

Abstract
Lack of high potency agonists has restricted analysis of the G protein–coupled receptor GPR35. Moreover, marked variation in potency and/or affinity of current ligands between human and rodent orthologs of GPR35 has limited their productive use in rodent models of physiology. Based on the reported modest potency of the antiasthma and antiallergic ligands cromolyn disodium and nedocromil sodium, we identified the related compounds lodoxamide and bufrolin as high potency agonists of human GPR35. Unlike previously identified high potency agonists that are highly selective for human GPR35, both lodoxamide and bufrolin displayed equivalent potency at rat GPR35. Further synthetic antiallergic ligands, either sharing features of the standard surrogate agonist zaprinast, or with lodoxamide and bufrolin, were also shown to display agonism at either human or rat GPR35. Because both lodoxamide and bufrolin are symmetric di-acids, their potential mode of binding was explored via mutagenesis based on swapping between the rat and human ortholog nonconserved arginine residues within proximity of a key conserved arginine at position 3.36. Computational modeling and ligand docking predicted the contributions of different arginine residues, other than at 3.36, in human GPR35 for these two ligands and were consistent with selective loss of potency of either bufrolin or lodoxamide at distinct arginine mutants. The computational models also suggested that bufrolin and lodoxamide would display reduced potency at a low-frequency human GPR35 single nucleotide polymorphism. This prediction was confirmed experimentally.
Introduction
Although poorly characterized, the seven transmembrane domain (TMD), G protein–coupled receptor (GPCR) GPR35 has attracted attention as a potential therapeutic target in disease areas ranging from pain to hypertension (MacKenzie et al., 2011; Milligan, 2011). Although GPR35 is indicated to be a receptor responsive to the endogenously produced tryptophan metabolite kynurenic acid (Wang et al., 2006), lack of convergence on this issue has resulted in a number of efforts to identify surrogate agonist ligands that might be used to help further define the roles of this receptor. Although a number of such ligands have been identified (Taniguchi et al., 2006; Jenkins et al., 2010; Zhao et al., 2010; Deng et al., 2012; Funke et al., 2013; Neetoo-Isseljee et al., 2013), many of these are either of modest potency and/or display markedly different potency at human and rodent orthologs of GPR35 (Jenkins et al., 2010; Funke et al., 2013; Neetoo-Isseljee et al., 2013). This has posed challenges both in efforts to define the orthosteric binding pocket of the receptor and to use rodents and cell lines derived from such animals to further explore the function of GPR35. It would, therefore, be of particular value to identify agonists with similar and high potency at the human and rodent orthologs of GPR35 and to better understand the basis of ligand selectivity between species. One common feature of human and rat GPR35 is the presence of an arginine residue in TMD III [position 3.36 in the Ballesteros and Weinstein (1995) residue numbering system] that when mutated to a nonbasic residue results in virtual complete loss of potency of kynurenic acid and a number of GPR35 surrogate agonist ligands (Jenkins et al., 2011). Importantly, arginine 3.36 is conserved in a number of other receptors, such as the lactate receptor GPR81 (Liu et al., 2009), that are activated by endogenously generated acids, suggesting that kynurenic acid and potentially other acidic ligands, interact with this residue via an ionic interaction.
The antiasthma and antiallergic agents cromolyn disodium (Jenkins et al., 2010; Yang et al., 2010) and nedocromil sodium (Yang et al., 2010) were recently shown to act as moderately potent agonists of GPR35. These ligands are both di-acids with marked mirror image symmetry (Fig. 1). On the basis of these findings, we considered whether a number of other symmetric di-acids might also be GPR35 agonists. The antiallergic mast cell stabilizers lodoxamide and bufrolin (also designated ICI 74917, 6-butyl-4,10-dihydroxy-1,7-phenanthroline-2,8-dicarboxylic acid) both display these characteristics (Fig. 1) and were shown to be the most potent agonists of both human and rat GPR35 yet reported. We then considered whether activation of GPR35 might provide a common mechanism of action of a broad group of mast cell stabilizers. This, however, was not the case. Zaprinast (Taniguchi et al., 2006; Jenkins et al., 2010, 2011) has become the standard surrogate agonist of GPR35 and, akin to zaprinast, a number of mast cell stabilizers also contain an acid bioisostere. Because zaprinast is an example of a ligand that displays marked variation in potency between rodent and human GPR35 (Taniguchi et al., 2006; Jenkins et al., 2010, 2011), this also encouraged us to also explore the contributions of a series of nonmaintained arginine residues within regions of the receptor species orthologs that are predicted to define the ligand binding pocket. Cross-species alterations of such residues provided novel insights into the binding pocket. Computational modeling and ligand docking studies provided strong rationale for the experimental data and further suggested that a specific nonsynonymous single-nucleotide polymorphism (SNP) variant in human GPR35 might show differences in ligand potency. This hypothesis was tested and shown to be valid, whereas ligand potency at other human SNP variants was unaffected.
Fig. 1.
Structures of key GPR35 ligands. The structures of key ligands used in the studies are shown.
Materials and Methods
Materials for cell culture were from Sigma-Aldrich (Gillingham, Dorset, UK), Life Technologies (Paisley, Strathclyde, UK), or PAA Laboratories Ltd (Yeovil, Somerset, UK). Polyethylenimine linear MW-25000 was from Polysciences, Inc. (Warrington, PA). Zaprinast was purchased from Tocris Bioscience (Bristol, UK). Amlexanox, cromolyn disodium, doxantrazole, ketitofen fumarate, pemirolast potassium, tranilast, and Hoechst 33258 were purchased from Sigma-Aldrich. Bufrolin, BRL10833 [5,6-dimethyl-2-nitro-1H-indene-1,3(2H)-dione], and nedocromil sodium were synthesized in house. Lodoxamide was a gift from Drs. Ed McIver and Debra Taylor (Medical Research Council Technology, London, UK). With the exception of pemirolast potassium, which was reconstituted in dH2O, ligands were initially dissolved in dimethylsulfoxide (DMSO) and then diluted in assay buffer.
Plasmids and Mutagenesis.
All novel plasmids employed expressed human (GPR35a or GPR35b) or rat GPR35 receptor constructs that contain an enhanced yellow fluorescent protein (eYFP) fused to the C terminus and an N-terminal FLAG epitope tag as previously described (Jenkins et al., 2010, 2011). Individual amino acid swap mutations between human and rat sequences were introduced into the FLAG-hGPR35a-eYFP or FLAG-rGPR35-eYFP constructs using the QuikChange method (Stratagene, La Jolla, CA). All mutations were confirmed by DNA sequencing.
Cloning of Human GPR35b.
Human GPR35b, containing a FLAG epitope (amino acid sequence DYKDDDDK) at the N terminus, was produced from cDNA generated from HT29 cells by polymerase chain reaction using the following primers: sense, 5′ACTCAAGCTTGCCACCATGGATTACAAGGATGACGACGATAAGCTGAGTGGTTCCCGGG 3′; and antisense: 5′ACTCGCGGCCGCAGGCGAGGGTCACGC 3′. The HinDIII and NotI restriction sites used for cloning are underlined. The resulting construct was cloned in-frame into the HinDIII/NotI sites of an eYFP-pcDNA5/FRT/TO plasmid and integrity of the fusion was confirmed by DNA sequencing.
Cell Culture and Transfection.
Flp-In TREx 293 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) without sodium pyruvate (Life Technologies), supplemented with 10% (v/v) fetal bovine serum, 1% penicillin/streptomycin mixture, and 10 μg/ml blasticidin. Human embryonic kidney (HEK) 293T cells were maintained in DMEM supplemented with 0.292 g/l l-glutamine, 10% (v/v) fetal bovine serum, and 1% penicillin/streptomycin mixture. Transient transfections using HEK293T cells were performed using polyethylenimine, with experiments carried out 48 hours after transfection (Jenkins et al., 2011). HT-29 human adenocarcinoma cells were purchased from the American Type Culture Collection (Gaithersburg, MD) and maintained in McCoy’s 5A (Modified) media containing 25 mM HEPES. PathHunter β-arrestin recruitment Chinese hamster ovary (CHO–K1) cells stably expressing human GPR35 and β-arrestin-2 (DiscoveRx, Fremont, CA) (Neetoo-Isseljee et al., 2013) were routinely passaged in complete growth medium (DMEM/F12 with GlutaMAX + 10% fetal bovine serum). All cells were maintained at 37°C and 5% CO2 in a humidified cell culture incubator.
GPR35–β-Arrestin-2 Interaction Assays.
We used two distinct methods. The PathHunter β-arrestin-2 recruitment assay was performed using the CHO–K1 cell line stably expressing human GPR35 and β-arrestin-2 (DiscoveRx) as previously described (Neetoo-Isseljee et al., 2013). The bioluminescence resonance energy transfer (BRET)–based β-arrestin-2 recruitment assay was performed using HEK293T cells transfected transiently to coexpress forms of human GPR35a, human GPR35b, or rat GPR35 along with β-arrestin-2, as described by Jenkins et al., (2011).
ArrayScan High Content Analysis of GPR35 Internalization.
FLAG-hGPR35-eYFP Flp-In T-REx 293 cells were seeded into poly(d-lysine)–coated black clear-bottom 96-well plates at a density of 80,000 cells/well. Receptor expression was induced via the addition of doxycycline (100 ng/ml) 6 hours after seeding. Twenty-four hours later, cells were washed twice with serum-free medium and incubated with ligand for 45 minutes at 37°C, before being fixed with paraformaldehyde (4% v/v). Cells were washed with phosphate-buffered saline and incubated with 10 μg/ml Hoechst nuclear stain at 37°C for 30 minutes to allow determination of cell number. Receptor internalization was quantified using an ArrayScan II high content imager (Cellomics, Boston, MA), which detected FLAG-hGPR35-eYFP receptor trafficking to endocytic recycling compartments.
Visualization of GPR35 Internalization.
FLAG-hGPR35-eYFP Flp-In T-REx 293 cells were cultured on poly(d-lysine)–coated glass coverslips and incubated for 24 hours before treatment with doxycycline (100 ng/ml) to induce receptor expression. Live cells were then imaged using a Zeiss VivaTome spinning disk confocal microscopy system (Carl Zeiss GmbH, Oberkochen, Germany). Images were taken prior to the addition of ligand, and every 15 minutes after ligand addition for a total of 45 minutes.
Epic Label-Free Dynamic Mass Redistribution Assays.
HT-29 cells were plated in complete medium at a density of 30,000 cells/well into fibronectin-coated Epic 384-well plates (Corning, Inc., Corning, NY), at a volume of 50 µl/well, prior to incubation for 24 hours at 37°C in a humidified atmosphere containing 5% CO2. At 95% confluence, the cells were washed three times with serum-free medium containing 25 mM HEPES using a Biomek FXP automated liquid handling system (Beckman Coulter, Brea, CA), before a final addition of serum-free medium containing 0.01% DMSO. Cells were then equilibrated for 90 minutes at 26°C in the Epic BT system (Corning), before acquisition of basal data for 2 minutes. Ligand, prepared in serum-free medium and normalized for DMSO concentration (0.01%), was then added using the Biomek FXP at a volume of 10 µl/well before measurement of dynamic mass redistribution (DMR) for 60 minutes at 26°C. Kinetic reads generated using the whole-cell scanning application (4 reads/well) were baseline corrected, and concentration-response curves plotted from data generated 5 minutes after ligand addition using GraphPad Prism (GraphPad Software, Inc., La Jolla, CA). Antagonist assays were performed as described for the agonist format, except that the antagonist was added 30 minutes prior to the addition of agonist, with the agonist response plotted from data generated at 35 minutes.
Enzyme-Linked Immunosorbent Assay.
PathHunter CHO–K1 cells stably expressing human GPR35 and β-arrestin-2 were seeded into clear 96-well plates at a density of 30,000 cells/well in complete growth medium. Receptor expression was induced via the addition of doxycycline (100 ng/ml). Twenty-four hours later, cells were washed with wash buffer (phosphate-buffered saline with 1% bovine serum albumin) and incubated with a 1:300 dilution of anti-FLAG antibody (Sigma-Aldrich) in complete growth medium for 120 minutes at 37°C. Medium-only controls were also run. Cells were washed with wash buffer and incubated with a 1:1000 dilution of anti-mouse horseradish peroxidase (Cell Signaling Technology, Danvers, MA) in complete growth medium at 37°C for 40 minutes. Cells were washed with wash buffer and incubated with ABTS [2,2′-azino-bis(3-ethylbenzothiazoline-6-sulphonic acid)] substrate (Peprotech, London, UK) for 40 minutes at room temperature. The plate was centrifuged at 3000g, 40μl of supernatant was transferred to a clear 96-well plate, and the plate was read on a Synergy plate reader (Biotek, Potton, Bedfordshire, UK) at 405 and 605 nm. The 605-nm values were subtracted from 405-nm values. Mean values for medium-only controls were subtracted from anti-FLAG values and the resulting values were divided by the mean wild-type value to give expression relative to wild type.
Computational Methods.
Modeler 9v8 (Martí-Renom et al., 2000) was used to model the TMD helices I–VII, and both intracellular and extracellular loops 1–3, of human and rat GPR35 using the structure of the protease-activated receptor 1 as the template (PDB ID 3VW7) (Zhang et al., 2012). Ligands were docked, by interactive computer graphics, into the receptor models with a negatively charged group interacting with Arg3.36. These ligand-receptor complexes were embedded in membrane bilayer and refined by energy minimization and molecular dynamics simulations with gromacs4.6 (Hess et al., 2008) using a previously described protocol (Cordomí et al., 2012). Modeling figures were generated using PyMol 1.5.3. (Schrödinger LLC, 2012).
Data Analysis.
All data presented represent the mean ± S.E. of at least three independent experiments. Data analysis and curve fitting were carried out using GraphPad Prism version 5.0. Concentration-response data were plotted on a log axis, where the untreated vehicle control condition was plotted at one log unit lower than the lowest test concentration of ligand and fitted to three-parameter agonist concentration response curves using standard approaches. Statistical analysis was carried out using one-way analysis of variance followed by Dunnett’s post-hoc test.
Results
Previous studies showed that the antiasthma and antiallergic agents cromolyn disodium (Jenkins et al., 2010; Yang et al., 2010) and nedocromil sodium (Yang et al., 2010) are modestly potent agonists of GPR35. This was confirmed in a PathHunter human GPR35a–β-arrestin-2 interaction and complementation assay (Table 1), with nedocromil sodium (EC50 = 0.97 ± 0.09 μM) being approximately 3-fold more potent than cromolyn disodium (EC50 = 2.98 ± 1.27 μM). Because GPR35 is expressed by human mast cells and levels are reportedly upregulated by exposure to the allergic stimulus IgE (Yang et al., 2010), we set out to investigate whether other antiallergic ligands and mast cell stabilizers might also act as agonists at GPR35 and, if so, potentially provide a common mode of action. Bufrolin (Fig. 1), a compound shown originally to be an effective inhibitor of passive cutaneous anaphylaxis in rats, a model of IgE-induced allergy, was a highly potent agonist of human GPR35a (EC50 = 2.9 ± 0.7 nM) (Table 1), whereas another antiallergic compound effective in the passive cutaneous anaphylaxis model, sodium nivimedone (BRL10833) (Lumb et al., 1979), although substantially less potent (EC50 = 1.9 ± 0.3 μM), also displayed agonism at human GPR35a (Table 1). Interestingly, lodoxamide (Fig. 1; Table 1), used topically for the treatment of allergic conjunctivitis, was also identified as a highly potent (EC50 = 1.6 ± 0.4 nM) agonist of GPR35a. Despite this overlap of GPR35 agonism and either clinical use or initial development as antiasthma or antiallergic medicines, this did not provide a common mode of action of mast cell stabilizing compounds. For example, ligands such as tranilast, which has been used to treat allergic disorders such as asthma, allergic rhinitis, and atopic dermatitis, displayed no agonist activity (Table 1).
TABLE 1.
A number of antiasthma and antiallergenics act as potent agonists of human GPR35a
PathHunter human GPR35a–β-arrestin-2 interaction assays were performed using a CHO–K1 cell line stably expressing human GPR35 and β-arrestin-2. Data are presented as the mean ± S.E.M. and are derived from three independent experiments.
| Compound | EC50 |
|---|---|
| nM | |
| Nedocromil sodium | 970 ± 87.0 |
| Cromolyn disodium | 2980 ± 127 |
| Bufrolin | 2.93 ± 0.71 |
| Lodoxamide | 1.61 ± 0.42 |
| Nivimedone sodium | 1930 ± 348 |
| Tranilast | No response |
To build on these initial studies, we also used a distinct human GPR35a–β-arrestin-2 interaction assay that is based on agonist-induced BRET (Jenkins et al., 2010, 2011, 2012). As noted previously (Neetoo-Isseljee et al., 2013), although this assay often displays slightly lower sensitivity than the above-described PathHunter protein complementation assay, both lodoxamide (EC50 = 3.6 ± 0.2 nM) and bufrolin (EC50 = 12.8 ± 0.7 nM) were again highly potent human GPR35a agonists (Fig. 2A), displaying approximately 200- to 700-fold greater potency than the current standard surrogate ligand, zaprinast (EC50 = 2.6 ± 0.1 μM) (Fig. 2A). Moreover, tranilast was again inactive in this assay format (data not shown), as were two further mast cell stabilizers, pemirolast (Fig. 2A) and ketotifen fumarate (data not shown). Furthermore, doxantrazole, another ligand trialed for the treatment of asthma, although displaying modest potency (EC50 = 3.4 ± 0.5 μM), was clearly a partial agonist (Fig. 2A), as was amlexanox (EC50 = 4.1 ± 0.5 μM) (Fig. 2A), a mast cell stabilizer, also recently described (Neetoo-Isseljee et al., 2013; Southern et al., 2013) as a GPR35 agonist.
Fig. 2.

Some, but not all, antiallergenics are GPR35 agonists. The ability of a range of compounds that have effectiveness as antiallergenics and mass cell stabilizers, including lodoxamide (●), bufrolin (□), amlexanox (▴), doxantrazole (◇), pemirolast (▿), and cromolyn disodium (○), to promote interactions between human GPR35a (A) or human GPR35b (B) and β-arrestin-2 was compared with the prototypic GPR35 agonist zaprinast (♦) in BRET-based assays. As noted in Results, the absolute signals obtained using GPR35b were substantially lower than when using GPR35a.
Although the above studies clearly demonstrated that GPR35a is activated by a number of antiallergic ligands, they did not suggest that this GPCR might provide a common target and mechanism of action for the broad class of ligands. However, human GPR35 exists as two distinct splice variant forms, whereby GPR35b differs from GPR35a by the presence of an additional 31 amino acids inserted into the extreme N-terminal, extracellular domain (Okumura et al., 2004; Milligan, 2011). To assess any potentially relevant differences in ligand pharmacology between these variants, we cloned GPR35b from human HT-29 colorectal adenocarcinoma cells. After in-frame addition of eYFP to the C-terminal tail of the receptor and introduction of a N-terminal FLAG epitope tag sequence to mimic the GPR35a construct, BRET-based GPR35b–β-arrestin-2 interaction assays were also performed in HEK293T cells. Although the absolute signal in these assays was substantially lower than when using GPR35a, the overall results and the measured potency and rank order of ligands including lodoxamide, bufrolin, zaprinast, and cromolyn disodium were indistinguishable from GPR35a, and both amlexanox and doxantrazole remained partial agonists (Fig. 2B).
A number of previously reported GPR35 agonists cause rapid and extensive internalization of the receptor (Jenkins et al., 2011). We therefore used this as a further endpoint to determine potential effects of these compounds on human GPR35a. Flp-In T-REx 293 cells stably harboring human FLAG-GPR35a-eYFP at the Flp-In T-REx locus were induced to express the receptor by the addition of the antibiotic doxycycline (Jenkins et al., 2011). Using high content analysis of the cellular distribution of eYFP, the effect and high potency of lodoxamide (EC50 = 5.4 ± 0.6 nM) and bufrolin (EC50 = 22.7 ± 2.0 nM) as GPR35a agonists were confirmed (Fig. 3). Moreover, as in the β-arrestin-2 interaction assay, these two compounds were substantially more potent than either zaprinast (EC50 = 4.1 ± 1.0 μM) or cromolyn disodium (EC50 = 3.7 ± 1.2 μM) (Fig. 3). Finally, because all the above assays used cells transfected with substantially modified forms of the human GPR35 variants, we also conducted “label-free” DMR experiments (Schröder et al., 2010; Deng et al., 2011, 2012) using HT-29 cells (Fig. 4). Lodoxamide, bufrolin, zaprinast, and cromolyn disodium all generated concentration-dependent modulation of the signal. Moreover, each ligand displayed both highly similar potencies and rank order of potency as in the more artificial β-arrestin-2 interaction and receptor internalization assays, consistent with the effects of the ligands in HT-29 cells being mediated by GPR35 (Fig. 4A). To further confirm that effects of lodoxamide reflected activation of GPR35, HT-29 cells were coincubated with lodoxamide and various fixed concentrations of the human GPR35–specific antagonist (Zhao et al., 2010; Jenkins et al., 2012) ML-145 (2-hydroxy-4-[4-[(5Z)-5-[(E)-2-methyl-3-phenylprop-2-enylidene]-4-oxo-2-sulfanylidene-1,3-thiazolidin-3-yl]butanoylamino]benzoic acid). This resulted in a parallel, surmountable, and rightward shift of the concentration-response curve for lodoxamide, consistent with competitive interactions of the ligands with GPR35 (Fig. 4B).
Fig. 3.

High content analysis of internalization of human GPR35a confirms agonism of certain antiallergenics at this receptor. (A) The capacity of varying concentrations of lodoxamide (●), bufrolin (□), zaprinast (♦), or cromolyn disodium (○) to promote internalization of human GPR35a-eYFP was assayed and quantified via high content analysis as number of endocytic recycling compartments (ERCs) per cell. (B) Representative images of the cellular distribution of human FLAG-GPR35a-eYFP at the times indicated after addition of maximally effective concentrations of lodoxamide (5 μM), bufrolin (100 nM), zaprinast (50 μM), and cromolyn disodium (100 μM) are displayed.
Fig. 4.

DMR studies indicate that ligand effects in HT-29 cells are mediated via GPR35. (A) The capacity of varying concentrations of lodoxamide (●), bufrolin (□), zaprinast (♦), and cromolyn disodium (○) to modulate DMR signals in HT-29 cells is shown. (B) Effects of defined concentrations of the human GPR35-specific antagonist ML-145 on the agonist effects of lodoxamide are shown.
We and others have reported that many compounds first identified as ligands at human GPR35, including zaprinast, display markedly different potency and/or activity at rodent orthologs (Taniguchi et al., 2006; Jenkins et al., 2010, 2012; Funke et al., 2013; Neetoo-Isseljee et al., 2013). We next examined the activity of a variety of mast cell stabilizers at the rat ortholog of GPR35 in the BRET-based β-arrestin-2 interaction assay. Lodoxamide (EC50 = 12.5 ± 0.6 nM) was also a high potency agonist at rat GPR35 (Fig. 5), and this was also true for bufrolin (EC50 = 9.9 ± 0.4 nM) (Fig. 5). As noted previously (Jenkins et al., 2010, 2011), zaprinast was substantially more potent (EC50 = 98.4 ± 3.7 nM) at rat GPR35 (Fig. 5). In addition, amlexanox, although remaining a partial agonist compared with zaprinast, was approximately 500-fold more potent at the rat ortholog (EC50 = 23.2 ± 3.3 nM) than at human GPR35a (EC50 = 4.1 ± 0.4 μM) (Fig. 5). As at the human receptor, tranilast and ketotifen fumarate displayed no significant activity (data not shown). However, in marked contrast to the human ortholog, pemirolast functioned as a high potency (94.8 ± 5.5 nM), full agonist at rat GPR35 (Fig. 5), and doxantrazole also markedly gained potency and efficacy (Fig. 5).
Fig. 5.
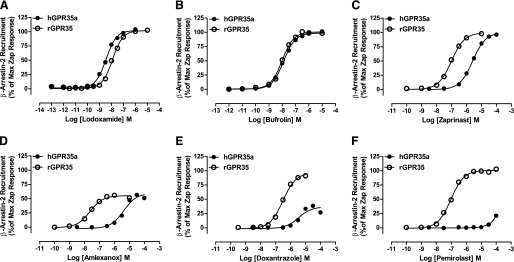
Lodoxamide and bufrolin are high potency agonists at both human and rat GPR35, whereas other ligands display substantial species ortholog selectivity. The ability of varying concentrations of lodoxamide (A), bufrolin (B), zaprinast (C) amlexanox (D), doxantrazole (E), and pemirolast (F) to promote interactions between β-arrestin-2 and either human GPR35a or rat GPR35 in BRET-based assays is shown.
Given both that the novel ligands identified in these studies are negatively charged and that most are characterized by a strong planarity due to a series of fused rings (bufrolin, amlexanox, doxantrazole, pemirolast), we then performed a series of studies designed to define the mode of binding of the most potent ligands (i.e., lodoxamide and bufrolin). Although each of these ligands showed high potency at both human and rat GPR35, we also aimed to determine whether there might be significant differences in the details of binding between the rat and human orthologs. As anticipated from previous work using zaprinast and kynurenic acid (Jenkins et al., 2011), replacement of Arg3.36 with Ala eliminated responsiveness to all of the ligands tested, confirming the importance of this residue (not shown). To extend these studies, initially we aligned the sequences of human GPR35a and rat GPR35 (Fig. 6A). Based on the fact that most GPR35 agonists are acids, acid bioisosteres, or even di-acids with marked mirror image symmetry (Fig. 1), we generated a series of reverse-swap mutations involving positively charged residues that vary between these species within regions of the TMDs and extracellular loops that recent comparative analysis of X-ray structures suggest may form parts of the general GPCR ligand binding pocket (Venkatakrishnan et al., 2013). Alteration in human GPR35a of Arg6.58 to Gln (Arg240Gln), as found in rat GPR35, resulted in modest reduction (1.5- to 6-fold) in potency of lodoxamide, zaprinast, and cromolyn disodium but a substantially larger (12-fold) reduction in potency for bufrolin (Fig. 6; Tables 2 and 3). The reverse mutation of Gln to Arg (Gln238Arg) in rat GPR35 resulted in no substantial changes in potency to these ligands (Fig. 6; Tables 2 and 3). More interestingly, alteration of Arg7.32 in human to the rat equivalent residue serine (Arg255Ser), as well as apparently reducing the overall efficacy response to ligands, resulted in a substantial decrease in potency to lodoxamide (41-fold) without a corresponding effect on the potency of bufrolin (2-fold), but also with an increase in potency to both zaprinast (2-fold) and cromolyn disodium (4-fold) (Fig. 6; Tables 2 and 3). The reverse alteration of Ser7.32 to Arg (Ser253Arg) in rat GPR35 resulted in a small increase in potency for lodoxamide (3-fold) without substantial effect on bufrolin and without changes in potency for zaprinast or cromolyn disodium (Fig. 6; Tables 2 and 3). Noticeably, however, as with the rat Gln6.58Arg mutation, this alteration also resulted in a marked increase in interactions between rat GPR35 and β-arrestin-2 in the absence of an agonist ligand, consistent with the introduction of a substantial level of constitutive activity (Fig. 6). We next combined the alterations in sequence at positions 6.58 and 7.32 to generate human Gln6.58, Ser7.32 (Arg240Gln, Arg255Ser) GPR35a. This resulted in no alteration in potency of cromolyn disodium, a modest 3-fold reduction in potency to zaprinast but a virtual complete loss of function for lodoxamide (at least 960-fold reduction), and also a large 100-fold reduction in potency for bufrolin (Fig. 6; Tables 2 and 3). By contrast, rat Arg6.58, Arg7.32 (Gln238Arg, Ser253Arg) GPR35 displayed equivalent potency for cromolyn disodium and bufrolin, a small decrease in potency for zaprinast, and a similarly small increase in potency for lodoxamide. Most notably, however, rat Arg6.58, Arg7.32 GPR35 now displayed very high levels of ligand-independent constitutive activity (Fig. 6). This feature was only observed at the rat ortholog; the reverse-swap mutations at human GPR35a had no significant effect on basal BRET signals (Fig. 6).
Fig. 6.
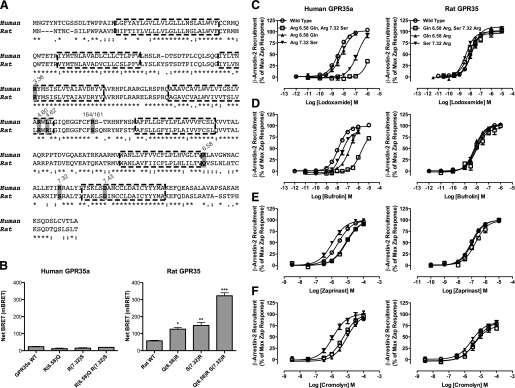
Swapping nonconserved arginine residues between human and rat GPR35a influences ligand potency. (A) Alignment of the sequences of human GPR35a and rat GPR35. Key residues that vary between the two orthologs that were swapped between species or otherwise modified are highlighted, as are the conserved arginine at position 3.36 that acts as a key ligand binding contact in both species and the aspartate at position 7.43, predicted to form an ionic interaction with arginine 3.36 in the absence of agonist. (B) Mutations altered the basal, ligand-independent activity at rat GPR35 but not at human GPR35a. Significantly different from wild type at *P < 0.05; **P < 0.01; ***P < 0.001. The ability of lodoxamide (C), bufrolin (D), zaprinast (E), and cromolyn disodium (F) to promote interactions between human GPR35a (left side) and rat GPR35 (right side) with β-arrestin-2 are shown for wild-type (WT) and ortholog residue swaps at positions 6.58, 7.32, and 6.58 plus 7.32.
TABLE 2.
Effects of arginine swap mutations on ligand pharmacology at human GPR35
Data are human GPR35a pEC50 values and ΔpEC50 compared with wild-type. All studies employed the BRET-based GPR35–β-arrestin 2 interaction assay. Potency estimates of <4 reflect lack of adequate data fit.
| Ligand | Wild-Type | Arg164Ser |
Arg4.60Met |
Leu4.62Arg |
Arg6.58Gln |
Arg7.32Ser |
Arg6.58Glu, Arg7.32Ser |
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| pEC50 | ΔpEC50 | pEC50 | ΔpEC50 | pEC50 | ΔpEC50 | pEC50 | ΔpEC50 | pEC50 | ΔpEC50 | pEC50 | ΔpEC50 | ||
| Zaprinast | 5.59 ± 0.01 | 5.45 ± 0.08 | −0.14 | <4 | <−1.59 | 6.13 ± 0.04*** | +0.54 | 5.11 ± 0.05*** | −0.48 | 5.99 ± 0.09*** | +0.4 | 5.08 ± 0.05*** | −0.51 |
| Lodoxamide | 8.44 ± 0.02 | 6.65 ± 0.09*** | −1.79 | 5.88 ± 0.04*** | −2.56 | 8.63 ± 0.09 | +0.19 | 8.19 ± 0.09 | −0.25 | 6.83 ± 0.01*** | −1.61 | <4 | <−4.44 |
| Bufrolin | 7.90 ± 0.04 | 7.36 ± 0.001* | −0.54 | 6.28 ± 0.05*** | −1.62 | 8.18 ± 0.05 | +0.28 | 6.82 ± 0.09*** | −1.08 | 7.61 ± 0.1 | −0.29 | 5.88 ± 0.23*** | −2.02 |
| Cromolyn | 5.20 ± 0.03 | 4.3 ± 0.13*** | −0.9 | <4 | <−1.20 | 5.72 ± 0.04*** | +0.52 | 5.05 ± 0.04 | −0.15 | 5.95 ± 0.15*** | +0.75 | 5.23 ± 0.08 | +0.03 |
| Amlexanox | 5.39 ± 0.06 | 5.28 ± 0.04 | −0.11 | 4.48 ± 0.09*** | −0.91 | 5.83 ± 0.09 | +0.44 | 5.54 ± 0.11 | +0.15 | 5.86 ± 0.15* | +0.47 | 5.74 ± 0.11 | +0.35 |
| Doxantrazole | 5.47 ± 0.07 | 5.27 ± 0.01 | −0.2 | 5.11 ± 0.06 | −0.36 | 5.69 ± 0.06 | +0.22 | 5.09 ± 0.12 | −0.38 | 5.87 ± 0.17 | +0.4 | 5.03 ± 0.11 | −0.44 |
| Pemirolast | <4 | <4 | N/A | <4 | N/A | <4 | N/A | <4 | N/A | <4 | N/A | <4 | N/A |
N/A, not applicable, as pemirolast lacked potency at wild-type human GPR35a.
P < 0.001; *P < 0.05: pEC50 significantly different from corresponding wild-type.
TABLE 3.
Effects of arginine swap mutations on ligand pharmacology at rat GPR35
Data are rat GPR35 pEC50 values and ΔpEC50 compared with wild type. All studies employed the BRET-based GPR35–β-arrestin 2 interaction assay. Potency estimates of <4 reflect lack of adequate data fit.
| Ligand | Wild-Type | Ser161Arg |
Arg4.60Met |
Arg4.62Leu |
Gln6.58Arg |
Ser7.32Arg |
Glu6.58Arg, Ser7.32Arg |
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| pEC50 | ΔpEC50 | pEC50 | ΔpEC50 | pEC50 | ΔpEC50 | pEC50 | ΔpEC50 | pEC50 | ΔpEC50 | pEC50 | ΔpEC50 | ||
| Zaprinast | 7.01 ± 0.02 | 6.07 ± 0.02*** | −0.94 | 5.03 ± 0.08*** | −1.98 | 5.96 ± 0.04*** | −1.05 | 6.97 ± 0.08 | −0.04 | 7.03 ± 0.06 | +0.02 | 6.77 ± 0.2 | −0.24 |
| Lodoxamide | 7.90 ± 0.02 | 7.47 ± 0.002** | −0.43 | 5.18 ± 0.13*** | −2.72 | 6.24 ± 0.07*** | −1.66 | 8.09 ± 0.08 | +0.19 | 8.25 ± 0.08** | +0.35 | 8.15 ± 0.2* | +0.25 |
| Bufrolin | 8.00 ± 0.02 | 8.08 ± 0.007 | +0.08 | 7.25 ± 0.26* | −0.75 | 7.11 ± 0.06** | −0.89 | 8.16 ± 0.1 | +0.16 | 8.1 ± 0.1 | +0.1 | 7.88 ± 0.28 | −0.12 |
| Cromolyn | 5.42 ± 0.03 | 4.32 ± 0.12*** | −1.1 | <4 | <−1.42 | <4 | <−1.42 | 5.33 ± 0.06 | −0.09 | 5.57 ± 0.10 | +0.15 | 5.48 ± 0.34 | +0.06 |
| Amlexanox | 7.63 ± 0.07 | 5.86 ± 0.05*** | −1.77 | 5.66 ± 0.27*** | −1.97 | 6.93 ± 0.06** | −0.7 | 7.50 ± 0.12 | −0.13 | 7.59 ± 0.15 | −0.04 | 7.23 ± 0.18 | −0.4 |
| Doxantrazole | 6.51 ± 0.02 | 5.35 ± 0.09*** | −1.16 | 5.28 ± 0.25*** | −1.23 | 4.98 ± 0.16*** | −1.53 | 6.37 ± 0.12 | −0.14 | 7.15 ± 0.13** | +0.64 | 6.70 ± 0.47 | +0.19 |
| Pemirolast | 7.02 ± 0.03 | 5.78 ± 0.08*** | −1.24 | 5.05 ± 0.46*** | −1.97 | 5.97 ± 0.04*** | −1.05 | 6.86 ± 0.09 | −0.16 | 7.49 ± 0.12** | +0.47 | 6.64 ± 0.29* | −0.38 |
N/A, not applicable, as pemirolast lacked potency at wild-type human GPR35a.
P < 0.05; **P < 0.01; ***P < 0.001: pEC50 significantly different from corresponding wild type.
The introduction of Arg instead of Leu at position 4.62 (Leu153Arg) in human GPR35a enhanced the potency of zaprinast and cromolyn disodium, without significantly affecting the potency of lodoxamide or bufrolin (Fig. 7; Tables 2 and 3). In contrast, the corresponding switch from Arg4.62 to Leu (Arg150Leu) in rat GPR35 resulted in a substantial reduction in potency for each of lodoxamide (46-fold) and cromolyn disodium (approximately 40-fold), with more limited effects for bufrolin (8-fold) and zaprinast (11-fold) (Fig. 7; Tables 2 and 3). Based on the markedly higher potency of both doxantrazole and pemirolast at rat GPR35 compared with human GPR35, we also assessed the activity and potency of these ligands at the above-described various species swap mutations, with the largest effects being at the rat Arg4.62Leu GPR35 mutation with between an 11- and 30-fold reduction in potency (Tables 2 and 3).
Fig. 7.

Alterations of further conserved and nonconserved residues also affect ligand potency at GPR35. Residue 4.60 is an arginine in both human and rat orthologs of GPR35 and was converted to methionine, whereas residue 4.62, leucine in human but arginine in rat was swapped between species. Arginine 164 in ECL2 of human GPR35a was converted to serine, whereas the equivalent residue, 161 in rat GPR35, was altered to arginine. These mutants were then compared with the wild-type sequences in BRET-based GPR35 β-arrestin-2 interaction studies to explore effects on the potency of lodoxamide (A), bufrolin (B), zaprinast (C), and cromolyn disodium (D). Human GPR35a is shown on the left side of the figure, whereas rat GPR35 is shown on the right.
To attempt to provide a conceptual framework for these data, we applied both further mutagenesis and homology modeling and ligand docking approaches to better understand the basis of ligand binding and species ortholog selectivity. Despite good overall structural similarity between the TMD helices of class A GPCRs crystallized to date (Venkatakrishnan et al., 2013), the extracellular side, particularly extracellular loop 2 (ECL2), shows a very low conservation of both primary and tertiary structures (Mason et al., 2012; Wheatley et al., 2012). This is not surprising because parts of the ECL2 are frequently involved in ligand binding selectivity and potentially in initial ligand recognition, as are the extracellular faces of the helices. Moreover, human GPR35a possesses an arginine residue at position 164 that is lacking in rat, located two amino acids beyond the highly conserved Cys residue of ECL2 that is routinely involved in formation of a disulfide bridge. Mutation of this residue to the equivalent residue, serine, in rat produced a 60-fold reduction in potency for lodoxamide, but only a 4-fold reduction in potency for bufrolin (Fig. 7), resulting in a reversal of the rank order of potency of these two ligands (Fig. 7). The reciprocal mutation, to generate Ser161Arg rat GPR35, was without significant effect on the potency of either lodoxamide or bufrolin (Fig. 7). Residue 4.60 is an arginine in both species (Fig. 6). Mutation to methionine in either human or rat GPR35 resulted in reduction in potency of between 100- and 500-fold for both lodoxamide and zaprinast (Fig. 7; Tables 2 and 3) whereas the low potency of cromolyn disodium at wild-type orthologs of GPR35 resulted in the loss of potency at Arg4.60Met being sufficient to prevent effective analysis of the data. In contrast, again at both species, this mutation had less effect on bufrolin, with loss of potency being only approximately 30-fold (Fig. 7; Tables 2 and 3).
The data appeared to favor a shared general binding site for lodoxamide and bufrolin, centered in the narrow space between TMD III and VII, with the conserved Arg3.36 at its base and Arg4.60 providing the principal interaction residues for a negatively charged group from the ligands. Moreover, for lodoxamide at the human ortholog, there appeared to be a potential, specific role for ECL2 and the mutational data indicated differences in detail of binding of lodoxamide and bufrolin with particular TMD arginine residues. Arg3.36 is potentially involved in an ionic interaction with Asp7.43 (Fig. 8) that is likely disrupted by agonist binding. Lodoxamide and bufrolin are both characterized by the presence of two carboxyl groups (Fig. 1). Modeling and docking studies, indeed, indicated a rather conserved location of binding within the two species but that varied in the detailed interactions with amino acids in the binding site (Fig. 8). In human GPR35, which possesses arginine residues at positions 6.58 and 7.32 and a further arginine at position 164 in ECL2, these residues appear to be directly but differentially involved in binding these di-acidic groups (Fig. 8). In agreement with data in Tables 2 and 3, lodoxamide binding to human GPR35 reflects interaction of one of the carboxyl groups with both Arg3.36 and Arg4.60, whereas the other binds to Arg7.32 and Arg164 (Fig. 8A). Consistent with such a model, a large reduction in potency for lodoxamide was observed when Arg7.32 was converted to serine, whereas a gain of potency was observed for lodoxamide when serine7.32 in rat was altered to arginine (Tables 2 and 3). Moreover, mutation of Arg164 resulted in a marked reduction in potency for lodoxamide (Fig. 7). Bufrolin binds to human GPR35 in a similar pose as lodoxamide (Fig. 8B). However, based on the different structure of this ligand, Arg164 is less involved in binding, interacting only with a noncharged carbonyl oxygen, whereas alongside Arg3.36 at the base of the binding pocket and Arg4.60, which again binds one of carboxyl groups, Arg6.58 is the residue mainly involved in the interaction with the second carboxyl group (Fig. 8B). The substantial reduction in potency of bufrolin at human Arg6.58Gln is consistent with this model, whereas potency of lodoxamide was less affected by this mutation. Interestingly, Arg7.32 in human GPR35 does not appear to be a substantial contributor to the binding of bufrolin. Removal of this residue had a very modest effect on potency, whereas introduction of an arginine in this position in rat also did not alter potency significantly but did produce a modest increase in potency for lodoxamide. Rat GPR35 is characterized by the lack of the two arginines in TMD VI and VII present in the human ortholog as well as that at position 161 (residue 164 in human ortholog, serine in rat) and by the presence of an arginine at position 4.62. As noted earlier, mutation of this Arg to the equivalent human residue, Leu, strongly affected the potency of all ligands, particularly lodoxamide. In the rat GPR35 model, Arg 4.62 acts as a surrogate of the human-specific Arg164 and indeed this residue contributes more to the binding of lodoxamide binding than brufolin (Tables 2 and 3). Docking of lodoxamide and brufolin to rat GPR35 predicts that Arg4.62 interacts with the second carboxyl group of these ligands, the first interacting with Arg3.36 and Arg4.60 (Fig. 8, C and D).
Fig. 8.

Homology modeling and ligand docking studies of human and rat GPR35a. Potential modes of binding of lodoxamide (A and C) and bufrolin (B and D) to human GPR35a (A and B) and rat GPR35 (C and D) are shown. These are consistent with mutational data shown in Figs. 6 and 7 and quantified in Tables 2 and 3.
A prediction of these docking poses is that both the cyano group of lodoxamide and the propyl appendage of bufrolin point toward Val2.60 in human GPR35 (Fig. 8, A and B). Human GPR35 is markedly polymorphic, with a number of variations reported via the 1000 Genomes sequencing project (Abecasis et al., 2010) that are predicted to be nonsynonymous and to alter the amino acid sequence of the encoded polypeptide. One of these is Val76Met (i.e., at position 2.60). Although uncommon, with minor allele frequency of 0.014, we generated this variant and assessed responsiveness to lodoxamide and bufrolin in the BRET-based β-arrestin-2 interaction assay. Both ligands displayed substantially lower (13- to 15-fold) potency at the Met containing variant (Fig. 9; Table 4). Moreover, a similar loss of potency to both zaprinast and cromolyn disodium was also observed at the Met containing variant (Fig. 9; Table 4). The effect of the Val76Met variant did not reflect issues with cell surface delivery: incorporation of an N-terminal FLAG epitope tag allowed enzyme-linked immunosorbent assay–based detection of effective cell surface delivery and this was not different from the wild-type receptor sequence (Table 4).
Fig. 9.
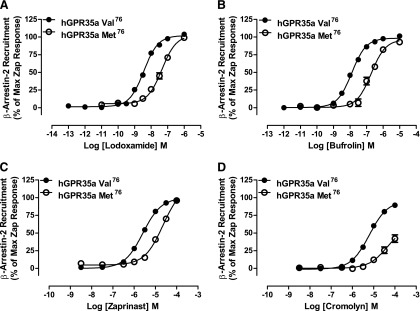
The polymorphic variant Val76Met reduces potency of a series of ligands at human GPR35a. The potencies of lodoxamide (A), bufrolin (B), zaprinast (C), and cromolyn disodium (D) were compared between human Val76 GPR35a and Met76 GPR35a using the BRET-based β-arrestin-2 interaction assay. In each case, data are presented as a percentage of the maximal effect of zaprinast at the appropriate variant.
TABLE 4.
Lack of effect of most polymorphic variants of human GPR35a on ligand potency
The possible effect of nonsynonomous SNP variation in human GPR35a on ligand potency was assessed for a number of previously reported examples (Abecasis et al., 2010). In each case, the minor allele amino acid (listed second) was introduced into otherwise wild-type human GPR35a and ligand potency was then assessed in PathHunter human GPR35a–β-arrestin-2 interaction assays as in Table 1. Only in the case of Val76Met was a significant alteration in potency recorded.
| Variant | Expressiona | Lodoxamide EC50 | Zaprinast EC50 | Cromolyn EC50 |
|---|---|---|---|---|
| nM | μM | μM | ||
| Ala25Thr | 0.97 ± 0.13 | 3.70 ± 1.76 | 1.84 ± 0.66 | 2.94 ± 0.48 |
| Val29Ile | 1.27 ± 0.15 | 3.58 ± 1.67 | 1.70 ± 0.49 | 3.70 ± 1.72 |
| Val76Met | 1.06 ± 0.08 | 33.38 ± 16.85* | 16.37 ± 5.84* | 20.26 ± 4.86* |
| Thr108Met | 0.67 ± 0.14 | 6.49 ± 3.80 | 3.12 ± 1.07 | 5.36 ± 1.83 |
| Arg125Ser | 1.32 ± 0.16 | 5.25 ± 2.80 | 1.55 ± 0.62 | 3.73 ± 1.43 |
| Thr253Met | 0.63 ± 0.18 | 2.50 ± 0.95 | 1.60 ± 0.75 | 1.86 ± 0.74 |
| Ser294Arg | 1.48 ± 0.51 | 3.07 ± 1.46 | 1.14 ± 0.48 | 1.95 ± 0.19 |
| Wild type | 1.00 ± 0.23 | 3.97 ± 1.77 | 2.29 ± 0.79 | 2.98 ± 1.27 |
Expression as fold relative to wild type.
P < 0.05.
Six other distinct single amino acids variants (Ala25Thr, Val29Ile, Thr108Met, Arg125Ser, The253Met, and Ser294Arg) of human GPR35a were generated, including those reported to have the highest minor allele frequency (Ser294Arg = 0.494; Thr108Met = 0.161; Thr253Met = 0.058; and Arg125Ser = 0.021) and those located within TMDs or extracellular segments of the receptor (Ala25Thr and Val29Ile). These were also assessed for effects on the potency of lodoxamide, zaprinast, and cromolyn disodium using the GPR35a–β-arrestin-2 interaction assays. Unlike the Val76Met variant, each of these was completely without effect on either the ligand potency or cell surface delivery (Table 4).
Discussion
Although the tryptophan metabolite kynurenic acid is able to activate GPR35 (Wang et al., 2006), the marked variation in potency between species orthologs and its very modest potency at the human receptor (Jenkins et al., 2011) have resulted in questions regarding the physiologic relevance of this ligand as a GPR35 agonist (at least in humans) (Milligan, 2011). This has also encouraged the search for other and more potent ligands to help define the functions of GPR35. This has included the suggestion that forms of lysophosphatidic acid might act as endogenous agonists (Oka et al., 2010). Among synthetic small molecule ligands, the antiasthma and antiallergenic drugs cromolyn disodium (Jenkins et al., 2010; Yang et al., 2010) and nedocromil sodium (Yang et al., 2010) have been shown to have agonist activity at GPR35. Although these drugs have modest potency, this encouraged us to explore the possibility that other drugs with similar functional pharmacology might also be GPR35 agonists. Although this did not prove to be a generic feature, the fact that the function of both kynurenic acid and cromolyn disodium is lost after mutation of Arg3.36 (Jenkins et al., 2010) and that related receptors that have an arginine at this position also respond to small acidic ligands (Liu et al., 2009) led us to look more closely at this feature. The dicarboxyl containing antiallergenics lodoxamide and bufrolin were shown to be the most potent agonists of GPR35 yet identified, whereas amlexanox, a carboxylate-containing tricyclic antiasthma drug was also a GPR35 agonist. Moreover, a number of other agonist ligands with antiasthma/antiallergenic pharmacology, including doxantrazole and pemirolast, although lacking a carboxyl function, clearly contain an acid bioisostere related to the triazole structure present in the standard GPR35 surrogate agonist zaprinast. These ligands, although also active at GPR35, displayed marked differences in potency between the rat and human orthologs, a feature previously noted for zaprinast (Taniguchi et al., 2006; Jenkins et al., 2010). The published literature indicates that certain GPR35 agonists such as pamoate (Jenkins et al., 2010), 4-{(Z)-[(2Z)-2-(2-fluorobenzylidene)-4-oxo-1,3-thiazolidin-5-ylidene]methyl}benzoic acid (Neetoo-Isseljee et al., 2013), and various 8-benzamidochromen-4-one-2-carboxylic acids (Funke et al., 2013) are highly selective for the human ortholog, whereas others such as pemirolast and amlexanox are highly selective for the rat ortholog. Although it is clearly speculative, it may be interesting to consider whether the inability to progress various antiallergics in the clinic may have a basis in this species selectivity. Based on the fact that pemirolast, zaprinast, and amlexanox were each markedly more potent in receptor–β-arrestin-2 interaction studies at the rat ortholog than at human GPR35, that the key residue Arg3.36 is conserved between species and because a number of GPR35 agonists, including lodoxamide and bufrolin, are di-acids we considered, therefore, the potential contribution to ligand recognition and potency of arginine residues in TMDs and near the top of the predicted TMD helix bundle that are not conserved between human and rat GPR35. This provided significant new insights. Furthermore, combinations of such mutations provided further insights. Although mutation of both Arg6.58 and Arg7.32 in human GPR35 had little effect on the potency of zaprinast, it resulted in a greater than 900-fold reduction in potency for lodoxamide, an effect greater than predicted from either alteration alone. Results such as these encouraged us to apply homology modeling and computational ligand docking approaches to further explore the basis of ligand binding and species ortholog selectivity.
Despite strong structural similarity between the TMD helices of class A GPCRs crystallized to date, the extracellular side, particularly ECL2, shows low conservation of both primary and tertiary structures (Wheatley et al., 2012; Venkatakrishnan et al., 2013). This is not surprising because parts of the ECL2 are frequently involved in ligand binding and potentially in initial ligand recognition, as are elements of the extracellular face of the helices. Human and rat GPR35 (71% sequence identity, 81% in the transmembrane helices) differ in both sequence and length of ECL2, potentially the most relevant differences being an arginine in rat GPR35 at position 4.62 (leucine in human), and a serine (arginine in human) and a glutamine (missing in human) that are located respectively two and four residues after the cysteine involved in the conserved disulfide bridge between ECL2 and TMD III. These species variations strongly affect the residues of ECL2 involved in ligand binding. Such differences, however, pose substantial challenges for homology modeling and ligand docking studies.
All GPR35 agonist ligands are negatively charged or possess acid bioisosteres and are frequently characterized by a strong planarity, either due to a series of fused rings (bufrolin, amlexanox, doxantrazole, pemirolast) or induced by intramolecular hydrogen bonds (zaprinast). In contrast, lodoxamide contains a single aromatic ring. The results of the mutational studies generally suggested a common binding site, centered between TMD III and TMD VII, with the core conserved Arg3.36, which forms an ionic interaction with Asp 7.43 in the basal state, at its base providing, together with Arg4.60, the principal interaction residues for a negatively charged group of the ligands. Lodoxamide and bufrolin are characterized by the presence of two carboxyl groups and, unusually for GPR35 ligands, are highly potent at both the human and rat orthologs. Although they bind in similar regions in different species orthologs distinct, species-specific arginines, all at the extracellular side of the binding cavity are involved in binding the second carboxyl group of both lodoxamide and brufolin.
A prediction of the docking pose in human GPR35 illustrated is that the cyano group of lodoxamide points toward Val2.60. Moreover, the propyl appendage of bufrolin is also predicted to orientate toward this residue. It is of particular interest, therefore, that Val2.60Met was the only human nonsynonymous SNP variant we assessed that modified the potency of these GPR35 agonists. It is therefore reasonable, based on this model and docking pose, to suggest that steric hindrance may be responsible for the potency decrease observed in the human GPR35 Val2.60Met variant. The loss of potency for both zaprinast and cromolyn disodium also hints at a similar general binding location as for lodoxamide and bufrolin and, as shown previously for zaprinast and cromolyn disodium (Jenkins et al., 2011), with all these ligands being “orthosteric” with respect to one another. However, because zaprinast and cromolyn disodium are far less potent than lodoxamide and bufrolin at the human receptor, this is challenging to model directly and there must be substantial differences in detail given the modest effect on the potency of these ligands of a number of the mutants we characterized.
There is considerable interest in the idea that nonsynonymous SNP variation in the open reading frame of receptor sequences may help define the basis of distinct responsiveness to drugs and may be important for the continuing efforts to personalize therapy and treatment. GPR35 is markedly polymorphic in the open reading frame but accumulating evidence suggests that many of these are infrequent alleles. However, because a number of the reported variants in human GPR35 are located either within TMDs or at the interface of the helix bundle and the connecting extracellular loops, we also explored the pharmacology of a number of other variants, including Thr108Met located in transmembrane domain III and Thr253Met located within extracellular loop III because these both are reported to occur with significant allelic frequency. However, despite rs3749171, a SNP associated in genome-wide association analyses as a risk locus for ulcerative colitis (Ellinghaus et al., 2013) and inflammatory bowel disease (Jostins et al., 2012), encompassing the GPR35 locus, at least in the receptor–β-arrestin-2 interaction assays employed, neither of these variations had any detectable effect on ligand pharmacology.
In conclusion, we report the identification of antiallergic compounds with low nanomolar potency for human GPR35. In contrast to the marked variation in potency of many previously described agonists of GPR35 between human and rodent orthologs, both lodoxamide and bufrolin are also highly potent at rat GPR35 with a predicted mode of binding similar to that of human GPCR35a. Remarkably, the ligand docking studies on these ligands suggested that an uncommon, nonsynonymous variant of this receptor in the human population might display marked variation in ligand potency and this was confirmed experimentally to be correct.
Acknowledgments
The authors thank Drs. Ed McIver and Debbie Taylor (Medical Research Council Technology) for provision of lodoxamide and Dr. John Souness (Sanofi) for insight and helpful discussions. The DMR assays were conducted at the laboratories of Medical Research Council Technology under the guidance of Craig Southern and Zaynab Neetoo-Isseljee.
Abbreviations
- ABTS
2,2′-azino-bis(3-ethylbenzothiazoline-6-sulphonic acid)
- BRET
bioluminescence resonance energy transfer
- BRL10833
5,6-dimethyl-2-nitro-1H-indene-1,3(2H)-dione
- CHO–K1
Chinese hamster ovary
- DMEM
Dulbecco’s modified Eagle’s medium
- DMSO
dimethylsulfoxide
- ECL2
extracellular loop 2
- DMR
dynamic mass redistribution
- eYFP
enhanced yellow fluorescent protein
- GPCR
G protein–coupled receptor
- HEK
human embryonic kidney
- ICI 74917 (bufrolin)
6-butyl-4,10-dihydroxy-1,7-phenanthroline-2,8-dicarboxylic acid
- http://dx.doi.org/10.1124/mol.113.089482ML-145
2-hydroxy-4-[4-[(5Z)-5-[(E)-2-methyl-3-phenylprop-2-enylidene]-4-oxo-2-sulfanylidene-1,3-thiazolidin-3-yl]butanoylamino]benzoic acid
- SNP
single-nucleotide polymorphism
- TMD
transmembrane domain
Authorship Contributions
Participated in research design: MacKenzie, Kent, Hudson, Nicklin, Charlton, Milligan.
Conducted experiments: MacKenzie, Caltabiano, Jenkins, McCallum, Hudson, Fawcett, Markwick.
Contributed new reagents or analytic tools: Kent, Charlton.
Performed data analysis: Mackenzie, Caltabiano, Kent, Hudson, Milligan.
Wrote or contributed to the writing of the manuscript: MacKenzie, Milligan.
Footnotes
This work was supported by the Biotechnology and Biosciences Research Council (BBSRC DTG FO1673511); Medical Research Council Technology [Industrial CASE Studentship to A.E.M.]; the British Heart Foundation (FS/09/052/28032) [Studentship Support to J.E.M.]; and the Spanish Ministry of Education, Culture, and Sport National Program on Mobility of Human Resources of National I+D Plan 2008–2011 [José Castillejo 2012 Fellowship to G.C.].
References
- Abecasis GR, Altshuler D, Auton A, Brooks LD, Durbin RM, Gibbs RA, Hurles ME, McVean GA, 1000 Genomes Project Consortium (2010) A map of human genome variation from population-scale sequencing. Nature 467:1061–1073 PubMed [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballesteros JA and Weinstein H (1995) Integrated methods for modeling G-protein coupled receptors. Methods Neurosci 366–428.
- Cordomí A, Caltabiano G, Pardo L. (2012) Membrane protein simulations using AMBER force field and Berger lipid parameters. J Chem Theory Comput 8:948–958 [DOI] [PubMed] [Google Scholar]
- Deng H, Hu H, He M, Hu J, Niu W, Ferrie AM, Fang Y. (2011) Discovery of 2-(4-methylfuran-2(5H)-ylidene)malononitrile and thieno[3,2-b]thiophene-2-carboxylic acid derivatives as G protein-coupled receptor 35 (GPR35) agonists. J Med Chem 54:7385–7396 PubMed [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng H, Hu H, Ling S, Ferrie AM, Fang Y. (2012) Discovery of natural phenols as G protein-coupled receptor-35 (GPR35) agonists. ACS Med Chem Lett 3:165–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellinghaus D, Folseraas T, Holm K, Ellinghaus E, Melum E, Balschun T, Laerdahl JK, Shiryaev A, Gotthardt DN, Weismüller TJ, et al. (2013) Genome-wide association analysis in Primary sclerosing cholangitis and ulcerative colitis identifies risk loci at GPR35 and TCF4. Hepatology 58:1074–1083 PubMed [DOI] [PubMed] [Google Scholar]
- Funke M, Thimm D, Schiedel AC, Müller CE. (2013) 8-Benzamidochromen-4-one-2-carboxylic acids: potent and selective agonists for the orphan G protein-coupled receptor GPR35. J Med Chem 56:5182–5197 PubMed [DOI] [PubMed] [Google Scholar]
- Hess B, Kutzner C, van der Spoel D, Lindahl E. (2008) GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J Chem Theory Comput 4:435–447 [DOI] [PubMed] [Google Scholar]
- Jenkins L, Alvarez-Curto E, Campbell K, de Munnik S, Canals M, Schlyer S, Milligan G. (2011) Agonist activation of the G protein-coupled receptor GPR35 involves transmembrane domain III and is transduced via Gα13 and β-arrestin-2. Br J Pharmacol 162:733–748 PubMed [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins L, Brea J, Smith NJ, Hudson BD, Reilly G, Bryant NJ, Castro M, Loza MI, Milligan G. (2010) Identification of novel species-selective agonists of the G-protein-coupled receptor GPR35 that promote recruitment of β-arrestin-2 and activate Gα13. Biochem J 432:451–459 PubMed [DOI] [PubMed] [Google Scholar]
- Jenkins L, Harries N, Lappin JE, MacKenzie AE, Neetoo-Isseljee Z, Souther C, McIver E, Nicklin SA, Taylor DL, Milligan G. (2012) Antagonists of GPR35 display high species ortholog selectivity and varying modes of action. J Pharmacol Exp Ther 343:683–695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, Lee JC, Schumm LP, Sharma Y, Anderson CA, et al. International IBD Genetics Consortium (IIBDGC) (2012) Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 491:119–124 PubMed [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Wu J, Zhu J, Kuei C, Yu J, Shelton J, Sutton SW, Li X, Yun SJ, Mirzadegan T, et al. (2009) Lactate inhibits lipolysis in fat cells through activation of an orphan G-protein-coupled receptor, GPR81. J Biol Chem 284:2811–2822 PubMed [DOI] [PubMed] [Google Scholar]
- Lumb EM, McHardy GJ, Kay AB. (1979) The orally administered anti-allergic agent, sodium nivimedone (BRL 10833); efficacy in bronchial asthma and effects on IgE, complement and eosinophils. Br J Clin Pharmacol 8:65–74 PubMed [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie AE, Lappin JE, Taylor DL, Nicklin SA, Milligan G. (2011) GPR35 as a novel therapeutic target. Front Endocrinol (Lausanne) 2:68 PubMed [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martí-Renom MA, Stuart AC, Fiser A, Sánchez R, Melo F, Sali A. (2000) Comparative protein structure modeling of genes and genomes. Annu Rev Biophys Biomol Struct 29:291–325 PubMed [DOI] [PubMed] [Google Scholar]
- Mason JS, Bortolato A, Congreve M, Marshall FH. (2012) New insights from structural biology into the druggability of G protein-coupled receptors. Trends Pharmacol Sci 33:249–260 PubMed [DOI] [PubMed] [Google Scholar]
- Milligan G. (2011) Orthologue selectivity and ligand bias: translating the pharmacology of GPR35. Trends Pharmacol Sci 32:317–325 PubMed [DOI] [PubMed] [Google Scholar]
- Neetoo-Isseljee Z, MacKenzie AE, Southern C, Jerman J, McIver EG, Harries N, Taylor DL, Milligan G. (2013) High-throughput identification and characterization of novel, species-selective GPR35 agonists. J Pharmacol Exp Ther 344:568–578 PubMed [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka S, Ota R, Shima M, Yamashita A, Sugiura T. (2010) GPR35 is a novel lysophosphatidic acid receptor. Biochem Biophys Res Commun 395:232–237 PubMed [DOI] [PubMed] [Google Scholar]
- Okumura S, Baba H, Kumada T, Nanmoku K, Nakajima H, Nakane Y, Hioki K, Ikenaka K. (2004) Cloning of a G-protein-coupled receptor that shows an activity to transform NIH3T3 cells and is expressed in gastric cancer cells. Cancer Sci 95:131–135 PubMed [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schröder R, Janssen N, Schmidt J, Kebig A, Merten N, Hennen S, Müller A, Blättermann S, Mohr-Andrä M, Zahn S, et al. (2010) Deconvolution of complex G protein-coupled receptor signaling in live cells using dynamic mass redistribution measurements. Nat Biotechnol 28:943–949 PubMed [DOI] [PubMed] [Google Scholar]
- Schrödinger LLC. (2012) The PyMOL Molecular Graphics System, version 1.5r3, Schrödinger LLC, Cambridge, MA [Google Scholar]
- Southern C, Cook JM, Neetoo-Isseljee Z, Taylor DL, Kettleborough CA, Merritt A, Bassoni DL, Raab WJ, Quinn E, Wehrman TS, et al. (2013) Screening β-arrestin recruitment for the identification of natural ligands for orphan G-protein-coupled receptors. J Biomol Screen 18:599–609 PubMed [DOI] [PubMed] [Google Scholar]
- Taniguchi Y, Tonai-Kachi H, Shinjo K. (2006) Zaprinast, a well-known cyclic guanosine monophosphate-specific phosphodiesterase inhibitor, is an agonist for GPR35. FEBS Lett 580:5003–5008 PubMed [DOI] [PubMed] [Google Scholar]
- Venkatakrishnan AJ, Deupi X, Lebon G, Tate CG, Schertler GF, Babu MM. (2013) Molecular signatures of G-protein-coupled receptors. Nature 494:185–194 PubMed [DOI] [PubMed] [Google Scholar]
- Wang J, Simonavicius N, Wu X, Swaminath G, Reagan J, Tian H, Ling L. (2006) Kynurenic acid as a ligand for orphan G protein-coupled receptor GPR35. J Biol Chem 281:22021–22028 PubMed [DOI] [PubMed] [Google Scholar]
- Wheatley M, Wootten D, Conner MT, Simms J, Kendrick R, Logan RT, Poyner DR, Barwell J. (2012) Lifting the lid on GPCRs: the role of extracellular loops. Br J Pharmacol 165:1688–1703 PubMed [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Lu JY, Wu X, Summer S, Whoriskey J, Saris C, Reagan JD. (2010) G-protein-coupled receptor 35 is a target of the asthma drugs cromolyn disodium and nedocromil sodium. Pharmacology 86:1–5 PubMed [DOI] [PubMed] [Google Scholar]
- Zhang C, Srinivasan Y, Arlow DH, Fung JJ, Palmer D, Zheng Y, Green HF, Pandey A, Dror RO, Shaw DE, et al. (2012) High-resolution crystal structure of human protease-activated receptor 1. Nature 492:387–392 PubMed [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao P, Sharir H, Kapur A, Cowan A, Geller EB, Adler MW, Seltzman HH, Reggio PH, Heynen-Genel S, Sauer M, et al. (2010) Targeting of the orphan receptor GPR35 by pamoic acid: a potent activator of extracellular signal-regulated kinase and β-arrestin2 with antinociceptive activity. Mol Pharmacol 78:560–568 PubMed [DOI] [PMC free article] [PubMed] [Google Scholar]