

Abstract
Prostaglandin E2 (PGE2) regulates numerous biological processes by modulating transcriptional activation, epigenetic control, proteolysis, and secretion of various proteins. Scar formation depends on fibroblast elaboration of matrix proteins such as collagen, and this process is strongly suppressed by PGE2 through activation of cAMP-dependent protein kinase A (PKA). However, the actual mechanism by which PGE2-PKA signaling inhibits collagen expression in fibroblasts has never been delineated, and that was the objective of this study. PGE2 unexpectedly induced a rapid reduction in procollagen I protein expression in adult lung fibroblasts, with a half-maximum effect at 1.5 h. This effect reflected its inhibition of translation rather than transcription. Global protein synthesis was also inhibited by PGE2. This action was mediated by PKA and involved both activation of ribosomal protein (rpS6) and suppression of mammalian target of rapamycin (mTOR). Similar effects of PGE2 were demonstrated in mouse peritoneal macrophages (PMs). These findings identify inhibition of translation as a new mechanism by which PGE2 regulates cellular function and a novel example of translational inhibition mediated by opposing actions on two distinct translational control pathways. Translational inhibition would be expected to contribute to dynamic alterations in cell function that accompany the changing PGE2 levels observed in disease states and with various pharmacotherapies.—Okunishi K., DeGraaf, A. J., Zasłona, Z., Peters-Golden, M. Inhibition of protein translation as a novel mechanism for prostaglandin E2 regulation of cell functions.
Keywords: protein kinase A, ribosomal protein S6, mammalian target of rapamycin, fibroblasts, macrophages
Fibrotic tissue remodeling is the consequence of an aberrant repair response to injury. Fibrotic diseases affect virtually all vital organs, impair normal organ function, are largely irreversible without any effective treatment, and are a leading cause of morbidity and mortality (1). New therapeutic strategies for these disorders are desperately needed and may be informed by a better understanding of how scar formation is regulated. Fibroblasts are the primary effector cells responsible for elaborating matrix proteins such as collagen I, the predominant type of collagen produced by lung fibroblasts (2), which comprise the scar. The suppression of collagen I production by fibroblasts is an attractive strategy for preventing or halting this process (1).
Prostaglandin E2 (PGE2) is a pleiotropic lipid mediator that is well known to suppress most fibroblast functions, including collagen production (3–12). Indeed, an impairment in lung synthesis of PGE2 is associated with and very likely facilitates the development of pulmonary fibrosis (13, 14). The suppressive actions of PGE2 in fibroblasts are mediated by its ability to increase intracellular cAMP production after binding to its Gαs-coupled receptors EP2 and EP4 (9, 11). cAMP activates 2 downstream effectors: protein kinase A (PKA) and guanine nucleotide exchange protein activated by cAMP (Epac). PKA and Epac have both redundant and nonredundant roles in PGE2's regulation of cell functions. For example, we have shown in lung fibroblasts that PKA is responsible for suppressing expression of collagen I and that Epac is responsible for inhibiting cell proliferation (10). However, the actual mechanism by which PGE2-PKA signaling reduces collagen I protein levels remains unknown.
Steady-state levels of any cellular protein are determined by the balance of the processes of transcription, translation, and degradation. There are conflicting reports regarding PGE2 or cAMP regulation of mRNA expression of collagen I. An early study showed a reduction, albeit modest, of collagen I mRNA expression in lung fibroblasts (15), and a subsequent one showed no or only minimal effects (16). Degradation of collagen can be mediated by matrix metalloproteinases (MMPs) that possess collagenase activity. Data regarding the effects of PGE2 on MMPs in fibroblasts are similarly conflicting. For example, PGE2 was reported to increase MMP-2 expression and activity in WI-38 lung fibroblasts (16), but to have no effect on MMPs in human fetal lung (HFL)-1 fibroblasts (17). These limited data therefore suggest that PGE2 regulation of collagen I protein expression in lung fibroblasts is independent of its effects on transcription or MMP-dependent extracellular degradation.
In the current study, we sought to clarify the mechanisms by which PGE2 regulates collagen I expression in lung fibroblasts. We report that both endogenous and exogenous PGE2, via PKA signaling, suppressed translation of collagen I and global protein synthesis. Protein translation is well known to be enhanced by mammalian target of rapamycin (mTOR; reviewed in ref. 18), but data are conflicting regarding its suppression by ribosomal protein S6 (rpS6; 19, 20). In the current study, PGE2 inhibited mTOR while activating rpS6, and both of these regulatory events contributed to inhibition of collagen synthesis. PGE2 had similar effects on these two translational control pathways and on translation itself in macrophages, the key effector cells of inflammatory and innate immune responses.
Inhibition of translation provides a novel means by which PGE2 can regulate diverse cellular functions, including fibrogenesis. Our findings also predict that changes in protein translation accompany alterations in tissue levels of PGE2 observed in disease states, such as fibrosis, infection, inflammation, and cancer, and in response to commonly used medications, such as glucocorticoids and nonsteroidal anti-inflammatory agents.
MATERIALS AND METHODS
Reagents
Primary antibodies (Abs) for immunoblot analysis were obtained from the following suppliers: anti-human type I collagen for immunoblot analysis from CedarLane Laboratories (Burlington, ON, Canada) and for immunoprecipitation from Abcam (Cambridge, MA, USA) (both Abs predominantly detect a band for procollagen I, as described in Results); anti-GAPDH from Santa Cruz Biotechnology (Dallas, TX, USA); anti-total and anti-phosphorylated rpS6 and S6 kinase 1 (S6K1), anti-total binding protein-1 of eukaryotic initiation factor 4E (4E-BP1), and anti-X-chromosome-linked inhibitor of apoptosis (XIAP) from Cell Signaling Technologies (Danvers, MA, USA); and anti-cystatin C from Merck Millipore (Billerica, MA, USA). Secondary anti-rabbit and anti-murine Abs for immunoblot analyses were from Cell Signaling Technologies. Nonspecific control rabbit IgGs for immunoprecipitation were obtained from Santa Cruz Biotechnology. Protein A Sepharose for immunoprecipitation was from GE Healthcare (Little Chalfont, UK). Myristoylated PKA inhibitory peptide 14–22 (PKI), cycloheximide (CHX), and the nonselective cyclooxygenase (COX) inhibitor aspirin [acetylsalicylic acid (ASA)] were purchased from Sigma-Aldrich (St. Louis, MO, USA). PGE2 and the EP2 antagonist PF-04418948 were obtained from Cayman Chemicals (Ann Arbor, MI, USA) and dissolved in DMSO. Control siRNA and siRNA targeting rpS6 were On-Target plus Smart pool products from Thermo Scientific (Waltham, MA, USA). Dulbecco's modified Eagle's medium (DMEM), methionine-free DMEM, RPMI 1640, Opti-MEM I reduced-serum medium, RNAiMAX for siRNA transfection, Click-IT l-azidohomoalanine (AHA) for nascent protein synthesis, and the Click-IT Biotin Protein Analysis Detection Kit were purchased from Life Technologies (Carlsbad, CA, USA). The PGE2 ELISA kit was obtained from Enzo Life Sciences (Farmingdale, NY, USA), the protease inhibitor from Roche (Basel, Switzerland), and the Phosphatase Inhibitor Cocktail Set I and II from Calbiochem-Novabiochem (Beeston, UK).
Human lung fibroblast culture
Human adult fibroblasts from normal lung (CCL210) and HFL fibroblasts (IMR-90) were purchased from American Type Culture Collection (Manassas, VA, USA). The cells were cultured at 37° in 5% CO2. The HFL fibroblasts were cultured in DMEM supplemented with 10% fetal bovine serum (FBS; Thermo Scientific) and 1% penicillin/streptomycin and were used in experiments at passages 6–10. No apparent change in fibroblast responsiveness to PGE2 was noted during cell passage. Human fibroblasts were plated in Falcon 6-well plates (BD Biosciences, San Jose, CA, USA) at 5 × 105 cells/well, or at 2–3 × 105 cells/well for RNA silencing. They were allowed to adhere for 8–10 h in serum-containing DMEM and then cultured in serum-free DMEM for 16–20 h. After the medium was removed, the cells were treated as indicated. Cell lysates were obtained with lysis buffer consisting of PBS with 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, 1× protease inhibitor, and 1:100 dilution of Phosphatase Inhibitor Cocktail Set I and II (21).
Animals
Naive C57BL/6 male mice (8–12 wk) were purchased from The Jackson Laboratory (Bar Harbor, ME, USA) and bred in the University of Michigan Unit for Laboratory Animal Medicine. The animals were treated according to U.S. National Institutes of Health guidelines for the care and use of experimental animals, with the approval of the University of Michigan Committee for the Use and Care of Animals.
Culture of mouse peritoneal macrophages (PMs)
Resident mouse PMs were isolated by lavage of the peritoneal cavity with 5 ml PBS (22). The PMs were incubated in RPMI 1640 (Life Technologies) supplemented with 10% FBS and 1% penicillin/streptomycin for 2 h at 4–6 × 106 cells/well and were allowed to adhere to plastic 6-well plates. The cells were washed twice with serum-free medium for removal of nonadherent cells and were then treated as indicated in serum-free medium.
RNA silencing
CCL210 human adult lung fibroblasts were incubated with 50 nM of rpS6-targeting or control siRNA plus 5 μl RNAiMAX in 2 ml Optimem I (Life Technologies) reduced-serum medium without antibiotics. After 72 h, cells were washed twice with serum- and antibiotic-free DMEM, then incubated in serum-free DMEM with antibiotics in the presence or absence of PGE2 at 500 nM for 4 h.
Quantitative PCR and RNA isolation
Cells were suspended in 1 ml Trizol (Life Technologies), and RNA was extracted (21). RNA was amplified by quantitative RT-PCR performed with a SYBR Green PCR kit (Life Technologies) on an ABI Prism 7300 thermocycler (Life Technologies). Relative gene expression was determined by the ΔCT method, with β-actin used as the reference gene. Primer sequences were as follows: COL1A1 forward, 5′-GGGCAAGACAGTGATTGAATA-3′, reverse, 5′-ACGTCGAAGCCGAATTCCT-3′); COL1A2 forward, 5′-TCTCTACTGGCGAAACCTGTA-3′, reverse, 5′-TCCTAGCCAGACGTGTTTCTT-3′ (23), and β-actin forward, 5′-GCCACGGCTGCTTCCA-3′, reverse, 5′-GAACCGCTCATTGCCATTG-3′ (9).
Immunoblot analysis
Protein (12–25 μg) in scraped cell lysates containing both intracellular and extracellular proteins was loaded for electrophoresis, and subsequent immunoblot and densitometric analyses were performed (21) with the primary Abs described earlier. Densitometric values of bands for procollagen I were normalized to GAPDH detected in the same membrane, and relative levels of procollagen I were expressed as defined in the figure legends. Based on the molecular mass of target proteins, 8, 10, or 12.5% acrylamide gels were used. For normalization of p-S6K1 or p-rpS6 to total S6K1 or total rpS6, respectively, an amount of lysate protein identical with that used for immunoblot against phosphorylated protein was loaded on another parallel gel for electrophoresis, and total protein expression was detected by immunoblot analysis.
Detection of nascent proteins
Newly synthesized proteins were detected by using the Click-IT method (Life Technologies) according to the manufacturer's instructions. In brief, cells were incubated in methionine-free DMEM for 30 min before being pulsed with the nonradioactive, azide-containing methionine analog AHA for 40 min. Click-IT chemistry was subsequently used to biotin conjugate the methionine analog incorporated in the proteins. After electrophoresis of cell lysates on 8 or 10% acrylamide gels and transfer of proteins onto the membranes, streptavidin-HRP was applied to the membranes to detect newly synthesized proteins that contained biotin-conjugated AHA.
Immunoprecipitation
For procollagen I immunoprecipitation, cell lysates were incubated overnight at 4°C with anti-collagen I Ab or control rabbit IgG (both at 2.5 μg/ml). Protein A-Sepharose was added and incubated for 3–4 h with rotation at 4°C, and immunoprecipitates were isolated and subjected to electrophoresis, as described previously (24). Total protein (80–120 μg) after Click-IT reaction was used for immunoprecipitation, and immunoprecipitates were subjected to electrophoresis and immunoblot analysis to detect both total procollagen I and newly synthesized procollagen I containing biotin-conjugated AHA.
Statistical analyses
Data are presented as means ± sem of values determined from ≥3 experiments or, in some instances, actual values from 2 experiments. Data analysis was performed with Prism software (GraphPad, San Diego, CA, USA), using Student's t test or ANOVA with Tukey's or Dunnett's multiple-comparison test, as appropriate, to determine significant differences between group means. In all instances, statistical significance was inferred from a value of P < 0.05.
RESULTS
Rapid suppression by PGE2 of procollagen I protein expression in adult lung fibroblasts
It should be noted that the anti-collagen I Ab used detects a single or a predominant band at a molecular mass between 150 and 250 kDa, which actually corresponds to the immature form of collagen I, termed procollagen I, and we have designated this band as procollagen I throughout the article. Procollagen I is synthesized intracellularly and forms a triple helix composed of two α1 chains and an α2 chain. After its secretion into the extracellular space, procollagen I undergoes removal of extension peptides to form mature collagen I and subsequently collagen fibrils. Procollagen I is the molecular species that is susceptible to regulation at the biosynthetic and degradative levels, and such regulation results in changes in mature collagen I levels (25).
We first examined the time course of PGE2-induced suppression of collagen I protein expression in CCL210 adult lung fibroblasts obtained from histologically normal lung. Surprisingly, PGE2 at 500 nM, a concentration that we previously reported leads to maximum suppression of collagen I expression after overnight incubation (18–20 h) in normal adult (9) or HFL (10) fibroblasts, unexpectedly decreased procollagen I protein expression very quickly, with a statistically significant effect observed as early as 1 h, a half-maximum effect at ∼1.5 h, and a maximum effect of ∼90% reduction within 8 h (Fig. 1A). We also confirmed the dose-dependent ability of PGE2 to elicit a rapid suppression of procollagen I protein expression at the 4 h time point (Fig. 1B). Effects of PGE2 at 4 h were detectable at 1–10 nM, half-maximum at ∼100 nM, and maximum at ∼250–1000 nM. The shape of this dose–response relationship paralleled that which we had observed for overnight incubations (9, 10). Since PGE2 exerted its maximum suppressive effect on procollagen I expression at 500 nM, we used this concentration in subsequent mechanistic experiments, unless otherwise indicated. As mentioned above, procollagen I is composed of two α1 chains and an α2 chain (26), and we therefore sought to clarify the effect of PGE2 on levels of mRNA encoding both procollagen I α1 (ColIA1) and α2 (ColIA2). PGE2 had no substantial inhibitory effects on mRNA expression of either ColIA1 or ColIA2 at time points ranging from 1 to 24 h (Fig. 1C), indicating that PGE2 reduces procollagen I protein expression at a posttranscriptional level. We speculate that the apparent increase in ColIA2 mRNA at 24 h represents an attempt to compensate for the reduction in its protein level.
Figure 1.
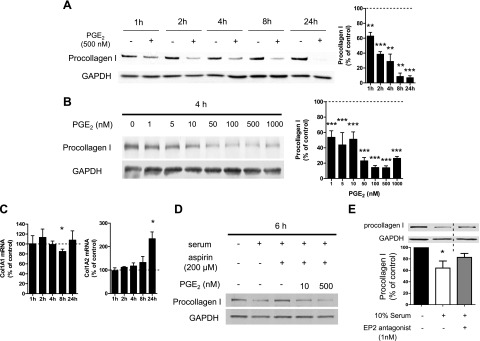
Rapid suppression by PGE2 of procollagen I protein expression in normal adult lung fibroblasts. A–C) After overnight serum starvation, cell culture medium was changed to fresh serum-free medium, and normal adult lung fibroblasts were then incubated with or without PGE2 (at 500 nM in A and at various concentrations in B). A, B) Time course (A) and dose dependency (B) of the effect of PGE2 on procollagen I protein expression. At each time point after addition of PGE2 at the indicated concentrations, cell lysates were harvested, and procollagen I levels in the lysates were determined by immunoblot analysis with densitometry. Left panel: representative immunoblot. Right panel: results of densitometric analysis of procollagen I levels from 3–5 experiments (A) or 3 experiments (B). After normalization to GAPDH, the procollagen I level in PGE2-treated cells was expressed relative to that in cells without PGE2 (expressed as 100%, dashed line) at each time point in each experiment. Data represent means ± sem. C) PGE2 did not suppress collagen I mRNA expression. At the indicated time points, ColIα1 (left panel) and ColIα2 (right) mRNA levels in cell lysates were determined by semiquantitative real-time RT-PCR and then were expressed relative to the level in the no-PGE2 control condition (expressed as 100%, dashed line) at each time point in each experiment. Data are expressed as means ± sem from 4–5 (left panel) or 3–4 (right panel) experiments. *P < 0.05, **P < 0.01, ***P < 0.001 vs. control fibroblasts without PGE2 treatment. D, E) Serum decreased procollagen I protein expression via the PGE2-EP2 pathway. After cells were serum starved overnight, serum-free medium was removed, and cells were incubated in serum-containing medium, with or without aspirin (200 μM) (or its vehicle DMSO at a final concentration of 0.2%) in the absence or simultaneous presence of PGE2 (10 or 500 nM; D), or with or without the EP2 antagonist PF-04418948 (1 nM; E) for 6 h. Then collagen I protein levels in the cell lysates were determined by immunoblot analysis. Immunoblot representative of 3 experiments is shown. Lanes separated by the vertical dashed line were from the same blot, but were not contiguous in the original gel. After normalization to GAPDH, the procollagen I level in PGE2-treated cells was expressed relative to that in cells treated without serum (expressed as 100%) in each experiment (E). Data represent means ± se. *P < 0.05 vs. control fibroblasts without serum treatment.
PGE2 is the predominant endogenous prostanoid product of normal lung fibroblasts (14), and we therefore evaluated whether endogenous PGE2 regulates procollagen I protein expression in a manner similar to the exogenously added prostanoid. Serum contains numerous growth factors that increase expression of inducible COX-2 and subsequent PGE2 production in fibroblasts (27). After overnight serum starvation, cells were incubated in fresh medium for various intervals, with or without 10% FBS, and in the presence or absence of the COX inhibitor ASA, and PGE2 concentrations in the supernatants and procollagen I protein expression in the cell lysates were examined. The PGE2 content of the supernatants was markedly increased by 6 h after addition of serum-containing medium (Supplemental Fig. S1A), and a parallel reduction in procollagen I protein expression was observed at 6 h (Fig. 1D and Supplemental Fig. S1B). Both the induction of PGE2 and the inhibition of procollagen I were completely reversed by simultaneous administration of ASA at 400 μM (Supplemental Fig. S1). ASA likewise inhibited serum suppression of procollagen I at the lower dose of 200 μM (Fig. 1D). The inhibitory effect of ASA on serum suppression of procollagen I was dose dependently overcome by exogenous PGE2 (Fig. 1D). There are 4 receptors for PGE2 [E prostanoid (EP) 1–4], and the suppressive effect of PGE2 on procollagen I protein levels is mainly mediated via ligation of the Gαs-coupled EP2 receptor in normal lung fibroblasts (9). To determine whether the effect of serum described above is also mediated by PGE2-EP2 signaling, we used a newly developed, potent, selective EP2 antagonist, PF-04418948 (28). This EP2 antagonist reversed serum suppression of procollagen I protein expression, as did ASA (Fig. 1E). These results demonstrate that endogenous PGE2 shares with exogenous PGE2 the ability to rapidly decrease procollagen I protein expression in adult lung fibroblasts.
PGE2 suppressed procollagen I protein synthesis by inhibiting its translation in adult lung fibroblasts
Posttranscriptional regulation of procollagen I protein by PGE2 could be directed at either translation or degradation steps. We assessed the effects of PGE2 on turnover of procollagen I protein in cells pretreated for 30 min with CHX, a well-known protein translation inhibitor. In the absence of PGE2, procollagen I protein decreased in CHX-treated fibroblasts with a half-life of ∼1.5 h, and turnover was essentially complete by 4.5 h (Fig. 2A). It should be noted that because CHX was added 30 min before PGE2, the time points indicated in Fig. 2A, B actually reflect an additional 30 min of incubation in CHX. Such a rapid turnover of procollagen I protein is in fact consistent with results reported previously (reviewed in refs. 29, 30). The addition of PGE2 to CHX-treated cells had no further effect on procollagen I turnover, indicating a failure of the prostanoid to directly influence degradation of this protein. We obtained similar results with another translation inhibitor, puromycin (Supplemental Fig. S2).
Figure 2.
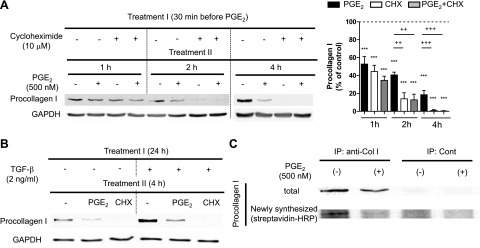
Inhibition by PGE2 of collagen I protein translation in normal adult lung fibroblasts. A) Rapid suppression of collagen I protein expression by CHX and PGE2. After overnight serum starvation, cells were cultured in serum-free medium with or without the translation inhibitor CHX (10 μM), PGE2 (500 nM), or both. CHX was added first, and 30 min later, PGE2 was added to the indicated wells without changing the medium. At the indicated time points after addition of PGE2, cell lysates were harvested, and collagen I levels in cell lysates were determined by immunoblot analysis with densitometry. Left panel: representative immunoblot. Right panel: results of densitometric analysis of collagen I levels from 5 experiments. After normalization to GAPDH, collagen I was expressed relative to that in control without PGE2 at each time point in each experiment. Data represent means ± se. ***P < 0.001 vs. control fibroblasts without PGE2 treatment; ++P < 0.01, +++P < 0.001 between indicated groups. B) Rapid suppression by PGE2 and CHX of TGF-β-enhanced collagen I protein expression. After overnight serum starvation, medium was changed, and cells were treated with or without TGF-β (2 ng/ml) for 24 h. Following removal of medium, cells were incubated in fresh serum-free medium with or without PGE2 (500 nM) or CHX (10 μM) for 4 h, after which cell lysates were harvested, and collagen I levels in cell lysates were determined by immunoblot analysis. Immunoblot representative of 2 experiments is shown. C) Suppression by PGE2 of translation of collagen I protein in normal adult lung fibroblasts. After overnight serum starvation, cells were washed once with methionine- and serum-free medium, and then were cultured in methionine- and serum-free medium with or without PGE2 (500 nM) for 30 min. Methionine analog AHA was added at final concentration 25 μM into each well, and cells were further incubated with AHA for 40 min. Cell lysates were harvested, and Click-iT reaction was performed on cell lysates to conjugate biotin to the AHA analog incorporated into newly synthesized proteins. After immunoprecipitation with anti-collagen I Ab, immunoprecipitates were electrophoresed, and total collagen I protein expression was first determined by immunoblot analysis (top panel). Then, anti-collagen I Ab was stripped from the membrane, and newly synthesized collagen I protein containing biotin-conjugated AHA was detected with streptavidin-HRP (bottom panel). Immunoblot is representative of 3 experiments.
Data presented thus far are limited to the regulation of basal expression of procollagen I, but fibroblasts are well known to up-regulate their production of extracellular matrix proteins in response to profibrotic growth factors, such as TGF-β. We therefore determined the effects of PGE2 and CHX on TGF-β-stimulated procollagen I protein expression. Under these conditions, both PGE2 and CHX elicited a marked reduction in procollagen I protein within 4 h (Fig. 2B). Together, these results strongly suggest that PGE2 suppresses collagen I protein expression via the same pathway as does CHX—namely, inhibition of translation.
We sought to confirm this finding directly by studying the effect of PGE2 on newly synthesized procollagen I protein. After a 30 min preincubation with or without PGE2 in methionine-free medium, fibroblasts were pulsed with the nonradioactive methionine analog AHA for 40 min, after which the cell lysates were harvested. A commercially available kit to detect protein synthesis (Click-iT; Invitrogen) was used to biotin conjugate the methionine analog incorporated into proteins and streptavidin-HRP, to detect proteins synthesized during the methionine analog pulse. Procollagen I protein in lysates was purified by immunoprecipitation with anticollagen I Ab. After electrophoresis of immunoprecipitates, total and newly synthesized procollagen I proteins were detected with anti-collagen I Ab or with streptavidin-HRP, respectively. As shown in Fig. 2C, PGE2 strongly suppressed incorporation of methionine analog into procollagen I protein, representing an inhibitory effect on neosynthesis of procollagen I during this brief period of pulse labeling. These data strongly support the conclusion that PGE2 suppresses procollagen I synthesis at the level of translation.
PGE2 suppressed global protein synthesis in fibroblasts
We next used the Click-iT system to examine the effect of PGE2 on global protein synthesis. CCL210 adult lung fibroblasts were treated as in Fig. 2C, with or without PGE2 or CHX, for 30 min, followed by a 40-min pulse with methionine analog. Cell lysates were harvested and subjected to gel electrophoresis, and newly synthesized proteins transferred to the membrane were detected with streptavidin-HRP and quantitated by densitometry of the entire lane (Fig. 3A). CHX strongly (∼75%) suppressed neosynthesis of total proteins, as expected, whereas the suppressive effect of PGE2 was of lesser magnitude (∼30%), but nonetheless significant. Whereas CHX uniformly reduced the density of all protein bands (Supplemental Fig. S3), the inhibitory effect of PGE2 on synthesis of individual protein bands was variable.
Figure 3.
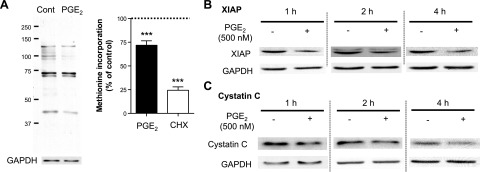
PGE2 inhibition of global protein synthesis in fibroblasts at the translation step. A) PGE2 inhibition of global protein synthesis. After overnight serum starvation, normal adult lung fibroblasts were washed once with methionine- and serum-free medium and then were cultured in methionine- and serum-free medium, with or without PGE2 (500 nM) or CHX (10 μM) for 30 min. Methionine analog AHA was added at final concentration of 25 μM into each well, and cells were further incubated with AHA for 40 min. Then, cell lysates were harvested, and a Click-iT reaction was performed on cell lysates to conjugate biotin to AHA analog incorporated into newly synthesized proteins. After electrophoresis of the cell lysates, newly synthesized proteins containing biotin-conjugated AHA were detected with streptavidin-HRP. Left panel: representative immunoblot. Right panel: results of densitometric analysis from 5–6 experiments. Densitometry of the entire lane is expressed relative to that in control without any treatment (expressed as 100%, dashed line) in each experiment. ***P < 0.001 vs. no-treatment control. B, C) Suppression by PGE2 of expression of short half-life proteins other than collagen I in normal lung fibroblasts. After overnight serum starvation, cells were incubated in fresh serum-free medium, with or without PGE2 (500 nM). At the indicated time points after addition of PGE2, the cell lysates were harvested, and XIAP (B) and cystatin C levels (C) were detected by immunoblot analysis. Immunoblot is representative of 3 experiments.
The effect of PGE2's ability to inhibit translation on steady-state protein levels would be expected to be magnified in proteins with rapid turnover, similar to procollagen I. We therefore also examined its effect on steady-state levels of two additional proteins known to have a short half-life: XIAP and cystatin C. As was the case for procollagen I, PGE2 decreased expression of XIAP (Fig. 3B) and cystatin C (Fig. 3C) within 1–2 h. A reduction in XIAP synthesis by CHX has been observed to be necessary for Fas ligand to induce apoptosis in fibroblasts (31). Taken together, these data demonstrate that PGE2 suppresses global protein synthesis in adult lung fibroblasts.
PGE2 suppressed the mTOR pathway and activated rpS6 in fibroblasts
Translation initiation is the rate-limiting step in protein synthesis, and one of its key controllers is mTOR. This serine/threonine kinase is the catalytic subunit of mTOR complexes (mTORCs) 1 and 2. Each complex has distinct roles in regulation of cell functions (18), with mTORC1 recognized as a key promoter of translation initiation via its phosphorylation of the substrates S6K1 and 4E-BP1. We first asked whether PGE2 suppresses mTORC1 activity, by assessing its effects on phosphorylation of S6K1 and 4E-BP1. Indeed, PGE2 reduced phosphorylation of both of these mTOR substrates within 5 to 10 min (Fig. 4A). Although the rate of dephosphorylation of both substrates was similar to that elicited by the well-established mTORC1 inhibitor rapamycin, the extent of dephosphorylation was slightly less (Fig. 4C). By contrast, PGE2 reduced collagen I protein expression at early time points to a greater extent than did rapamycin (Fig. 4D). Moreover, the inhibitory effects of PGE2 on procollagen I protein expression were additive to those of rapamycin at 4 h (Fig. 4D, right panel). These comparative results with rapamycin suggest that, although PGE2 inhibits mTORC1, this action is unlikely to fully explain its inhibition of procollagen I synthesis.
Figure 4.
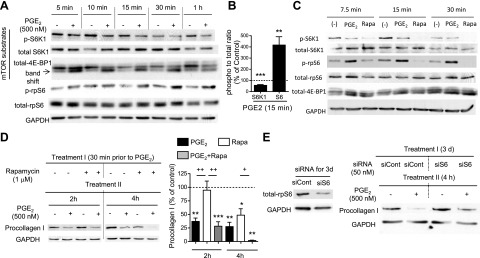
PGE2 suppressed phosphorylation of mTOR substrates while enhancing phosphorylation of rpS6 in normal adult lung fibroblasts. A–C) PGE2-induced suppression of the mTOR pathway along with enhancement of phosphorylation of rpS6. After overnight serum starvation, serum-free medium was changed, and cells were incubated with or without PGE2 (500 nM) or rapamycin (1 μM). At the indicated time points, cell lysates were harvested, and total and phosphorylated S6K1, 4E-BP1, and rpS6 levels in cell lysates were determined by immunoblot analysis. A) Representative immunoblot for PGE2 regulation of the mTOR pathway and rpS6. B) Densitometric ratios of phospho:total S6K1 and rpS6 at 15 min after addition of PGE2, expressed relative to that in control fibroblasts without PGE2 treatment. Dashed line indicates 100%. Data were obtained from 5 experiments and are expressed as means ± sem. **P < 0.01, ***P < 0.001 vs. control fibroblasts without PGE2. C) Comparison of regulation by PGE2 vs. rapamycin (Rapa) of mTOR substrates and rpS6. Immunoblot for phosphorylation of mTOR substrates and rpS6 representative of 3 experiments is shown. D) Stronger suppression of collagen I protein expression by PGE2 than by rapamycin. After overnight serum starvation, medium was changed, and cells were cultured in serum-free medium, with or without rapamycin (1 μM) and PGE2 (500 nM). Rapamycin was added first; 30 min later, PGE2 was added where indicated without changing the medium. At the indicated time points after addition of PGE2, cell lysates were harvested, and collagen I levels in the cell lysates were determined by immunoblot analysis with densitometry. Left panel: representative immunoblot. Right panel: results of densitometric analysis of collagen I levels in 3–5 experiments. After normalization to GAPDH, collagen I was measured relative to that in the no-PGE2 control at each time point in each experiment. Data represent means ± se. *P < 0.051, **P < 0.01,***P < 0.001 vs. control fibroblasts without PGE2 treatment; +P < 0.05, ++P < 0.01 between indicated groups. E) Suppression by PGE2 of collagen I protein levels in normal adult lung fibroblasts was blunted by siRNA that targets rpS6. After incubation of cells in Optimem serum-free medium with control siRNA or siRNA targeting rpS6 for 3 d, cells were either harvested to determine the efficiency of silencing the target protein or were further incubated in serum-free DMEM, with or without PGE2 (500 nM), for 4 h. Cell lysates were collected, and collagen I levels in the lysates were determined by immunoblot analysis. Left panel: reduction of rpS6 protein expression with siRNA specific for rpS6. Right panel: effect of silencing of rpS6 on PGE2-induced suppression of collagen I protein expression. Immunoblot is representative of 3 experiments.
Active S6K1 phosphorylates and activates 3 distinct downstream substrates that influence protein translation. Among these, eukaryotic initiation factor 4B (eIF4B) and eukaryotic elongation factor 2 (eEF2) promote protein translation, whereas rpS6 inhibits it (reviewed in refs. 19, 20). Thus, the net effect of S6K1 on protein translation can be defined by the balance between eIF4B and eEF2 vs. rpS6 pathways. S6K1 dephosphorylation would be expected to concomitantly reduce phosphorylation of its substrate rpS6, and this was indeed the observed effect of rapamycin (Fig. 4C). By contrast, PGE2 caused a marked increase in phosphorylation of rpS6, despite the inhibited activation of S6K1 (Fig. 4A–C). Enhancement of rpS6 phosphorylation by PGE2 was dose dependent (Supplemental Fig. S4A) over the same concentration range at which procollagen I was suppressed and lasted for more than 8 h (Supplemental Fig. S4B). We concluded that this distinctive ability of PGE2 to phosphorylate and thereby activate rpS6 could represent an mTORC1-independent mechanism contributing to PGE2 inhibition of procollagen I synthesis. To evaluate this possibility, rpS6 was knocked down with siRNA. The ∼50% knockdown of rpS6 protein abrogated the inhibitory effect of PGE2 on procollagen I levels (Fig. 4E). Activation of the translation suppressor rpS6 is thus a mechanism for PGE2 inhibition of procollagen I synthesis that is not shared by rapamycin and one that can explain how PGE2 potentiates rapamycin's effects. These data demonstrate that PGE2 suppresses procollagen I protein synthesis by both inhibiting mTORC1 and activating rpS6.
PKA mediated PGE2-induced suppression of mTORC1 as well as activation of rpS6
We previously identified PKA as the primary cAMP effector that mediates PGE2-induced suppression of procollagen I expression in normal lung fibroblasts (10). In the current study, we confirmed that a PKA inhibitor attenuates the rapid (within 4 h) suppression by PGE2 of procollagen I expression (Fig. 5A), just as we had reported it to attenuate the suppression of procollagen I after 20 h incubation with PGE2 (10). We next sought to clarify the role of PKA in PGE2 regulation of the pathways controlling protein synthesis, as described in Fig. 4, and found that a PKA inhibitor likewise attenuated both the PGE2-induced suppression of mTOR activity (as assessed by monitoring phosphorylation of S6K1; Fig. 5B) and its enhancement of phosphorylation of rpS6 (Fig. 5C). These data implicate PKA as the primary effector downstream of PGE2, which is responsible for inhibiting protein translation through both of these control pathways.
Figure 5.
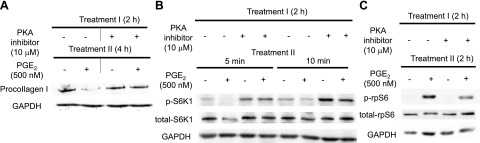
Actions of PGE2 in lung fibroblasts were attenuated by a PKA inhibitor. After overnight serum starvation, serum-free medium was replaced, and cells were preincubated with or without the PKA inhibitor myr-PKI (10 μM; refs. 14–22) for 2 h. PGE2 (500 nM) was added, and cell lysates were harvested at the indicated time points after addition of PGE2. Levels of collagen I (A), phospho-S6K1 (B), and phospho-rpS6 (C) in cell lysates at the indicated time points were determined by immunoblot analysis. PKA inhibitor interfered with PGE2-induced suppression of collagen I (A) and phospho-S6K1 (B) levels, as well as enhancement of phorpho-rpS6 (C). Immunoblot is representative of 2 experiments.
PGE2 suppressed global protein synthesis in macrophages
Macrophages have pleiotropic roles in both homeostasis and inflammation (32), and, like fibroblasts, many of their activation functions are suppressed by PGE2 (reviewed in refs. 33, 34). We therefore asked whether PGE2 has inhibitory effects on protein synthesis in macrophages similar to those in fibroblasts. Indeed, PGE2 significantly suppressed new protein synthesis (Fig. 6A) in mouse PMs, and this effect was associated with suppression of mTORC1 activity and enhancement of rpS6 phosphorylation (Fig. 6B). However, the magnitude of these PGE2 effects was more modest than observed in fibroblasts. Thus, these results suggest that regulation by PGE2 of pathways controlling protein translation is not limited to fibroblasts and is likely to be generalizable, to some degree; however, cell specificity may pertain in the magnitude of these effects.
Figure 6.
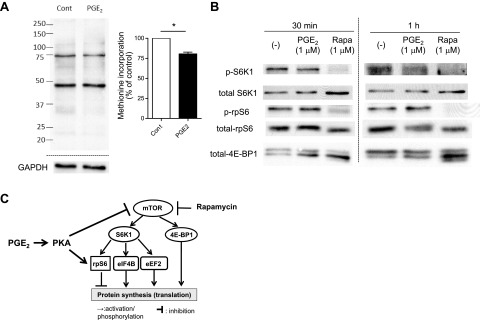
PGE2 inhibited global protein synthesis, suppressed the mTOR pathway, and enhanced activation of rpS6 in mouse PMs. PMs were harvested by lavage from naive C57BL/6 mice and were plated in serum-containing RPMI medium at 3–5 × 106 cells/ml for 2 h. A) PGE2-induced suppression of global protein synthesis in mouse PMs. After 2 h incubation in serum-containing medium, mouse PMs were washed twice with methionine- and serum-free medium and then were cultured for 1 h in the medium, with or without PGE2 (1 μM). Methionine analog AHA was added to each well at a final concentration of 25 μM, and cells were further incubated with AHA for 40 min. Then, cell lysates were harvested, and the Click-iT reaction was performed on the cell lysates to conjugate biotin to AHA analog incorporated into newly synthesized proteins. After electrophoresis of cell lysates, newly synthesized proteins containing biotin-conjugated AHA were detected with streptavidin-HRP. Left panel: representative immunoblot. Right panel: densitometry results from 3 experiments. Densitometry of the entire lane was expressed relative to that in control without any treatment (expressed as 100%, dashed line) in each experiment. *P < 0.05 vs. control without PGE2 treatment. B) PGE2 suppressed the mTOR pathway while enhancing activation of rpS6 in mouse PMs. After 2 h incubation in serum-containing medium, cells were washed twice with serum-free medium to remove nonadherent cells and then were incubated in the medium, with or without PGE2 (1 μM) or rapamycin (Rapa, 1 μM). At the indicated time points, cell lysates were harvested, and levels of phosphorylated S6K1 and rpS6 in the lysates were determined by immunoblot analysis. Immunoblot representative of 3 experiments is shown. C) Summary of the findings. PGE2, via PKA, activates rpS6 while suppressing the mTORC1 pathway; both actions contribute to inhibition of protein translation
DISCUSSION
In our study, PGE2, the most abundant and well studied of the prostanoid family of COX-derived lipid mediators, inhibited translation of proteins in fibroblasts and macrophages. This action was mediated by PKA and involved divergent regulation of two distinct pathways controlling translation initiation: suppression of the permissive mTORC1 pathway and activation of the inhibitory rpS6 pathway (as depicted in Fig. 6C). These findings provide a novel paradigm for understanding how this ubiquitous prostanoid regulates diverse cell functions.
mTORC1 is recognized as a master regulator of protein translation. Of interest, the suppressive effects of the well-known mTORC1 inhibitor rapamycin on steady-state levels (Fig. 4D) of procollagen I protein were weaker than those of PGE2. These findings suggested that PGE2 inhibits protein synthesis by modulating an additional translational control pathway other than mTORC1. In this regard, we found that PGE2 strongly enhanced phosphorylation (and hence activation) of rpS6 (Fig. 4A–C). Moreover, knockdown of rpS6 interfered with PGE2-induced reduction of procollagen I (Fig. 4E). These results suggest that activation of rpS6 is more important for PGE2-mediated reduction of procollagen I protein expression in lung fibroblasts than is suppression of the mTOR pathway. The role of rpS6 in the control of protein translation has been somewhat controversial. Although rpS6 was once thought to facilitate translation, recent data suggest instead that it functions as a negative regulator of translation, serving to restrain energy waste in certain settings of mTORC1 activation (20). Our findings lend support to rpS6's functioning in a negative regulatory role in this experimental system.
S6K1 is a critical regulator of phosphorylation of rpS6, and generally, phosphorylation/activation of rpS6 parallels that of S6K1. This pattern is the same as that observed with rapamycin treatment. However, PGE2 paradoxically enhanced phosphorylation of rpS6 while suppressing that of S6K1. To our knowledge, PGE2 is the first example of a single substance's modulating phosphorylation of rpS6 and S6K1 in opposing directions. PKA was implicated in both of these actions of PGE2 (Fig. 5A, B), and PKA has notably been shown to be capable of phosphorylating rpS6 independent of S6K1 (35). Indeed, the program GPS 2.0 (http://gps.biocuckoo.org/down.php) indicates that the S235/S236 residues of rpS6 (whose phosphorylation we assayed in this study) are candidates for direct phosphorylation by PKA. Such a direct role for PKA in our system remains to be established.
The conclusion that PGE2 suppresses mTORC1 was based on its ability to suppress phosphorylation of two mTORC1 substrates: S6K1 and 4E-BP1. Studies of a PKA inhibitor implicated PKA in these PGE2 effects. Since mTORC1 is a well-known inhibitor of autophagy (18), suppression of mTORC1 by the PGE2-PKA axis would be predicted to promote autophagy, reflecting yet another novel mechanism for the regulation of cell function by PGE2. Although PGE2 and cAMP have been reported in some experimental systems to increase activation of mTORC1 (36, 37), the suppressive actions observed in our system are consistent with other reports of inhibition of mTORC1 by cAMP (38, 39) and by PKA (39, 40) in cell types other than the fibroblasts and macrophages used in the current study.
It is unclear whether the suppression of mTORC1 reflects a direct or indirect action of PKA. PKA regulatory subunit Iα has been reported to colocalize with mTOR (40), and cAMP-PKA has been noted to cause dissociation of mTORC1 (39). Both of these actions were associated with suppression of mTORC1 activity. Alternatively, it is possible that PGE2-PKA indirectly opposes mTORC1 activity via phosphatase activation. For example, we have shown that PGE2 enhances fibroblast activity of phosphatase and tensin homologue deleted on chromosome 10 (PTEN; refs. 8, 11), a phosphatase that suppresses fibroblast survival and activation by opposing the PI3 kinase/Akt pathway. Akt is an upstream activator of mTORC1 (18), and it is possible that PGE2 indirectly suppresses mTORC1 activity by PTEN-mediated suppression of Akt signaling. A similar outcome could result from PGE2-PKA activation of Src homology domain 2–containing phosphotyrosine phosphatase (SHP)-2, since SHP-2 is activated by PKA (41) and also down-regulates mTORC1 activity, possibly through a well-known negative regulator of mTORC1, 5′-AMP-activated protein kinase (42).
PGE2 appeared to differentially inhibit translation of individual proteins, as evidenced by the electrophoretic patterns observed in Fig. 3A and by the fact that procollagen I protein was reduced to a greater degree than was total protein synthesis. Differential modulation of translation of specific mRNAs is consistent with emerging themes in translational control (43–45), but little is known about how such differences are manifested. On the basis of our results, we speculate that mTORC1 and rpS6 checkpoints, which are modulated differently by rapamycin vs. PGE2, control translation of distinct groups of transcripts.
PGE2 influences a myriad of functions of diverse target cells. The ability of PGE2 to regulate translation extends its repertoire of regulatory actions, which already includes modulation of transcription factor expression and activation (46, 47), of DNA methylation (12, 48), and of protein secretion (49). Several lines of evidence point to the importance of translational control in the evolution of fibrotic disease (43, 50). The findings described in this report provide a new paradigm for understanding the role of PGE2 as a brake on, and a potential treatment for, fibrotic diseases. Furthermore, in view of the pleiotropic actions of mTOR in controlling cellular energy balance and metabolism, its inhibition by the mediator PGE2, a mediator whose synthesis is itself altered in numerous pathophysiologic processes and by commonly used medications, opens new avenues for understanding cell function in a clinical context.
Supplementary Material
Acknowledgments
The authors thank Scott H. Wettlaufer for technical contributions, Teresa M. Murphy and Sally Przybranowski for general technical assistance, and members of the laboratory of M.P.-G. for constructive input.
This work was supported by an American Lung Association Senior Research Training Fellowship (K.O.), Japanese Grants-in-Aid for Scientific Research (K.O.), Deutsche Forschungsgemeinschaft (German Research Foundation; Z.Z.), and U.S. National Institutes of Health grant HL094311 (M.P.-G.).
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
- 4E-BP1
- binding protein-1 of eukaryotic initiation factor 4E
- Ab
- antibody
- AHA
- l-azidohomoalanine
- ASA
- acetylsalicylic acid
- CHX
- cycloheximide
- ColIA1/2
- procollagen I α1/2
- COX
- cyclooxygenase
- DMEM
- Dulbecco's modified Eagle's medium
- eEF2
- eukaryotic elongation factor 2
- eIF4B
- eukaryotic initiation factor 4B
- EP
- E prostanoid
- Epac
- guanine nucleotide exchange protein activated by cAMP
- FBS
- fetal bovine serum
- HFL
- human fetal lung
- MMP
- matrix metalloproteinase
- mTOR
- mammalian target of rapamycin
- mTORC
- mTOR complex
- PGE2
- prostaglandin E2
- PKA
- protein kinase A
- PKI
- myristoylated PKA inhibitory peptide 14-22
- PM
- peritoneal macrophage
- PTEN
- phosphatase and tensin homolog deleted on chromosome 10
- rpS6
- ribosomal protein S6
- S6K1
- S6 kinase 1
- XIAP
- X-chromosome-linked inhibitor of apoptosis
REFERENCES
- 1. Wynn T. A. (2007) Common and unique mechanisms regulate fibrosis in various fibroproliferative diseases. J. Clin. Invest. 117, 524–529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hetzel M., Bachem M., Anders D., Trischler G., Faehling M. (2005) Different effects of growth factors on proliferation and matrix production of normal and fibrotic human lung fibroblasts. Lung 183, 225–237 [DOI] [PubMed] [Google Scholar]
- 3. Elias J. A., Rossman M. D., Zurier R. B., Daniele R. P. (1985) Human alveolar macrophage inhibition of lung fibroblast growth: a prostaglandin-dependent process. Am. Rev. Respir. Dis. 131, 94–99 [DOI] [PubMed] [Google Scholar]
- 4. Bitterman P. B., Wewers M. D., Rennard S. I., Adelberg S., Crystal R. G. (1986) Modulation of alveolar macrophage-driven fibroblast proliferation by alternative macrophage mediators. J. Clin. Invest. 77, 700–708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fine A., Poliks C. F., Donahue L. P., Smith B. D., Goldstein R. H. (1989) The differential effect of prostaglandin E2 on transforming growth factor-beta and insulin-induced collagen formation in lung fibroblasts. J. Biol. Chem. 264, 16988–16991 [PubMed] [Google Scholar]
- 6. Kohyama T., Ertl R. F., Valenti V., Spurzem J., Kawamoto M., Nakamura Y., Veys T., Allegra L., Romberger D., Rennard S. I. (2001) Prostaglandin E(2) inhibits fibroblast chemotaxis. Am. J. Physiol. Lung Cell. Mol. Physiol. 281, L1257–L1263 [DOI] [PubMed] [Google Scholar]
- 7. Kolodsick J. E., Peters-Golden M., Larios J., Toews G. B., Thannickal V. J., Moore B. B. (2003) Prostaglandin E2 inhibits fibroblast to myofibroblast transition via E. prostanoid receptor 2 signaling and cyclic adenosine monophosphate elevation. Am. J. Respir. Cell Mol. Biol. 29, 537–544 [DOI] [PubMed] [Google Scholar]
- 8. White E. S., Atrasz R. G., Dickie E. G., Aronoff D. M., Stambolic V., Mak T. W., Moore B. B., Peters-Golden M. (2005) Prostaglandin E(2) inhibits fibroblast migration by E-prostanoid 2 receptor-mediated increase in PTEN activity. Am. J. Respir. Cell Mol. Biol. 32, 135–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Huang S., Wettlaufer S. H., Hogaboam C., Aronoff D. M., Peters-Golden M. (2007) Prostaglandin E(2) inhibits collagen expression and proliferation in patient-derived normal lung fibroblasts via E prostanoid 2 receptor and cAMP signaling. Am. J. Physiol. Lung Cell. Mol. Physiol. 292, L405–413 [DOI] [PubMed] [Google Scholar]
- 10. Huang S. K., Wettlaufer S. H., Chung J., Peters-Golden M. (2008) Prostaglandin E2 inhibits specific lung fibroblast functions via selective actions of PKA and Epac-1. Am. J. Respir. Cell Mol. Biol. 39, 482–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Huang S. K., White E. S., Wettlaufer S. H., Grifka H., Hogaboam C. M., Thannickal V. J., Horowitz J. C., Peters-Golden M. (2009) Prostaglandin E(2) induces fibroblast apoptosis by modulating multiple survival pathways. FASEB J. 23, 4317–4326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Huang S. K., Scruggs A. M., Donaghy J., McEachin R. C., Fisher A. S., Richardson B. C., Peters-Golden M. (2012) Prostaglandin E2 increases fibroblast gene-specific and global DNA methylation via increased DNA methyltransferase expression. FASEB J. 26, 3703–3714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Borok Z., Gillissen A., Buhl R., Hoyt R., Hubbard R., Ozaki T., Rennard S., Crystal R. (1991) Augmentation of functional prostaglandin E levels on the respiratory epithelial surface by aerosol administration of prostaglandin E. Am. Rev. Respir. Dis. 144, 1080–1084 [DOI] [PubMed] [Google Scholar]
- 14. Wilborn J., Crofford L. J., Burdick M. D., Kunkel S. L., Strieter R. M., Peters-Golden M. (1995) Cultured lung fibroblasts isolated from patients with idiopathic pulmonary fibrosis have a diminished capacity to synthesized prostaglandin E2 and to express cyclooxygenase-2. J. Clin. Invest. 95, 1861–1868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fine A., Matsui R., Zhan X., Poliks C. F., Smith B. D., Goldstein R. H. (1992) Discordant regulation of human type I collagen genes by prostaglandin E2. Biochim. Biophys. Acta 1135, 67–72 [DOI] [PubMed] [Google Scholar]
- 16. Liu X., Ostrom R. S., Insel P. A. (2004) cAMP-elevating agents and adenylyl cyclase overexpression promote an antifibrotic phenotype in pulmonary fibroblasts. Am. J. Physiol. Cell Physiol. 286, C1089–1099 [DOI] [PubMed] [Google Scholar]
- 17. Zhu Y., Liu X., Sköld C. M., Wang H., Kohyama T., Wen F. Q., Ertl R. F., Rennard S. I. (2001) Collaborative interactions between neutrophil elastase and metalloproteinases in extracellular matrix degradation in three-dimensional collagen gels. Respir. Res. 2, 300–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zoncu R., Efeyan A., Sabatini D. M. (2011) mTOR: from growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 12, 21–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ruvinsky I., Meyuhas O. (2006) Ribosomal protein S6 phosphorylation: from protein synthesis to cell size. Trends Biochem. Sci. 31, 342–348 [DOI] [PubMed] [Google Scholar]
- 20. Meyuhas O., Dreazen A. (2009) Ribosomal protein S6 kinase from TOP mRNAs to cell size. Prog. Mol. Biol. Transl. Sci. 90, 109–153 [DOI] [PubMed] [Google Scholar]
- 21. Bauman K. A., Wettlaufer S. H., Okunishi K., Vannella K. M., Stoolman J. S., Huang S. K., Courey A. J., White E. S., Hogaboam C. M., Simon R. H., Towes G. B., Sisson T. H., Moore B. B., Peters-Golden M. (2010) The antifibrotic effects of plasminogen activation occur via prostaglandin E2 synthesis in humans and mice. J. Clin. Invest. 120, 1950–1960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Canetti C., Aronoff D. M., Choe M., Flamand N., Wettlaufer S., Toews G. B., Chen G. H., Peters-Golden M. (2006) Differential regulation by leukotrienes and calcium of Fcγ receptor-induced phagocytosis and Syk activation in dendritic cells versus macrophages. J. Leukoc. Biol. 79, 1234–1241 [DOI] [PubMed] [Google Scholar]
- 23. Poulalhon N., Farge D., Roos N., Tacheau C., Neuzillet C., Michel L., Mauviel A., Verrecchia F. (2006) Modulation of collagen and MMP-1 gene expression in fibroblasts by the immunosuppressive drug rapamycin: a direct role as an antifibrotic agent? J. Biol. Chem. 281, 33045–33052 [DOI] [PubMed] [Google Scholar]
- 24. Serezani C. H., Aronoff D. M., Jancar S., Mancuso P., Peters-Golden M. (2005) Leukotrienes enhance the bactericidal activity of alveolar macrophages against Klebsiella pneumoniae through the activation of NADPH oxidase. Blood 106, 1067–1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jensen L. T., Høst N. B. (1997) Collagen: scaffold for repair or execution. Cardiovasc. Res. 33, 535–539 [DOI] [PubMed] [Google Scholar]
- 26. Hulmes D. J. (2002) Building collagen molecules, fibrils, and suprafibrillar structures. J. Struct. Biol. 137, 2–10 [DOI] [PubMed] [Google Scholar]
- 27. Gilroy D. W., Saunders M. A., Wu K. K. (2001) COX-2 expression and cell cycle progression in human fibroblasts. Am. J. Physiol. Cell Physiol. 281, C188–C194 [DOI] [PubMed] [Google Scholar]
- 28. af Forselles K. J., Root J., Clarke T., Davey D., Aughton K., Dack K., Pullen N. In vitro and in vivo characterization of PF-04418948, a novel, potent and selective prostaglandin EP2 receptor antagonist. (2011) Br. J. Pharmacol. 164, 1847–1856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rennard S.I., Stier L.E., Crystal R.G. (1982) Intracellular degradation of newly synthesized collagen. J. Invest. Dermatol. 79(Suppl 1), 77S–82S [DOI] [PubMed] [Google Scholar]
- 30. Laurent G. J. (1987) Dynamic state of collagen: pathways of collagen degradation in vivo and their possible role in regulation of collagen mass. Am. J. Physiol. 252, C1–C9 [DOI] [PubMed] [Google Scholar]
- 31. Tanaka T., Yoshimi M., Maeyama T., Hagimoto N., Kuwano K., Hara N. (2002) Resistance to Fas-mediated apoptosis in human lung fibroblast. Eur. Respir. J. 20, 359–368 [DOI] [PubMed] [Google Scholar]
- 32. Gordon S. (2007) The macrophage: past, present and future. Eur. J. Immunol. 37(Suppl 1), S9–S17 [DOI] [PubMed] [Google Scholar]
- 33. Peters-Golden M. (2009) Putting on the brakes: cyclic AMP as a multipronged controller of macrophage function. Sci. Signal. 2, pe37. [DOI] [PubMed] [Google Scholar]
- 34. Kalinski P. (2012) Regulation of immune responses by prostaglandin E2. J. Immunol. 188, 21–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Moore C. E., Xie J., Gomez E., Herbert T. P. (2009) Identification of cAMP-dependent kinase as a third in vivo ribosomal protein S6 kinase in pancreatic beta-cells. J. Mol. Biol. 389, 480–494 [DOI] [PubMed] [Google Scholar]
- 36. Misra U. K., Pizzo S. V. (2009) Epac1-induced cellular proliferation in prostate cancer cells is mediated by B-Raf/ERK and mTOR signaling cascades. J. Cell. Biochem. 108, 998–1011 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 37. Kim H. W., Ha S. H., Lee M. N., Huston E., Kim D. H., Jang S. K., Suh P. G., Houslay M. D., Ryu S. H. (2010) Cyclic AMP controls mTOR through regulation of the dynamic interaction between Rheb and phosphodiesterase 4D. Mol. Cell. Biol. 30, 5406–5420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Scott P. H., Lawrence J. C., Jr. (1998) Attenuation of mammalian target of rapamycin activity by increased cAMP in 3T3-L1 adipocytes. J. Biol. Chem. 273, 34496–34501 [DOI] [PubMed] [Google Scholar]
- 39. Xie J., Ponuwei G. A., Moore C. E., Willars G. B., Tee A. R., Herbert T. P. (2011) cAMP inhibits mammalian target of rapamycin complex-1 and -2 (mTORC1 and 2) by promoting complex dissociation and inhibiting mTOR kinase activity. Cell. Signal. 23, 1927–1935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mavrakis M., Lippincott-Schwartz J., Stratakis C. A., Bossis I. (2006) Depletion of type IA regulatory subunit (RIalpha) of protein kinase A (PKA) in mammalian cells and tissues activates mTOR and causes autophagic deficiency. Hum. Mol. Genet. 15, 2962–2971 [DOI] [PubMed] [Google Scholar]
- 41. Rocchi S., Gaillard I., van Obberghen E., Chambaz E. M., Vilgrain I. (2000) Adrenocorticotrophic hormone stimulates phosphotyrosine phosphatase SHP2 in bovine adrenocortical cells: phosphorylation and activation by cAMP-dependent protein kinase. Biochem. J. 352(Pt 2), 483–490 [PMC free article] [PubMed] [Google Scholar]
- 42. Zito C.I., Qin H., Blenis J., Bennett A. M. (2007) SHP-2 regulates cell growth by controlling the mTOR/S6 kinase 1 pathway. J. Biol. Chem. 282, 6946–6953 [DOI] [PubMed] [Google Scholar]
- 43. Larsson O., Diebold D., Fan D., Peterson M., Nho R. S., Bitterman P. B., Henke C. A. (2008) Fibrotic myofibroblasts manifest genome-wide derangements of translational control. PLoS One 3, e3220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Thoreen C. C., Chantranupong L., Keys H. R., Wang T., Gray N. S., Sabatini D. M. (2012) A unifying model for mTORC1-mediated regulation of mRNA translation. Nature 485, 109–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hsieh A. C., Liu Y., Edlind M. P., Ingolia N. T., Janes M. R., Sher A., Shi E. Y., Stumpf C. R., Christensen C., Bonham M. J., Wang S., Ren P., Martin M., Jessen K., Feldman M. E., Weissman J. S., Shokat K. M., Rommel C., Ruggero D. (2012) The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature 485, 55–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Valdez P. A., Vithayathil P. J., Janelsins B. M., Shaffer A. L., Williamson P. R., Datta S. K. (2012) Prostaglandin E2 suppresses antifungal immunity by inhibiting interferon regulatory factor 4 function and interleukin-17 expression in T cells. Immunity 36, 668–679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ji R., Sanchez C. M., Chou C. L., Chen X. B., Woodward D. F., Regan J. W. (2012) Prostanoid EP1 receptors mediate up-regulation of the orphan nuclear receptor Nurr1 by cAMP-independent activation of protein kinase A, CREB and NF-κB. Br. J. Pharmacol. 166, 1033–1046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Xia D., Wang D., Kim S. H., Katoh H., DuBois R. N. (2012) Prostaglandin E2 promotes intestinal tumor growth via DNA methylation. Nat. Med. 18, 224–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Schweda F., Klar J., Narumiya S., Nüsing R. M., Kurtz A. (2004) Stimulation of renin release by prostaglandin E2 is mediated by EP2 and EP4 receptors in mouse kidneys. Am. J. Physiol. Renal Physiol. 287, F427–F433 [DOI] [PubMed] [Google Scholar]
- 50. Goc A., Choudhary M., Byzova T. V., Somanath P. R. (2011) TGFbeta- and bleomycin-induced extracellular matrix synthesis is mediated through Akt and mammalian target of rapamycin (mTOR). J. Cell. Physiol. 226, 3004–3013 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.