

Abstract
The basic helix-loop-helix (bHLH) transcription factor TWIST1 is essential to embryonic development, and hijacking of its function contributes to the development of numerous cancer types. It forms either a homodimer or a heterodimeric complex with an E2A or HAND partner. These functionally distinct complexes display sometimes antagonistic functions during development, so that alterations in the balance between them lead to pronounced morphological alterations, as observed in mice and in Saethre–Chotzen syndrome patients. We, here, describe the structures of TWIST1 bHLH–DNA complexes produced in silico through molecular dynamics simulations. We highlight the determinant role of the interhelical loops in maintaining the TWIST1–DNA complex structures and provide a structural explanation for the loss of function associated with several TWIST1 mutations/insertions observed in Saethre–Chotzen syndrome patients.
An animated interactive 3D complement (I3DC) is available in Proteopedia at http://proteopedia.org/w/Journal:JBSD:27
Keywords: TWIST, dimerization, DNA interaction, homology models, 3D-models, molecular dynamics, embryonic transcription factors, bHLH
Introduction
The basic helix-loop-helix (bHLH) nuclear transcription factors belong to an evolutionarily conserved superfamily of nearly 125 proteins playing pivotal roles in numerous essential cellular functions, including adaptation to micro-environmental changes, specification, differentiation, proliferation, and apoptosis. Although they were previously classified on the basis of a combination of functional features (Atchley & Fitch, 1997), unbiased sequence comparisons across proteins of numerous species have led to their classification into five clades (Stevens, Roalson, & Skinner, 2008). For information, the TWIST, HAND, and NEUROD proteins belong to clade A, the E2A proteins to clade B, and MYOD1 to clade C (Skinner, Rawls, Wilson-Rawls, & Roalson, 2010).
The bHLH structure comprises a motif of mainly basic residues (the b-domain), allowing protein binding to 5′-CANNTG-3′ cis-regulatory elements called E-boxes (De Masi et al., 2011; Murre, 1999), and a dimerization motif of 50 residues forming two amphipathic α-helices separated by an interhelical loop (the HLH domain) (Voronova & Baltimore, 1990). Post-translational regulations, including protein modifications and competition between partners for binding, are described to affect complex formation and the resulting bHLH transcription factor activities. The regulation of TWIST1 during embryonic development and its alteration in human syndromes are classical examples of this complexity. TWIST1 was originally identified in Drosophila (the Twi gene) as a zygotic developmental gene crucial to the establishment of the ventral furrow, a prerequisite to the development of all mesoderm-derived internal organs (Thisse, el Messal, & Perrin-Schmitt, 1987). In mice, Twist1 haplo-insufficiency produces viable offspring but leads to abnormal craniofacial structures and polydactyly on the hind limb, a phenotype reminiscent of the dominantly inherited Saethre–Chotzen syndrome (SC) in humans, where TWIST1 is mutated (Bourgeois et al., 1998). TWIST1-depleted mice die at E10.5–11 because of numerous defects reflecting a role of this transcription factor in neural crest cell migration and determination and in mesoderm differentiation (Hebrok, Wertz, & Fuchtbauer, 1994; Lee, Lowe, Strong, Wergedal & Glackin, 1999; Spicer, Rhee, Cheung, & Lassar, 1996). TWIST1 has been found to dimerize, either with itself or with an E2A (E12 or E47) or HAND (HAND1 or HAND2) partner, and TWIST1 dimer selection has been shown to determine both the regulation of limb development (Firulli et al., 2005; Firulli, Redick, Conway, & Firulli, 2007) and cranial suture patterning and fusion (Connerney et al., 2006, 2008). Mutations in TWIST1, as observed in SC patients, favor homodimerization, either by reducing the level of the protein (monoallelic nonsense mutations) or by disrupting the Serl23 phosphorylation that dictates partner selection (El Ghouzzi et al., 2000, 2001; Firulli et al., 2005; Vichalkovski, Gresko, Hess, Restuccia, & Hemmings, 2010).
While undetectable in most differentiated adult cells, TWIST genes (TWIST1 and TWIST2) are aberrantly reactivated in numerous human cancer types, including carcinoma (breast, lung, and colon), sarcoma, melanoma, glioma, and retinoblastoma (Ansieau, Morel, Hinkal, Bastid & Puisieux, 2010). Because it promotes the epithelial-to-mesenchymal transition (EMT), TWIST1 was originally depicted as a prometastatic factor (Yang, Mani, & Weinberg, 2006). TWIST proteins actually display additional oncogenic properties. They abolish the activation of failsafe programs (senescence and apoptosis) in response to an oncogenic insult by turning down both RB- and p53-dependent oncosuppressive pathways (Ansieau et al., 2008; Kwok, Ling, Yuen, Wong, & Wang, 2007; Maestro et al., 1999; Valsesia-Wittmann et al., 2004), by inducing the AKT2 serine/threonine kinase-dependent pathway (Cheng et al., 2007), and by promoting expression of anti-apoptotic BH3-only proteins (Kwok et al., 2005; Wang et al., 2004; Zhuo, Wang, Zhuo, Zhang, & Chen, 2008). They thereby cooperate with mitogenic proteins, such as RAS and ERBB2, in promoting cell transformation in vitro and tumor development in vivo (Morel et al., 2012; Tran et al., 2012). Both intrinsic properties (Pham et al., 2007) and associated EMT (Kajiyama et al., 2007; Li et al., 2009; Yang et al., 2006) additionally lead to cell chemoresistance. Lastly, cell commitment to EMT is associated with reacquisition of stem-cell-like properties, including self-renewal potential, an important prerequisite for tumor development (Mani et al., 2008; Morel et al., 2008; Vesuna, Lisok, Kimble, & Raman, 2009). Furthermore, EMT associated reprogramming facilitates cell transformation in vitro (Morel et al., 2012). TWIST proteins, thus, display pleiotropic oncogenic and prometastatic properties favoring tumor initiation, development, and dissemination.
When TWIST proteins are depleted in vitro or in vivo in cancer cells, senescence and apoptotic programs are restored (Ansieau et al., 2008; Kwok et al., 2007; Tran et al., 2012). This and the accelerated senescence and decreased lifespan of SC patient osteoblasts as compared to control cells (Cakouros et al., 2012) suggest that TWIST proteins may constitute appealing therapeutic targets. In the present study, we have established, on the basis of homology with the X-ray structure of the bHLH domains of the murine E47/NEUROD1 complex, homology models of the human TWIST1 homodimer and heterodimers (formed with either the El2 or HAND1 dimerization partner) bound to their target DNA sequences. Although the protein backbone is similar for all of our in silico-generated models, the TWIST complexes differ substantially in their interhelical loops. Alterations of these loops, as observed in SC patients, significantly alter the stability of TWIST dimer–DNA complexes by inducing conformational changes. This suggests that these structures may constitute prime targets for pharmacological inhibitors.
Materials and methods
Selection of X-ray structure and model building
The primary sequences of human TWIST1 (Q15672), TWIST2 (Q8WVJ9), E12 (P15923-1), HAND1 (O906004) HAND2 (P61296), NEUROD1 (Q13562), E47 (P15923-2), and MYOD1 (P15172) were downloaded from the UniProtKB/Swiss-Prot website. The mouse NEUROD1 and E47 sequences were obtained from the 2ql2 PDB files. Primary sequences, restricted to the bHLH domains, were aligned with the ClustalW 2.0 software (Thompson, Higgins, & Gibson, 1994).
The X-ray structures of the human MYC/MAX dimer (PDB code lnkp) (Nair & Burley, 2003), mouse MYOD1/MYOD1 complex (PDB code lmdy) (Ma, Rould, Weintraub, & Pabo, 1994), and mouse NEUROD1/E47 complex (PDB code 2ql2) (Longo, Guanga, & Rose, 2008) were downloaded from the RCSB protein data bank. Two X-ray structures of mouse NEUROD1/E47 have been reported (2ql2A/2ql2B; 2ql2C/2ql2D), which slightly diverge (root-mean-square deviation, RMSD). The calculated RMSD (based on align seed residue and list distance parameters) on C-chains is weak and evaluated at 0.618 Å, suggesting that the two models can be considered similar.
Sequence alignments were submitted to the SWISS-MODEL Workspace (Arnold, Bordoli, Kopp, & Schwede, 2006) to generate models for homodimeric and heterodimeric TWIST1 complexes by homology with the NEURODl(2ql2D)/E47(2ql2C) or NEUROD1(2ql2B)/E47(2ql2A) template, respectively, as described in (Maia, da Silva, Mencalha, Caffarena, & Abdelhay, 2012). The RMSDs between the models generated by Swiss Model Workspace and the folds are, respectively, 1.065/0.069 Å and 1.60/0.064 Å for TWIST1/E12 (modeled reference 2ql2B/A) and TWIST1/TWIST1 (modeled with the reference 2ql2D/C). The choice between the two homology models was finally made by calculating the RMSD of each dimer with respect to its reference. Homology modeling was performed with the same DNA sequence as used in the reported NEUROD1/E47 X-ray structure. The dynamics simulations represent series of possible conformational states of the DNA-bound complexes.
To gain further insight into TWIST1 dimer function and specificity, we generated additional models with two functionally active TWIST1 variants expressed in SC patients. These variants result from a 21-bp tandem repeat insertion into the TWIST1 gene, leading to the aberrant presence of seven extra amino acids in the interhelical loop at position 135 or 139 (Ins-135 or Ins-139, Figure S5) (El Ghouzzi et al., 1997). We, thus, built homology models of the following complexes: TWIST1/TWIST1, TWIST1 Ins-135/TWIST1 Ins-135, TWIST1 Ins-139/TWIST1 Ins-139, TWIST1/E12, TWIST1 Ins-135/E12, and TWIST1 Ins-139/E12.
Minimizations (>10,000 steps with the conjugated gradient algorithm) were carried out with the Sybyl-X 1.1 software package, elaborated by the Tripos company. We applied the Tripos force field with the Gasteiger–Marsilli partial charges and a dielectric constant of 80 to simulate an implicit water phase (the dielectric constant of water is 20.10 at 20 °C). No restrains was applied to our models. This step principally refines and corrects the positions of residue side chains. The effects of minimization on the homology models were estimated approximately by calculating their RMSDs before and after the minimization step. In the heterodimer, the RMSDs of the E12 and TWIST1 peptides were 1.58 and 1.97 Å, respectively. In the homodimer, the RMSDs of the two TWIST1 peptides were 3.21 and 1.69 Å. The E-loop, thus, seems more disordered that the T-loop in the homodimeric complex, in agreement with the NeuroDl/E47 X-ray structure (Longo et al., 2008). Nonetheless, the RMSD calculation during the minimization step showed that the E-loop structure converges to a stable conformation, which could be used in dynamic simulations.
Molecular dynamics analysis
The established homology models (including DNA sequences) were visualized with the VMD1.9.1 software (Humphrey, Dalke & Schulten, 1996; Jorgensen, Chandrasekhar, Madura, Impey & Klein, 1983). The resulting model was inserted into a parapipedic TIP3P solvent box by means of the add solvation box module of the VMD 1.9.1 software. A distance of 15 Å was set between the surfaces of the protein and the limit of the solvent box.
Conditions were computed to reach neutral charges before adding sodium and chloride to concentrations corresponding to physiological conditions. The model was minimized with the NaMD 2.8 bl software for 1000 steps before the molecular dynamics simulations (Phillips et al., 2005). It was computed on a 144 xeon core CPU cluster supercomputer (SGI Altix). Simulations were carried out at constant temperature (300 K) and pressure (1 atm) and by implementing the widely used CHARMM 27 force fields. The time step was set at 1 fs and Langevin dynamics was performed with a target piston pressure of 1.01325 bar and a damping coefficient of 5ps−−1. There is no coupling of the Langevin temperature with hydrogen. The PME algorithms were applied with a grid extended by 10 Å from the periodic boundary condition size (Darden, Perera, Li, & Pedersen, 1999). The electrostatic cut-off was set at 14 Å. A conformation was sampled every 10 ps. As the solvent was described, the dielectric constant was set at 1. To identify steady conformations, 2D-RMSD calculations were carried out on 100 conformations selected, with a stride of 10, from the 1000 conformations produced during the 10-ns simulation. The equilibrium state was reached around 30-ps for all studied models.
Results
Generation of 3D models of TWIST1 by homology with the NEUROD1/E47 X-ray structure
The X-ray structures of the NEUROD1/E47 and MYOD1/MYOD1 bHLH domains and MYC/MAX bHLH-LZ domains bound to the corresponding E-box sequences have been reported (Longo et al., 2008; Ma et al., 1994; Nair & Burley, 2003). To determine the suitable template structure for establishing by homology the structures homodimeric and heterodimeric TWIST1 complexes, primary sequence alignments were carried out. Use of the MYC/MAX structure was excluded, as the identity scores of TWIST1 and E12 vs. MYC and MAX are low (23 and 18% for TWIST1 and 17 and 9% for E12, respectively). Despite higher identity scores (37, 30, 35, 37, and 33% identity between MYOD1 and E47, E12, TWIST1, HAND2, and HAND1, respectively, Figure 1(A)), the MYOD/MYOD X-ray structure was also excluded, as the E-box sequence within it (5′-CAGCTG-3′) is not recognized by TWIST complexes (De Masi et al., 2011) and as MYOD1 belongs to a different clade of bHLH proteins (Skinner et al., 2010). Strikingly, the mouse NEURODl/E47 complex (2ql2 pdb file) meets all the criteria for constituting an appropriate template. In their bHLH domains, human E12 and E47 are 85 and 91% identical, respectively, to the murine E47 protein and TWIST1 is 48 and 29% identical, respectively, to the murine NEUROD1 and E47 proteins (Figure 1(A)). Moreover, the 5′-CATCTG-3′ E-box sequence included in this X-ray structure (Figure 1(B)) has been reported as a TWIST-responsive element (Connerney et al., 2006; De Masi et al., 2011). Although the C-terminal sequences adjacent to the bHLH domain, known as the TWIST-box, constitute a protein interaction site essential for TWIST function (Bialek et al., 2004; Spicer et al., 1996), we reasoned that a structural model restricted to the bHLH domains should provide additional insights into the TWIST–DNA interface and highlight functional differences between TWIST1 complexes. Moreover, the extreme conservation (89% identity) between the two TWIST proteins (Figure 1) suggests that the results of the present work can most certainly be extrapolated to the TWIST2 protein.
Figure 1.
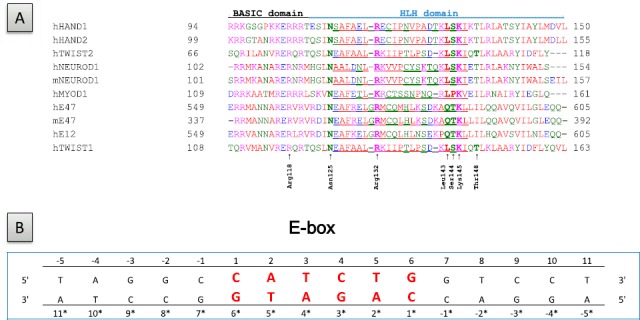
Primary sequences of the proteins and DNA sequence employed in the present study. (A) Primary sequence alignment of the bHLH domains of the NEUROD1, TWIST, HAND, and E2A proteins. TWIST1 residues identified as essential to the structure of the interhelical loops or to their binding to DNA are indicated. Residues within the interhelical loops are underlined, h and m stand for human and murine, respectively. (B) Sequence of the cis-regulatory element of the insulin promoter included in the NEUROD1/E47 X-ray structure and employed to establish the TWIST homology models.
To facilitate comparison of the DNA-binding properties of the dimers, numbering of the residues in each strand begins with the first base of the 5′-CATCTG-3′ E-box core (uppercase bold). Consequently, the paired bases located on the opposite strand are differently numbered. The strands containing the CAT and CAG half-sites are bound to TWIST1 and El2, respectively.
Following the dynamics simulations, 2D-RMSD calculations taking the constraints of the template as a reference were carried out on all models. Analysis of the 2D-RMSD variations led to identifying stable conformations under physiological conditions. For each complex, three different steady states are achieved (Figure 1S, labeled 1–3). On the 2D-RMSD plots, these steady states are represented as the centers of yellow squares (Figure S1). As it took only 30-ps for each model to appear having achieved equilibrium, the 100-ps time point was excluded from the next analysis.
The resulting structural models highlight an interhelical loop between the two amphipathic α-helices, likely involved in protein–DNA complex stabilization and in specifying the target DNA sequence (Figure 2). This assumption is supported by the observation that in the TWIST1/E12 heterodimer, the highly conserved residues Asn125, 1ysl42, and Lysl45 of the interhelical loop of TWIST1 and the corresponding residues Asn566, Lys584, and Lys588, as well as the residue Gln586 of the interhelical loop of E12, are all located in the vicinity of the DNA and likely contribute to DNA binding through hydrogen bonds (H-bonds) or allosteric effects (Figure 2(A)).
Figure 2.
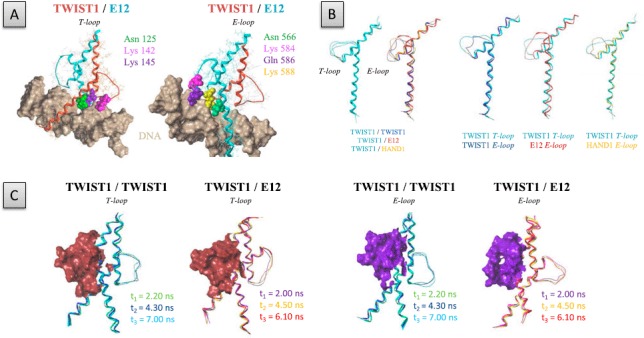
Comparison of the T- and E-loop structures in TWIST1 complexes. (A) Visualization of key residues within the T-loop and E-loop of the TWIST1/E12 heterodimeric complex, located at the vicinity of DNA. The protein structures are represented as ribbons. (B) Left panels: superposition of the T-loop and E-loop structures of the TWIST1/TWIST1, TWIST1/E12, and TWIST1/HAND1 dimers, as indicated at the bottom. Right panels: superposition of the TWIST1 T-loop with the E-loops of TWIST1, E12, and HAND1, as indicated at the bottom. Images correspond to the second steady states of the dynamics simulations. C) Superposition of the E- and T-loop structures (represented as ribbons) of the different TWIST1 complexes as detected at the third steady state of the dynamics simulation. E- and T-loop surfaces are colored in violet and brown, respectively.
We next compared the dimer structures. For convenience, we arbitrarily called ‘T-loop’ the interhelical loop of the monomer modeled on the basis of the structure of murine NEUROD1 and ‘E-loops’ those of monomers modeled on the basis of the E47 structure. While the backbone of the T-loop showed similar conformations in the homodimer and different heterodimers, the backbone of the E-loop was found to diverge significantly between dimers (Figure 2(B)), because the amino acids composing the sequences of the different TWIST1 partners differ as regards length, polarity, charge, and hydrophobicity (Figure 1). Significant divergence was found between the T-loop and E-loop of the TWIST1 homodimer (Figure 2(B)). This divergence may be largely attributable to the use of a heterodimeric complex as a template. The E-loop of mE47 is quite disordered in the X-ray structure, but the 10-ns dynamics simulations showed its stability in space and time. The loops of the TWIST1/TWIST1 complex reach equilibrium at 5.5 ns (Figure S1(B)). This stabilization is likely due largely to interaction with DNA, as described below. We, then, investigated whether the conformation of the lateral loops might depend on the strand of DNA associated with the monomer, as E-boxes are asymmetrical. For this, we inverted the orientation of the DNA in the TWIST1/TWIST1 model, and carried out an additional 10-ns dynamics simulation. This inversion was found to affect the loop conformations only slightly, as symmetrical interactions between Arg118 and the DNA were found (Figure S2). We conclude that the divergence between T- and E-loops within the TWIST homodimeric complex is unlikely due to sequence difference between the two DNA strands.
Taken together, our data demonstrate that TWIST1 complexes differ significantly in the conformation of their interhelical loops. Remarkably, unlike the T- and E-loops of the TWIST1 homodimer, the E-loop of the heterodimer was found to form a deep cavity that might potentially be targeted in an effort to select specific inhibitors (Figure 2(C)).
Structural determinants of the interhelical loops
We next aimed to identify residues essential for interhelical loop conformation. Although these loops do not have a structural organization strictly speaking, H-bond formation can stiffen their structure. By limiting our study to H-bonds with an occupancy exceeding 5% of the total simulation and by excluding H-bonds between residues of different protein partners or involved in α-helix structures, we were led to focus on four TWIST1 residues: Lysl45, Serl44, Argl32, and Argll8.
Our results show that in the T-loop and E-loop of the TWIST1 homodimer, the –NZ2 functional group of Lysl45 interacts with the oxygen atom of the –OD1 group of Asnl25 residue (Figure 3(A)), while it fails to do so in the T-loop of the heterodimer (Figure 4). Interestingly, the functional groups of Lysl45 and or Asnl25 additionally establish H-bonds with oxygen groups of DNA bases (Figure 3). In both the T-loop and E-loop of the homodimer, the Lysl45 residue interacts alternatively with the oxygen of the Asnl25 residue and with the Ade (+2) nucleotide of the E-box during the 4 first ns of the simulation, and sporadically during the rest of the simulation (Figure 3). In parallel, a second H-bond is established between the Lysl45 residue and the Cyt(+1) base (Figure 3). In addition to the Lysl45–Cyt(+1) interaction detectable in both the T-loop and the E-loop, the Asnl25–Ade(+2) interaction is additionally established in the T-loop (Figure 3(A)).
Figure 3.

Hydrogen bonds involving the Asnl25, Lysl42, and Lysl45 residues in the TWIST1 lateral loops.
Left panels: positions of residues Asnl25, Lysl42, and Lysl45 within the T-loop (A) and E-loop (B) of the TWIST1 homodimeric complex. Right panels: distances between residues and/or bases during the dynamics simulation are depicted as plots of distance from donor to acceptor atom (A) versus frame number (total time, 100 frames per ns). H-bonds are established when the two functional groups are less than 2 Å apart.
Figure 4.

Hydrogen bonds involving the Asn125, Lys142, and Lysl45 residues of TWIST1 and the Asn566 and Lys588 residues of E12 within the lateral loops of the TWIST1/E12 heterodimeric complex.
Left panels: positions of the Asnl25, Lysl42, Lysl45, Asn566, and Lys588 residues within the T-loop (A) and E-loop (B) of the TWIST1/E12 heterodimeric complex. Right panels: distances between residues and/or bases during the dynamics simulation are depicted as plots of distance from donor to acceptor atom (Å) versus frame number (total time, 100 frames per ns). H-bonds are established when the two functional groups are less than 2 Å apart.
Both the Lysl45 and the Asnl25 residues within the T-loop of the TWIST1/E12 heterodimer also establish H-bonds with DNA bases (Cyt(+1) and Ade(+2), respectively) (Figure 4). It is worth noting that the Lysl42–Ade (+2) interaction established in the first part of the simulation is partially transferred to the Asn125 residue in the course of the dynamics simulation (Figure 4). Similar interactions between the corresponding Lys588 and Asn566 residues of E12 and the Ade(+2) and Cyt(+1) nucleotides also take place sporadically within the E-loop (Figure 4). In summary, these observations show that Asnl25 and Lysl45 residues play a determining role in stabilizing TWIST1 complexes on the DNA, by contributing to the establishment of the interhelical loop conformation and by directly interacting with oxygen atoms of DNA bases. In support of this view, the K145E mutation in TWIST1 abolishes the stable binding both the TWIST1 homodimer and the TWIST1/E12 heterodimer (Figure S3, (El Ghouzzi et al., 2001; Maia et al., 2012)).
In both the homodimeric and heterodimeric TWIST1 complexes, the Serl44 residue of TWIST1 and its counterpart Thr587 in E12 or Serl31 in HAND1 are invariably located along the bottom edge of a T- or E-loop (Figures 5 and 6 and S4). These residues form H-bonds with either Thrl48 of TWIST1 (Figure 5), Leu590 (counterpart of Glnl47) of E12 (Figure 6), or Lysl34 and Thrl35 (counterparts of Glnl47 and Thrl48) of HAND1 (Figure S4). While the Serl44-Thrl48 interaction is fairly stable throughout the dynamics simulations in all homodimeric and heterodimeric complexes (Figures 5 and 6), the Serl44-Glnl47 interaction appears to be less stable and even absent in the TWIST1/HAND1 heterodimer (Figure S4).
Figure 5.
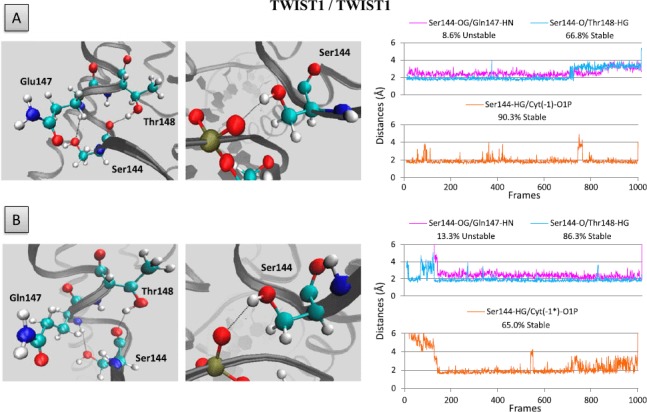
Hydrogen bonds involving the Ser144 residue of TWIST1.
Left panels: position of Ser144 residue within the T-loop (A) and E-loop (B) of the homodimeric TWIST1 complex. Right panels: distance between residues and/or bases during the dynamics simulation are depicted as plots of distance from donor to acceptor atom (A) versus frame number (total time, 100 frames per ns). H-bonds are established when the two functional groups are less than 2 Å apart.
Figure 6.
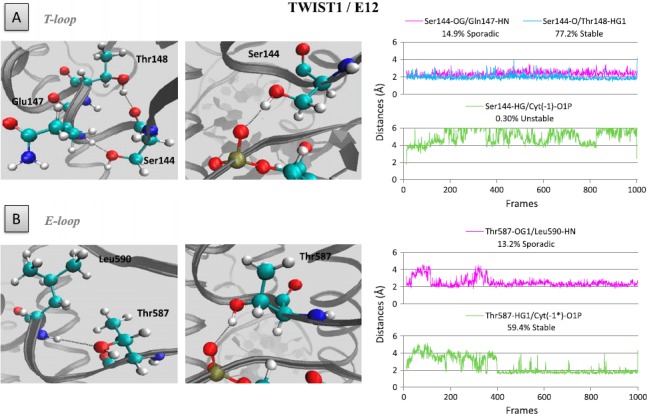
Hydrogen bonds involving residue Serl44 of and its counterpart Thr587 in E12.
Left panels: position of the Serl44 residues within the T-loop (A) and E-loop (B) of the TWIST1/E12 heterodimeric complex. Right panels: distances between residues and/or bases during the dynamics simulation are depicted as plots of distance from donor to acceptor atom (A) versus frame number (total time, 100 frames per ns). H-bonds are established when the two functional groups are less than 2 Å apart.
Interestingly, in both the T-loop and E-loop of the homodimer as well as in the E-loop of the TWIST1/E12 heterodimer, the HG group of Serl44 residue additionally establishes stable H-bonds with the O1P group of Cyt (—l) nucleotide (Figure 5). Surprisingly, the Serl44–Cyt (—1) and Serl31–Cyt(—l) H-bonds are not conserved in the TWIST1/HAND 1 heterodimer (Figure S4). Taken together, these data demonstrate that H-bonds are determinant in maintaining interhelical loop conformation and contribute to determining the affinity of TWIST1 complexes for E-boxes. In support of this view, the S144R mutation in TWIST1 abolishes the stable binding to DNA of both the TWIST1 homodimer and the TWIST1/E12 heterodimer (Figure S3) (El Ghouzzi et al., 2001; Maia et al., 2012)).
Interestingly, in both the T- and E-loops of the TWIST1/TWIST1 and TWIST1/E12 dimers, the highly conserved residue Arg118 (Figure 1) additionally establishes stable H-bonds with both the O2P group of Cyt (+4) and the O2P group of Ade(+4∗) bases (Figure 7). The fourth base of the E-box is, thus, important in stabilizing TWIST1 complex binding to DNA. When the DNA is placed in the opposite orientation in the TWIST1 homodimeric model, the H-bonds between residue Arg118 and the fourth base of the E-box (Ade(+4∗) or Thy(+4)) are conserved (Figure S2(B)). We conclude that E-box asymmetry most likely does not determine either the sequence targeted or the stability of’ the protein/DNA complex. Maintenance of these interactions largely explains the stability of T- and E- loop conformations in the TWIST1 homodimeric complex (Figure S2(A)).
Figure 7.
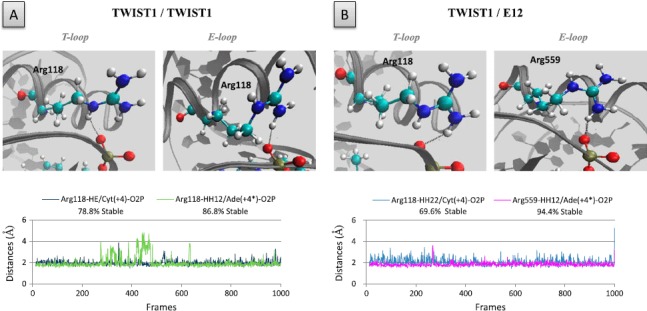
Hydrogen bonds involving the Arg1l8 residue of TWIST1 and its Arg559 counterpart in E12.
The role of residues, Arg1l8 and Arg559, was examined in the context of both the TWIST1 homodimer (A) and the TWIST1/E12 heterodimer (B). Upper panels: positions of the Argll8 residues within the homodimeric TWIST1 complex (left panels) and of the Arg1l8 and Arg559 residues within the heterodimeric TWISTI/E12 complex (right panels). Lower panels: distances between residues and/or bases during the dynamics simulations are depicted as plots of distance from donor to acceptor atom (A) versus frame number (total time, 100 frames per ns). H-bonds are established when the two functional groups are less than 2 Å apart.
In all TWIST1 complexes examined, the Arg132 residue is invariably oriented towards the inside of the T-loop (Figure 8). In both the homodimer and the TWIST1/E12 heterodimer, interestingly, this residue establishes H-bonds with the oxygen atoms of both Leul43 and Thrl48, although the configuration adopted and the timing of bond formation differs according to the complex. In the homodimer, Arg132 interacts concomitantly with both residues during the first 2.88-ns of the dynamic simulation, i.e. in the first steady state identified, and both interactions are lost in the next two steady states. The T-loop, thus, evolves from a closed to an open conformation (Figure 8(A)). In the TWIST1/E12 heterodimer, Arg132 interacts alternatively with the two residues, the Arg132–Leu143 interaction being favored with time. The T-loop, thus, evolves from an open to a closed conformation (Figure 8(B)). In the TWIST1/ HAND1 complex, Arg132 interacts concomitantly with both Thrl48 and Leul43, with a preference for Thrl48 over time (Figure S4). Despite these differences, Arg132 is likely to be determinant in establishing the T-loop conformation in all TWIST1 complexes. These H-bonds are not formed in the E-loop of the homodimer, or the bond with Serl40 forms only transiently (Figure 8); as a result, the two helices come close together and reduce the space inside the E-loop. In support of the hypothesis that Arg132 plays a structural role in the T-loop only, the orientation of the side chain of its counterpart in E12 (Arg574) excludes an involvement in H-bonds. In contrast, in the E-loop of the TWIST1/HAND1 heterodimer, Arg118 (counterpart of Arg132) establishes an H-bond with Asnl23 (Figure S5).
Figure 8.
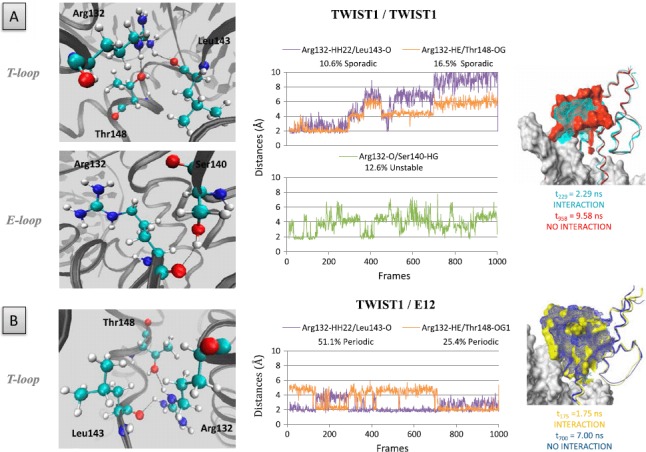
Hydrogen bonds involving residue Argl32 of TWIST1.
The role of the residue was examined in the context of the TWIST1 homodimer (A) and TWIST1/E12 heterodimer (B). Left panels: positions of the Argl32 residues within the complexes. Central panels: distances between residues and/or bases during the dynamics simulations are depicted as plots of distance from donor to acceptor atom (A) versus frame number (total time, 100 frames = l ns). H-bonds are established when the two functional groups are 2 Å apart. Right panels: superposition of the complex structures as detected at different time points of the dynamics simulations.
Insertions in the interhelical loops of TWIST1, as observed in SC patients, affect both their structure and interaction with DNA
To gain further insights into the role of the interhelical loops in TWIST1 dimer function and specificity, we next focused on additional Saerthe–Chotzen associated TWIST1 variants with aberrant insertion of seven amino acids into the interhelical loop at position 135 or 139 (Ins-135 or Ins-139, Figure S5) due to the presence of a 21-bp tandem repeat in the TWIST1 gene (El Ghouzzi et al., 1997). Using a homology strategy similar to that described above, in silico models based on the murine NEUROD1/E47 X-ray structure were established and subjected to dynamics simulations. From the 2D-RMSD calculations, three steady states were selected as previously (Figure S6). The results show, as expected, that both insertions increase the length and surface of the interhelical loops and significantly move the T-loop towards the second helix of the HLH, this effect being more pronounced in the Ins-139 variant (Figure S5). Many interactions between loop residues are modified in complexes containing an Ins-variant of TWIST1 (e.g. the Arg132-Thr148 interact is disrupted in the T-loops of homodimers containing either Ins-variant) (Table 1).
Table 1.
Main H-bonds established within the interhelical loops of the various TWIST1 complexes. Interactions are either established (% occupancy is indicated) or absent (—), over the time course of the dynamics simulations minus the first 100 ps. The percentage of occupancy represents the time during which the H-bond is established. Stable, periodic, and sporadic interactions are labeled in dark grey, light grey, white respectively.
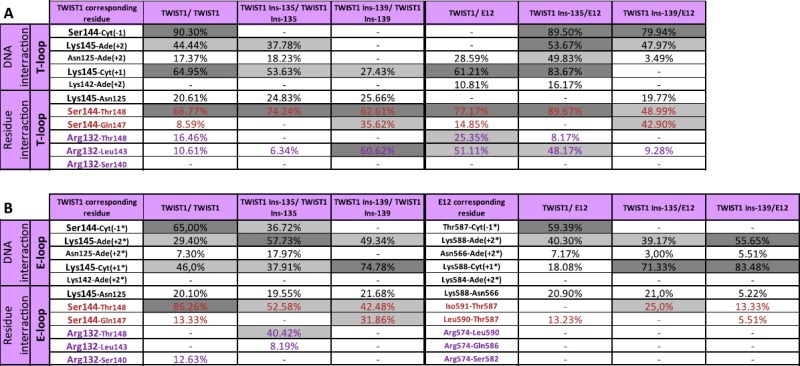
It is noteworthy that the two insertions have somewhat different effects (e. g. the Arg132–Leu143 interaction is disrupted in the T-loop of the Ins-135 homodimer and is fully stabilized in that of the Ins-139 homodimer) (Table 1). Conversely, the insertions create some novel concomitant H-bonds (Met575-His593, Gln577-Gln594, and Gln577-Glu583 in the E-loop of the TWIST1 Insl39/E12 heterodimer, Figure 9), Lysl45–Thrl48 in the T-loop and Argl39–Aspl57 in the E-loop of the TWIST1 Ins-135 homodimer, (Figure 10), stiffening the structures of these loops. In the TWIST1 Ins-139 homodimer, some newly established H-bonds involve aberrantly inserted residues (Lys (Ins + 1)–Aspl41, Lys (Ins + l)–Pro (Ins + 7)) (Figure 10).
Figure 9.

Amino acid insertions within the interhelical loops of the TWIST1/E12 heterodimer (Ins-135 and Ins-139), as observed in SC patients, generate novel H-bonds.
Left panels: positions of the aberrantly inserted residues involved in novel H-bonds within the interhelical loops in the Ins-135 TWIST1/E12 (upper panel) and Ins-139 TWIST1/E12 (lower panel) heterodimeric complexes. Right panels: distances between residues and/or bases during the dynamics simulations are depicited as plots of distance from donor to acceptor atom (A) versus frame number (total time 100 frames per 1 ns). H-bonds are established when the two functional groups are less than 2 Å apart.
Figure 10.
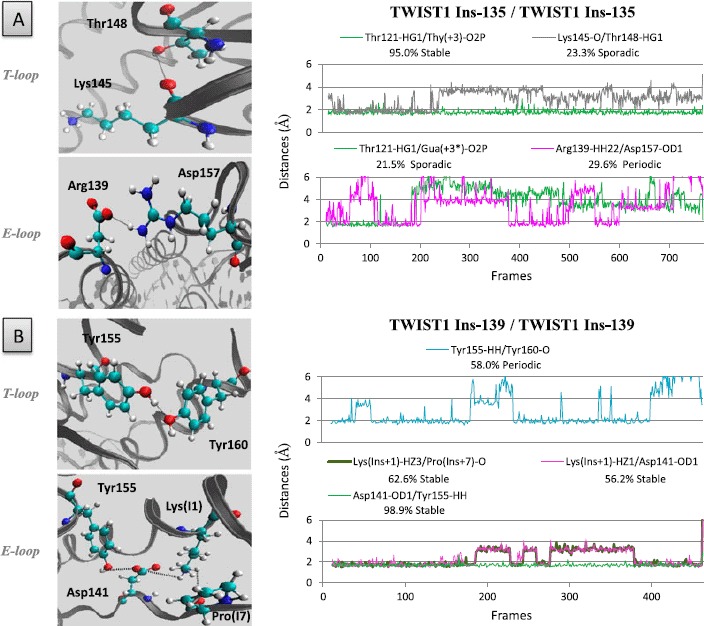
Amino acid insertions within the interhelical loops of the TWIST1/TWIST1 homodimer (Ins-135 and Ins-139), as observed in SC patients, generate novel H-bonds.
Left panels: positions of the aberrantly inserted residues involved in novel H-bonds within the interhelical loops of the Ins-135 (A) and Ins-139 (B) homodimeric TWIST complexes. Right panels: distances between residues and/or bases during the dynamics simulations are depicted as plots of distance from donor to acceptor atom (A) versus frame number (total time, 100 frames per 1 ns) H-bonds are established when the two functional groups are less than 2 Å apart.
The insertions additionally alter certain residue-DNA interactions (e. g. Serl44–Cyt(—1) in the T-loops of the homodimers containing either insertion variant) and create novel ones (e.g. Serl44–Cyt(—l) in the T-loops of the heterodimers containing either insertion variant) (Table 1). Taken together, these data suggest that both insertions distort the interhelical loops, thus, greatly affecting protein/DNA complex stability.
Discussion
As a regulator of cell migration and specification, the TWIST1 transcription factor is essential to embryonic development in multiple species (El Ghouzzi et al., 1997). As a reminiscence of its embryonic functions, its aberrant reactivation observed in numerous cancer types was originally associated with cancer cell dissemination (Yang et al., 2004). Further investigations have led to attributing to TWIST1 additional functions in malignant conversion and tumor progression, mainly involving failsafe program inhibition and EMT induction (Ansieau et al., 2010). Interestingly, TWIST1 depletion in numerous cancer cell types is sufficient to reactivate senescence or apoptosis programs (Ansieau et al., 2008; Maestro et al., 1999; Tran et al., 2012; Valsesia-Wittmann et al., 2004), suggesting that TWIST1 inhibition may constitute a novel anti-cancer therapeutic route.
Beyond the difficulty of targeting nuclear factors, an additional complexity of TWIST1 activity arises from its ability to associate with different partners, i.e. with itself or with E12/E47 or HAND, to form functionally different homo- or heterodimers. The balance between these complexes determines TWIST1 function, and altered dimer selection has dramatic consequences, as demonstrated by the altered phenotype of mice and SC patients with TWIST1 haplo-insufficiency (Connerney al., 2006; Firulli et al., 2005, 2007) and mice ectopically expressing TWIST1-tethered dimers (Connerney et al., 2008).
On the basis of this information, we have sought to compare the structures of different protein–DNA complexes to gain further insights into the functional consequences of the TWIST1 mutations/insertions encountered in SC patients, and potentially to identify structures that might be targeted to impair TWIST1 function.
Although the N- and C-terminal sequences of TWIST1 also play a role in TWIST1 functions, we have restricted our analysis to the bHLH domain, taking advantage of the available X-ray structure of the murine NEUROD1/E47 protein complex associated with its DNA target sequence (Longo et al., 2008). The high identity scores computed for the two clade-A proteins, TWIST1 and NEUROD1, and for the two E2A splice variants, E12 and E47, along with the fact that the E-box selected has been identified as a functional TWIST1-responsive cis-regulatory element in the promoting sequences of several of its target genes (Connerney et al., 2006; De Masi et al., 2011), argue strongly in favor of the reliability of the established TWIST1/E12 homology models. Although TWIST1 and HAND1 diverge more markedly from E47, the identity scores remain high enough to generate trustworthy homodimeric and TWIST1/HAND1 heterodimeric complexes.
Structural analysis highlights, between the two amphipathic α-helices, an interhelical loop whose conformation differs between the monomers forming a given TWIST1-containing dimer and according to the partner of TWIST1 in the dimer (Figure 2). We demonstrate that these structural divergences notably reflect differences in residue composition, as they affect the formation of H-bonds involving either residues or DNA bases. Interestingly, the E-loop of the TWIST1/E12 heterodimer forms a unique cavity that might reasonably be targeted in efforts to select specific intercalating molecules for pharmacological purposes. Preliminary docking analysis of the NCI library supports this hypothesis (data not shown).
Residues located in the loops of all of the homodimers and heterodimers studied interact with nucleotides – 1 to +4 of the E-box sequence (Figures 3–8), known to be important in bHLH binding (De Masi et al., 2011; Ohno, Sadeh, Blatt, Brengman, & Engel 2003). In the light of the importance of these interactions, we have assessed the consequences of insertions within the interhelical loops (Ins-135 and Ins-139) (El Ghouzzi et al., 1997) observed in SC patients on TWIST1 complex structure. As expected, both insertions significantly increase the surface and strongly modify the conformations of the interhelical loops (Figures 9 and 10, S5 and S6). They notably abolish certain H-bonds between T-loop residues and promote the formation of novel ones (Table 1). The Ins-139 insertion additionally disrupts many interactions of the T-loop with DNA, especially within the homodimer (Figures 9 and 10 and Table 1). This observation corroborates the reported reduced DNA-binding affinity of the TWIST1 Ins-139/E12 heterodimeric complex (El Ghouzzi et al., 2001).
Remarkably, the two SC-associated TWIST1 alterations are not equivalent, as the Ins-135 insertion disrupts much fewer contacts between interhelical loops and DNA (Table 1).
The relevance of our models is additionally supported by the analysis of two additional TWIST1 variants found in SC patients, characterized, respectively, by the point mutations K145E and S144R (El Ghouzzi et al., 2001). Residues 145 and 144 are located, respectively, at the center of the dimer and along the bottom edge of the interhelical loop, at the interface with the DNA (Figure S3). Both mutations appear to increase complex flexibility (Maia et al., 2012). The K145 substitution strongly impairs TWIST1 binding to DNA and only slightly reduces dimerization (El Ghouzzi et al., 2001). Lysl45 actually interacts with the second, determinant nucleotide of the E-box. The K145E substitution probably abolishes this interaction through repulsion between the carbonyl group of Glu and the phosphate group on the DNA base. In addition, Serl44 in the T-loops of both the TWIST homodimer and the TWIST1/E12 heterodimer and its counterpart Thr587 in the E-loop of the latter are in close contact with Cyt(—l) (Figures 5 and 6). In support of the view that Serl44 plays a role in DNA binding, the SC-associated point mutation S144R was also found to abolish TWIST1-DNA binding, likely by destabilizing the DNA–protein complex (El Ghouzzi et al., 2001; Maia et al., 2012). Interestingly, the Ins-135 and Ins-139 insertions strongly alter the capacity of Serl44 (or Thr587) to interact with Cyt(—1) in both loops of both the homodimeric and the heterodimeric complex (Table 1). This provides an explanation for the reported reduced DNA affinity of the TWIST1 Ins-l39/E12 heterodimer (El Ghouzzi et al., 2001). It is worth noting that in the TWIST1/HAND1 heterodimer, neither Serl44 in the TWIST1 T-loop nor its counterpart Serl31 in the HAND1 E-loop interacts with Cyt(—1) of the E-box. This highlights a functional divergence between TWIST1 complexes (Figure S4).
Taken together, our data support the view that interhelical loops within the bHLH play a determining role in maintaining TWIST1 –DNA complex structures and provide a structural explanation for the loss of function associated with several TWIST1 mutations/insertions observed in SC patients.
Supporting Information
Supplementary material for this paper is available online at http://dx.doi.[10.1080/07391102.2012.762722].
Supplementary material dealing with the “Interhelical loops within the bHLH domain are determinant in maintaining TWIST1–DNA complexes” study is available from authors directly by e-mail transmission.
Author contributions
All authors have given approval to the final version of the manuscript. The manuscript was written through contributions of all authors. CB, RT, JH, JAC, and LP carried out experiments. RT, AP, SA, and LP designed the experimental strategy, and SA and LP wrote the manuscript.
Funding sources
Our team is labeled by the Ligue Nationale contre le Cancer. This work was additionally supported by institutional grants from the LabEX DEVweCAN (ANR-10-LABX-61) and from the LyRIC (Lyon Recherche Integree en Cancerologie, Institut National Contre le Cancer, INCa_4664).
Abbreviations
- SC
Saethre–Chotzen syndrome
- bHLH
basic helix-loop-helix
- EMT
epithelial-to-mesenchymal transition
Acknowledgment
We thank all members of the laboratory for insightful discussions and Dr Kathleen Broman for critical reading of the manuscript.
References
- Ansieau S., Bastid J., Doreau A., Morel A. R., Bouchet B. R., Thomas C., Puisieux A. Induction of EMT by twist proteins as a collateral effect of tumor-promoting inactivation of premature senescence. Cancer Cell. 2008;14:79–89. doi: 10.1016/j.ccr.2008.06.005. [DOI] [PubMed] [Google Scholar]
- Ansieau S., Morel A. P., Hinkal G., Bastid J., Puisieux A. TWISTing an embryonic transcription factor into an oncoprotein. Oncogene. 2010;29:3173–3184. doi: 10.1038/onc.2010.92. [DOI] [PubMed] [Google Scholar]
- Arnold K., Bordoli L., Kopp J., Schwede T. The Swiss-model workspace: A web-based environment for protein structure homology modelling. Bioinformatics. 2006;22:195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- Atchley W. R., Fitch W. M. A natural classification of the basic helix-loop-helix class of transcription factors. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:5172–5176. doi: 10.1073/pnas.94.10.5172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bialek P., Kern B., Yang X., Schrock M., Sosic D., Hong N., Karsenty G. A twist code determines the onset of osteoblast differentiation. Developmental Cell. 2004;6:423–435. doi: 10.1016/s1534-5807(04)00058-9. [DOI] [PubMed] [Google Scholar]
- Bourgeois P., Bolcato-Bellemin A. L., Danse J. M., Bloch-Zupan A., Yoshiba K, Stoetzel C., Perrin-Schmitt F. The variable expressivity and incomplete penetrance of the twist-null heterozygous mouse phenotype resemble those of human Saethre-Chotzen syndrome. Human Molecular Genetics. 1998;7:945–957. doi: 10.1093/hmg/7.6.945. [DOI] [PubMed] [Google Scholar]
- Cakouros D., Isenmann S., Cooper L., Zannettino A., Anderson P., Glackin C., Gronthos S. Twist-1 induces recruitment regulating histone methylation along the Ink4A/Arf locus in mesenchymal stem cells. Molecular and Cell Biology. 2012;32:1433–1441. doi: 10.1128/MCB.06315-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng G. Z., Chan J., Wang Q., Zhang W., Sun C. D., Wang L. H. Twist transcriptionally up-regulates AKT2 in breast cancer cells leading to increased migration, invasion, and resistance to paclitaxel. Cancer Research. 2007;67:1979–1987. doi: 10.1158/0008-5472.CAN-06-1479. [DOI] [PubMed] [Google Scholar]
- Connerney J., Andreeva V., Leshem Y., Mercado M. A., Dowell K., Yang X., Lindner V., Spicer D. B. Twist1 homodimers enhance FGF responsiveness of the cranial sutures and promote suture closure. Developmental Biology. 2008;318:323–334. doi: 10.1016/j.ydbio.2008.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connerney J., Andreeva V., Leshem Y., Muentener C., Mercado M. A., Spicer D. B. Twist1 dimer selection regulates cranial suture patterning and fusion. Developmental Dynamics. 2006;235:1345–1357. doi: 10.1002/dvdy.20717. [DOI] [PubMed] [Google Scholar]
- Darden T., Perera L., Li L., Pedersen L. New tricks for modelers from the crystallography toolkit: The particle mesh Ewald algorithm and its use in nucleic acid simulations. Structure. 1999;7:R55–R60. doi: 10.1016/s0969-2126(99)80033-1. [DOI] [PubMed] [Google Scholar]
- De Masi R., Grove C. A., Vedenko A., Alibes A., Gisselbrecht S. S., Serrano L., Bulyk, Walhout A. J. Using a structural and logics systems approach to infer bHLH-DNA binding specificity determinants. Nucleic Acids Research. 2011;39:4553–4563. doi: 10.1093/nar/gkr070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Ghouzzi V., Le Merrer M., Perrin-Schmitt R., Lajeunie E., Benit R., Renier D., Bonaventure J. Mutations of the TWIST gene in the Saethre-Chotzen syndrome. Nature Genetics. 1997;15:42–46. doi: 10.1038/ng0197-42. [DOI] [PubMed] [Google Scholar]
- El Ghouzzi V., Legeai-Mallet L., Aresta S., Benoist C., Munnich A., de Gunzburg J., Bonaventure J. Saethre-Chotzen mutations cause TWIST protein degradation or impaired nuclear location. Human and Molecular Genetics. 2000;9:813–819. doi: 10.1093/hmg/9.5.813. [DOI] [PubMed] [Google Scholar]
- El Ghouzzi V., Legeai-Mallet L., Benoist-Lasselin C., Lajeunie E., Renier D., Munruch A., Bonaventure J. Mutations in the basic domain and the loop-helix II junction of TWIST abolish DNA binding in Saethre-Chotzen syndrome. FEBS Letters. 2001;492:112–118. doi: 10.1016/s0014-5793(01)02238-4. [DOI] [PubMed] [Google Scholar]
- Firulli B. A., Krawchuk D., Centonze V. E., Vargesson N., Virshup D. M., Conway S. J., Firulli A. B. Altered Twist1 and Hand2 dimerization is associated with Saethre-Chotzen syndrome and limb abnormalities. Nature Genetics. 2005;37:373–381. doi: 10.1038/ng1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firulli B. A., Redick B. A., Conway S. J., Firulli A. B. Mutations within helix I of Twist1 result in distinct limb defects and variation of DNA binding affinities. Journal of Biological Chemistry. 2007;282:27536–27546. doi: 10.1074/jbc.M702613200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebrok M., Wertz K., Fuchtbauer E. M. M-twist is an inhibitor of muscle differentiation. Developmental Biology. 1994;165:537–544. doi: 10.1006/dbio.1994.1273. [DOI] [PubMed] [Google Scholar]
- Humphrey W., Dalke A., Schulten K. VMD: Visual molecular dynamics. Journal of Molecular Graphics. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. 27–38. [DOI] [PubMed] [Google Scholar]
- Jorgensen W. L., Chandrasekhar J., Madura J. D., Impey R. W., Klein M. L. Comparison of simple potential functions for simulating liquid water. Journal of Chemical Physics. 1983;79:926–935. [Google Scholar]
- Kajiyama H, Shibata K., Terauchi M., Yamashita M., Ino K., Nawa A., Kikkawa F. Chemoresistance to paclitaxel induces epithelial-mesenchymal transition and enhances metastatic potential for epithelial ovarian carcinoma cells. International Journal of Oncology. 2007;31:277–283. [PubMed] [Google Scholar]
- Kwok W. K., Ling M. T., Lee T. W., Lau T. C., Zhou C., Zhang X., Wang X. Up-regulation of TWIST in prostate cancer and its implications as a therapeutic target. Cancer Research. 2005;65:5153–5162. doi: 10.1158/0008-5472.CAN-04-3785. [DOI] [PubMed] [Google Scholar]
- Kwok W. K., Ling M. T., Yuen H. F., Wong Y. C., Wang X. Role of pl4ARF in TWIST-mediated senescence in prostate epithelial cells. Carcinogenesis. 2007;28:2467–2475. doi: 10.1093/carcin/bgm185. [DOI] [PubMed] [Google Scholar]
- Lee M. S., Lowe G. N., Strong D. D., Wergedal J. E., Glackin C. A. TWIST, a basic helix-loop-helix transcription factor, can regulate the human osteogenic lineage. Journal of Cellular Biochemistry. 1999;75:566–577. doi: 10.1002/(sici)1097-4644(19991215)75:4<566::aid-jcb3>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- Li Q. Q., Xu J. D., Wang W. J., Cao X. X., Chen Q., Tang F., Xu Z. D. Twist1-mediated adriamycin-induced epithelial-mesenchymal transition relates to multi-drug resistance and invasive potential in breast cancer cells. Clinical Cancer Research. 2009;15:2657–2665. doi: 10.1158/1078-0432.CCR-08-2372. [DOI] [PubMed] [Google Scholar]
- Longo A., Guanga G. P., Rose R. B. Crystal structure of E47-NeuroDl/beta2 bHLH domain-DNA complex: Heterodimer selectivity and DNA recognition. Biochemistry. 2008;47:218–229. doi: 10.1021/bi701527r. [DOI] [PubMed] [Google Scholar]
- Ma P. C., Rould M. A., Weintraub H., Pabo C. O. Crystal structure of MyoD bHLH domain-DNA complex: Perspectives on DNA recognition and implications for transcriptional activation. Cell. 1994;77:451–459. doi: 10.1016/0092-8674(94)90159-7. [DOI] [PubMed] [Google Scholar]
- Maestro R., Dei Tos A. P., Hamamori Y., Krasnokutsky S., Sartorelli V., Kedes L., Hannon G. J. Twist is a potential oncogene that inhibits apoptosis. Genes and Development. 1999;13:2207–2217. doi: 10.1101/gad.13.17.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maia A. M., da Silva J. H., Mencalha A. L., Caffarena E. R., Abdelhay E. Computational modeling of the bHLH domain of the transcription factor TWIST1 and R118C, S144R and K145E mutants. BMC Bioinformatics. 2012;13:184. doi: 10.1186/1471-2105-13-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mani S. A., Guo W., Liao M. J., Eaton E. N., Ayyanan A., Zhou A. Y., Weinberg R.A. The epithelial- mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704–715. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morel A. P., Hinkal G. W., Thomas C, Fauvet F., Courtois-Cox S., Wierinckx A, Puisieux A. EMT inducers catalyze malignant transformation of mammary epithelial cells and drive tumorigenesis towards claudin-low tumors in transgenic mice. PLoS Genetics. 2012;8:e1002723. doi: 10.1371/journal.pgen.1002723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morel A. P., Lievre M., Thomas C., Hinkal G., Ansieau S., Puisieux A. Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS ONE. 2008;3:e2888. doi: 10.1371/journal.pone.0002888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murre C. Role of helix-loop-helix proteins in lymphocyte development. Cold Spring Harbor Symposia on Quantitative Biology. 1999;64:39–44. doi: 10.1101/sqb.1999.64.39. [DOI] [PubMed] [Google Scholar]
- Nair S. K., Burley S. K. X-ray structures of Myc-Max and Mad-Max recognizing DNA. Molecular bases of regulation by proto-oncogenic transcription factors. Cell. 2003;112:193–205. doi: 10.1016/s0092-8674(02)01284-9. [DOI] [PubMed] [Google Scholar]
- Ohno K., Sadeh M., Blatt I., Brengman J. M., Engel A. G. E-box mutations in the RAPSN promoter region in eight cases with congenital myasthenic syndrome. Human Molecular Genetics. 2003;12:739–748. doi: 10.1093/hmg/ddg089. [DOI] [PubMed] [Google Scholar]
- Pham C. G., Bubici C., Zazzeroni E., Knabb J. R., Papa S., Kuntzen C., Franzoso G. Upregulation of Twist-1 by NF-kappaB blocks cytotoxicity induced by chemotherapeutic drugs. Molecular and Cellular Biology. 2007;27:3920–3935. doi: 10.1128/MCB.01219-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips J. C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Schulten K. Scalable molecular dynamics with NAMD. Journal of Computational Chemistry. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinner M. K., Rawls A., Wilson-Rawls J., Roalson E. H. Basic helix-loop-helix transcription factor gene family phylogenetics and nomenclature. Differentiation. 2010;80:1–8. doi: 10.1016/j.diff.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spicer D. B., Rhee J., Cheung W. L., Lassar A. B. Inhibition of myogenic bHLH and MEF2 transcription factors by the bHLH protein Twist. Science. 1996;272:1476–1480. doi: 10.1126/science.272.5267.1476. [DOI] [PubMed] [Google Scholar]
- Stevens J. D., Roalson E. H., Skinner M. K. Phylogenetic and expression analysis of the basic helix-loop-helix transcription factor gene family: Genomic approach to cellular differentiation. Differentiation. 2008;76:1006–1022. doi: 10.1111/j.1432-0436.2008.00285.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thisse B., el Messal M., Perrin-Schmitt F. The twist gene: Isolation of a Drosophila zygotic gene necessary for the establishment of dorsoventral pattern. Nucleic Acids Research. 1987;15:3439–3453. doi: 10.1093/nar/15.8.3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson J. D., Higgins D. G., Gibson T. J. ClustalW: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Research. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran P. T., Shroff E. H., Burns T. F., Thiyagarajan S., Das S. T., Zabuawala T, Felsher D. W. Twist1 suppresses senescence programs and thereby accelerates and maintains mutant Kras-induced lung tumorigenesis. PLoS Genetics. 2012;8:e1002650. doi: 10.1371/journal.pgen.1002650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valsesia-Wittmann S., Magdeleine M., Dupasquier S., Garin E., Jallas A. C., Combaret V., Puisieux A. Oncogenic cooperation between H-Twist and N-Myc overrides failsafe programs in cancer cells. Cancer Cell. 2004;6:625–630. doi: 10.1016/j.ccr.2004.09.033. [DOI] [PubMed] [Google Scholar]
- Vesuna E., Lisok A., Kimble B., Raman V. Twist modulates breast cancer stem cells by transcriptional regulation of CD24 expression. Neoplasia. 2009;11:1318–1328. doi: 10.1593/neo.91084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vichalkovski A., Gresko E., Hess D., Restuccia D. E., Hemmings B. A. PKB/AKT phosphorylation of the transcription factor Twist-1 at Ser42 inhibits p53 activity in response to DNA damage. Oncogene. 2010;29:3554–3565. doi: 10.1038/onc.2010.115. [DOI] [PubMed] [Google Scholar]
- Voronova A., Baltimore D. Mutations that disrupt DNA binding and dimer formation in the E47 helix-loop-helix protein map to distinct domains. Proceedings of the National Academy of Sciences of the United States of America. 1990;87:4722–4726. doi: 10.1073/pnas.87.12.4722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Ling M. T., Guan X. Y., Tsao S. W., Cheung H. W., Lee D. T., Wong Y. C. Identification of a novel function of TWIST, a bHLH protein, in the development of acquired taxol resistance in human cancer cells. Oncogene. 2004;23:474–482. doi: 10.1038/sj.onc.1207128. [DOI] [PubMed] [Google Scholar]
- Yang J., Mani S. A., Donaher J. L., Ramaswamy S., Itzykson R. A., Come C., Weinberg R. A. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004;117:927–939. doi: 10.1016/j.cell.2004.06.006. [DOI] [PubMed] [Google Scholar]
- Yang J., Mani S. A., Weinberg R. A. Exploring a new twist on tumor metastasis. Cancer Research. 2006;66:4549–4552. doi: 10.1158/0008-5472.CAN-05-3850. [DOI] [PubMed] [Google Scholar]
- Zhuo W. L., Wang Y., Zhuo X. L., Zhang Y. S., Chen Z. T. Short interfering RNA directed against TWIST, a novel zinc finger transcription factor, increases A549 cell sensitivity to cisplatin via MAPK/mitochondrial pathway. Biochemical and Biophysical Research Communications. 2008;369:1098–1102. doi: 10.1016/j.bbrc.2008.02.143. [DOI] [PubMed] [Google Scholar]