcium; bubbled with 5% carbon dioxide, 95% nitrogen gas) to remove excess blood and anesthetic and fixed overnight in 4% paraformaldehyde. Procedures were performed in accordance with the ethics guidelines of the National Health and Medical Research Council of Australia, with approval from the South Eastern Sydney and Illawarra Area Health Service Human Research Ethics Committee and the University of New South Wales Human Research Ethics Committee.
Immunohistochemistry and immunofluorescence
Vessels fixed in PLP were embedded in polyester wax (polyethylene glycol 400 distearate; Polysciences, Warrington, PA, USA), whereas those fixed in NBF and paraformaldehyde were embedded in paraffin wax. Sections (5 μm) were cut and mounted on SuperFrost PLUS slides (Menzel-Gläser, Braunschweig, Germany). Procedures for brightfield immunohistochemistry are detailed in Vodstrcil et al. (3). Rat tissues were incubated overnight at 4°C with 3 μg/ml rat RXFP1 antiserum (#107, raised against amino acid residues 107–119 of the rat RXFP1 protein) or preimmune serum (rabbit IgG), kindly supplied by Prof. O. David Sherwood (Department of Physiology and Biophysics, University of Illinois, Urbana, IL, USA). An additional negative control was primary antiserum (3 μg/ml) preabsorbed overnight with 3 μg/ml rat relaxin. Immunoreactivity was detected using the MACH 2 system (Biocare Medical, Concord, CA, USA) and 3, 3′ diaminobenzidine (DAB) as the chromagen substrate (Vector Laboratories, Burlingame, CA, USA). Cross-reactivity of the rat RXFP1 antibody with another relaxin/insulin-like family peptide receptor, RXFP2, was tested on sections of rat gubernaculum testis (kindly provided by Prof, John Hutson, Royal Children's Hospital, University of Melbourne) which only expresses RXFP2 (29). Human arteries were incubated in Background Sniper (Biocare Medical) for 20 min to block nonspecific binding and then with 0.36 μg/ml monoclonal anti-human RXFP1 antiserum (H00059350-M01; Abnova, Novus Biologicals, Littleton, CO, USA) or mouse IgG (diluted to 0.36 μg/ml) overnight at 4°C. Antibody specificity for RXFP1 was determined in previous studies (30). Immunoreactivity was detected with the MACH 4 Universal HRP detection kit (Biocare Medical) and DAB (Biocare Medical). Relative RXFP1 immunostaining was semiquantified by analyzing the percentage area occupied by DAB in each vessel. Using Photoshop CS6 (Adobe Systems, San Jose, CA, USA), relative pixel density as the proportion of area occupied as brown pixels (reflecting DAB label) was taken to reflect the intensity of labeling according to a method modified from Tse et al. (31). In brief, uniform color range was set, and areas of intima and media of the same size were selected at random, and the intensity of signal was recorded using the integrated pixel density function. Equivalent regions of intima and media from negative control tissue were taken as baseline. Comparisons were made between cell types in each vessel (n=6 replicates).
All procedures for immunofluorescence were the same as described above, except different secondary antibodies and detection systems were used (3). To identify rat RXFP1, goat anti-rabbit Dylight 488 (1:100) was applied for 1 h. Slides were briefly rinsed in 1× TBS containing 1% Tween-20 (TTBS) and then a 1:1000 dilution of monoclonal antibody against α-SMA conjugated to Cy5 (Sigma-Aldrich Pty. Ltd. Sydney, NSW, Australia) was applied for 40 min. Control samples were incubated with a 1:1000 dilution of anti-mouse IgG conjugated to Cy5. Nuclei were counterstained with DAPI (Invitrogen, Life Technologies, Grand Island, NY, USA). Sections were coverslipped using fluorocare mounting medium (Biocare Medical) and imaged under 488 and 594 wavelengths on a Zeiss imager D1 with AxioCam mrC5 camera (Carl Zeiss Pty. Ltd., North Ryde, NSW, Australia).
Effects of chronic infusion of serelaxin on vascular function in male rats
Twelve-week old male Wistar rats were implanted with a 7-d Alzet osmotic minipump (Model 2001; Bioscientific, Gymea, NSW, Australia) to infuse recombinant human H2 relaxin (serelaxin; Novartis Pharma AG, Basel, Switzerland; n=23) or placebo (20 mM sodium acetate, pH 5.0; n=21) subcutaneously. The animals were anesthetized using isofluorane, and the osmotic minipumps were inserted under the skin between the shoulder blades using aseptic techniques. Serelaxin was infused at a dose of 13.33 μg/h/kg. This dose was predicted to yield concentrations of circulating serelaxin similar to those measured on gestational d 12–14 in pregnant rats (20–40 ng/ml; ref. 32). Between 09:00 and 12:00 on d 5 after pump insertion, rats were anesthetized with isofluorane and euthanized by cervical dislocation. Cardiac puncture was performed to collect 1 ml blood to verify plasma serelaxin levels. The femoral arteries and veins (diameter ≈ 240 μm and 370 μm, respectively), and small second order mesenteric arteries and veins (diameter ≈200 and 250 μm, respectively) were dissected from each animal and placed in sterile 0.1 M PBS solution on ice to clear fat and loose connective tissue. These four vessels were selected because they represent arteries and veins in different vascular beds. A section of each vessel was used either for the in vitro analysis of passive mechanical wall properties or vascular reactivity studies. Sections of arteries were also snap frozen for additional analyses (see below). The plasma concentration of serelaxin was measured using the Human Relaxin-2 Quantinine ELISA Kit (R&D Systems, Minneapolis, MN, USA) following manufacturer's instructions. Plasma samples were diluted 1:500 in the assay, with a 15.6 pg/ml limit of detection.
Passive mechanical wall properties
Mesenteric and femoral arteries and veins from serelaxin- and placebo-treated rats were transferred to a Ca2+-free PSS (14.9 mM NaCl, 4.7 mM KCl, 1.7 mM NaHCO3, 1.2 mM KH2PO4, 1.7 mM MgSO4, 5 mM glucose, 10 mM HEPES, and 2 mM EGTA). Leak-free segments of vessels were mounted on a pressure myograph system (Living Systems Instrumentation, Burlington, VT, USA), and arterial data [vessel length, outer diameter (OD), inner diameter (ID), and wall thickness (WT)], as well as wall stress and strain were acquired and calculated as described previously (3, 33). For normalization of ID and OD, values were expressed as (value at pressure – value at baseline)/(value at baseline). Volume compliance was calculated for each pressure increment using the following calculation: volume compliance = (Δ volume)/(Δ pressure), where Δ volume = (Δ cross sectional area) × (Δ length), and cross sectional area = (π × ID2)/4. Because wall thickness could not be defined in veins (it was below the limit of detection for the magnification we used), only OD was measured, and vascular stiffness was evaluated by calculating change in OD/pressure.
Collagen and elastin assay
Collagen content was measured in abdominal aorta, mesenteric, and femoral arteries using the Sircol collagen dye binding assay (Biocolor, Carrickfergus, UK) as described previously (21). Elastin content was only determined in mesenteric and femoral arteries (Fastin elastin assay kit; Biocolor). Arteries were lyophilized, and elastin was solubilized using the hot oxalic acid digestion method. Samples and standards (ranging from 0–50 μg) were then incubated in elastin precipitating reagent at 4°C overnight. Final pellets were resuspended in 250 μl of dye dissociation reagent. Absorbance was determined at 540 nm. and relative elastin content was quantified as the ratio of elastin concentration (μg/ml) to dry weight (mg).
Vascular reactivity
Rings (1–2 mm in length) of mesenteric and femoral arteries and veins from serelaxin (n=15) and placebo (n=14) treated rats were mounted on a four-channel Mulvany-Halpern myograph (model 610M; Danish Myo Technology, Aarhus, Denmark). The vessels were then stretched in increments to a tension equivalent to ∼70 mmHg for arteries and ∼20 mmHg for veins. Vascular smooth muscle and endothelial cell function were tested as described previously (33, 34). Blood vessels were submaximally (50–60% maximal constriction) preconstricted with phenylephrine (PE; ∼10−9 to 10−6 M), and endothelium-dependent relaxation was assessed with increasing concentrations of acetylcholine (ACh; 10−10 to 10−5 M; all arteries and veins) or bradykinin (BK; 10−10 to 10−6 M; mesenteric arteries and veins because BK caused contraction in the other vessels) applied cumulatively. Responses to ACh and BK were recorded before and after NOS and cyclooxygenase blockade with Nω-nitro-l-arginine methyl ester (L-NAME; 2×10−4 M) and/or indomethacin (Indo; 10−6 M), respectively. The relaxation remaining in the presence of both inhibitors was attributed to EDH (35). Smooth muscle contraction was examined in vessels exposed to increasing concentrations of PE (10−9 to 10−4 M) applied cumulatively. The relative contribution of NO, PGI2, and EDH to relaxation evoked by BK was determined by analyzing the area under curve (AUC) of the BK-response curves. The responses attributed to each component were calculated using the following equations: PGI2 contribution = AUC (L-NAME alone) − AUC (L-NAME+Indo); NO contribution = AUC (no inhibitors) – AUC (L-NAME alone); EDH contribution = AUC (L-NAME+Indo).
Quantitative PCR
RNA was extracted from individual arteries, and cDNA was synthesized from 0.5 μg of RNA in a 20 μl reaction containing random hexamers (50 ng/μl) and 200 U of Superscript III (Invitrogen, Mulgrave, VIC, Australia; ref. 3). The comparative cycle threshold (2−ΔCt) method of quantitative real-time polymerase chain reaction (qPCR) was used to quantify eNOS III (Nos3) gene expression in the mesenteric arteries and veins of serelaxin and placebo treated male rats. Rat-specific forward/reverse primers and 6-carboxyl fluorescein (FAM)-labeled TaqMan probes were designed and purchased from Biosearch Technologies (Novato, CA, USA). qPCR was performed on the Opticon 3 PCR machine (Bio-Rad, West Ryde, NSW, Australia) using 96-well reaction plates with 20-μl reactions in triplicate containing SensiMix (Bioline, Alexandria, NSW, Australia) and 10 μM of primers and FAM-labeled probe. Ribosomal 18S (Rn18s) was the reference gene. Negative template controls substituting cDNA with water or RT negative controls substituting the reverse transcriptase in the cDNA synthesis, were included on each plate. For each sample, the mean Rn18s CT triplicate value was subtracted from the mean gene of interest triplicate CT value to normalize gene of interest expression to the reference gene. These normalized data were then presented as a relative value (means±se).
Statistical analysis
Data are expressed as means ± se with n representing the number of rats. The stress-strain and volume compliance data were analyzed with repeated measures 2-way ANOVA (treatment vs. strain) with Bonferroni post hoc analysis using GraphPad Prism 5 (GraphPad Software, San Diego, CA, USA). Independent t tests assessed statistical differences in collagen and elastin concentrations between placebo- and serelaxin-treated rats. Concentration-response curves were constructed using GraphPad Prism and sigmoidal curves were fitted using the least squares method and analyzed using repeated measures 2-way ANOVA (treatment vs. concentration). Contractions to PE were expressed as percentage contraction to high KPSS. ACh- and BK-evoked relaxation was expressed as percentage of precontraction evoked by PE. The concentration of drug required to evoke a half-maximal response (EC50) was determined, and the pD2 (−log EC50), maximum contraction (Emax), and maximum relaxation (Rmax) were compared using independent t tests.
RESULTS
Localization of RXFP1 in arteries and veins
Immunoreactive RXFP1 was localized in small renal arteries, abdominal aorta, vena cava, large and small pulmonary arteries, mesenteric and femoral arteries, and veins of rats, but the cellular distribution of RXFP1 differed between vessel types (Figs. 1 and 2 and Table 1). In the kidney, RXFP1 was predominantly localized to the small renal artery and renal tubules (Fig. 1A, D). There was no RXFP1 staining in the preimmune serum negative controls (Fig. 1B) or in sections that were incubated with antiserum preabsorbed with rat relaxin (data not shown). It was important to demonstrate specificity to RXFP1, as the Rxfp2 gene is expressed in blood vessels (26). The absence of RXFP1 immunostaining in rat gubernaculum testis (Fig. 1C), which only expresses RXFP2 (29), is evidence that the rat RXFP1 antibody used in this study did not cross-react with RXFP2. Double-labeling immunofluorescence confirmed colocalization of RXFP1 with α-SMA in vascular smooth muscle cells within the media of small renal arteries and not veins (Fig. 1D, G). When one of the primary antisera was replaced with preimmune serum, there was only positive staining for RXFP1 (Fig. 1E, H) or α-SMA (Fig. 1F). The negative controls with preimmune rabbit serum revealed little background fluorescence (Fig. 1E, F; insets). Endothelial cells were difficult to identify in these small renal artery cross-sections but at higher magnification, there appeared to be RXFP1 immunostaining in cells lining the lumen that did not stain positive for α-SMA (Fig. 1I).
Figure 1.

Localization of RXFP1 protein in rat small renal arteries by immunohistochemistry (A–C) and immunofluorescence (D–I). Immunoreactive RXFP1 is predominantly localized to the vascular smooth muscle of the media (A). Immunofluorescent RXFP1 (E, H) and α-SMA (F), with the overlay (D, G, I) demonstrate colocalization. Higher magnification illustrates RXFP1 in endothelial cells that did not stain positive for α-SMA (I, arrow). There is no RXFP1 staining in the preimmune serum negative controls (B and C, E, F, insets) and rat gubernaculum testis (GB; C). VSMC, vascular smooth muscle cell, EC, endothelial cell. Scale bars =50 μm (A); 500 μm (C). Note the high level of autofluorescence in the internal elastic lamina (IEL).
Figure 2.
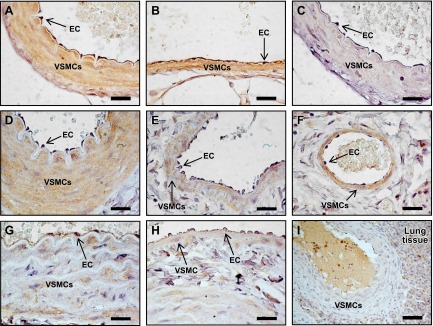
Localization of RXFP1 protein by immunohistochemistry in endothelial cells (ECs) and vascular smooth muscle cells (VSMCs) in the mesenteric artery (A), mesenteric vein (B), and abdominal aorta (G). In the femoral artery (D) and vein (E) and small pulmonary artery (F), RXFP1 is predominantly localized to the VSMC. RXFP1 staining in the vena cava (H) and large pulmonary artery (I) is considerably lower than other blood vessels. There is no RXFP1 staining in the preimmune serum negative control (C). Scale bars = 20 μm (A–H); 50 μm (I).
Table 1.
Quantification (% area of DAB labeling) of RXFP1-positive staining in the intima and media of the aorta, vena cava, femoral artery and vein, mesenteric artery and veins, and large and small pulmonary arteries
| Artery or vein | Endothelium | Smooth muscle |
|---|---|---|
| Aorta | 32.9 ± 0.3* | 22.3 ± 0.3 |
| Femoral artery | 3.2 ± 0.4 | 39.7 ± 1.7* |
| Femoral vein | 3.2 ± 0.6 | 34.6 ± 1.7* |
| Mesenteric artery | 84.3 ± 1.1* | 56.5 ± 1.0 |
| Mesenteric vein | 78.2 ± 3.4* | 68.4 ± 1.7 |
| Pulmonary artery (large) | 4.2 ± 0.8 | 5.4 ± 0.6 |
| Pulmonary artery (small) | 11.6 ± 1.0 | 66.1 ± 4.1* |
| Vena cava | 25.4 ± 2.1* | 4.6 ± 0.3 |
Data are means ± se of 6 uniform areas for endothelium and smooth muscle.
P < 0.05 vs. other cell type.
Comparisons between endothelial and vascular smooth muscle cells revealed differences in relative RXFP1 immunostaining in rat arteries and veins (Fig. 2 and Table 1). In the mesenteric artery and vein, RXFP1 was localized to both endothelial cells and the smooth muscle (Fig. 2A, B) but with significantly (P<0.05) higher immunostaining in endothelial cells compared with smooth muscle cells (Table 1). There was no RXFP1 staining in the preimmune serum negative controls (Fig. 2C). RXFP1 immunostaining in endothelial cells was also significantly (P<0.05) higher in the aorta and vena cava (Table 1, Fig. 2G, H). In the femoral artery and vein and small pulmonary artery, RXFP1 was predominantly localized to smooth muscle (Fig. 2D–F) and significantly (P<0.05) higher compared with endothelial cells (Table 1). RXFP1 immunostaining was relatively low in the large pulmonary artery (Fig. 2I).
In human mesenteric arteries, immunoreactive RXFP1 was clearly localized in endothelial cells, with less intense immunostaining in the vascular smooth muscle and adventitia (Fig. 3). There was no RXFP1 staining in the negative control with mouse IgG (Fig. 3, insets).
Figure 3.

A) Localization of RXFP1 protein by immunohistochemistry in a human mesenteric artery (representative of arteries from all 3 patients). B) RXFP1 immunostaining is detected in endothelial cells (ECs) and vascular smooth muscle cells (VSMCs) but not in the mouse IgG negative controls (insets). Scale bars = 100 μm (A), 20 μm (B).
Effects of chronic infusion of serelaxin on passive mechanical wall properties
Rats treated with serelaxin had significantly increased plasma recombinant human serelaxin concentrations (39.93±4.06 ng/ml) compared with placebo-treated rats, in which no human serelaxin was detected. There was no significant difference in WT, OD, and ID in any vessel analyzed at baseline (5 mmHg; Tables 2 and 3). Over the pressurization range, ID of mesenteric arteries increased to a significantly greater extent (P=0.03) in the serelaxin-treated animals compared with controls (Fig. 4A). There was no significant difference in OD and WT responses to increasing pressure between the two groups (Fig. 4C, E). There was a significant shift to the right of the stress-strain curve of mesenteric arteries from serelaxin-treated rats (F1,10=5.55, P=0.04) compared with controls (Fig. 5A), indicative of an overall reduction in passive wall stiffness. Furthermore, overall volume compliance was significantly (F1,10=5.04, P=0.03) increased in the mesenteric arteries of serelaxin-treated rats compared with controls (Fig. 6A).
Table 2.
Mesenteric artery and vein dimensions at baseline (5 and 2 mmHg, respectively)
| Parameter | Mesenteric artery |
Mesenteric vein |
||||||
|---|---|---|---|---|---|---|---|---|
| Placebo | n | Serelaxin | n | Placebo | n | Serelaxin | n | |
| Stress-strain (K) | 7.09 ± 1.67 | 6 | 6.08 ± 0.53 | 8 | N/M | – | N/M | – |
| Baseline OD (μm) | 150.4 ± 6.8 | 6 | 149.4 ± 9.5 | 8 | 251.3 ± 37.2 | 7 | 251.3 ± 34.59 | 6 |
| Baseline ID (μm) | 123.5 ± 5.3 | 6 | 116.6 ± 9.2 | 8 | N/M | – | N/M | – |
| Baseline WT (μm) | 13.44 ± 1.37 | 6 | 16.39 ± 0.81 | 8 | N/M | – | N/M | – |
| Hi K Emax (mN/mm) | 9.59 ± 0.37 | 13 | 9.95 ± 0.55 | 13 | 3.79 ± 0.35 | 14 | 3.88 ± 0.48 | 13 |
| PE pD2 | 5.93 ± 0.13 | 8 | 5.81 ± 0.14 | 8 | N/D | 10 | N/D | 8 |
| PE Emax (% Hi K) | 99.74 ± 4.01 | 8 | 97.85 ± 4.37 | 8 | 60.38 ± 7.39 | 10 | 54.83 ± 7.22 | 8 |
| ACh pD2 | 7.97 ± 0.09 | 8 | 8.00 ± 0.06 | 8 | 7.15 ± 0.52 | 8 | 6.71 ± 0.22 | 8 |
| ACh Rmax (%) | 101.57 ± 0.78 | 8 | 99.85 ± 0.82 | 8 | 84.06 ± 12.71 | 8 | 84.06 ± 10.07 | 8 |
| ACh (+L-NAME+Indo) pD2 | 7.25 ± 0.09 | 6 | 7.49 ± 0.13 | 6 | N/D | 5 | N/D | 7 |
| ACh (+L-NAME+Indo) Rmax (%) | 99.17 ± 2.40 | 6 | 99.92 ± 2.50 | 6 | 17.67 ± 10.82 | 5 | 16.98 ± 8.52 | 7 |
| BK pD2 | 7.73 ± 0.10 | 9 | 9.24 ± 0.61* | 9 | 8.88 ± 0.14 | 8 | 8.65 ± 0.12 | 9 |
| BK Rmax (%) | 92.87 ± 2.28 | 9 | 98.45 ± 1.51 | 9 | 109.1 ± 1.66 | 8 | 105.5 ± 1.62 | 9 |
| BK (+L-NAME) pD2 | 7.45 ± 0.25 | 5 | 7.22 ± 0.44 | 7 | 8.15 ± 0.21 | 5 | 8.15 ± 0.24 | 7 |
| BK (+L-NAME) Rmax (%) | 85.11 ± 7.62 | 7 | 92.94 ± 6.15 | 8 | 55.99 ± 4.21 | 7 | 59.08 ± 5.48 | 9 |
| BK (+L-NAME+Indo) pD2 | 7.46 ± 0.17 | 4 | 7.20 ± 0.15 | 6 | 8.23 ± 0.26 | 6 | 7.69 ± 0.24 | 7 |
| BK (+L-NAME+Indo) Rmax (%) | 85.84 ± 7.32 | 7 | 92.94 ± 9.49 | 6 | 58.87 ± 9.21 | 7 | 59.06 ± 5.21 | 7 |
OD, outer diameter; ID, inner diameter; WT, wall thickness. Vascular reactivity data: PE, phenylephrine; ACh, acetylcholine; BK, bradykinin; Hi K, 100 mM potassium PSS; L-NAME, nitric oxide synthase inhibitor; Indo, indomethacin; Emax, maximum response; Rmax, maximum relaxation; pD2, sensitivity (−log EC50); N/M, not measurable, N/D, not defined. All values are expressed as mean ± se; n = sample size.
P < 0.05 vs. placebo controls.
Table 3.
Femoral artery and vein dimensions at baseline (5 mmHg)
| Parameter | Femoral artery |
Femoral vein |
||||||
|---|---|---|---|---|---|---|---|---|
| Placebo | n | Serelaxin | n | Placebo | n | Serelaxin | n | |
| Stress-strain (K) | 3.81 ± 0.52 | 6 | 3.34 ± 0.45 | 8 | N/M | – | N/M | – |
| Baseline OD (μm) | 243.3 ± 16.3 | 6 | 234.2 ± 13.5 | 8 | 373.3 ± 26.9 | 7 | 375.3 ± 31.5 | 6 |
| Baseline ID (μm) | 169.2 ± 16.4 | 6 | 157.2 ± 16.8 | 8 | N/M | – | N/M | – |
| Baseline WT (μm) | 37.08 ± 1.23 | 6 | 44.83 ± 5.80 | 8 | N/M | – | N/M | – |
| Hi K Emax (mN/mm) | 15.07 ± 0.97 | 13 | 15.01 ± 0.87 | 12 | 3.56 ± 0.11 | 14 | 3.30 ± 0.23 | 13 |
| PE pD2 | 5.72 ± 0.12 | 8 | 5.63 ± 0.12 | 8 | 6.03 ± 0.15 | 11 | 6.05 ± 0.20 | 10 |
| PE Emax (% Hi K) | 95.57 ± 2.96 | 8 | 98.79 ± 4.54 | 8 | 54.07 ± 3.31 | 11 | 50.39 ± 5.75 | 10 |
| ACh pD2 | 6.98 ± 0.09 | 10 | 6.85 ± 0.13 | 9 | 7.26 ± 0.15 | 11 | 7.14 ± 0.21 | 8 |
| ACh Rmax (%) | 91.91 ± 3.40 | 10 | 91.15 ± 5.94 | 9 | 96.95 ± 2.24 | 11 | 95.16 ± 3.75 | 8 |
| ACh (+L-NAME+Indo) pD2 | N/D | 9 | N/D | 8 | 6.34 ± 0.32 | 9 | 5.72 ± 0.31 | 7 |
| ACh (+L-NAME+Indo) Rmax (%) | 66.86 ± 10.09 | 9 | 89.09 ± 5.08 | 8 | 45.31 ± 10.70 | 9 | 48.93 ± 11.15 | 7 |
OD, outer diameter; ID, inner diameter; WT, wall thickness. Vascular reactivity data: PE, phenylephrine; ACh, acetylcholine; Hi K, 100 mM potassium PSS; L-NAME, nitric oxide synthase inhibitor; Indo, indomethacin; Emax, maximum response; pD2, sensitivity (−log EC50); N/M , not measurable; N/D, not defined. All values are expressed as mean ± se; n = sample size.
Figure 4.
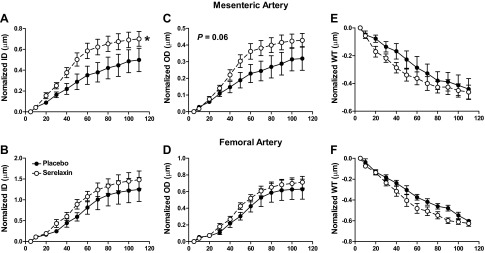
Normalized passive ID (A, B), (OD; C, D), and WT (E, F) against intraluminal pressures in the mesenteric (A, C, E) and femoral arteries (B, D F) from placebo-treated (solid circles, n=6) and serelaxin-treated (open circles, n=8) rats. Values are means ± se. *P < 0.05 vs. placebo controls.
Figure 5.

Stress-strain relationships in mesenteric (A) and femoral (B) arteries, and pressure-OD relationships in mesenteric (C) and femoral (D) veins from from placebo-treated (solid circles, n=6–7) and serelaxin-treated (open circles, n=7–8) male rats. Values are means ± se. *P < 0.05 vs. placebo controls.
Figure 6.

A, B) Volume compliance throughout the pressure range (5–110 mmHg) in the mesenteric (A) and femoral (B) arteries from placebo-treated (solid circles, n=6–7) and serelaxin-treated (open circles, n=7–8) rats. Values are means ± se. *P < 0.05 vs. placebo controls. C, D) Quantitative analysis of total soluble collagen (C) and elastin (D) content in the thoracic aorta (TA), mesenteric artery (MA) and femoral artery (FA) of placebo-treated (solid squares) and serelaxin-treated (open squares) rats. Values are expressed as mean ± se ratio of total dry weight. Sample sizes are indicated in parentheses.
In femoral arteries, treatment with serelaxin had no significant effect on normalized OD, ID, and WT (Fig. 4B, D, F) or wall stiffness compared with controls (Fig. 5B). Furthermore, there was no significant difference in volume compliance across the pressurization range. Serelaxin treatment also had no significant effect on the OD of the mesenteric and femoral veins when pressurized between 5 and 15 mmHg (Fig. 5C, D), suggesting no change in vascular remodeling.
We then assessed total soluble collagen and elastin content to establish whether the reduction in wall stiffness in serelaxin-treated rats was associated with compositional remodeling. There was no significant effect of serelaxin treatment on either collagen or elastin content (Fig. 6C, D) or on the collagen: elastin ratio (data not shown) in the mesenteric and femoral arteries compared with controls. In the thoracic aorta, there was a decrease in collagen content, but it did not reach significance (P=0.051).
Effects of chronic infusion of serelaxin on vascular function
Smooth muscle function
PE evoked concentration-dependent (10−9 to 10−4 M) contraction in mesenteric and femoral arteries and veins but there was no significant difference in the sensitivity (pD2) or maximal response (Emax) between serelaxin-treated rats and controls (Fig. 7 and Tables 2 and 3). Serelaxin treatment had no effect on 100 mM potassium PSS-induced contraction in any blood vessel examined (Tables 2 and 3).
Figure 7.

Concentration-response curves to PE (A, C, E, G) and ACh (B, D, F, H) in endothelium-intact mesenteric arteries (A, B), mesenteric veins (C, D), femoral arteries (E, F), and femoral veins (G, H) isolated from placebo-treated (circles) and serelaxin-treated (squares) rats (n=8–11/group). ACh-evoked relaxation was compared in the absence (continuous line) and presence (dotted line) of L-NAME + Indo. Values are means ± se.
Endothelium-dependent relaxation
Stimulation of the endothelium with ACh resulted in concentration-dependent (10−10 to 10−5 M) relaxation in mesenteric and femoral arteries and veins but serelaxin treatment had no significant effect on either the sensitivity (pD2) or Rmax compared with controls (Fig. 7 and Tables 2 and 3). The effects of serelaxin treatment on EDH-mediated relaxation evoked by ACh were also evaluated; 75% of femoral and mesenteric veins, but not arteries, developed spontaneous tone after L-NAME + Indo incubation, but this did not differ between serelaxin and placebo-treated rats. L-NAME + Indo shifted the concentration-relaxation curves to ACh to the right in all blood vessels, indicating the involvement of both NO and/or vasodilator prostanoids (Fig. 7). In the femoral artery, blockade with L-NAME + Indo reduced maximum relaxation to ACh (to 10.91±5.08%) in serelaxin-treated rats compared with controls (to 33.14±10.09%), but this was not significant (P=0.07; Fig. 7F). In all other vessels examined, the EDH-mediated relaxation evoked by ACh was unaffected by serelaxin treatment (Fig. 7).
We expanded our analysis of endothelium-dependent relaxation in the mesenteric arteries and veins using BK. In mesenteric arteries of serelaxin-treated rats the concentration-dependent relaxation curve was significantly augmented compared with controls (P<0.001; Fig. 8A). Post hoc analysis revealed that 10−9 and 10−8 M BK caused significantly (P<0.01) greater relaxation in serelaxin-treated animals compared with controls. Sensitivity (pD2) to BK was also significantly (P=0.03) greater in serelaxin-treated animals (9.24±0.6) compared with controls (7.73±0.10) (Fig. 8A, Table 2). Analysis of the area under the curve indicated that this was due to a significant (P=0.02) increase in the component attributed to NO. Conversely, in the presence of L-NAME and L-NAME + Indo there was no significant difference in BK-mediated relaxation between serelaxin and placebo-treated rats (Fig. 8A), indicating the absence of a PGI2 component. There was no significant difference in BK-evoked relaxation in mesenteric veins of serelaxin and placebo-treated rats (Fig. 8B, Table 2).
Figure 8.
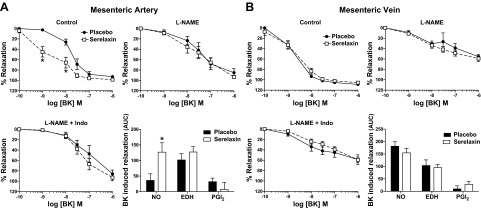
Concentration-response curves for BK in endothelium-intact mesenteric arteries (A) and veins (B) isolated from placebo-treated (solid circles) and serelaxin-treated (open squares) rats (n=6–9/group). BK-evoked relaxation was compared between groups in the presence of L-NAME alone and L-NAME + Indo. AUC of BK response curves were analyzed to reveal the relative contribution of NO, EDH, and PGI2. Values are means ± se. *P < 0.05 vs. placebo controls.
To investigate whether the serelaxin-enhanced BK-mediated vasodilation involves an increase in total eNOS, we analyzed Nos3 gene expression in mesenteric arteries and veins. There was no significant difference in Nos3 expression between placebo and serelaxin-treated animals in these two vessels (Supplemental Fig. 1).
DISCUSSION
The major findings of this study were that receptors for relaxin, RXFP1, were differentially localized in endothelial and smooth muscle cells of arteries and veins. RXFP1 was predominantly expressed in endothelial cells in the mesenteric artery and vein, and also in the aorta and vena cava. In contrast, the femoral artery and vein and small pulmonary arteries had higher RXFP1 immunostaining in the vascular smooth muscle. Chronic infusion with serelaxin for 5 d reduced vessel wall stiffness in the mesenteric arteries but not in the other vessels examined. This was primarily associated with vascular remodeling and changes in geometrical properties, and not compositional changes in the extracellular matrix (ECM). Serelaxin treatment had no effect on ACh-mediated relaxation. However, BK-mediated relaxation in mesenteric arteries, but not veins, was enhanced following serelaxin treatment. This was due to an increase in the NO component of BK-mediated relaxation and not to changes in EDH or vasodilator prostanoids. Overall, our data demonstrate increases in compliance and BK-mediated relaxation but only in the mesenteric artery of healthy serelaxin-treated rats.
Previous studies have shown Rxfp1 gene expression in small renal and mesenteric arteries, as well as thoracic aortae isolated from mice and rats (26, 27) and in human subcutaneous arteries (18). In the rat uterine artery, RXFP1 protein is predominantly localized to the vascular smooth muscle. Our data now demonstrate RXFP1 in endothelial cells of intact mesenteric arteries of rats and humans, and veins, aorta and the vena cava of rats. Importantly, the large pulmonary and femoral artery and femoral vein had few RXFP1-positive endothelial cells illustrating differences between vessel types. These data represent a significant advance in our understanding of serelaxin's vascular actions because they illustrate the diversity in RXFP1 expression between arteries and veins, and suggest that the effects of serelaxin vary across the vasculature.
To test our hypothesis that region-specific vascular responses to serelaxin are due, in part, to differential localization of RXFP1 in endothelial and vascular smooth muscle cells, we first analyzed passive mechanical wall properties in mesenteric and femoral arteries and veins because the cellular distribution of RXFP1 differed between these two vascular beds. In the mesenteric artery, serelaxin treatment reduced passive arterial wall stiffness, which is consistent with previous studies (7, 21). This was associated with a significant increase in ID, without a change in WT. Serelaxin treatment also increased volume compliance in the low pressure range, which reflects the ability of the artery to lengthen. Conversely, there were no changes in circumferential wall stiffness or volume compliance in the femoral artery. We suggest that this is not related to RXFP1 localization because both vessel types have RXFP1 in the vascular smooth muscle. An alternative explanation for the lack of a response in the femoral artery is that the dose of serelaxin and duration of infusion may not have been sufficient to reach threshold levels to induce substantial vascular remodeling. The media of the femoral artery is thicker and consists of more ECM than a resistance artery, so it is also possible that a greater extent of arterial remodeling is required to influence wall stiffness and compliance. Another hypothesis is related to the known effects of shear stress on the vascular wall, which activates vascular remodeling pathways (36). Relaxin decreases vascular resistance (37) and increases blood flow velocity (3). It is therefore possible that the differential effects of serelaxin on vascular remodeling between resistance and capacitance arteries are related to differences in blood flow (or shear stress). Only one study has reported improvements in elasticity in a large capacitance artery (carotid artery) after relaxin treatment but this was in senescent, spontaneously hypertensive rats (25). The duration of relaxin treatment in that study was also considerably longer (2 wk) and the dose higher (≈20 μg/kg/h). Consistent with previous studies (17), serelaxin treatment had no effect on wall stiffness in mesenteric and femoral veins. However, this was not explained by the absence of RXFP1 in the vascular smooth muscle, and is more likely related to differences in venous wall structure or the relationship between shear stress and vascular remodeling.
The consensus view is that relaxin promotes vascular remodeling and compositional changes in the ECM of arteries. Our study showed increases in pressurized ID, but no significant change in OD or WT of mesenteric arteries in serelaxin-treated rats. Relaxin treatment increased ID and WT of brain parenchymal arterioles (23) and increased unpressurized wall area, wall-to-lumen area ratio and smooth muscle cell density in small renal arteries (21). Conversely, Xu et al. (25) reported decreases in the OD, ID and media thickness in the aorta of relaxin-treated, aged hypertensive rats. Vascular wall compliance is also dependent on the relative contribution and organization of collagen and elastin (38). In mice administered relaxin, the increase in small renal artery compliance was associated with a decrease in collagen but no change in elastin (21). Similarly, in aged relaxin-treated hypertensive rats there was a reduction in collagen content, but not elastin, in the aorta resulting in an increase in elastin:collagen ratio (25). This is thought to be a key factor in altering vascular stiffness. In our study, there was no change in total soluble collagen or elastin, or the elastin:collagen ratio in the mesenteric or femoral arteries of serelaxin-treated rats. However, we did observe a trend toward a reduction in collagen in the aorta, which suggests that serelaxin may act on some arteries to induce compositional changes in the ECM.
We also investigated the potential differential effects of 5 d in vivo serelaxin administration on smooth muscle function and endothelium-dependent relaxation in arteries and veins. As shown by others (13, 17), we found no change in PE-evoked contraction in arteries and veins from serelaxin-treated rats. Other studies in female rats reported a reduction in the sensitivity to PE in mesenteric arteries after in vivo relaxin treatment (14, 15) but the arteries were bathed in PSS containing 30 ng/ml relaxin in the organ bath and plasma relaxin levels were 60–70 ng/ml. Therefore, it is possible that differences between studies are associated with different concentrations of relaxin in the mesenteric vessel bed. We found that 5 d in vivo serelaxin had no effect on ACh-evoked relaxation in arteries and veins. Previous studies have shown that ACh-mediated relaxation of rat aortic rings was impaired by treatment with tumor necrosis factor α (TNF-α), and subsequently improved by coincubation with relaxin in vitro for 48 h (39). This study also demonstrated that in vivo serelaxin treatment enhanced BK-evoked relaxation in mesenteric arteries, and involved NO pathways.
The contribution of NO to relaxin-mediated dilation in small renal, mesenteric and human subcutaneous arteries has been demonstrated previously by endothelium removal and NOS inhibitors (13, 14, 18). Further analysis in cultured human aortic and coronary artery endothelial cells revealed that relaxin rapidly stimulates phosphorylation of Akt (Ser473) and eNOS (Ser1177) through activation of PI3K, resulting in generation of NO (18). Similarly, 48 h of relaxin treatment had no effect on eNOS protein expression but augmented eNOS activity via enhanced phosphorylation at Ser1177 and Ser633 and dephosphorylation at Thr495 in rat aortic rings with endothelial dysfunction after exposure to TNF-α (39). We confirmed no effects of serelaxin on eNOS expression and suggest that serelaxin enhanced BK-mediated vasodilation through phosphorylation of eNOS which increases NO production in endothelial cells.
Previous studies on mesenteric veins showed that chronic administration of relaxin had no effect on myogenic reactivity but addition of relaxin to the perfusate resulted in a concentration-dependent relaxation in veins preconstricted with PE (17). Relaxin was also reported to antagonize adrenergic and cholinergic vascular contractions in human saphenous veins (40). Although there are relaxin receptors in rat mesenteric and femoral veins, our pharmacological approach showed that serelaxin treatment did not alter ACh-mediated endothelium-dependent relaxation or PE-evoked contraction in either vein. Unlike the mesenteric artery, serelaxin also had no effect on total BK-mediated relaxation in mesenteric veins.
In summary, RXFP1 is differentially localized to both endothelial and vascular smooth muscle cells in arteries and veins. Specifically, the aorta, vena cava, mesenteric artery, and vein had significantly higher RXFP1 immunostaining in endothelial cells compared with vascular smooth muscle, whereas the femoral artery and vein and small pulmonary arteries had higher RXFP1 immunostaining in the vascular smooth muscle. All measures of vascular function (arterial stiffness and volume compliance, BK-mediated relaxation) were significantly improved in mesenteric arteries after serelaxin treatment, demonstrating that this major resistance vascular bed is a key target for serelaxin. The serelaxin-enhanced BK-evoked relaxation involved a significant contribution from NO but not EDH or vasodilatory prostanoids. Despite the localization of RXFP1 in femoral arteries and veins, and mesenteric veins, we showed no significant functional effects of serelaxin in these blood vessels.
Supplementary Material
Acknowledgments
The authors thank Dr. Dennis Stewart, the coordinator of the research partnership (Novartis Pharma AG, Basel, Switzerland) for helpful advice with the experimental design and critical analysis of the data. The authors also thank Dr. Elaine Unemori for helpful suggestions and manuscript review. The authors are grateful to Dr. Jill Verlander, Professor John Hutson, and Dr. Marc Mazzuca for their valuable contributions to this work, and Professor O. David Sherwood (University of Illinois, Urbana, IL, USA) for his generous gift of the RXFP1 antibody.
The authors disclose that this project was partially funded by Novartis, which also provided the serelaxin, as conditions of an Australian Research Council Linkage grant. The research was funded by Australian Research Council Linkage grant LP110200543 (L.J.P., M.T., H.C.P., and M.E.W.), U.S. National Institutes of Health grant HL067937 (K.P.C. and L.J.P.) and an Australian and New Zealand Medical Research and Technology in Victoria grant (L.J.P.). M.J. received an Australian Postgraduate Award.
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
- α-SMA
- α-smooth muscle actin
- ACh
- acetylcholine
- Akt
- protein kinase B
- AUC
- area under curve
- BK
- bradykinin
- COX
- cyclooxygenase
- DAB
- 3,3′-diaminobenzidine
- ECM
- extracellular matrix
- EDH
- endothelium-derived hyperpolarization
- EC50
- half-maximal effective concentration
- eNOS
- endothelial nitric oxide synthase
- Emax
- maximum contraction
- FAM
- 6-carboxy fluorescein
- ID
- inner diameter
- Indo
- indomethacin
- L-NAME
- Nω-nitro-l-arginine synthase; methyl ester
- NBF
- neutral buffered formalin
- NO
- nitric oxide
- NOS
- nitric oxide Nos3, endothelial nitric oxide synthase III
- NBF
- neutral buffered formalin
- OD
- outer diameter
- PBS
- phosphate-buffered saline
- pD2
- −log EC50
- PE
- phenylephrine
- PGI2
- prostacyclin
- PI3K
- phosphatidylinositol-3-kinase
- PLP
- paraformaldehyde-lysine-periodate
- PSS
- physiological saline solution
- Rmax
- maximum relaxation
- Rn18S
- ribosomal 18S
- RXFP1
- relaxin/insulin-like family peptide receptor 1
- RXFP2
- relaxin/insulin-like family peptide receptor 2
- WT
- wall thickness
REFERENCES
- 1. Jeyabalan A., Shroff S. G., Novak J., Conrad K. P. (2007) The vascular actions of relaxin. Adv. Exp. Med. Biol. 612, 65–87 [DOI] [PubMed] [Google Scholar]
- 2. Conrad K. P. (2011) Maternal vasodilation in pregnancy: the emerging role of relaxin. Am. J. Physiol. 301, R267–R275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Vodstrcil L. A., Tare M., Novak J., Dragomir N., Ramirez R. J., Wlodek M. E., Conrad K. P., Parry L. J. (2012) Relaxin mediates uterine artery compliance during pregnancy and increases uterine blood flow. FASEB J. 26, 4035–4044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Teerlink J. R., Metra M., Felker M. G., Ponikowoski P., Voors A. A., Weatherley B. D., Marmor A., Katz A., Grzybowski J., Unemori E., Teichman S. L., Cotter G. (2009) Relaxin for the treatment of patients with acute heart failure (Pre-RELAX-AHF): a multicentre, randomised, placebo-controlled, parallel-group, dose-finding phase IIb study. Lancet 373, 1429–1439 [DOI] [PubMed] [Google Scholar]
- 5. Dschietzig T., Teichman S., Unemori E., Wood S., Boehmer J., Richter C., Baumann G., Stangl K. (2009) Clinical trial: intravenous recombinant human relaxin in compensated heart failure: a safety, tolerability, and pharmacodynamic trial. J. Card. Fail. 15, 182–190 [DOI] [PubMed] [Google Scholar]
- 6. Teerlink J. R., Cotter G., Davison B. A., Felker G. M., Filippatos G., Greenberg B. H., Ponikowski P., Unemori E., Voors A. A., Adams K. F. J., Dorobantu M. I., Grinfeld L. R., Jondeau G., Marmor A., Masip J., Pang P. S., Werdan K., Teichman S. L., Trapani A., Bush C. A., Saini R., Schumacher C., Severin T. M., Metra M. (2013) Serelaxin, recombinant human relaxin-2, for treatment of acute heart failure (RELAX-AHF): a randomised, placebo-controlled trial. Lancet 381, 29–39 [DOI] [PubMed] [Google Scholar]
- 7. Conrad K. P., Debrah D. O., Novak J., Danielson L. A., Shroff S. G. (2004) Relaxin modifies systemic arterial resistance and compliance in conscious, nonpregnant rats. Endocrinology 145, 3289–3296 [DOI] [PubMed] [Google Scholar]
- 8. Debrah D. O., Conrad K. P., Jeyabalan A., Danielson L. A., Shroff S. G. (2005) Relaxin increases cardiac output and reduces systemic arterial load in hypertensive rats. Hypertension 46, 745–750 [DOI] [PubMed] [Google Scholar]
- 9. Sasser J. M., Molnar M., Baylis C. (2011) Relaxin ameliorates hypertension and increases nitric oxide metabolite excretion in angiotensin II but not N-omega-nitro-L-arginine methyl ester hypertensive rats. Hypertension 58, 197–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. St-Louis J., Massicotte G. (1985) Chronic decrease of blood pressure by rat relaxin in spontaneously hypertensive rats. Life Sci. 37, 1351–1357 [DOI] [PubMed] [Google Scholar]
- 11. Bani-Sacchi T., Bigazzi M., Bani D., Mannaioni P. F., Masini E. (1995) Relaxin-induced increased coronary flow through stimulation of nitric oxide production. Br. J. Pharmacol. 116, 1589–1594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Danielson L. A., Sherwood O. D., Conrad K. P. (1999) Relaxin is a potent renal vasodilator in conscious rats. J. Clin. Invest. 103, 525–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Novak J., Ramirez R. J., Gandley R. E., Sherwood O. D., Conrad K. P. (2002) Myogenic reactivity is reduced in small renal arteries isolated from relaxin-treated rats. Am. J. Physiol. 283, R349–355 [DOI] [PubMed] [Google Scholar]
- 14. Van Drongelen J., Ploemen I. H. J., Pertijs J., Gooi J. H., Sweep F. C. G. J., Lotgering F. K., Spaanderman M. E. A., Smits P. (2011) Ageing attenuates the vasodilator response to relaxin. Am. J. Physiol. 300, H1609–H1615 [DOI] [PubMed] [Google Scholar]
- 15. Van Drongelen J., van Koppen A., Pertijs J., Gooi J. H., Parry L. J., Sweep F. C. G. J., Lotgering F. K., Smits P., Spaanderman M. E. A. (2012) Impaired vascular responses to relaxin in diet-induced overweight female rats. J. Applied. Physiol. 112, 969–969 [DOI] [PubMed] [Google Scholar]
- 16. Massicotte G., Parent A., St-Louis J. (1989) Blunted responses to vasoconstrictors in mesenteric vasculature but not in portal vein of spontaneously hypertensive rats treated with relaxin. Proc. Soc. Exp. Biol. Med. 190, 254–259 [DOI] [PubMed] [Google Scholar]
- 17. Li Y., Brookes Z. L. S., Kaufman S. (2005) Acute and chronic effects of relaxin on vasoreactivity, myogeinc reactivity and compliance of the rat mesenteric arterial and venous vasculature. Regul. Pept. 132, 41–46 [DOI] [PubMed] [Google Scholar]
- 18. McGuane J. T., Debrah J. E., Sautina L., Jarajapu Y. P. R., Novak J., Rubin J. P., Grant M. B., Segal M., Conrad K. P. (2011) Relaxin induces rapid dilation of rodent small renal and human subcutaneous arteries via PI3 kinase and nitric oxide. Endocrinology 152, 2786–2796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fisher C., MacLean M., Morecroft I., Seed A., Johnston F., Hillier C., McMurray J. (2002) Is the pregnancy hormone relaxin also a vasodilator peptide secreted by the heart? Circulation 106, 292–295 [DOI] [PubMed] [Google Scholar]
- 20. Sandoo A., Veldhuijzen van Zanten J. J. C. S., Metsios G. S., Caroll D., Kitas G. D. (2010) The endothelium and its role in regulating vascular tone. Open Med. J. 4, 302–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Debrah D. O., Debrah J. E., Haney J. L., McGuane J. T., Sacks M. S., Conrad K. P., Shroff S. G. (2011) Relaxin regulates vascular wall remodeling and passive mechanical properties in mice. J. Applied Physiol. 111, 260–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gooi J. H., Richardson M. L., Jelinic M., Girling J. E., Wlodek M. E., Tare M., Parry L. J. (2013) Enhanced uterine artery stiffness in aged pregnant relaxin mutant mice is reversed with exogenous relaxin treatment. Biol. Reprod 89, 18; 10.1095/biolreprod.113.108118 [DOI] [PubMed] [Google Scholar]
- 23. Chan S. L., Cipolla M. J. (2011) Relaxin causes selective outward remodeling of brain parenchymal arterioles via activation of peroxisome proliferator-activated receptor-gamma. FASEB J. 25, 3229–3239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chan S. L., Sweet J. G., Cipolla M. J. (2013) Treatment for cerebral small vessel disease: effect of relaxin on the function and structure of cerebral parenchymal arterioles during hypertension. [E-pub ahead of print] FASEB J. 10.1096/fj.13-230797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Xu Q., Chakravorty A., Bathgate R. A., Dart A. M., Du X. J. (2010) Relaxin therapy reverses large artery remodeling and improves arterial compliance in senescent spontaneously hypertensive rats. Hypertension 55, 1260–1266 [DOI] [PubMed] [Google Scholar]
- 26. Novak J., Parry L. J., Matthews J. E., Kerchner L. J., Indovina K., Hanley-Yanez K., Doty K. D., Debrah D. O., Shroff S. G., Conrad K. P. (2006) Evidence for local relaxin ligand-receptor expression and function in arteries. FASEB J. 20, 2352–2362 [DOI] [PubMed] [Google Scholar]
- 27. Ferreira V. M., Gomes T. S., Reis L. A., Ferreira A. T., Razvickas C. V., Schor N., Boim M. A. (2009) Receptor-induced dilatation in the systemic and intrarenal adaptation to pregnancy in rats. PLoS One 4, e4845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Failli P., Nistri S., Quattrone S., Mazzetti L., Bigazzi M., Sacchi T. B., Bani D. (2002) Relaxin up-regulates inducible nitric oxide synthase expression and nitric oxide generation in rat coronary endothelial cells. FASEB J. 16, 252–254 [DOI] [PubMed] [Google Scholar]
- 29. Kubota Y., Temelcos C., Bathgate R. A. D., Smith K. J., Scott D., Zhao C., Hutson J. M. (2002) The role of insulin-3, testosterone Mullerian inhibiting substance and relaxin in rat gubernacular growth. Mol. Cell. Endocrinol. 8, 900–905 [DOI] [PubMed] [Google Scholar]
- 30. Kern A., Bryant-Greenwood G. D. (2009) Characterization of relaxin receptor (RXFP1) desensitization and internalization in primary human decidual cells and RXFP1-transfected HEK293 cells. Endocrinology 150, 2419–2428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tse G. H., Marson L. P. (2013) A comparative study of 2 computer-assisted methods of brightfield microscopy images. [E-pub ahead of print] Appl. Immunohistochem. Mol. Morphol. 10.1097/PAI.0b013e31827ba0e1 [DOI] [PubMed] [Google Scholar]
- 32. Sherwood O. D., Crnekovic V. E., Gordon W. L., Rutherford J. E. (1980) Radioimmunoassay of relaxin throughout pregnancy and during parturition in the rat. Endocrinology 107, 691–698 [DOI] [PubMed] [Google Scholar]
- 33. Holobotovskyy V., Manzur M., Tare M., Burchell J., Bolitho E., Viola H., Hool L. C., Arnolda L. F., McKitrick D. J., Ganss R. (2013) Regulator of G protein signaling 5 controls blood pressure homeostasis and vessel wall remodeling. Circulation 112, 781–791 [DOI] [PubMed] [Google Scholar]
- 34. Mazzuca M. Q., Tare M., Parkington H. C., Dragomir N. M., Parry L. J., Wlodek M. E. (2012) Uteroplacental insufficiency programmes vascular dysfunction in non-pregnant rats: compensatory adaptations in pregnancy. J. Physiol. 590, 3375–3388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tare M., Parkington H. C., Coleman H. A. (2000) EDHF, NO and a prostanoid: hyperpolarization-dependent and -independent relaxation in guinea-pig arteries. Br. J. Pharmacol. 130, 605–618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gao Y. J., Yang L. F., Stead S., Lee R. M. (2008) Flow-induced vascular remodelling in the mesenteric artery of spontaneously hypertensive rats. Can. J. Physiol. Pharmacol. 86, 737–744 [DOI] [PubMed] [Google Scholar]
- 37. Conrad K. P., Novak J. (2004) Emerging role of relaxin in renal and cardiovascular function. Am. J. Physiol. 287, R250–R261 [DOI] [PubMed] [Google Scholar]
- 38. Zieman S. J., Melenovsky V., Kass D. A. (2005) Mechanisms, pathophysiology and therapy of arterial stiffness. Arterioscler. Thromb. Vasc. Biol. 25, 932–942 [DOI] [PubMed] [Google Scholar]
- 39. Dschietzig T., Brecht A., Bartsch C., Baumann G., Stangl K., Alexiou K. (2012) Relaxin improves TNF-α-induced endothelial dysfunction: the role of glucocorticoid receptor and phosphatidylinositol 3-kinase signalling. Cardiovasc. Res. 95, 97–107 [DOI] [PubMed] [Google Scholar]
- 40. Adams J., Schott S., Bern A., Renz M., Ikenberg K., Garbe C., Busch C. (2012) A novel role for relaxin-2 in the pathogenesis of primary varicosis. PLoS ONE 7, e39021. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.