

Abstract
It has been reported that Mitofusin2 (Mfn2) inhibits cell proliferation when overexpressed. We wanted to study the role of endogenous Mfn2 in cell proliferation, along with the structural features of Mfn2 that influence its mitochondrial localization and control of cell proliferation. Mfn2-knockdown clones of a B-cell lymphoma cell line BJAB exhibited an increased rate of cell proliferation. A 2-fold increase in cell proliferation was also observed in Mfn2-knockout mouse embryonic fibroblast (MEF) cells as compared with the control wild-type cells, and the proliferative advantage of the knockout MEF cells was blocked on reintroduction of the Mfn2 gene. Mfn2 exerts its antiproliferative effect by acting as an effector molecule of Ras, resulting in the inhibition of the Ras-Raf-ERK signaling pathway. Furthermore, both the N-terminal (aa 1–264) and the C-terminal (aa 265–757) fragments of Mfn2 blocked cell proliferation through distinct mechanisms: the N-terminal-mediated inhibition was due to its interaction with Raf-1, whereas the C-terminal fragment of Mfn2 inhibited cell proliferation by interacting with Ras. The inhibition of proliferation by the N-terminal fragment was independent of its mitochondrial localization. Collectively, our data provide new insights regarding the role of Mfn2 in controlling cellular proliferation.—Chen, K.-H., Dasgupta, A., Ding, J., Indig, F. E., Ghosh, P., Longo, D. L. Role of Mitofusin 2 (Mfn2) in controlling cellular proliferation.
Keywords: HSG, Ras, Raf, ERK
A perfectly balanced and well-coordinated regulation of cell proliferation plays an important role in maintenance of tissue homeostasis. Disturbances in this delicate equilibrium due to alterations in genes involved in this highly regulated system are crucial events for neoplastic cell growth and other hyperproliferative diseases. Numerous genes govern normal cell cycle progression; understanding their mechanisms of action has provided insights about the deregulation of cell behavior in hyperproliferative diseases.
Mfn2 is a protein that localizes to the mitochondrial outer membrane and possesses an essential role in mitochondrial fusion, thus regulating mitochondrial morphology and function in mammalian cells, yeasts, and flies (1, 2). Apart from its major involvement in mitochondrial fusion, dysfunction of this gene is also associated with a variety of pathological conditions, including Charcot-Marie-Tooth (CMT) disease type 2A (3, 4), diabetes mellitus type 2 (5, 6), obesity (7), atherosclerosis, and hypertension (8, 9), suggesting the diverse functions of this gene. Mfn2 is also known as hyperplasia suppressor gene (HSG), and our previous studies using an overexpression system mediated by adenoviral infection in vascular smooth muscle cells (VSMCs) and various cancer cell lines have demonstrated that this gene possesses major roles in controlling cell proliferation and apoptosis both in vitro and in vivo (8, 10–12).
Although our previous study has demonstrated the inhibitory capacity of overexpressed Mfn2, the role of endogenous Mfn2 in controlling cell proliferation and the functional domains of Mfn2 mediating this activity were not completely understood. In our present work, we have systematically explored the role of endogenous Mfn2 in controlling cell proliferation. Our results demonstrate that endogenous Mfn2 plays a role in controlling cell growth by inhibiting the Ras-Raf–extracellular signal-regulated kinase (ERK) 1/2 signaling pathway via interaction with both Ras and Raf-1. We also report that Mfn2 is a novel Ras effector molecule and both the N-terminal (aa 1–264) and C-terminal (aa 265–757) fragments of Mfn2 can inhibit cell proliferation by at least two mechanisms: the N-terminal-mediated inhibition is mediated by its interaction with Raf-1, whereas the C-terminal fragment of Mfn2 inhibits cell proliferation by interacting with Ras.
MATERIALS AND METHODS
Cells and tissue cultures
Two B-cell lymphoma cell lines, BJAB (EBV-negative Burkitt's lymphoma) and RL (diffuse large B-cell lymphoma), with stable expression of adenoviral receptors were maintained in RPMI 1640 supplemented with 10% fetal bovine serum (FBS; Life Technologies, Rockville, MD), 100 U/ml penicillin, 100 μg/ml streptomycin, and 2 mM glutamine. HEK293T, HEK293A, and mouse embryonic fibroblasts (MEFs) from SV 40-transformed wild-type (WT) and Mfn2−/− mice (gifts from Dr. David C. Chan, California Institute of Technology, Pasadena, CA, USA) were cultured in Dulbecco's modified Eagle's medium (DMEM) containing 10% FBS, 100 U/ml penicillin, 100 μg/ml streptomycin, and 2 mM glutamine and passaged every 3 d. Transient transfections were done using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the supplier's instruction.
Reagents and antibodies
Unless otherwise indicated, all chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA). The anti-Mfn2 (M-6444) polyclonal and monoclonal antibodies were purchased from Sigma-Aldrich and Abcam (Cambridge, MA, USA), respectively. The anti-Mfn1 (ab104274) polyclonal and Drp1 monoclonal antibodies were purchased from Abcam. The anti-OPA1 monoclonal antibody was purchased from BD Transduction Laboratories (San Jose, CA, USA). The anti-phospho-ERK1/2, anti-ERK1 and 2, anti-Raf-1, and anti-PGC-1 antibodies and ImmunoCruz immunoprecipitation (IP)/Western blot (WB) Optima system were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The anti-β-tubulin was obtained from Abcam. Expression Arrest GIPZ lentiviral shRNAmirs of Mfn2 were purchased from Open Biosystems (Huntsville, AL, USA). Anti-Pan-Ras monoclonal antibody was purchased from Calbiochem (San Diego, CA, USA). The anti-Pan-Ras polyclonal and anti-phospho-Raf-1 antibodies were from Cell Signaling (Beverly, MA, USA). The anti-Flag monoclonal antibody M2 was from Sigma-Aldrich. WST-1 reagent and anti-hemagglutinin (HA) monoclonal antibody clone 12CA5 were from Roche Applied Science (Indianapolis, IN, USA). MitoTracker Red CMXRos, anti-mouse IgG Alexa Fluor 488, and ProLong Gold antifade reagent were from Molecular Probes (Eugene, OR, USA). TnT Quick Coupled Transcription and Translation (T3) kit was from Promega (Madison, WI, USA).
WB and IP
Whole-cell lysates were prepared as described previously (8). For WB analysis, 25 μg of cell lysates was analyzed on 4–12% NuPAGE gel (Invitrogen). The proteins were electrotransferred to PVDF membrane (Invitrogen), and detection of specific proteins was carried out with indicated antibodies and ECL-Plus Western blotting Detection System (GE Healthcare, Piscataway, NJ, USA). IP was performed using the appropriate ImmunoCruz IP/WB Optima system following the manufacturer's instruction. After being washed, immunoprecipitates were eluted and separated by SDS-PAGE.
Cloning and constructs
pCGN HA vectors harboring WT K-Ras (WT-K-Ras), constitutive active K-Ras (CA-K-Ras), and CA-K-Ras with a mutation at the effector binding region (CA-K-Ras-E37G) were generous gifts from Geoffrey J. Clark (University of Louisville, Louisville, KY, USA). For in vitro transcription and translation, the complete open reading frames for these Ras variant cDNAs were recloned by PCR into the BamHI and XhoI sites of pCMV-3Tag-1A vector (Stratagene, La Jolla, CA, USA) and in frame with N-terminal 3xFlag peptide epitopes. CA-H-Ras and CA-H-Ras with a mutation at the effector binding region (CA-H-Ras-E37G) were purchased from Addgene and recloned by PCR into the BamHI and XhoI sites of pCMV-3Tag-1A vector and in frame with N-terminal 3xFlag peptide epitopes. Human Mfn21–756 was generated by PCR using the forward primer (primer1) containing BamHI site 5′-ATGACGGATCCTCCCTGCTCTTCTCTCGATGCAACTCTATCG-3′ and the reverse primer (primer2) containing XhoI site 5′-CCCCCCCTCGAGTCTGCTGGGCTGCAGGTACTGGT-3′; Mfn21–264 was generated with primer1 and the reverse primer containing XhoI site 5′-CCCCCCCTC GAGGGCAGATGCATCCCAGCGGTT-3′; and Mfn2265–756 was generated with the forward primer containing BamHI site 5′-GATGACGGATCCTCAGAGCCCGAGTACATG GAGGA-3′ and primer2. These PCR products were digested with BamHI and XhoI, introduced into pCMV-3Tag-1A vector and in frame with N-terminal 3xFlag tag sequence. All constructs were verified by DNA sequencing to ensure that they were correct. The pCMV-Raf-1 vector that expresses the human WT v-Raf-1 protein was purchased from Clontech. For in vitro transcription and translation, the complete open reading frame of pCMV-Raf-1 was recloned by PCR into the BamHI and XhoI sites of pCMV-3Tag-2A vector. Human Mfn1 cDNA in pCMV 6-XL5 vector was purchased from OriGene (Rockville, MD, USA) and recloned by PCR into the BamHI and XhoI sites of pCMV-3Tag-1A vector. The primers used to reclone human Mfn1 c-DNA into the vector were the forward primer containing BamHI site 5′-GATGACGGATCCGCAGAACCTGTTTCTG-3′ and the reverse primer containing XhoI site 5′-CCCGGACTCGAGGGATTCTTCTTCATTGCT-3′. WT B-Raf mammalian expression construct was a kind gift from Dr. Martin McMahon (University of California, San Francisco, CA, USA).
In vitro transcription and translation
In vitro synthesis of flag-tagged K-Ras variants (WT-K-Ras, CA-K-Ras, and CA-K-Ras-E37G) and flag-tagged Mfn21–756 were conducted by using the rabbit reticulocyte Lysate TNT Coupled Transcription/Translation System (Promega) according to the protocol provided. The products were allowed to interact first and then subjected to IP assays as described above.
Lentiviral production
The standard protocol for lentivirus production using calcium phosphate transfections in HEPES-buffered saline (HBS) was performed using a previous report as a reference (13). The HEK293T cells were seeded and cultured in 10-cm dishes. The amount of plasmid DNA was scaled-up proportionally to the surface area of the 10-cm dish, as described above. Ethanol-precipitated plasmid DNAs were diluted in 0.5 ml of 2.0 M calcium chloride solution. The diluted plasmid DNAs were added to an equal volume of 2× HBS (pH 7.12) dropwise while being vortexed. The solution was incubated for 30 min at room temperature and then added to the cells. The cells were incubated for 24 h at 37°C in 5% CO2, the transfection medium was changed, and the viral supernatant was collected as described above. The collected supernatants were spun down at 2000 rpm for 5 min, filtered through 0.45-μm filter, and stored at −80°C.
Adenoviral infection
MEFs were plated in 6-well plates and incubated overnight. When the cells reached the optimal confluency, the culture medium was aspirated, and the cells were counted and infected with the indicated viruses at 37°C at the indicated multiplicity of infection. The cells were then analyzed after 24 to 48 h of infection. BJAB cells were counted and infected with the indicated viruses.
Generation of stable cell lines
To generate BJAB cells with stable expression of Mfn2-specific shRNAmirs (V2LHS255771, V2LHS255949, V2LHS90079, and V2LHS90083), BJAB cells were infected with the recombinant lentiviral vectors containing Mfn2-specific shRNAmirs or nonsilencing shRNAmir and subjected to puromycin selection (2–4 μg/ml). After a 4-wk selection, nonsilencing shRNAmir-containing cells and cells stably transfected with Mfn2-specific shRNAmirs from 3 independently derived clones were examined for the level of Mfn2 expression by Western blotting.
Cell proliferation assay
Cell proliferation assay was performed in quadruplicate using a cell proliferation reagent, WST-1 (Roche Applied Science), or cell counting. For the WST-1 assay, cells were seeded in 96-well plates and noninfected or infected with the indicated adenoviruses at 100 plaque-forming units (pfu). Assays were performed by addition of WST-1 reagent directly to the culture wells and incubation for 2 h at 37°C. Plates were then read by a scanning multiwell spectrophotometer by measuring the absorbance of the dye with a wavelength of 450 nm and a reference wavelength of 655 nm. Three different experiments were performed for each experimental condition. For direct cell counting, MEFs were seeded in 6-well plates (10,000 cells/well) and cultured overnight. Cells were then harvested and counted at intervals of 24 h for 3 d. Experiments were performed in triplicate, and each experiment was repeated 3 times.
Cell cycle analysis
Mfn2−/− MEFs were infected with indicated adenoviruses cultured in 2% FBS/DMEM for 48 h. The cells were then cultured in 10% FBS/DMEM for 24 h. Cells were harvested, resuspended in 300 μl 1× phosphate-buffered saline (PBS), and fixed in 70% ethanol. The fixed cells were washed again with PBS twice and treated with 500 μl of PI/RNase staining buffer in darkness for 1 h at room temperature. The samples were analyzed through flow cytometry using a fluorescence-activated cell sorter manufactured by BD Biosciences (San Jose, CA, USA).
Flow cytometry analysis of apoptosis
Mfn2−/− MEFs were seeded in 100-mm dishes. After 24 h, cells were counted and infected with indicated adenoviruses at 30 pfu/cell. BJAB cells were infected directly at 50 pfu/cell. Cells were collected at 24, 48, and 72 h and stained with the PE Annexin V Apoptosis Detection kit (BD Biosciences) following the manufacturer's instruction. Detection and quantification of apoptotic cells were obtained by flow cytometry analysis (Becton Dickinson, Franklin Lakes, NJ, USA).
Oxygen consumption measurement
Basal oxygen consumption rate (OCR) was measured with Extracellular Flux Analyzer (XF24; Seahorse Bioscience, North Billerica, MA, USA) according to the manufacturer's protocol.
Confocal microscopy
MEFs were plated in 2-chamber slides and transfected with Lipofectamine 2000 reagent (Life Technologies) and the indicated flag-tagged expression constructs of Mfn2, according to the manufacturer's instructions. Nontransfected MEFs served as a control. After 24 h of transfection, viable cells were incubated with 200 nM MitoTracker Red CMXRos (Molecular Probes) at 37°C for 1 h, washed 3 times with PBS, and fixed with 4% formaldehyde/PBS for 30 min. Cells were permeabilized for 15 min with 0.2% Triton X-100 in PBS and blocked for 1 h with PBS containing 5% FBS. Cells were then incubated with mouse anti-FLAG antibody overnight at 4°C and subsequently incubated with anti-mouse IgG Alexa Fluor 350 for 3 h at room temperature. Cells were then mounted in ProLong Gold antifade reagent (Molecular Probes) and examined with a confocal microscope.
RESULTS
Overexpression of Mfn2 inhibits proliferation of B-cell lymphoma cell lines BJAB and RL
It has been reported that the overexpression of Mfn2 suppressed serum-stimulated VSMC proliferation in culture (8). To determine whether this overexpression can also inhibit cell proliferation of B-cell lymphoma cells, we overexpressed Mfn2 by adenovirus-mediated infection in 2 different B-cell lymphoma cell lines, BJAB and RL, which stably expressed adenoviral receptors. WB analysis was first carried out to confirm the expression of exogenous Mfn2. Exogenous Mfn2 expression was found to be significantly higher in BJAB cells infected with Adv-Mfn2 (100 pfu) compared with controls, i.e., cells infected with or without Adv-LacZ (100 pfu) (Fig. 1A). Further, cell proliferation assay showed that the overexpression of Mfn2 substantially inhibited cell proliferation (Fig. 1B and Supplemental Fig. S1A). Noninfected and Adv-LacZ-infected cells did not show significant differences in their proliferation pattern. Similar results were also observed in RL cell line infected with Adv-Mfn2 (Fig. 1C, D). The growth suppression was not due to apoptotic cell death (Supplemental Fig. S1B). We have also shown that the increase in growth suppression with time was due to the time-dependent increase in Mfn2 expression (Supplemental Fig. S1C). To determine the effect of Mfn2 overexpression on other mitochondrial proteins, BJAB cells were infected with either Adv-LacZ or Adv-Mfn2 for 48 h, and the cell lysates were analyzed by WB analysis. As shown in Supplemental Fig. S1D, while PGC1 level was increased, the levels of Mfn1, Drp1, and OPA1 were decreased on Mfn2 overexpression, suggesting a complex relationship between Mfn2 and other mitochondrial proteins during growth suppression. Our results suggested that Mfn2 overexpression has antiproliferative effects.
Figure 1.
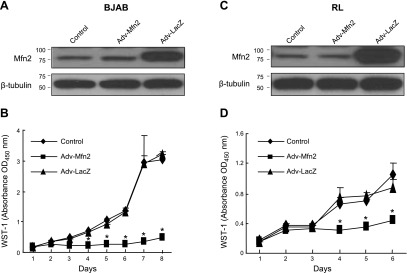
Overexpression of Mfn2 inhibits proliferation of B lymphoma cell lines BJAB (A, B) and RL (C, D). Overexpression of Mfn2 by adenovirally mediated gene transfer in BJAB cells (A) and RL (C) cells, shown by WB (25 μg cell lysates) with an anti-Mfn2 antibody. β-Tubulin was used as a loading control. Inhibitory effect of Mfn2 on BJAB growth (B) and RL growth (D) was examined by WST-1 assay (n=3, performed in quadruplicate). *P < 0.01.
Knockdown of Mfn2 results in increased cell proliferation
If Mfn2 participates in controlling cell proliferation, knocking down this gene should enhance cell proliferation. To test this hypothesis, we stably knocked down the Mfn2 gene in the BJAB cell line using the RNA interference technique. Three independent clones were analyzed by WB to check the knockdown efficiency compared with the nonsilencing shRNAmir-transfected cells. As shown in Fig. 2A, the expression of Mfn2 in these 3 stable clones was decreased ∼60% compared with the control. Proliferation assay revealed an increase in cell proliferation in the Mfn2-knockown clones as compared with the control (Fig. 2B), supporting a role of endogenous Mfn2 in controlling cell proliferation. To further confirm the role of Mfn2 in cell proliferation, we tested MEFs derived from Mfn2-knockout (Mfn2−/−) and WT mice. The status of Mfn2 expression in these 2 cell types was confirmed by WB analysis (Fig. 3A). As shown in Fig. 3B, the proliferation rate of Mfn2−/− MEFs was found to be markedly higher (2-fold) than the WT cells. Moreover, when Mfn2 was introduced into Mfn2−/− MEFs by adenovirus-mediated gene transfer, a striking decline in cell proliferation was observed (Fig. 3C, D, and Supplemental Fig. S2A). The growth suppression was not due to the apoptotic cell death (Supplemental Fig. S2B). Collectively, these data demonstrated that endogenous Mfn2 plays an important role in controlling cell proliferation.
Figure 2.
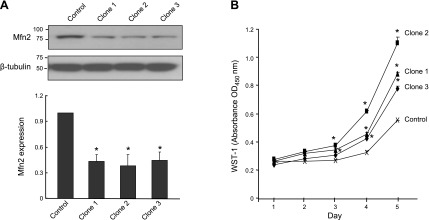
Mfn2-knockdown BJAB cells grow faster than the control cells. A) BJAB cells were transduced with lentiviral particles expressing Mfn2-specific or nonsilencing shRNAmirs. Top panel: residual expression levels of Mfn2 in 3 stable Mfn2-silencing clones relative to the nonsilencing control. Bottom panel: quantitative analysis of Mfn2 protein abundance in these stable cell lines (n=3 independent experiments). *P < 0.01 vs. control group. B) Growth curve of the 3 stable Mfn2 silencing clones and the nonsilencing control, assayed by the cleavage of the tetrazolium salt WST-1 (n=3). *P < 0.01 vs. control group.
Figure 3.
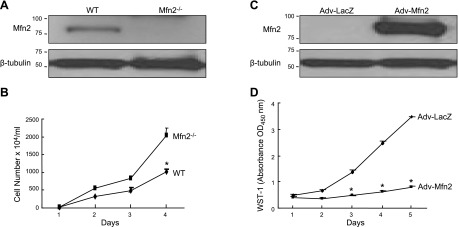
Mfn2 involved in controlling cell proliferation. A) WB of total cell lysates (30 μg) from Mfn2−/− and WT MEFs probed with an anti-Mfn2 antibody. B) Growth curve of MEFs from WT or Mfn2−/− mice determined by cell counting (n=3, performed in triplicate). *P < 0.01. C) Level of Mfn2 protein in Mfn2−/− MEFs infected with either Adv-LacZ or Adv-Mfn2 at 100 pfu/cell. D) Inhibitory effect of Mfn2 on serum-induced Mfn2−/− MEFs proliferation (n=3, performed in quadruplicate). *P < 0.01.
Mfn2 inhibits phosphorylation of ERK1/2 and Raf-1
Next, we wanted to investigate the underlying mechanism of Mfn2-mediated growth suppression. It has been shown previously that overexpressed Mfn2 blocks cell proliferation by interacting with Ras and inhibiting the downstream Ras-Raf-ERK signaling pathway (8). Therefore, we hypothesized that endogenous Mfn2 may also be involved in blocking the Ras-Raf-ERK signaling pathway. To test our hypothesis, WB analysis was performed to check the phosphorylation status of ERK1/2 and Raf-1 in Mfn2−/− MEFs. As shown in Fig. 4A, C, Ras downstream signaling kinases Raf-1 and ERK1/2 were hyperphosphorylated in cells lacking Mfn2 compared with the WT MEFs. Moreover, when the Mfn2 gene was introduced into the Mfn2−/− MEFs by adenovirus-mediated gene transfer, the phosphorylation of Raf-1 and ERK1/2 was markedly inhibited in a dose-dependent manner (Fig. 4B, D). Taken together, these data suggested that the mechanism of growth suppression by Mfn2 might be due to the inhibition of the upstream kinase of Raf-1 and ERK1/2.
Figure 4.
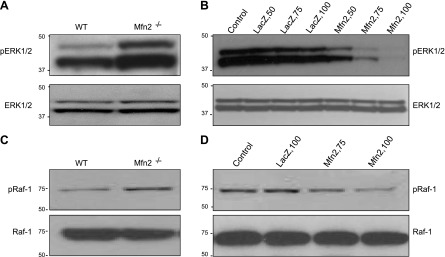
Introduction of Mfn2 into Mfn2−/− MEF cells inhibits serum-induced phosphorylation of ERK1/2 and Raf-1. A) WB of total cell lysates (25 μg) from Mfn2−/− and WT MEFs for phosphorylated (p-ERK1 and p-ERK2) and total ERK1/2. B) Total and phosphorylated ERK1/2 in Mfn2−/− MEFs infected with different titers (pfu) of Adv-LacZ or Adv-Mfn2. C) WB of total cell lysates (25 μg) from Mfn2−/− and WT MEFs for phosphorylated (p-Raf-1) and total Raf-1. D) Total and phosphorylated Raf-1in Mfn2−/− MEFs infected with different titers of Adv-LacZ or Adv-Mfn2.
Mfn2 interacts with Ras
To investigate whether Mfn2 interacts with Ras, a flag-tagged Mfn2 plasmid was transfected into the Mfn2−/− MEFs, and the cell lysates were used for IP assay with a Ras monoclonal antibody. As shown in Fig. 5A, Mfn2 was coimmunoprecipitated with Ras, as demonstrated by the WB analysis of the immunocomplexes using anti-Flag antibody (top panel). Ras immunoprecipitated by Ras monoclonal antibody was further corroborated by probing the same membrane with an anti-Ras polyclonal antibody (Fig. 5A, bottom panel). These data suggested that a small percentage of the total Mfn2 pool is associated with Ras. To gain further insight into the endogenous Mfn2-Ras interaction, we prepared whole-cell lysates from WT and Mfn2−/− MEFs. IP analyses were carried out with an anti-Ras antibody. As shown in Fig. 5B, Ras was only coimmunoprecipitated with Mfn2 in lysates from WT but not from Mfn2−/− MEFs. Collectively, these data suggested that Mfn2 caused growth suppression by inhibiting the Ras signaling pathway.
Figure 5.
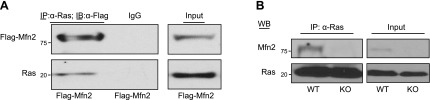
Mfn2 associates with Ras protein. A) Cell lysates (500 μg) from Mfn2−/− MEFs transfected with Flag-Mfn2 construct were immunoprecipitated with anti-Ras monoclonal antibody (2 μg) and immunoblotted with either anti-Flag antibody (top panel) or anti-Pan Ras polyclonal antibody (bottom panel). Total cell extracts (25 μg) were used as positive control for expression of Flag-tagged Mfn2 and Ras. B) Whole-cell lysates (500 μg) from WT or Mfn2−/− MEFs were immunoprecipitated with anti-Ras antibody and immunoblotted with either anti-Mfn2 or anti-Ras antibody. Total extracts (25 μg) were used as the control for the expression of Mfn2 and Ras.
Mfn2 is a Ras effector molecule
Most biological functions resulting from the interaction between Ras and its effector molecules have stimulatory effects on the cells, like inducing cell proliferation, remodeling of the actin cytoskeleton, etc. (14, 15). Recently, a small family of genes termed Ras association domain family (RASSF) has been described, the members of which are characterized by the presence of a Ras association (RA) domain (16–20). RASSF family members appear to behave like Ras effector molecules by preferentially binding to the guanosine triphosphate (GTP-bound) or active form of Ras and function like tumor suppressors by exhibiting antiproliferative and/or proapoptotic effects when overexpressed in tumor cell lines (18). Sequence comparison study between Mfn2 and NORE1A, one of the most studied members of the RASSF, revealed that the C-terminal region of Mfn2 (aa 490–757) shares 57% homology with the C-terminal region of NORE1A (aa 191–413) containing RA and Salvador, Rassf, Hippo (SARAH) domains (Fig. 6A). Based on these structural similarities, we wanted to determine whether Mfn2 is a Ras effector molecule. To be a Ras effector molecule, candidate proteins usually fulfill the following criteria: it has to bind strongly to the GTP-bound or active form of Ras but not the GDP-bound form, and it must interact poorly with a form of Ras mutated at the effector-binding site (21). To determine whether Mfn2 is a Ras effector, we cotransfected Mfn2−/− MEFs with a flag-tagged Mfn2 plasmid along with different HA-tagged K-Ras plasmids: WT-K-Ras, CA-K-Ras, (a Ras construct carrying glycine to valine mutation at codon 12, resulting in a constitutively active GTP-bound conformation; refs. 22, 23); and mutant CA-K-Ras-E37G (constitutively active Ras carrying glutamic acid to glycine mutation at codon 37 in the effector domain; refs. 22, 23). Cell lysates were used for IP assay with anti-HA antibody, and the immunocomplexes were analyzed by WB analysis. As shown in Fig. 6B, Mfn2 was found to interact more strongly with CA-K-Ras than with WT Ras and to have much less binding to the effector mutant Ras construct. Unexpectedly, the expression levels of both Mfn2 and Ras were higher in the cells cotransfected with Mfn2 and CA-K-Ras plasmids compared with the cells cotransfected with either Mfn2/vector or Mfn2/CA-K-Ras-E37G plasmids, whereas cells transfected with individual Ras plasmids had comparable levels of expression (Fig. 6C). From these data, we cannot conclude about the preferential binding of Mfn2 to constitutively active Ras. Interestingly, the increase in the expression of each interacting partner due to the binding of Mfn2 to the active form of Ras was not specific for K-Ras, since the same phenomenon was also observed in case of H-Ras (Fig. 6D). The mechanism of this observation is under investigation. Furthermore, the effect of Ras on Mfn2 was specific, since we did not observe a similar effect when we coexpressed another Ras effector molecule, Raf-1, along with either WT-K-Ras, CA-K-Ras, or mutant Ras (Fig. 6E). These data suggested that on interaction with each other, Mfn2 and Ras increase each other's expression depending on the conformation of Ras protein.
Figure 6.

Mfn2 is a Ras effector molecule. A) Sequence similarity between the C-terminal region of human Mfn2 (aa 490–757) and the C-terminal region of NORE1A (aa 191–413) containing RA and SARAH domains. Red box indicates the RA domain of NORE1A (aa 191–363); black box indicates the SARAH domain of NORE1A (aa 366–413). Asterisks indicate identity; colons indicate strong conservation; periods indicate weak conservation. B) Lysates (500 μg) from Mfn2−/− MEFs cotransfected with flag-tagged Mfn21–756 along with different HA-tagged K-Ras plasmids, WT-K-Ras, CA-K-Ras and CA-K-Ras-E37G, were immunoprecipitated with anti-HA monoclonal antibody (2 μg) and immunoblotted with either anti-Flag antibody (top panel) or anti-HA antibody (bottom panel). Total cell extracts (25 μg) were used to monitor the expression of flag-tagged Mfn2 and HA-tagged Ras. Similar results were obtained in 3 independent experiments. C) Expression levels of individual Ras constructs (WT-K-Ras, CA-K-Ras, and CA-K-Ras-E37G) were comparable. Mfn2−/− MEFs were cotransfected with empty vector and indicated HA-tagged Ras constructs, and 25 μg of total cell lysates was analyzed by WB analysis. D) Lysates (25 μg) from Mfn2−/− MEFs cotransfected with a flag-tagged Mfn2 plasmid together with H-Ras construct were analyzed by immunoblotting analysis. β-Tubulin was used as loading control. E) Total cell lysates (25 μg) from Mfn2−/− MEFs cotransfected with WT-Raf-1 together with indicated HA-tagged K-Ras constructs were analyzed by immunoblotting using anti-Raf-1 and anti-HA antibodies. β-Tubulin was used as loading control. F) Flag-tagged Mfn21-757 plasmid and indicated flag-tagged K-Ras constructs were expressed separately using in vitro transcription and translation system. Mfn2 protein was allowed to interact separately with the indicated Ras proteins first and then immunoprecipitated with anti-Ras antibody and immunoblotted with anti-Flag antibody. Similar results were obtained in 3 independent experiments. G, H) Mfn1 associates with Ras. Flag-tagged Mfn1 plasmid and HA-tagged CA-K-Ras plasmid were coexpressed in Mfn2−/− MEFs. Cell lysates (500 μg) were immunoprecipitated with either α-Flag monoclonal antibody (G) or α-HA monoclonal antibody (H), and the immunocomplexes were analyzed by WB analysis; 50 μg of total cell extracts was used as input.
To test the nature of the interaction between Ras and Mfn2 in a cell-free system, we used an in vitro transcription and translation system to express Mfn2 and different forms of Ras constructs separately. Different forms of K-Ras and Mfn2 proteins were allowed to interact first before IPs were performed using anti-Ras antibody, and the immunoprecipitated complexes were analyzed by WB analysis. As shown in Fig. 6F, Mfn2 was found to be strongly associated with CA-K-Ras, and this association was significantly decreased in case of effector mutant Ras, CA-K-Ras-E37G. Interestingly, while we observed some interaction between WT-K-Ras and Mfn2 in transfected cells (Fig. 6B), no such interaction was observed in cell-free system. This may be due to the fact that cells transfected with Mfn2 and WT-K-Ras constructs were cultured in serum-containing medium, which may mediate conversion of a certain percentage of WT-K-Ras to the active GTP-bound form. On the other hand, WT-K-Ras was not able to be activated in the cell-free system. Collectively, these data strongly suggested that Mfn2 is a Ras effector molecule and the interaction between Ras and Mfn2 increases the expression of each protein.
Since Mfn2 protein shares a high degree of homology with Mfn1 (81%; ref. 24), and the C-terminal region of Mfn1 (aa 469–741) shares 47% homology with the C-terminal region of NORE1A (aa 191–413; Supplemental Fig. S3A), we wanted to determine whether Mfn1 also interacts with constitutively active Ras. Mfn2−/− MEFs were transfected with a Flag-tagged Mfn1 plasmid along with the HA-tagged CA-K-Ras plasmid, the constitutively active form of Ras. Cell lysates were immunoprecipitated with anti-Flag antibody, and the immunocomplexes were analyzed by anti-HA antibody. As shown in Fig. 6G, constitutively active Ras was found to interact with Mfn1. This interaction was also observed in the reversed IP where anti-HA antibody successfully brought down Mfn1 (Fig. 6H). This interaction between Mfn1 and constitutively active Ras was also observed when IP assay was performed using in vitro transcribed and translated forms of Mfn1 and CA-K-Ras (Supplemental Fig. S3B). Next, we wanted to see whether the expression of Mfn1 and Ras was increased when Mfn1 and constitutively active Ras were coexpressed, as has been observed for Mfn2 (Fig. 6B, right panel). As shown in Supplemental Fig. S3C, Mfn1 and Ras increased each other's expression level depending on the conformation of Ras protein. These data suggested that Mfn1 may also be a Ras effector molecule.
Both N- and C-terminal fragments of Mfn2 cause growth suppression
Next, we attempted to determine the Ras-interacting domain of Mfn2. As shown in Fig. 7A, Mfn2 protein has an N-terminal p21ras signature motif, a GTP-binding domain, 2 coiled-coil regions, and 2 transmembrane domains spanning the mitochondrial outer membrane (2, 3, 8). We constructed 2 truncated flag-tagged constructs of Mfn2: an N-terminal construct (aa 1–264) that includes the p21ras signature motif and the GTP-binding domain, and a C-terminal construct (aa 265–757). The recombinant adenoviruses containing full-length Mfn2 (Adv-Mfn21–757), the N-terminal (Adv-Mfn21–264), and the C-terminal (Adv-Mfn2265–757) fragments of Mfn2 were constructed and used to infect Mfn2−/− MEFs. Cell proliferation assays showed an inhibition of proliferation in the cells infected with either adenovirus as compared with the LacZ infected and uninfected controls (Fig. 7B and Supplemental Fig. S4A). Interestingly, both truncated fragments had stronger growth-suppressive effects compared with the full-length Mfn2. To determine the stage of the cell cycle at which Mfn2 exerts its growth suppressive effect, we performed cell cycle analysis with Mfn2−/− MEFs infected with different Mfn2 constructs. As shown in Fig. 7C, Mfn2-mediated growth suppression was observed at G2/M phase irrespective of the different Mfn2 constructs.
Figure 7.

Both N- and C-terminal fragments of Mfn2 inhibit cell proliferation. A) Schematic representation of Mfn2 protein showing different domains. B) Mfn2−/− MEFs were infected with either nothing (control) or 100 pfu/cell of Adv-LacZ, adenovirus containing a full-length Mfn2 (Adv-Mfn21–757), N-terminal fragment of Mfn2 (Adv-Mfn21–264), or C-terminal fragment of Mfn2 (Adv-Mfn2265–757). Cell proliferation assay was performed in quadruplicate by WST-1 assay (n=3 independent experiments). *P < 0.01 vs. control groups. C) Cell cycle distribution of Mfn2−/− MEFs infected with recombinant adenoviruses (30 pfu) carrying different fragments of Mfn2 gene (n=3). D) Expression of different mitochondrial proteins after overexpression of different fragments of Mfn2. Mfn2−/− MEFs were transfected with either control vector or Flag-tagged Mfn21–757, Flag-tagged Mfn21–264, or Flag-tagged Mfn2265–757 plasmids for 48 h. Total cell extracts (35 μg) were used for WB analysis to check the expression of the indicated mitochondrial proteins. Right panel shows quantitation of the bands normalized to γ-tubulin. E) Antiproliferative capacity of Mfn2 protein is partially independent of its mitochondrial localization. WT MEFs (i), Mfn2−/− MEFs (ii), and Mfn2−/− MEfs transfected with flag-tagged Mfn21–757 (iii, iv), Mfn21–264 (v, vi), or Mfn2265–757 (vii, viii) were incubated with 200 nM MitoTracker Red CMXRos and then incubated with mouse anti-Flag antibody, subsequently incubated with anti-mouse IgG Alexa Fluor 350. Cells were then examined with a confocal microscope.
To determine the effect of different Mfn2 fragments on mitochondrial function, Mfn2−/− MEFs infected with different Mfn2 constructs were used to determine the rate of mitochondrial oxygen consumption. As shown in Supplemental Fig. S4B, the rate of mitochondrial oxygen consumption of Mfn2−/− MEFs was increased by the different Mfn2 constructs in comparison to LacZ-infected cells. These data suggested that the growth-suppressive property of Mfn2 is independent of mitochondrial respiration. To determine the effect of different Mfn2 constructs on the expression of other mitochondrial proteins, cell lysates from Mfn2−/− MEFs infected with different Mfn2 constructs were analyzed by WB analysis. As shown in Fig. 7D, while the level of PGC1 was increased, the level of Drp1 was decreased by the different Mfn2 constructs. Interestingly, the level of Mfn1 was increased both by the N- and the C-terminal fragments of Mfn2 but not by the full-length Mfn2. These data suggested that in Mfn2−/− MEFs, the overexpression of different forms of Mfn2 affected the expression of other mitochondrial proteins differentially.
Since both the N- and C-terminal fragments of Mfn2 successfully inhibited cell proliferation, we wanted to examine whether the antiproliferative effect of the Mfn2 protein was dependent on its mitochondrial localization. Immunostaining was carried out with an anti-Flag antibody after transfecting the Mfn2−/− MEFs with the flag-tagged plasmid containing full-length Mfn2 gene (Mfn21–757), N-terminal fragment of Mfn2 (Mfn21–264), or C-terminal fragment of Mfn2 (Mfn2265–757). Confocal microscopic studies of the WT MEFs exhibited elongated mitochondria, in contrast to the fragmented mitochondria observed in the Mfn2−/− MEFs (Fig. 7Ei, ii). Mitochondrial morphology of Mfn2−/− MEFs transfected with full-length Mfn21–757 plasmid showed elongated morphology, and the protein was localized in the mitochondria (Fig. 7Eiii, iv). On the other hand, the cells transfected with the N-terminal Mfn21–264 fragment showed fragmented mitochondrial morphology along with a small percentage of fusion, and the N-terminal fragment was localized only in the cytosol (Fig. 7Ev, vi). Fragmented mitochondrial morphology along with some elongated morphology was also observed when the Mfn2 −/− MEFs were transfected with the C-terminal Mfn2265–757 fragment, and the fragment was localized in the mitochondria (Fig. 7Evii, viii). Despite its localization in mitochondria, the appearance of fragmented mitochondrial morphology of the C-terminal fragment-transfected cells could be due to the fact that the C-terminal fragment lacks the GTPase domain, which is responsible for mitochondrial fusion (25). Degree of fusion/fission was not measured here, and the nonmitochondrial targeted fragment may nonetheless promote partial fusion. Taken together, these data demonstrated that the antiproliferative effect of Mfn2 was, at least, partially independent of its mitochondrial localization.
While the N-terminal fragment of Mfn2 interacts with Raf-1, the C-terminal fragment interacts with Ras
Next, we explored the possibility of Ras association with these fragments, since both the N- and C-terminal fragments of Mfn2 successfully inhibited cell proliferation. We therefore transfected the Mfn2−/− MEFs with either the flag-tagged N- or C-terminal constructs of Mfn2 and performed coIP assays with an anti-Ras monoclonal antibody. As shown in Fig. 8A, the C-terminal fragment of Mfn2 interacted with Ras, whereas very little or no interaction was observed with the N-terminal fragment. This interaction between C-terminal fragment of Mfn2 and Ras was also observed when both of these proteins were synthesized using an in vitro transcription and translation system, followed by a coIP experiment as described in Fig. 6F (Fig. 8B). Taken together, these observations suggested that the inhibitory capacity of the N-terminal fragment of Mfn2 is independent of Ras interaction, whereas the inhibitory property of the C-terminal fragment of Mfn2 is possibly due to its Ras association.
Figure 8.

While the N-terminal fragment of Mfn2 interacts with Raf-1, the C-terminal fragment interacts with Ras. A) Lysates (500 μg) from Mfn2−/− MEFs transfected with either control vector or plasmids containing Flag-tagged Mfn21–264 or Flag-tagged Mfn2265–757 fragments were immunoprecipitated with α-Ras monoclonal antibody and immunoblotted with anti-Flag antibody (top panel) and an anti-Pan Ras polyclonal antibody (bottom panel). B) Flag-tagged Mfn21–757 and Mfn2265–757 plasmids and Flag-tagged CA-K-Ras plasmid were expressed separately using an in vitro transcription and translation system. Mfn2 proteins were allowed to interact separately with the CA-K-Ras protein first and then immunoprecipitated with α-Mfn2 polyclonal antibody and immunoblotted with α -Flag monoclonal antibody. C) Sequence comparison between Mfn2 N-terminal protein (aa 97–325) and Ras like GTPase superfamily. Asterisks indicate identity; colons indicate strong conservation; periods indicate weak conservation. D) Mfn2−/− MEFs were cotransfected with WT-Raf-1 and indicated Flag-tagged Mfn2 plasmids. Cell lysates (500 μg) were immunoprecipitated with either α-Flag antibody or normal mouse IgG and immunoblotted with either α-Raf-1 antibody (top panel) or α-Flag antibody (bottom panel). Total extracts (25 μg) were used to monitor the expression of Flag-tagged Mfn21–264, Flag-tagged Mfn21–757, and Raf-1. E) Status of phosphorylated and total ERK1/2 in cells cotransfected with WT-Raf-1/empty vector or WT-Raf-1/Flag-tagged Mfn21–264 plasmids. F) Mfn2 associates with endogenous c-Raf protein. Cell lysates (500 μg) of cell lysates from 293A were immunoprecipitated with anti-Mfn2 polyclonal antibody and immunoblotted with either anti-Raf-1 antibody (top panel) or anti-Mfn2 monoclonal antibody (bottom panel); 50 μg of total extract was used as an input. G) Flag-tagged Mfn21–757, Flag-tagged Mfn21–264, and Raf-1 constructs were expressed separately using an in vitro transcription and translation system. Mfn2 proteins were allowed to interact separately with the Raf-1 protein first and then immunoprecipitated with α-Raf-1 monoclonal antibody and immunoblotted with α -Flag monoclonal antibody (top panel) and α Raf-1 polyclonal antibody (bottom panel). H) Mfn2 does not interact with B-Raf; 500 μg of lysates from Mfn2−/− MEFs transfected with either control vector or plasmids containing Flag-tagged Mfn21–264 or Flag-tagged Mfn21–757 were immunoprecipitated with α-Flag antibody and immunoblotted with α-B-Raf antibody (top panel) and an α-Flag antibody (bottom panel); 50 μg of total cell extracts was used to monitor the expression of Flag-Mfn21–264, Flag-Mfn21–757, and B-Raf.
The members of the Ras superfamily are identified by the GTP-binding site, which is made up of several characteristic sequence motifs: switch I, switch II, and several G-box motifs (26). All of these characteristic motifs are present in the N-terminal fragment of Mfn2 protein (aa 1–264) as analyzed by both sequence and structure analyses (Fig. 8C). Since the N-terminal fragment of Mfn2 has a striking similarity with the Ras molecule, we hypothesized that this fragment might interact with Raf-1 and sequester it away from Ras interaction. To evaluate this hypothesis, the flag-tagged N-terminal fragment of Mfn2 and full-length Raf-1 were coexpressed in Mfn2−/− MEFs, and the cell lysates were used for IP assays with an anti-Flag antibody. As shown in Fig. 8D, the N-terminal fragment successfully brought down Raf-1, as was also observed with the full-length Mfn2. Moreover, this interaction was functional, since coexpression of Raf-1 and N-terminal fragment of Mfn2 resulted in the inhibition of ERK1/2 phosphorylation compared with cells transfected with Raf-1 and the empty vector control (Fig. 8E). To determine the endogenous interaction between Mfn2 and Raf1, cell lysates from HEK293A cells were immunoprecipitated with anti-Mfn2 antibody, and the immunocomplexes were blotted with anti-Raf1 antibody. As shown in Fig. 8F, a small portion of total Mfn2 was found to be associated with Raf-1. To further investigate the interaction between Mfn2 and Raf-1, we used in vitro transcription and translation system to express Raf-1 and different forms of Flag-tagged Mfn2 constructs (full-length and N-terminal fragment). These proteins were then allowed to interact first before IPs were performed using anti-Raf-1 antibody. As shown in Fig. 8G, anti-Raf-1 antibody successfully brought down both the full-length and the N-terminal fragment of Mfn2. To determine the specificity of the interaction between Mfn2 and Raf-1, we investigated the interaction between Mfn2 and B-Raf. Cell lysates from Mfn2−/− MEFs transfected with either empty vector or plasmids containing either Flag-tagged full-length or N-terminal fragment of Mfn2 along with B-Raf expression vector were immunoprecipitated with anti-Flag antibody and immunoblotted with anti-B-Raf antibody. As shown in Fig. 8H, no interaction was observed between either the full-length or the N-terminal fragment of Mfn2 and B-Raf, indicating the interaction between the N-terminal fragment of Mfn2 and Raf-1 is specific. Taken together, our data suggest that Mfn2 is a novel Ras effector molecule with a negative effect on Ras-Raf-ERK signaling pathway and exerts growth inhibitory effects both by binding activated Ras and binding Raf-1. The Raf-1-interacting N-terminal fragment does not need to localize to the mitochondria to exert its growth inhibition.
Since Mfn2 protein shares a high degree of homology with Mfn1 (24), the N-terminal region of Mfn1 also carries all the characteristic motifs of the Ras-like GTPase superfamily (Supplemental Fig. S5A). We then investigated whether Mfn1 interacts with Raf-1, as it has been seen in case of Mfn2 (Fig. 8D). Mfn2−/− MEFs were transfected with Flag-tagged Mfn1 plasmid along with Raf-1 plasmid, the cell lysates were used for IP with anti-Flag antibody, and the immunocomplexes were analyzed by WB analysis. As shown in Supplemental Fig. S5B, Mfn1 was found to be associated with Raf-1. To verify this interaction, we used in vitro transcription and translated Mfn1 and Raf-1 proteins as described in Fig. 8G. In this system also, we observed the interaction between Mfn1 and Raf-1 (Supplemental Fig. S5C). These data suggested that like Mfn2, Mfn1 is also capable of interacting with Raf-1. Since Mfn1 shares all the interacting partners of Mfn2 responsible for Mfn2-mediated growth suppression, we wanted to investigate whether Mfn1 is capable of growth suppression in an overexpression system. HEK293A cells were transfected either with an empty vector or an Mfn1 expression vector, and the cell counts were performed at different time points. As shown in Supplemental Fig. S5D, Mfn1 also caused growth suppression when it was overexpressed. Collectively, these data suggested that like Mfn2, Mfn1 can also exert growth inhibitory effects both by binding activated Ras and binding Raf-1.
DISCUSSION
The current study provides the following new information regarding the mechanism of action of Mfn2: Mfn2 fulfilled all the criteria to be a Ras effector molecule in that it binds only to GTP-bound Ras, and this binding is sensitive to Ras effector domain mutation; structure-function analysis studies reveal that the N-terminal fragment of Mfn2 (aa 1–264) shares sequence similarity with the Ras-like GTPase superfamily members, whereas the C-terminal fragment of Mfn2 (aa 265–757) shares a striking similarity with another Ras effector molecule and a tumor suppressor protein, NORE1A; Both the N-terminal and C-terminal fragments of Mfn2 inhibit cell proliferation, and each acts by a distinct mechanism: the N-terminal mediated inhibition was due to its interaction with and inhibition of Raf-1, whereas the C-terminal fragment of Mfn2 inhibited cell proliferation by interacting with Ras; and the inhibition of proliferation by the N-terminal fragment is independent of its mitochondrial localization. Collectively, our data provide new insights regarding the role of Mfn2 in controlling cellular proliferation.
Although it has been shown previously that Mfn2 exerts its antiproliferative effect through inhibition of Ras-Raf-ERK signaling pathway, these studies were done in overexpression systems. The role of endogenous Mfn2 in controlling cell proliferation and the functional domains of Mfn2 mediating this activity were not completely understood. We have demonstrated here the role of endogenous Mfn2 in controlling cellular proliferation in 2 different systems: first, Mfn2 knockdown clones of a B-cell lymphoma cell line BJAB exhibited an increased rate of cell proliferation; and second, Mfn2-knockout MEF cells had 2-fold increase in cell proliferation as compared with the control WT cells, and the proliferative advantage of the knockout MEF cells was blocked on reintroduction of the Mfn2 gene. Collectively, these data demonstrated that endogenous Mfn2 plays an important role in controlling cell proliferation.
In this study, we have also reported for the first time that Mfn1 interacts with the same interacting partners as Mfn2 and causes growth suppression when overexpressed. This is not surprising, since Mfn1 and Mfn2 have a high degree of homology (81%; ref. 24). The role of endogenous Mfn1 on cellular proliferation is under investigation.
With regard to the structure-function relationship of Mfn2, a sequence comparison study reveals that the C-terminal region of Mfn2 (aa 490–757) shares 57% homology with the C-terminal region of NORE1A (aa 191–413), one of the most studied members of the tumor suppressor protein family RASSF (Fig. 6A), and was found to interact with Ras and was capable of inhibiting cell proliferation (Figs. 8A and 7B). Moreover, Mfn2 was found to interact more strongly with CA-K-Ras than with the WT Ras and exhibited much less binding to the CA-K-Ras-E37G construct carrying an effector domain mutation, thus fulfilling the criteria of a Ras effector molecule (Fig. 6F and ref. 21).
One of the most interesting features of Mfn2 is its N-terminal p21Ras signature motif followed by a GTP-binding domain. Based on the sequence and structural analyses, the N-terminal fragment of Mfn2 (aa 1–264) has the same characteristic sequence motifs as observed in the members of the Ras superfamily (Fig. 8C). Interestingly, this N-terminal fragment of Mfn2 does not interact with Ras but interacts with Raf-1 and causes growth suppression by inhibiting ERK phosphorylation (Fig. 8D, E). The Raf-1-interacting N-terminal fragment does not need to localize to the mitochondria to exert its growth inhibition. Our data suggested that inhibition of Ras-Raf-ERK pathway by Mfn2 was mediated by at least two mechanisms: direct interaction with Ras through its C terminus, and sequestration of Raf-1 via its N terminus. It is conceivable that Mfn2 sequesters Ras and Raf-1 away from the canonical activation site, thus implementing the growth-suppressive function of Mfn2. The detail of the model is currently under investigation.
Supplementary Material
Acknowledgments
The authors are grateful to Dr. David C. Chan (Division of Biology, California Institute of Technology, Pasadena, CA, USA) for WT and Mfn2-knockout MEFs. The authors thank Drs. Carl Sasaki and Jessica Curtis for technical assistance and Rachel Munk for help in editing the paper. The authors also thank the Flow Cytometry facility at the National Institute on Aging for help in cell cycle analysis.
This research was supported (in part) by the Intramural Research Program of the National Institutes of Health, National Institute on Aging.
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
- DMEM
- Dulbecco's modified Eagle's medium
- ERK
- extracellular signal-regulated kinase
- FBS
- fetal bovine serum
- GTP
- guanosine triphosphate
- HA
- hemagglutinin
- IP
- immunoprecipitation
- MEF
- mouse embryonic fibroblast
- PBS
- phosphate-buffered saline
- pfu
- plaque-forming unit
- RA
- Ras association
- RASSF
- Ras association domain family
- SARAH
- Salvador, Rassf, Hippo
- VSMC
- vascular smooth muscle cell
- WB
- Western blot
- WT
- wild type
REFERENCES
- 1. De Brito O. M., Scorrano L. (2008) Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456, 605–610 [DOI] [PubMed] [Google Scholar]
- 2. De Brito O. M., Scorrano L. (2009) Mitofusin-2 regulates mitochondrial and endoplasmic reticulum morphology and tethering: the role of Ras. Mitochondrion 9, 222–226 [DOI] [PubMed] [Google Scholar]
- 3. Zuchner S., Mersiyanova I. V., Muglia M., Bissar-Tadmouri N., Rochelle J., Dadali E. L., Zappia M., Nelis E., Patitucci A., Senderek J., Parman Y., Evgrafov O., Jonghe P. D., Takahashi Y., Tsuji S., Pericak-Vance M. A., Quattrone A., Battaloglu E., Polyakov A. V., Timmerman V., Schroder J. M., Vance J. M. (2004) Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat. Genet. 36, 449–451 [DOI] [PubMed] [Google Scholar]
- 4. Misko A. L., Sasaki Y., Tuck E., Milbrandt J., Baloh R. H. (2012) Mitofusin2 mutations disrupt axonal mitochondrial positioning and promote axon degeneration. J. Neurosci. 32, 4145–4155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hernandez-Alvarez M. I., Thabit H., Burns N., Shah S., Brema I., Hatunic M., Finucane F., Liesa M., Chiellini C., Naon D., Zorzano A., Nolan J. J. (2010) Subjects with early-onset type 2 diabetes show defective activation of the skeletal muscle PGC-1α/mitofusin-2 regulatory pathway in response to physical activity. Diabetes Care 33, 645–651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pich S., Bach D., Briones P., Liesa M., Camps M., Testar X., Palacin M., Zorzano A. (2005) The Charcot-Marie-Tooth type 2A gene product, Mfn2, up-regulates fuel oxidation through expression of OXPHOS system. Hum. Mol. Genet. 14, 1405–1415 [DOI] [PubMed] [Google Scholar]
- 7. Bach D., Pich S., Soriano F. X., Vega N., Baumgartner B., Oriola J., Daugaard J. R., Lloberas J., Camps M., Zierath J. R., Rabasa-Lhoret R., Wallberg-Henriksson H., Laville M., Palacin M., Vidal H., Rivera F., Brand M., Zorzano A. (2003) Mitofusin-2 determines mitochondrial network architecture and mitochondrial metabolism. A novel regulatory mechanism altered in obesity. J. Biol. Chem. 278, 17190–17197 [DOI] [PubMed] [Google Scholar]
- 8. Chen K. H., Guo X., Ma D., Guo Y., Li Q., Yang D., Li P., Qiu X., Wen S., Xiao R. P., Tang J. (2004) Dysregulation of HSG triggers vascular proliferative disorders. Nat. Cell Biol. 6, 872–883 [DOI] [PubMed] [Google Scholar]
- 9. Chien K. R., Hoshijima M. (2004) Unravelling Ras signals in cardiovascular disease. Nat. Cell Biol. 6, 807–808 [DOI] [PubMed] [Google Scholar]
- 10. Guo X., Chen K. H., Guo Y., Liao H., Tang J., Xiao R. P. (2007) Mitofusin 2 triggers vascular smooth muscle cell apoptosis via mitochondrial death pathway. Circ. Res. 101, 1113–1122 [DOI] [PubMed] [Google Scholar]
- 11. Shen T., Zheng M., Cao C., Chen C., Tang J., Zhang W., Cheng H., Chen K. H., Xiao R. P. (2007) Mitofusin-2 is a major determinant of oxidative stress-mediated heart muscle cell apoptosis. J. Biol. Chem. 282, 23354–23361 [DOI] [PubMed] [Google Scholar]
- 12. Wu L., Li Z., Zhang Y., Zhang P., Zhu X., Huang J., Ma T., Lu T., Song Q., Li Q., Guo Y., Tang J., Ma D., Chen K. H., Qiu X. (2008) Adenovirus-expressed human hyperplasia suppressor gene induces apoptosis in cancer cells. Mol. Cancer Ther. 7, 222–232 [DOI] [PubMed] [Google Scholar]
- 13. Naldini L., Blomer U., Gage F. H., Trono D., Verma I. M. (1996) Efficient transfer, integration, and sustained long-term expression of the transgene in adult rat brains injected with a lentiviral vector. Proc. Natl. Acad. Sci. U. S. A. 93, 11382–11388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Drosten M., Dhawahir A., Sum E. Y., Urosevic J., Lechuga C. G., Esteban L. M., Castellano E., Guerra C., Santos E., Barbacid M. (2010) Genetic analysis of Ras signalling pathways in cell proliferation, migration and survival. EMBO J. 29, 1091–1104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Leblanc V., Tocque B., Delumeau I. (1998) Ras-GAP controls Rho-mediated cytoskeletal reorganization through its SH3 domain. Mol. Cell. Biol. 18, 5567–5578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Van der Weyden L., Adams D. J. (2007) The Ras-association domain family (RASSF) members and their role in human tumourigenesis. Biochim. Biophys. Acta 1776, 58–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Richter A. M., Pfeifer G. P., Dammann R. H. (2009) The RASSF proteins in cancer; from epigenetic silencing to functional characterization. Biochim. Biophys. Acta 1796, 114–128 [DOI] [PubMed] [Google Scholar]
- 18. Avruch J., Xavier R., Bardeesy N., Zhang X. F., Praskova M., Zhou D., Xia F. (2009) Rassf family of tumor suppressor polypeptides. J. Biol. Chem. 284, 11001–11005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Park J., Kang S. I., Lee S.-Y., Zhang X. F., Kim M. S., Beers L. F., Lim D.-S., Avruch J., Kim H.-S., Lee S. B. (2010) Tumor suppressor Ras association domain family 5 (RASSF5/NORE1) mediates death receptor ligand-induced apoptosis. J. Biol. Chem. 285, 35029–35038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sherwood V., Recino A., Jeffries A., Ward A., Chalmers A. D. (2010) The N-terminal RASSF family: a new group of Ras-association-domain-containing proteins, with emerging links to cancer formation. Biochem. J. 425, 303–311 [DOI] [PubMed] [Google Scholar]
- 21. Joneson T., Bar-Sagi D. (1997) Ras effectors and their role in mitogenesis and oncogenesis. J. Mol. Med. 75, 587–593 [DOI] [PubMed] [Google Scholar]
- 22. Eckfeld K., Hesson L., Vos M. D., Bieche I., Latif F., Clark G. J. (2004) RASSF4/AD037 is a potential ras effector/tumor suppressor of the RASSF family. Cancer Res. 64, 8688–8693 [DOI] [PubMed] [Google Scholar]
- 23. Vos M. D., Ellis C. A., Elam C., Ulku A. S., Taylor B. J., Clark G. J. (2003) RASSF2 is a novel K-Ras-specific effector and potential tumor suppressor. J. Biol. Chem. 278, 28045–28051 [DOI] [PubMed] [Google Scholar]
- 24. Rojo M., Legros F., Chateau D., Lombes A. (2002) Membrane topology and mitochondrial targeting of mitofusins, ubiquitous mammalian homologs of the transmembrane GTPase Fzo. J. Cell Sci. 115, 1663–1674 [DOI] [PubMed] [Google Scholar]
- 25. Ishihara N., Eura Y., Mihara K. (2004) Mitofusin 1 and 2 play distinct roles in mitochondrial fusion reactions via GTPase activity. J. Cell Sci. 117, 6535–6546 [DOI] [PubMed] [Google Scholar]
- 26. Marchler-Bauer A., Lu S., Anderson J. B., Chitsaz F., Derbyshire M. K., DeWeese-Scott C., Fong J. H., Geer L. Y., Geer R. C., Gonzales N. R., Gwadz M., Hurwitz D. I., Jackson J. D., Ke Z., Lanczycki C. J., Lu F., Marchler G. H., Mullokandov M., Omelchenko M. V., Robertson C. L., Song J. S., Thanki N., Yamashita R. A., Zhang D., Zhang N., Zheng C., Bryant S. H. (2011) CDD: a conserved domain database for the functional annotation of proteins. Nucleic Acids Res. 39, D225–D229 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.