

Abstract
During mammalian palatogenesis, palatal shelves initially grow vertically from the medial sides of the paired maxillary processes flanking the developing tongue and subsequently elevate and fuse with each other above the tongue to form the intact secondary palate. Pathological palate-mandible or palate-tongue fusions have been reported in humans and other mammals, but the molecular and cellular mechanisms that prevent such aberrant adhesions during normal palate development are unknown. We previously reported that mice deficient in Jag2, which encodes a cell surface ligand for the Notch family receptors, have cleft palate associated with palate-tongue fusions. In this report, we show that Jag2 is expressed throughout the oral epithelium and is required for Notch1 activation during oral epithelial differentiation. We show that Notch1 is normally highly activated in the differentiating oral periderm cells covering the developing tongue and the lateral oral surfaces of the mandibular and maxillary processes during palate development. Oral periderm activation of Notch1 is significantly attenuated during palate development in the Jag2 mutants. Further molecular and ultrastructural analyses indicate that oral epithelial organization and periderm differentiation are disrupted in the Jag2 mutants. Moreover, we show that the Jag2 mutant tongue fused to wildtype palate shelves in recombinant explant cultures. These data indicate that Jag2-Notch1 signaling is spatiotemporally regulated in the oral epithelia during palate development to prevent premature palatal shelf adhesion to other oral tissues and to facilitate normal adhesion between the elevated palatal shelves.
Keywords: epithelial differentiation, maxillary-mandibular fusion, periderm, palate development, palatal fusion, palate-tongue fusion, synechiae, Jag2, Notch1
Introduction
Cleft palate is a common birth defect, occurring in approximately 1 in 2000 live births worldwide (Wilkie and Morris-Kay, 2001; Gorlin, 2001). The frequent occurrence and significant medical and economic burden of such birth defects underscore the importance of understanding the etiology and molecular pathogenic processes underlying cleft palate formation. The primary embryological reason why cleft palate is so common is because the secondary palate initiates as two separate outgrowths from the oral sides of the paired maxillary processes during embryogenesis. In mammals, the bilateral palatal processes initially grow vertically downward flanking the developing tongue. At a precise developmental time, the palatal shelves elevate to the horizontal position above the tongue and subsequently adhere and fuse with each other at the midline to form the intact palate. Disturbance of palatal shelf growth, elevation, adhesion, or fusion results in cleft palate (reviewed in Ferguson, 1988).
During mammalian palate development, the initially vertically oriented palatal shelves are often in direct contact with but do not normally adhere to the mandible or the developing tongue (Shapiro and Sweney, 1969; Mato and Uchiyama, 1975; Ferguson et al., 1984). Pathological palate-mandible and palate-tongue fusions, however, have been reported in humans, mice and rats (Humphrey, 1970; Mato and Uchiyama, 1975; Shah, 1977; Jiang et al., 1998; Din, 2003; Alappat et al., 2005), suggesting that the competence for oral and palatal shelf adhesion must be spatiotemporally regulated. Pourtois (1966) investigated the ability of different pre-fusion stage palatal shelves of rat embryos to fuse in explant cultures in vitro and concluded that a process of cellular differentiation occurs in the palatal shelves to acquire fusion competence during the stage of palatal shelf elevation in vivo. Electron microscopy studies showed that the pre-fusion palatal epithelium is composed of two layers of cells (Shapiro and Sweney, 1969; Fitchett and Hay, 1989). The surface layer consists of flat periderm cells, similar to the rest of the oral epithelia, and the basal layer consists of cuboidal shaped ectoderm cells. Soon after palatal shelf elevation, periderm cells covering the medial edge epithelia (MEE) of the palatal shelves bulge from the surface (Fitchett and Hay, 1989; Martinez-Alvarez et al., 2000), extend filopodia and lamellipodia (Waterman et al., 1973; Meller et al., 1980; Schupbach et al., 1983; Taya et al., 1999), and undergo programmed cell death (Shapiro and Sweney, 1969; Fitchett and Hay, 1989). Periderm cell death was observed specifically in the MEE before palatal shelves contact each other and was suggested to facilitate adherence of the exposed basal epithelial cells during palatal contact (Waterman et al., 1973; Fitchett and Hay, 1989, reviewed in Nawshad et al., 2004). Moreover, a glycoconjugate coat appears specifically over the MEE just prior to palatal shelf contact (Greene and Kochhar, 1974; Souchon, 1975; Meller and Barton, 1978; Zschabitz et al., 1994). Martinez-Alvarez et al. (2000) demonstrated that this MEE-specific surface coat contained chondroitin sulphate proteoglycan (CSPG). Gato et al. (2002) showed that disruption of the CSPG coat on the palatal MEE prevented palatal shelf fusion.
The MEE-specific changes prior to palatal fusion correlate with spatiotemporally regulated expression of Tgfβ3 in the MEE (Fitzpatrick et al., 1990; Pelton et al., 1990; Gato et al., 2002; reviewed in Nawshad et al., 2004). Targeted disruption of Tgfβ3 in mice causes cleft palate (Kaartinen et al., 1995; Proetzel et al., 1995). Palatal shelves in Tgfβ3-/- mutant mouse embryos grow, elevate and approach normally but fail to adhere properly (Proetzel et al., 1995; Martinez-Alvarez et al., 2000). Compared with normal developing palatal shelves, the Tgfβ3-/- mutant palatal shelves had reduced MEE periderm bulging and filopodial activity as well as reduced CSPG coat on the MEE (Taya et al., 1999; Martinez-Alvarez et al., 2000; Gato et al., 2002). Addition of Tgfβ3 to cultures of Tgfβ3-/- mutant mouse palatal shelf explants or chick palatal shelf explants, which normally do not express Tgfβ3 and do not fuse, stimulates secretion of CSPG and causes palatal shelf fusion in vitro (Gato et al., 2002). These data suggest that the oral periderm normally prevents the palatal shelves from adhering to other oral structures and that spatiotemporally specific Tgfβ3 expression in the MEE is required for normal palatal shelf fusion. However, the molecular mechanisms of oral periderm differentiation and the molecular and cellular etiology of pathological palate-tongue and palate-mandible fusion have not been characterized.
We reported previously that mice homozygous for a targeted null mutation in the Jag2 gene had cleft palate and aberrant fusion of the palatal shelves with the developing tongue (Jiang et al., 1998). Jag2 encodes one of five related cell surface ligands for the Notch family receptors in mice (Shawber et al., 1996; Lissemore and Starmer, 1999). Notch signaling has been shown to influence many developmental processes by controlling cell proliferation, cell differentiation, and cell-cell interactions (reviewed in Artavanis-Tsakonas et al., 1999; Kadesh, 2004; Lai, 2004). In this report, we show that Jag2 is normally expressed in the developing oral epithelia and is required for activation of the Notch1 receptor during oral periderm differentiation. We show that Jag2 mutants exhibit disorganized oral epithelia and that the Jag2 mutant tongue fuses to wildtype palatal shelves in explant cultures. Our data indicate that Jag2-Notch1 signaling plays an important role in normal palatogenesis by regulating oral epithelial differentiation.
Results
Jag2 plays a primary role in craniofacial development
We reported previously that a targeted null mutation in the Jag2 gene, Jag2ΔDSL, caused cleft palate and aberrant fusion of the palatal shelves to the lateral sides of the developing tongue in homozygous mutants (Jiang et al., 1998). However, a spontaneous missense mutation in the Jag2 gene, Jag2sm, was shown to cause syndactyly in homozygous mutants but no craniofacial defects were reported (Sidow et al., 1997). This raised the question whether the cleft palate phenotype in Jag2ΔDSL/ΔDSL mutants reflected a true function for Jag2 in craniofacial development. To address this question, we crossed Jag2ΔDSL/+ mice with the Jag2sm/sm mice and found that most of the trans-heterozygous mutants exhibited cleft palate (data not shown). Furthermore, after backcrossing to the C57BL/6J inbred strain, a normally non-cleft strain, for more than three generations, we found that many Jag2sm/sm homozygous mutants exhibited cleft palate associated with adhesion between the palate and tongue (Fig. 1B), although most Jag2sm/sm mutants maintained in its original stock background survive postnatally. We have so far backcrossed the Jag2sm/+ mice to C57BL/6J inbred mice for ten generations. In this congenic background, approximately 60% of Jag2sm/sm homozygous mutants were born with cleft palate (data not shown). In addition, the Jag2sm/sm mutants frequently displayed aberrant adhesions between the maxilla and mandible with or without cleft palate (Fig. 1B, C). Aberrant adhesions between the maxillary and mandibular epithelia were also observed in all Jag2ΔDSL/ΔDSL mutants examined by histological sections (Fig. 2). These data indicate that Jag2 function is indeed required for normal palate development and confirm that the Jag2sm mutation is a hypomorphic allele.
Fig. 1.
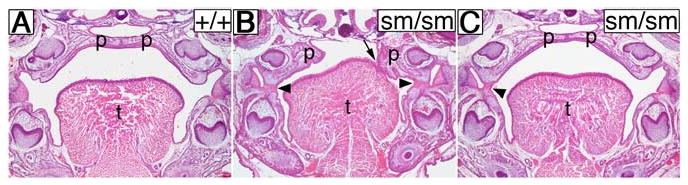
Jag2sm/sm homozygous mutants exhibit cleft palate and aberrant oral epithelial adhesions. Frontal sections of neonatal wildtype (A) and homozygous mutant (B, C) littermates from an intercross of Jag2+/sm heterozygotes. In the wildtype neonate the palatal shelves have fused to form the intact palate (A). In contrast, a Jag2sm/sm homozygous mutant had cleft palate with one palatal shelf adhered to the tongue (B, arrow) and bilateral adhesions between the maxillary and mandibular epithelia (B, arrowheads). Another mutant littermate showed an intact palate with a unilateral maxillary-mandibular adhesion (C, arrowhead). p, palatal shelf; t, tongue.
Fig. 2.
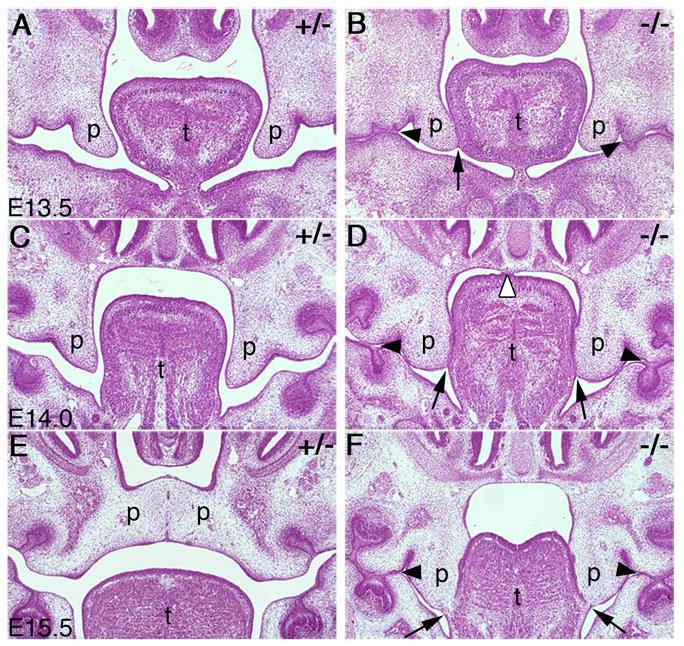
Palate development in the Jag2ΔDSL/ΔDSL mutants. Frontal sections of E13.5 to E15.5 heterozygous (A, C, E) and homozygous (B, D, F) embryos are shown. (A, B) At E13.5, the heterozygous and homozygous mutant embryos had similar sized palatal shelves but palate-tongue adhesion (B, closed arrow) and maxillary-mandibular adhesions (B, arrowheads) are beginning to form in the mutants. (C, D) At E14.0, palate-tongue adhesion in the mutants is often observed bilaterally and is more pronounced (D, arrows). The adhesion between the maxillary and mandibular epithelia is most pronounced in the regions of the mandibular molar tooth buds (D, closed arrowheads). The mutant shown in panel D also had aberrant adhesion between the tongue and the nasal septum (D, open arrowhead). (E, F) At E15.5, the wildtype and heterozygous embryos showed fusion of the palatal shelves and disappearance of the midline epithelial seam (E) whereas the homozygous mutant embryos exhibited bilateral palate-tongue fusion with degeneration of the most ventral aspect of the palate-tongue seam (F, arrows) while the maxillary-mandibular adhesion seam remained intact (F, arrowheads). p, palatal shelf; t, tongue.
To better understand the role of Jag2 in palate development, we carried out detailed histological analyses of wildtype and Jag2ΔDSL/ΔDSL mutant embryos throughout different stages of palatogenesis. No obvious differences in palatal shelf development were observed between mutant and wildtype embryos until E13.5, when the palatal shelves had extended vertically along the lateral sides of the developing tongue. At this stage, we often observed one or both palatal shelves contacting the sides of the tongue in mutant embryos (Fig. 2A, B). This initial contact involved the tip of the palatal shelf and the tongue epithelium immediately adjacent and first occurred at the midpoint of the antero-posterior axis of the palate. Also at this stage, we observed aberrant maxillary-mandibular adhesions forming independently of the palate-tongue adhesion (Fig. 2B). At E14.0, the palate-tongue adhesion was more pronounced and extended along the middle one third of the lateral sides of the tongue (Fig. 2D). The maxillary-mandibular adhesions persisted and occurred in the regions of epithelium overlying the molar tooth buds of the mandible (Fig. 2D) as well as in the lateral regions of the oral cavity (data not shown). In a few cases, we also observed adhesion between the dorsal surface of the tongue and the nasal septum (Fig. 2D). By E15.5, the palatal shelves had elevated to the horizontal position above the tongue and nearly completed fusion with each other in wildtype embryos (Fig. 2E). In contrast, palatal shelves in the mutant littermates remained vertical and attached to the lateral sides of the tongue, with partial loss of the palate-tongue epithelial seam (Fig. 2F). At this stage, the maxillary-mandibular adhesions in the mutants were extensive, involving the epithelium overlying the molar tooth buds (Fig. 2F). These aberrant attachments persist and prevent proper palatal shelf elevation, resulting in cleft palate at birth.
Jag2 is expressed in the oral epithelium and activates the Notch1 receptor during oral periderm differentiation
To better understand the pleiotropic effects of the Jag2 mutations on orofacial development, we examined Jag2 gene expression in the orofacial region by using radioactive in situ hybridization of serial frontal sections of E12.5, E13.5, and E14.5 embryos. At E12.5, Jag2 mRNA was abundantly expressed in the tongue epithelium and the oral epithelium of the mandible whereas weaker expression was observed in the maxillary and palatal epithelia (Fig. 3A). The same pattern of Jag2 mRNA expression in the oral regions was observed at E13.5 (Fig. 3B). By E14.5, the palatal shelves had elevated and initiated fusion. Jag2 mRNA was abundantly expressed throughout the oral epithelium except in the epithelial seam between the fusing palatal shelves (Fig. 3C).
Fig. 3.
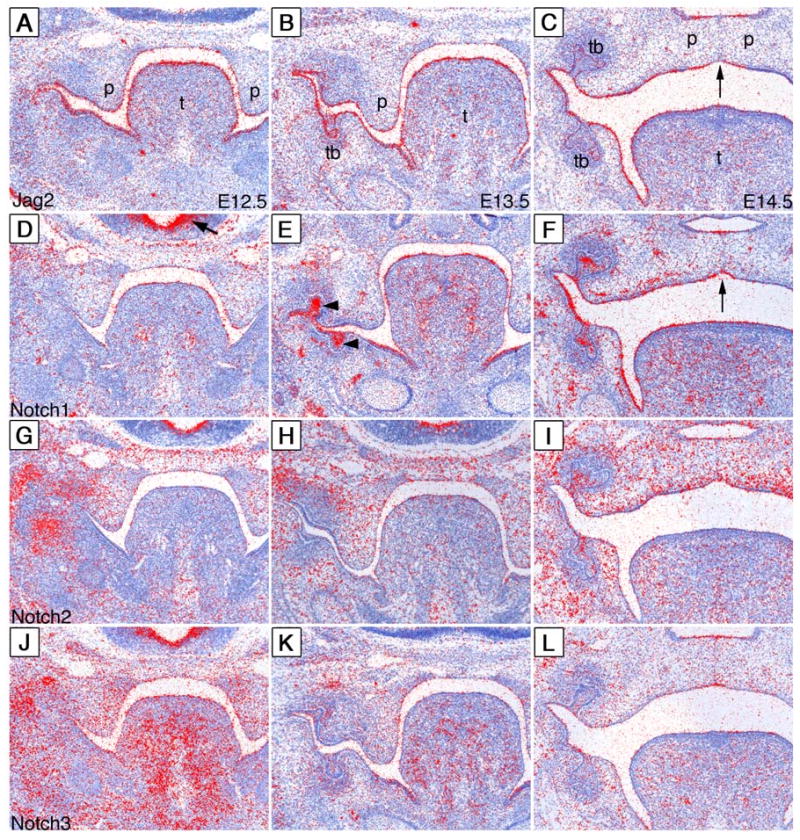
Expression patterns of Jag2, Notch1, Notch2 and Notch3 mRNA during palate development. Radioactive in situ hybridization of E12.5-E14.5 embryos with cRNA probes to Jag2 (A-C) Notch1 (D-F), Notch2 (G-I), and Notch3 (J-L). mRNA signals are show in red color with the samples counterstained blue. (A) At E12.5, Jag2 mRNA is detected throughout the oral epithelium with the highest level of expression in the tongue epithelium. (B) Jag2 mRNA shows a similar distribution in the oral epithelium of E13.5 embryos with the additional expression in the developing tooth bud epithelium. (C) At E14.5, Jag2 mRNA is still detected in the tongue, mandibular and maxillary epithelia. Jag2 mRNA is also present in the oral and nasal epithelia of the palate but is absent from the epithelial seam between the fusing palatal shelves (arrow). (D) At E12.5, low levels of Notch1 mRNA are detected in the tongue and palatal epithelia, whereas strong Notch1 mRNA expression is detected in the ventricular zone of the brain (arrow). (E) At E13.5, Notch1 mRNA is highly expressed in the tongue epithelium, the tooth bud epithelium (arrowheads), mandibular epithelium and the lateral maxillary epithelia. Little Notch1 mRNA expression is detected in the palatal epithelium at this stage. (F) At E14.5, embryos continue to show significant amounts of Notch1 mRNA expression in the tongue and throughout the lateral oral epithelium. The epithelial seam between the fusing palatal shelves shows little Notch1 expression (arrow). (G-I) Notch2 mRNA is expressed throughout the craniofacial mesenchyme from E12.5 to E14.5 with the oral epithelial expression restricted to the tooth buds (I). (J) Notch3 mRNA is expressed abundantly in the craniofacial mesenchyme at E12.5 and is weakly detected in the oral epithelium at this stage (J). (K) By E13.5, Notch3 expression is dramatically reduced in mesenchyme and little expression is observed in the oral epithelium. (L) At E14.5, low levels of Notch3 mRNA are detected in the tooth bud and palatal epithelium. p, palatal shelf; t, tongue; tb, tooth bud.
Jag2 function in the oral epithelium is likely determined by the distribution of its endogenous receptors during oral development. Thus, we examined the expression of all four Notch receptors in the orofacial region from E12.5 to E14.5. At E12.5, Notch1 mRNA was detected at low levels throughout the oral epithelium whereas it was strongly expressed in the ventricular zone of the brain (Fig. 3D). By E13.5, Notch1 mRNA was expressed at increased levels in the tooth bud epithelium, the lateral oral epithelium and the tongue epithelium (Fig. 3E). Notch1 mRNA expression in the palatal and nasal epithelia was still low but detectable (Fig. 3E). By E14.5, Notch1 mRNA was expressed abundantly throughout the oral epithelia except in the epithelial seam between the fusing palatal shelves where there was little Notch1 mRNA expression (Fig. 3F). Notch2 mRNA was expressed in the craniofacial mesenchyme but was not significantly expressed in the oral epithelium during these stages except in the stratified tooth bud epithelium at E14.5 (Fig. 3G-I), consistent with earlier reports (Mustonen et al., 2002). Notch3 mRNA was highly expressed in the craniofacial mesenchyme at E12.5 and weakly expressed in the oral epithelium (Fig. 3J). At E13.5, Notch3 expression in the craniofacial mesenchyme was greatly reduced and there was little to no expression of Notch3 in the oral epithelium (Fig. 3K). At E14.5, low levels of Notch3 mRNA were detected in the oral and nasal epithelia of the fusing palatal shelves (Fig. 3L). We did not detect any Notch4 mRNA expression in the oral epithelium during these stages (data not shown). These data indicate that Notch1 is the predominant receptor for Jag2 in the oral epithelium during these stages of orofacial development.
To determine whether the Notch1 receptor is activated during these stages of oral epithelial differentiation, we performed immunostaining of wildtype embryos from E11.5-E14.5 using an antibody specific for the activated form of Notch1. This antibody, raised against a peptide corresponding to the amino terminal residues of the activated Notch1 intracellular domain (NICD) (Schroeter et. al., 1998), has been used previously to examine Notch signaling in epithelial development (Lin and Kopan, 2003). A number of activated Notch1-positive nuclei were detected in the newly formed periderm layer at the oral surface of the tongue and maxillary epithelia at E11.5 (Fig. 4A, E). By E12.5, increased nuclear accumulation of activated Notch1 protein was observed in the surface layer covering the mandible and tongue whereas the palatal epithelium had lower levels of nuclearly localized Notch1 (Fig. 4B, F-H). Notch1 activation remained strong in the mandibular and tongue epithelia at E13.5 (Fig. 4C, J). Interestingly, nuclear accumulation of activated Notch1 protein appeared in a lateral to medial gradient in the maxillary oral epithelium at this stage, with the lateral regions exhibiting densely populated, highly positive surface and suprabasal cells similar to the mandibular epithelium while the medial palatal epithelium had fewer positive cells with lower levels of nuclear Notch1 staining (Fig. 4C, I, J). This distribution pattern of activated Notch1 protein is similar throughout the antero-posterior axis of the developing palate at this stage as revealed by staining of multiple serial sections (data not shown). Immunostaining of frontal sections through the anterior regions of the elevated pre-fusion palatal shelves at E14.5 showed a similar distribution pattern of activated Notch1 with palatal epithelium containing only a few cells with low levels of nuclear accumulation of activated Notch1 protein (Fig. 4D, K, L). Close examination of the oral epithelium showed that activated Notch1 was largely restricted to the nuclei of periderm and supra-basal layers of the oral epithelium, while a small fraction of cells in the basal layer exhibited moderate levels of activated Notch1 protein in their nuclei (Fig. 4I, J). These data indicate that Notch1 is activated in oral periderm and supra-basal cells from a very early stage in palate development and the adhesion-competent regions of the oral epithelium, the MEE, are also the regions with the lowest Notch1 activation.
Fig. 4.
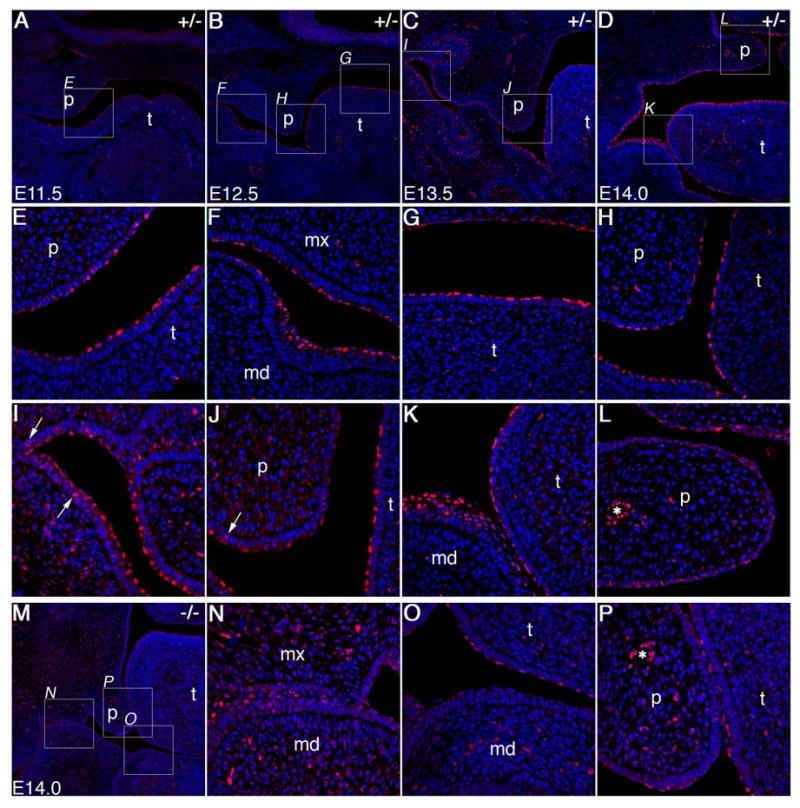
Spatiotemporal patterns of Notch1 activation in the oral epithelium during palate development in Jag2 heterozygous and homozygous mutant embryos. Activated Notch1 staining is shown in red, with DAPI staining of nuclei shown in blue. (A) At E11.5, acitvated-Notch1 is detected specifically in the outer layer cells of oral epithelium covering the tongue primordium in wildtype and heterozygous embryos. A few periderm cells at the oral surface of the maxillary processes are also positively stained. The boxed region is shown in higher magnification in panel E. (B) At E12.5, activated-Notch1 staining is observed throughout the periderm layer of the tongue and mandibular oral epithelia. Significantly lower but detectable levels of activated Notch1 protein are also observed in the outer layer of the maxillary and palatal epithelia. Boxed areas are shown at higher magnification in panels F-H. (C) At E13.5, embryos show abundant activated-Notch1 protein in the nuclei of the periderm and suprabasal cells of lateral oral epithelium, tongue epithelium and lateral maxillary epithelium. Activated-Notch1 is detected in one to two cell layers of the lateral oral and tooth bud epithelium but only in a single cell layer of the tongue, the periderm layer. Boxed regions are shown at higher magnification in panels I and J. Cells in the basal cell layer are occasionally positive for activated-Notch1 (arrows in I and J). In contrast to the rest of the oral epithelia, very few periderm cells in the medial and distal epithelium of the palatal shelves are positive for activated Notch1 protein (J). (D) At E14.0, the pattern of activated-Notch1 staining is largely the same with strong staining in the periderm and suprabasal cell layers of the lateral oral epithelium and in the periderm cells of the tongue. Higher magnification micrographs of boxed regions are shown in panels K and L. The elevated pre-fusion palatal shelves had little activated Notch1 staining in the surface epithelium compared with the vascular endothelial cells (L, asterisk) or other regions of the oral epithelium which are highly positive (K). (M) Low magnification view of a frontal section through the anterior oral cavity of an E14.0 Jag2ΔDSL/ΔDSL mutant embryo showing significantly reduced levels of activated-Notch1 throughout the oral epithelium. Higher magnification views of the regions boxed in panel M reveals that cells at the surface of lateral oral epithelia (N), of the tongue (O, P), and of mandible (N, P) exhibited either low levels or no staining for activated Notch1 in the nuclei, although endothelial cells lining blood vessels in the mutant palate had strong staining for activated Notch1 comparable to that in the heterozygous palate (asterisks in L and P). md, mandible; mx, maxilla; p, palatal shelf; t, tongue.
We next examined whether the pattern of Notch1 activation during orofacial development was altered in the Jag2ΔDSL/ΔDSL mutants and found that nuclear accumulation of activated Notch1 was absent or dramatically reduced throughout the oral epithelia in the Jag2ΔDSL/ΔDSL mutants compared with wildtype controls (Fig. 4M-P). In the tongue and lateral oral epithelia of the mutant embryos, there were significantly fewer nuclei stained positive for activated Notch1 and most of the positively stained nuclei showed reduced staining intensity compared with the wildtype embryos (Fig. 4M-P, compare with D and I-L). In contrast, other cell types, such as the vascular endothelium, had similar levels of nuclearly localized activated Notch1 staining in mutant and control embryos (Fig. 4L, P).
To investigate further whether Jag2-Notch1 signaling is involved in the early stage of oral periderm differentiation, we compared the patterns of Notch1 activation in the oral epithelia of wildtype and of Jag2ΔDSL/ΔDSL mutant embryos at E12.0. As shown in Fig. 5, Notch1 activation is dramatically reduced in the oral epithelia of the mutant embryos compared with that of the wildtype embryos (Fig. 5A, C, E, G). DAPI counterstaining highlighted the simple bi-layered nature of the wildtype tongue epithelium at this stage with the periderm cells at the surface of the epithelium displaying highly flattened nuclei (Fig. 5B, F). The mutant tongue epithelium was also bi-layered at this stage but the cells at the surface lacked the flattened nuclear morphology characteristic of normal periderm cells (Fig. 5D, H). These data indicate that Jag2 is the major endogenous ligand for Notch1 activation in the oral epithelium and strongly suggest that Jag2-Notch1 signaling is required for proper oral periderm differentiation.
Fig. 5.
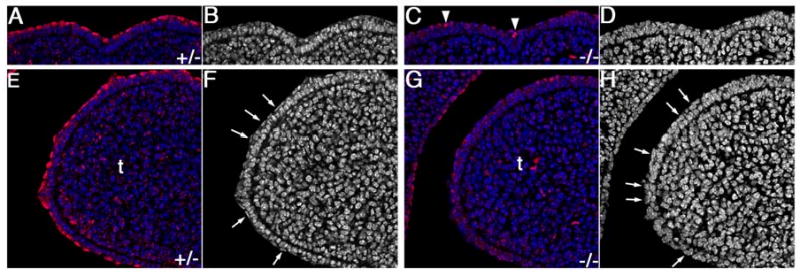
Jag2 is required for Notch1 activation and proper periderm differentiation in the oral epithelium. Confocal micrographs of dorsal (A-D) and lateral (E-H) regions of the developing tongue in heterozygous (A, B, E, F) and homozygous mutant (C, D, G, H) embryos at E12. Even at this early stage of oral epithelial differentiation, Jag2ΔDSL/ΔDSL mutant embryos show dramatically reduced activated Notch1 staining in the tongue epithelium (C, G), compared with that in heterozygous littermates (A, E). Very few cells in the homozygous mutant tongue epithelium exhibited high levels of activated Notch1 staining (arrowheads in C) whereas almost all surface layer cells in the wildtype and heterozygous tongue showed high levels of nuclear staining for activated Notch1 (A, E). (B, D) Black-white images of the same fields shown in A and E, respectively, with nuclear DAPI staining shown in white. The wildtype and heterozygous mutant tongue epithelium at this stage consists of two layers with the surface layer being the flat periderm cells. (D, H) Black-white images of the same fields shown in C and G, respectively, with nuclear DAPI staining shown in white. The homozygous mutant tongue epithelium at this stage also consists of two layers but the nuclei of the surface layer cells are irregular and do not resemble the flat nuclei characteristic of the wildtype periderm cells (arrows in F and H). t, tongue.
Disruption of oral epithelial organization in the Jag2 mutants
Notch signaling has been shown to regulate different cellular processes, such as cell differentiation and cell proliferation, in other developmental contexts (reviewed in Artavanis-Tsakonas et al., 1999; Kadesh, 2004; Lai, 2004). To investigate further the role of Jag2-Notch1 signaling in oral epithelial development, we carefully compared the mutant and wildtype oral epithelial structures by thin sections and transmission electron microscopy. Semi-thin sections of dissected E13.0 tongues showed that wildtype tongue epithelium had a regular thickness whereas the mutant tongue epithelium exhibited patchy stratification in the dorsal and lateral regions (Fig. 6A, B). Examination by transmission electron microscopy showed, whereas the wildtype tongue epithelium consisted of a single layer of cuboidal cells (under- and lateral sides of tongue) or columnar-shaped cells (top of tongue) overlaid by a thin layer of flat squamous periderm cells (Fig. 6C, and data not shown), that the mutant tongue epithelium had patches of disorganized, regionally thickened or thinned epithelium (Fig. 6D, and data not shown). The stratifications were as many as 4 to 5-cell layers thick compared to 2-cell layers in the wildtype tongue epithelium. Moreover, surface cells overlying the thickened areas in the mutant tongue epithelium appeared to lose contact with each other and lacked the long cellular processes and highly flattened nuclei characteristic of normal periderm cells (Fig. 6D). We also examined the oral epithelium of E13.5 Jag2sm/sm homozygous mutant embryos by transmission electron microscopy and found similar changes in the morphology of the tongue epithelium (data not shown). In contrast, comparison of mutant and control palatal epithelium did not reveal any differences (Fig. 6E, F).
Fig. 6.
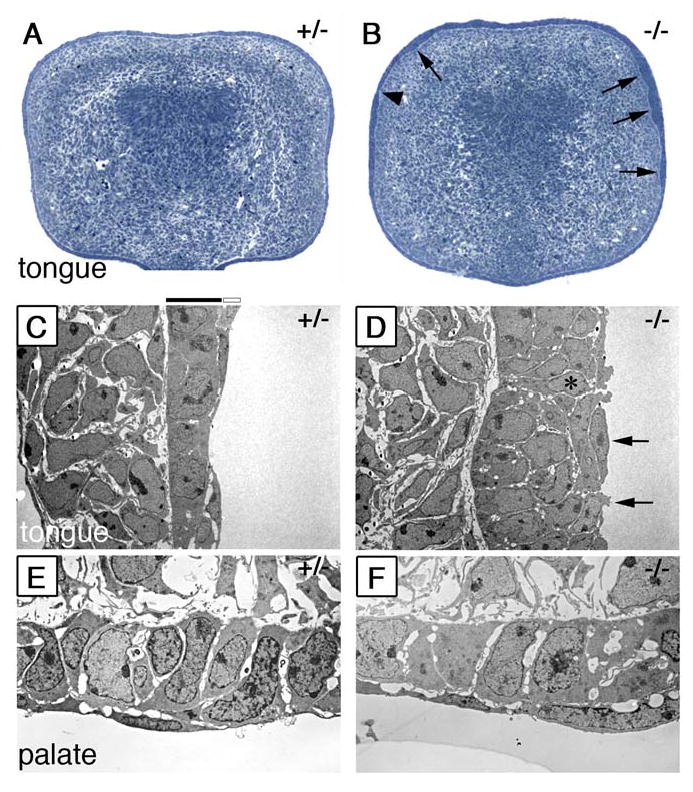
Defects in epithelial organization in the Jag2ΔDSL/ΔDSL mutants. (A, B) Semi-thin (2μm) sections of E13.0 wildtype (A) and Jag2ΔDSL/ΔDSL mutant (B) tongue stained with toluidine blue. The wildtype tongue epithelium shows an even thickness over the entire circumference of the tongue. In contrast the mutant tongue shows uneven thickness with patches of thick (B, arrows) and thin epithelia (B, arrowhead). (C) Electron micrographs of the lateral tongue epithelium show that the wildtype tongue epithelium has a basal layer of cuboidal shaped cells covered by a continuous sheet of flattened periderm cells. (D) Jag2ΔDSL/ΔDSL mutant tongue epithelium is multiple cell layers thick in some regions and appears disorganized. The periderm layer of the mutant tongue appears discontinuous as some of these cells appear less flattened and lack long cellular processes (D, arrows). In some cases cells attached to the basal lamina are exposed to the surface in the mutant tongue epithelium (D, asterisk). (E, F) Electron micrographs of the epithelia at the tip of the palatal shelves showing no significant differences between the wildtype (E) and Jag2ΔDSL/ΔDSL mutant (F) palatal epithelia.
To investigate further the disruption in oral epithelial organization in the Jag2 mutants, we analyzed expression of molecular markers of epithelial differentiation in the oral epithelia of wildtype and mutant embryos. E-cadherin is a cell adhesion molecule and a general marker for epithelial cells. Although no dramatic difference was observed in the overall level of E-cadherin protein expression between wildtype and Jag2-mutant oral epithelia (Fig. 7A, B), careful examination showed that there was disorganization in the mutant tongue epithelium with some basal cells exposed to the surface and regions of multi-layered cells with very little cell surface E-cadherin between them (Fig. 7C, D). We next analyzed the expression of keratin-17, a marker for periderm cells (McGowan and Coulombe, 1998). As shown in Fig. 7E, keratin-17 was present at low levels in the oral periderm layer covering the maxillary processes, palatal shelves and future nasal cavity but not in the tongue epithelium of wildtype embryos at E13.5. Interestingly, intense keratin-17 staining was observed throughout the oral surface cell layer, especially in the tongue epithelium of Jag2ΔDSL/ΔDSL mutant littermates (Fig. 7F). Similarly, increased keratin-17 staining was observed in the oral epithelium of Jag2sm/sm mutants at this stage (data not shown). In contrast, immunostaining with a pan-cytokeratin monoclonal antibody mixture that recognizes nine other epithelial keratins (see Materials and Methods) showed similar levels of expression of other keratins in the oral epithelia in wildtype and Jag2ΔDSL/ΔDSL mutant littermates (Fig. 7G, H). These data indicate that the epithelial cells at the oral surface of the Jag2 mutants were programmed to express at least some of the periderm differentiation markers although they lack the characteristic flattened morphology of the normal periderm cells.
Fig. 7.
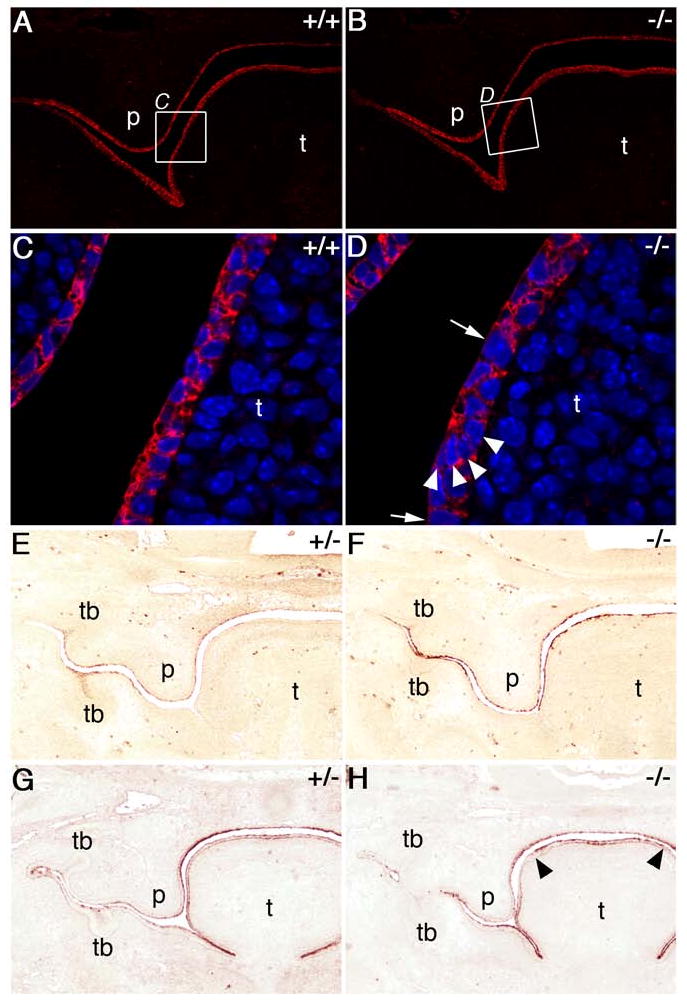
Analysis of epithelial differentiation markers in Jag2ΔDSL/ΔDSL homozygotes. Wildtype (A, C, E, G) and mutant (B, D, F, H) embryo sections immunostained for E-cadherin (A-D) or keratin-17 (E-H). A, B) At E13.0 E-cadherin is abundant throughout the oral epithelium of both wildtype and Jag2ΔDSL/ΔDSL-mutant embryos. C, D) Higher magnification views of the regions boxed in panels A and B showing that E-cadherin is detected in all cells of the oral epithelium but some regions of the mutant epithelium are one cell layer thick (D, arrows) while others are multiple cell layers thick with reduced E-cadherin staining between cells (D, arrowheads). E-H) E13.5 embryos fixed with bouin's fixative and immunostained for keratin-17 (E, F) or pan-cytokeratin (G, H). Wildtype embryos (E) show some keratin-17 expression in the stellate reticulum of the tooth buds, the periderm flanking the mandibular and maxillary tooth buds, and the periderm layer of the palate and nasal epithelium. At this stage the wildtype tongue epithelium is only weakly stained. In comparison, homozygous mutant littermates (F) show much stronger staining for keratin-17 throughout the oral epithelium, especially the tongue. Additionally, the mutant tongue epithelium appears irregular in thickness compared to wildtype tongue epithelium. Pan-cytokeratin staining of wildtype (G) and mutant (H) embryos shows that general keratinization is unaltered throughout much of the mutant oral epithelium except for the periderm cell layer on the dorsal side of the tongue which shows stronger staining compared to wildtype embryos (H, arrowheads). p, palate; t, tongue; tb, tooth bud.
Jag2 function is required in the tongue epithelium to prevent palate-tongue adhesion
The fact that the level of activated Notch1 protein is normally significantly lower in the palatal epithelium than in the rest of oral epithelia, together with the tongue epithelial defects and aberrant palate-tongue fusion in the Jag2ΔDSL/ΔDSL mutants, suggests that epithelial differentiation in the tongue and mandible regulated by Jag2-Notch1 signaling normally prevents inappropriate palatal adhesion in the oral cavity. To test this hypothesis, we used recombinant explant culture assays to investigate whether loss of Jag2 function in the tongue alone would be sufficient to cause it to adhere to the wildtype palatal shelves. After three days of explant culture, paired wildtype and mutant palatal shelves, respectively, from E13.5 embryos completed fusion to each other (Fig. 8A, B). Under the same conditions, paired wildtype and mutant palatal shelves also completely fused with each other (data not shown). We then modified the palate culture technique by placing wildtype or mutant tongue adjacent to wildtype or mutant palatal shelf explants. After five days in culture, the epithelial layers remained intact between the wildtype palate and wildtype tongue explants whereas the epithelial seam between the mutant palate-tongue pair appeared irregular and disrupted (Fig. 8C, D). Recombination of mutant palate with wildtype tongue did not result in disruption of the epithelial seam, whereas pairing wildtype palate with the mutant tongue resulted in an irregular and disrupted epithelial seam, similar to the mutant palate-mutant tongue pair (Fig. 8E, F). Thus, loss of Jag2 in the tongue alone appears sufficient to cause improper epithelial adhesion between the palate and tongue, indicating that Jag2-Notch signaling plays an important role in normal palate development by preventing palatal adhesion to other oral structures.
Fig. 8.
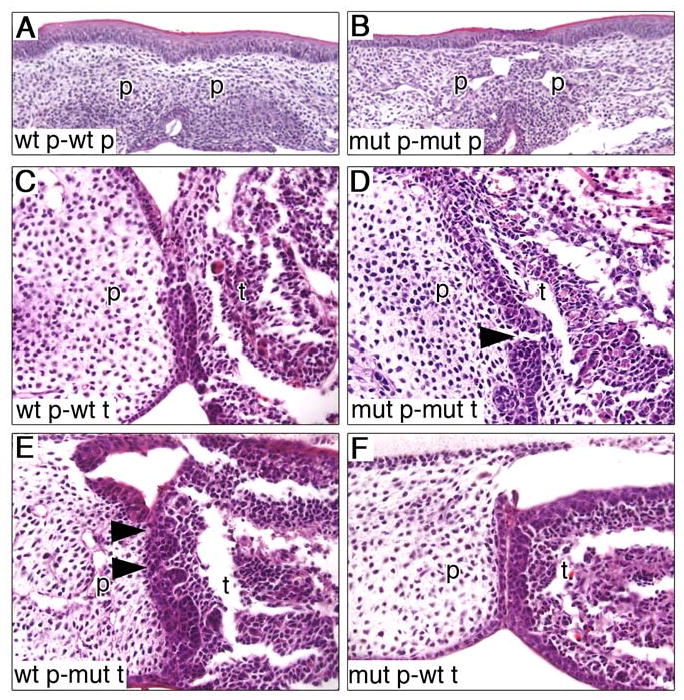
Palate-tongue recombination organ culture assays. (A, B) Pairings of wildtype (A) and Jag2ΔDSL/ΔDSL homozygous mutant (B) palatal shelves both showed complete fusion and disappearance of the epithelial seam after 3 days in culture. (C-F) In different pairings of palate and tongue tissues all pairings show association of palate and tongue epithelia but only the mutant palate-mutant tongue (D) and wildtype palate-mutant tongue (E) pairings showed an irregular and discontinuous epithelial seam (arrowheads in D and E). mut, mutant; p, palatal shelf; t, tongue; wt, wildtype.
Analysis of the expression patterns of Fgf10, Fgfr2b and Tgfβ3 in the Jag2 mutants
The palate-tongue fusion phenotype of Jag2 mutants is very similar to that reported in the Fgf10-/- mutant mice and Jag2 mRNA expression was shown to be downregulated in the oral epithelia of Fgf10-/- mutant embryos (Alappat et al., 2005). We therefore examined the expression of Fgf10 and its epithelial receptor Fgfr2b in wildtype and Jag2ΔDSL/ΔDSL mutant embryos at E13.5 and E14.5 (Fig. 9). Fgf10 mRNA is expressed similarly in the palatal and tongue mesenchyme at these stages in both wildtype and Jag2ΔDSL/ΔDSL mutant embryos (Fig. 9A, B). Fgfr2b mRNA is expressed throughout the oral epithelium and in the medial region of the palatal mesenchyme in wildtype embryos (Rice et al., 2004; Fig. 9C, E). Whereas the palatal mesenchyme expression of Fgfr2b is unaltered in the Jag2ΔDSL/ΔDSL mutants, Fgfr2b mRNA expression in the oral epithelium shows a slight but consistent decrease in E13.5 and E14.5 embryos, particularly in the tongue epithelium (Fig. 9D, F). Thus, whereas Jag2 is a possible downstream target of Fgf10-Fgfr2b signaling (Alappat et al., 2005), Jag2-Notch signaling may feedback to maintain Fgfr2b expression in the oral epithelium.
Fig. 9.
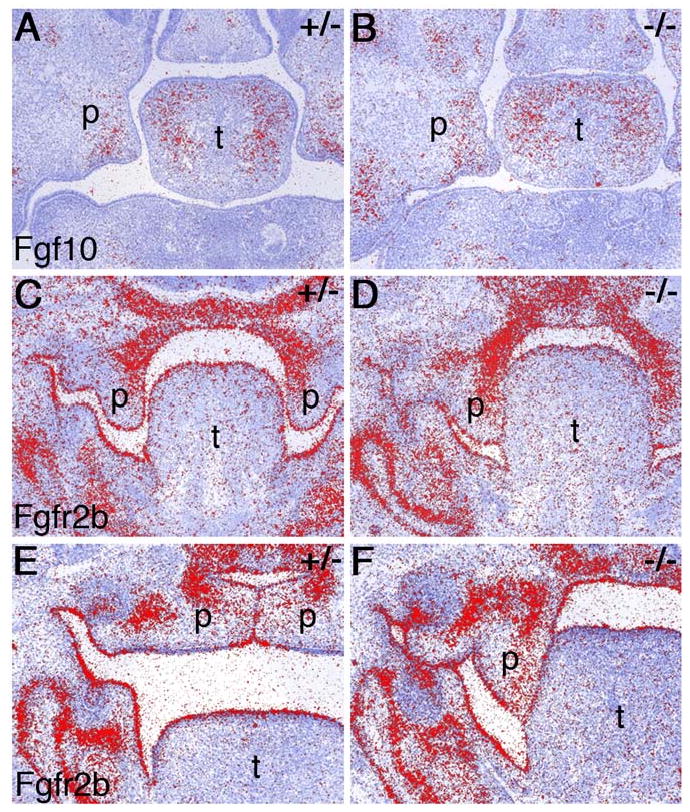
Expression analysis of Fgf10 and Fgfr2b in Jag2ΔDSL/ΔDSL mutant embryos. mRNA signals are shown in red on sections counterstained blue. (A, B) Jag2ΔDSL/+ heterozygous and Jag2ΔDSL/ΔDSL homozygous mutant littermates show comparable levels of Fgf10 mRNA expression in palatal and tongue mesenchyme at E13.5. (C-F) Fgfr2b mRNA is highly expressed throughout the oral epithelium of wildtype and Jag2ΔDSL/+ heterozygous embryos at E13.5 (C) and E14.5 (E) whereas its levels appear consistently reduced in the dorsal tongue epithelium of Jag2ΔDSL/ΔDSL homozygous mutants at these developmental stages (D, F). p, palatal shelf; t, tongue.
During palate development, Tgfβ3 expression in the oral epithelium is normally restricted to the MEE cells of the palatal shelves during palate development and has been shown to be critical for normal palatal fusion (Kaartinen et al., 1995; Proetzel et al., 1995). In the Fgf10-/- mutant mouse embryos, which had aberrant palate-tongue and palate-mandible fusions, it was shown that Tgfβ3 mRNA was ectopically expressed in the lateral palatal epithelium (Alappat et al., 2005). We examined Tgfβ3 mRNA expression in Jag2ΔDSL/ΔDSL mutant embryos to determine if it is ectopically expressed at the sites of oral adhesions. We found that Tgfβ3 mRNA expression remained localized in the MEE of palatal shelves in the Jag2ΔDSL/ΔDSL mutant embryos and did not expand to the lateral oral epithelium where aberrant adhesions occur (Fig. 10A, B). We also examined the expression pattern of Mmp13, a known downstream target of Tgfβ3 signaling (Blavier et al., 2001), in wildtype and Jag2ΔDSL/ΔDSL mutant embryos. Mmp13 mRNA expression is restricted to the palatal MEE in the oral epithelium of wildtype embryos at E14.5 (Fig. 10C). In Jag2ΔDSL/ΔDSL mutant littermates, Mmp13 mRNA is still restricted to the MEE cells at the tip of the palatal shelves (Fig. 10D). Chondroitin sulfate proteoglycan (CSPG) has been shown to be a critical factor for Tgfβ3-induced palatal fusion (Gato et al., 2002). We examined CSPG distribution in the oral epithelia by immunostaining but did not detect any differences between wildtype and Jag2ΔDSL/ΔDSL mutant embryos (data not shown). Thus, both Tgfβ3 expression and response to Tgfβ3 signaling are unaltered in the oral epithelium in Jag2ΔDSL/ΔDSL mutants, indicating that the aberrant oral adhesions in the Jag2 mutants result from a primary defect in the differentiation and organization of the oral epithelium rather than alteration in Tgfβ3 signaling.
Fig. 10.

Analysis of Tgfβ3 signaling in Jag2ΔDSL/ΔDSL mutant embryos. (A, B) At E14.5, Tgfβ3 mRNA expression in the oral epithelia is restricted to the MEE of the palatal shelves in both Jag2ΔDSL/+ heterozygous and Jag2ΔDSL/ΔDSL homozygous mutant embryos, although the palatal shelves adhered to the lateral sides of the tongue (arrow in B) in the homozygous mutant. No Tgfβ3 mRNA expression was detected at the site of maxillary-mandibular adhesion (B, arrowhead) in the homozygous mutant. (C, D) Mmp13 mRNA expression overlaps with that of Tgfβ3 mRNA in the MEE of the palatal shelves in both Jag2ΔDSL/+ heterozygous and Jag2ΔDSL/ΔDSL homozygous mutant palatal shelves (D, arrow). Mmp13 is not detected at the site of maxillary-mandibular adhesion in the Jag2ΔDSL/ΔDSL mutant embryo (D, arrwohead). p, palatal shelf; t, tongue.
Analysis of Jag2 mutants provides support for the role of Tgfβ3 signaling in programmed cell death during palatal epithelial seam breakdown
There is strong evidence that disruption of the palatal epithelial seam during palatal fusion results at least in part from apoptosis (Cecconi et al., 1998; Cuervo and Covarrubias, 2003; Martinez-Alvarez et al., 2004; Vaziri Sani et al., 2005). We carried out TUNEL analysis to investigate the mechanism involved in the disruption of the epithelial seam between the palate and tongue in the Jag2ΔDSL/ΔDSL mutants and found that the palate-tongue seam contained numerous apoptotic cells, similar to the wildtype palate-palate seam during palatal fusion (Fig. 11A, B). In contrast, the epithelial adhesions in the lateral regions of the oral cavity in the Jag2 mutants did not contain apoptotic cells (Fig. 11C). Thus, apoptosis in the wildtype and mutant epithelial seams correlates with Tgfβ3 expression and activation of downstream target genes such as Mmp13 (Fig. 10). Taken together, these data indicate that Jag2-Notch signaling regulates oral periderm differentiation to prevent palatal shelves from adhering to other oral tissues and Tgfβ3 signaling induces palatal fusion through programmed cell death during normal palate development.
Fig. 11.
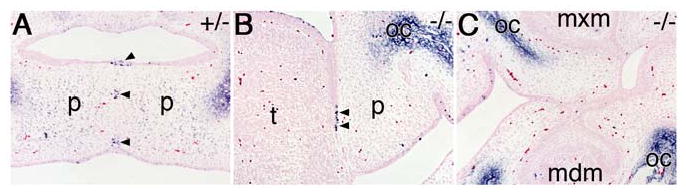
Cell death is detected in the palate-tongue epithelial seam but not in the maxillary-mandibular epithelial seam in the Jag2ΔDSL/ΔDSL mutant embryos. (A) In the Jag2ΔDSL/+ heterozygous embryo, a number of apoptotic cells are detected in the epithelial seam between the fusing palatal shelves (arrowheads). (B, C) Jag2ΔDSL/ΔDSL mutant embryos show numerous apoptotic cells in the palate-tongue epithelial seam (B, arrowheads) but not the maxillary-mandibular seam (C). Regions of ossification are stained non-specifically. mdm, mandibular molar; mxm, maxillary molar; oc, ossification center; p, palatal shelf; t, tongue.
Discussion
Requirement for Jag2-Notch1 signaling in normal palatogenesis
The original description of the Jag2ΔDSL/ΔDSL mutant phenotype included the observation that these mice had cleft palate and aberrant fusion between the palatal shelves and tongue (Jiang et al., 1998). In this report, we have confirmed that this phenotype reflects a requirement for Jag2-Notch signaling in orofacial development by demonstrating that mice homozygous for a spontaneous single missense mutation in the Jag2 gene, Jag2sm, also exhibit cleft palate and palate-tongue fusion. Furthermore, we showed that other regions of the oral epithelium also adhere improperly in both Jag2ΔDSL/ΔDSL and Jag2sm/sm mutants. These data, together with the report that Fgf10-/- mutant mice, which have aberrant palate-tongue and palate-mandible fusions, exhibited downregulation of Jag2 expression in the oral epithelia during palate development (Alappat et al., 2005), indicate that Jag2-mediated Notch signaling plays a unique role in normal palate development.
What is the primary function of Jag2 in palate development? We showed that Jag2 is expressed throughout the oral epithelium and is required for Notch1 activation during oral periderm differentiation. Relatively little is known about the development and function of the periderm. Existing studies have largely focused on the periderm of the epidermis. During mouse embryogenesis, periderm cells first appear at late E9 on the forelimb buds and over the next 3-4 days periderm cells emerge from the single-layered epithelium at many locations to cover the entire embryo (Holbrook and Odland, 1975; M'Boneko and Merker, 1988). This cell layer is transient and undergoes apoptosis late in gestation (Holbrook and Odland, 1975; Polakowska et al., 1994). Several functions have been proposed for this cell layer including protection from the environment (Hayward, 1983), regulation of underlying mesenchyme (Scott et al., 1987), and contribution to cornified envelope formation (Akiyama et al., 1999), but all of these functions have yet to be proven.
We found that Notch1 is selectively activated in oral periderm cells as well as in a few cells in the basal layer of the oral epithelia that may be in the process of differentiating into periderm cells. The best understood function of the Notch signaling pathway is to mediate cell differentiation via a lateral inhibition mechanism in which the cells receiving higher Notch activation remain undifferentiated and the signaling cells follow a differentiation program (reviewed in Artavanis-Tsakonas et al., 1999). The mammalian Jag2-Notch1 ligand-receptor pair has been implicated in mediating lateral inhibition during sensory hair cell differentiation in the developing inner ear (Lanford et al, 1999). Although lateral inhibition is a plausible mechanism for the specification of oral periderm fate in the initially single-layered oral ectoderm, our data that Notch1 is selectively activated in the differentiating periderm cells are inconsistent with Jag2-Notch1 signaling in mediating lateral inhibition during periderm formation. On the contrary, Jag2-Notch1 signaling appears unnecessary for the initial stratification of the oral epithelium since the bi-layered oral epithelium is formed in the Jag2 mutants. Recently, Lechler and Fuchs (2005) showed that the stratification of the mouse embryonic skin from a single-layered epidermis arises through asymmetric cell divisions with the epithelial cells dividing perpendicular to the basement membrane. It is likely that the stratification of the oral epithelium is also through such perpendicular cell divisions. If this is the mechanism for oral periderm differentiation, our data suggest that Jag2-Notch1 signaling is not required for the proper orientation of the mitotic spindles. However, it is possible that Jag2-Notch1 signaling is involved in the asymmetric distribution of cellular contents during such cell divisions because the newly formed suprabasal cells at oral surface of the Jag2 mutants have round instead of flattened nuclear morphology (Fig. 5). Alternatively, Jag2-Notch1 signaling may act to control oral periderm differentiation immediately following oral epithelial stratification. It is interesting that expression of keratin-17, a marker of periderm differentiation (McGowan and Coulombe, 1998), is upregulated in the oral epithelium of the Jag2 mutants. It is possible that increased keratin-17 expression is secondary to aberrant differentiation of the oral periderm of the Jag2 mutants. It is also possible that Jag2-Notch signaling may directly regulate the levels of keratin-17 expression during oral periderm differentiation. Further characterization of the mutant phenotype with more periderm-specific markers, when they become available, will provide a clearer understanding of the role of Jag2-Notch1 signaling in oral epithelial differentiation.
Molecular and cellular mechanisms underlying the aberrant oral and palatal adhesion in the Jag2 mutants
Pathological palate-tongue and palate-mandible fusions have been reported in humans and other mammals (Humphrey, 1970; Mato and Uchiyama, 1975; Shah, 1977; Jiang et al., 1998; Din, 2003; Alappat et al., 2005). The causes of these aberrant oral adhesions in humans are unknown. Certain drugs, including meclozine hydrochloride and chlorcyclizine have been shown to induce palate-tongue fusions in rat embryos (King, 1963; Koziol and Steffek, 1969). Mato and Uchiyama (1975) investigated the cellular basis of palate-tongue fusion in rats induced by meclozine hydrochloride by histological and ultrastructural analyses and suggested that the cause of palate-tongue fusion was due to aberrant periderm cell death in the tongue epithelium. In mice, targeted disruption of the Fgf10 gene resulted in cleft palate associated with palate-tongue and palate-mandible fusions (Alappat et al., 2005). Rice et al. (2004) detected aberrant apoptotic cells in the epithelia of the palate, tongue and the floor of the mouth in Fgf10-/- and Fgfr2b-/- mutant embryos and suggested that Fgf10-Fgfr2b signaling acts as an epithelial survival factor. Interestingly, Fgf10-/- mutants also exhibited down-regulation of Jag2 mRNA expression in the oral epithelium before palatal shelf elevation (Alappat et al., 2005). Thus, the aberrant palate-tongue and palate-mandible fusions in the Fgf10-/- mutants are likely caused by a combination of disruption of Jag2-Notch signaling and aberrant oral epithelial death. The Jag2 mutants exhibited a slight reduction in Fgfr2b expression in the oral epithelium, but we did not detect any significant oral epithelial apoptosis except in the palate-tongue fusion sites. It is possible that the reduced Fgfr2b mRNA expression is an indirect result of disrupted tongue epithelial development in the Jag2 mutants. Alternatively, Jag2-Notch signaling may be required to maintain Fgfr2b expression in the tongue epithelium.
During mouse palate development, Tgfβ3 mRNA is strongly expressed in the palatal MEE cells before palatal shelf elevation and this expression persists until palatal fusion in completed (Fitzpatrick et al., 1990; Pelton et al., 1990). In Tgfβ3-/- null mutant mouse embryos, palatal shelves grow and elevate normally but fail to adhere properly, resulting in cleft palate (Kaartinen et al., 1995; Proetzel et al., 1995). Further, characterization of Tgfβ3-/- null mutants showed a dramatic decrease in palatal MEE cell activity, including bulging, filopodial processes, and CSPG surface coat (Taya et al., 1999; Martinez-Alvarez et al., 2000; Gato et al., 2002). Alappat et al. (2005) showed that in Fgf10-/- mutant embryos Tgfβ3 mRNA expression was expanded in the lateral epithelium of the vertically oriented palatal shelves and suggested that this expanded Tgfβ3 expression rendered the mutant palatal shelves competent to fuse with the mandible. However, it is known that during normal palate development the MEE of the palatal shelves, which express Tgfβ3, are often in direct contact with but do not adhere to the tongue and mandible. Thus, it is unlikely that Tgfβ3 expression alone is sufficient to induce epithelial adhesion. Our data suggest that Tgfβ3 signaling must be coordinated with Jag2-Notch1 signaling, which can inhibit adhesion, to promote palate closure.
Jag2 mutants provide a unique model for understanding the role of Tgfβ3 in palate fusion. The process of palatal fusion has been studied extensively but there is still disagreement over the relative contributions of epithelial-mesenchymal transformation versus apoptosis to the disappearance of the epithelial seam [Fitchett and Hay, 1989; Cuervo and Covarrubias, 2004; reviewed in Nawshad et al., 2004]. Those favoring epithelial-mesenchymal transformation as the major mechanism propose that apoptosis is restricted to the periderm layer and suggest that this is important for the initial adhesion of the palatal shelf epithelia. Tgfβ3 has been proposed to induce epithelial-mesenchymal transformation of the palatal MEE (Kaartinen et al., 1997; Sun et al., 1998; reviewed in Nawshad et al., 2004). However, recent fate mapping of the mouse palatal MEE cells using the Cre/loxP-mediate in vivo lineage tracing method has ruled out MEE contribution to the palatal mesenchyme (Vaziri Sani et al., 2005). On the other hand, the fusing palatal epithelial seam cells have been shown to undergo apoptosis both in vivo and in vitro (Cuervo and Covarrubias, 2004; Vaziri Sani et al., 2005). Tgfβ3 has been shown to promote apoptosis in other developmental processes (Nguyen and Pollard, 2000; Opperman et al., 2000; Dunker et al., 2002). It is possible that Tgfβ3 induces palatal epithelial seam breakdown through apoptosis. In the Jag2ΔDSL/ΔDSL homozygous mutants, a significant amount of apoptosis was detected in the palate-tongue epithelial seam but not in the maxillary-mandibular epithelial seam. Since the palate-tongue seam is the only epithelial seam where Tgfβ3 expression is detected in Jag2ΔDSL/ΔDSL mutants, this suggests that Tgfβ3 expression in the palatal MEE contributes to the epithelial seam breakdown. Similarly, analyses of Fgf10-/- mutant mice showed that apoptosis in the aberrant palate-mandibular fusion seam correlates with Tgfβ3 expression (Alappat et al., 2005). Together, these data indicate that spatiotemporally regulated Tgfβ3 expression in the palatal MEE induces palatal fusion through programmed cell death of the epithelial seam cells.
Materials and Methods
Animals
Mice carrying the targeted Jag2ΔDSL mutant allele were maintained by breeding heterozygous males to C57BL/6J females and heterozygous offspring identified by PCR genotyping as described previously (Jiang et al., 1998). Heterozygous mice were intercrossed to generate homozygous Jag2ΔDSL/ΔDSL mutant embryos.
Mice carrying the spontaneous Jag2sm mutant allele (Sidow et al., 1997) were obtained from the Jackson laboratory Mutant Mouse Resources and bred to C57BL/6J inbred mice to establish a colony. Heterozygous mice were identified by PCR genotyping with primers SM1 (5′-GCT GAC CAT CTC GGC CAG TG-3′) and SM2 (5′-GAC GCA CGT ACC GTC GAC AC-3′), which amplify a 263 bp product from both wildtype and mutant alleles. The Jag2sm allele is distinguished from the wildtype counterpart by PvuII digestion, which specifically cleaves the mutant fragment into two fragments of 59 bp and 204 bp, respectively. Heterozygous mutants were backcrossed to C57BL/6J inbred mice for colony maintenance and intercrossed to generate homozygous mutant embryos. Embryos were genotyped by PCR using yolk sac or tail DNA.
Histology and in situ hybridization
For histology, embryos were dissected from timed pregnant heterozygous female mice and fixed for 1-2 days at room temperature in Bouin's fixative. Organ cultures were fixed for 1-2 hours at room temperature in 4% paraformaldehyde. Fixed embryos and organ cultures were dehydrated through a graded series of ethanol and embedded in paraffin wax. Serial sections of 5-7μm thickness were stained with hematoxylin and eosin for histology analyses. For in situ hybridization, embryos were fixed in 4% paraformaldehyde overnight at 4°C and processed for paraffin sections as described above. In situ hybridization of sections was performed as described previously (Lan et al., 2001). All micrographs were assembled using Adobe Photoshop 6.0.
Electron microscopy
Tissues from staged embryos were fixed in phosphate buffered 2.5% glutaraldehyde (pH 7.4) and post-fixed in phosphate buffered 1% osmium tetroxide solution. Fixed tissues were dehydrated through a graded series of ethanol to 100%, infiltrated with liquid Spurr epoxy resin and thin-sectioned with a diamond knife onto copper grids. The sections were treated with aqueous uranyl acetate for 10 minutes, lead citrate for 15 minutes, and imaged with a Hitachi 7100 transmission electron microscope.
Explant culture
Palatal shelves and tongues were microdissected from late E12.5 or early E13.5 embryos. Freshly dissected tissues were placed together on trapezoid-shaped type AA 0.8μm nylon filter papers [Millipore] with anterior aspects of the tissues toward the “top” of the trapezoid for orientation purposes. Recombination experiments were performed by positioning the palatal shelf from one embryo on a filter with the palate, tongue or palate and tongue from a second embryo. Filter papers were placed on wire mesh supports (Flynn and Enslow) in Trowell-type culture dishes (Corning) and cultured in BGJ-b serum free medium (Invitrogen) supplemented with 4 mM L-glutamine and ascorbic acid for 3-5 days at 37°C with 5% CO2. Medium was replaced every other day and tissues were fixed for histology after three to five days of culture. Genotypes of the explant tissues were determined by allele-specific PCR analysis of yolk sac DNA. Each tissue pairing was replicated 3-5 times.
Immunohistochemistry and TUNEL assay
Immunohistochemical localization of E-cadherin and activated-Notch1 was performed largely as described by Lin and Kopan (2003) with minor modifications. Briefly, paraformaldehyde fixed tissues were embedded in paraffin and serially sectioned at 5μm thickness. Sections were deparaffinized in xylene, rehydrated with graded ethanols, boiled for five minutes in a pressure cooker in Trilogy reagent (Cell Marque) for antigen retrieval and rinsed in dH2O. Sections were then treated with 3% H2O2 in methanol to block endogenous peroxidase activity, blocked with serum and then incubated overnight at 4°C with the E-cadherin antibody [1:500, Zymed Laboratories, Inc.] or the Val1744 activated-Notch1 antibody [1:1000, Cell Signaling Technology] diluted in PBS containing 0.2% skim milk and 0.3% Triton X-100. After washing with PBS containing 0.05% Tween-20, sections were incubated with biotinylated anti-rabbit secondary antibody (Zymed Laboratories Inc.), streptavidin-HRP (Zymed Laboratories Inc.), biotinylated tyramide diluted 1:200 (Perkin Elmer), another round of streptavidin-HRP incubation, and then developed using Cy3-conjugated tyramide substrate (Perkin Elmer). Washes were performed at each step after the primary antibody incubation using PBS containing 0.05% Tween-20.
General keratin expression was examined using a pan-cytokeratin mouse monoclonal antibody mixture for keratins 1, 4, 5, 6, 8, 10, 13, 18, and 19 (Sigma-Aldrich). Keratin-17 was detected using a peptide antibody generated by McGowan and Coulombe (1998b). Bouin's fixed tissues embedded in paraffin were sectioned, deparaffinized, rehydrated. For staining the pan-cytokeratin antibody mixture, sections were treated using the M.O.M. kit according to manufacturer's instructions (Vector Laboratories inc.). Sections were incubated with a 1:400 dilution of pan-cytokeratin antibody or 1:1000 dilution of keratin-17 antibody overnight at 4°C followed by detection using the Zymed Histostain Plus kit (Zymed Laboratories Inc.). Washes were carried out as described above.
Apoptosis was assayed by TUNEL assay of paraformaldehyde fixed tissue sections using the In Situ Cell Death Detection Kit following the manufacturer's instructions (Roche).
Acknowledgments
We are very grateful to Dr. Pierre Coulombe (Johns Hopkins) for the keratin-17 antibody, YiPing Chen and Brian Schutte for cDNA probes, and Karen Bentley (University of Rochester Medical Center, Electron Microscope Research Core) for TEM assistance. We thank other members of the Jiang Lab for helpful comments and discussions. This work was supported by NIH grants R01DE013681 to RJ and R01NS036437 to TG. LMC was supported by NIH training grant T32DE07202.
Grant Sponsor: NIH; Grant numbers: R01DE013681, R01NS036437, T32DE07202.
References
- Akiyama M, Smith LT, Yoneda K, Holbrook KA, Hohl D, Shimizu H. Periderm cells form cornified cell envelope in their regression process during human epidermal development. J Invest Dermatol. 1999;112:903–909. doi: 10.1046/j.1523-1747.1999.00592.x. [DOI] [PubMed] [Google Scholar]
- Alappat SR, Zhang Z, Suzuki K, Zhang X, Liu H, Jiang R, Yamada G, Chen Y. The cellular and molecular etiology of the cleft secondary palate in Fgf10 mutant mice. Dev Biol. 2005;277:102–113. doi: 10.1016/j.ydbio.2004.09.010. [DOI] [PubMed] [Google Scholar]
- Artavanis-Tsakonas S, Rand MD, Lake RJ. Notch signaling: cell fate control and signal integration in development. Science. 1999;284:770–776. doi: 10.1126/science.284.5415.770. [DOI] [PubMed] [Google Scholar]
- Blavier L, Lazaryev A, Groffen J, Heisterkamp N, DeClerck YA, Kaartinen V. TGF-beta3-induced palatogenesis requires matrix metalloproteinases. Mol Biol Cell. 2001;12:1457–1466. doi: 10.1091/mbc.12.5.1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cecconi F, Alvarez-Bolado G, Meyer BI, Roth KA, Gruss P. Apaf1 (CED-4 homolog) regulates programmed cell death in mammalian development. Cell. 1998;94:727–737. doi: 10.1016/s0092-8674(00)81732-8. [DOI] [PubMed] [Google Scholar]
- Cuervo R, Covarrubias L. Death is the major fate of medial edge epithelial cells and the cause of basal lamina degradation during palatogenesis. Development. 2004;131:15–24. doi: 10.1242/dev.00907. [DOI] [PubMed] [Google Scholar]
- Din SU. Atypical tongue-tie due to congenital tongue-palate fusion. J Coll Physicians Surg Pak. 2003;13:459–460. [PubMed] [Google Scholar]
- Dunker N, Schmitt K, Krieglstein K. TGF-beta is required for programmed cell death in interdigital webs of the developing mouse limb. Mech Dev. 2002;113:111–120. doi: 10.1016/s0925-4773(02)00015-1. [DOI] [PubMed] [Google Scholar]
- Fitchett JE, Hay ED. Medial edge epithelium transforms to mesenchyme after embryonic palatal shelves fuse. Dev Biol. 1989;131:455–474. doi: 10.1016/s0012-1606(89)80017-x. [DOI] [PubMed] [Google Scholar]
- Fitzpatrick DR, Denhez F, Kondaiah P. Differential expression of TGF-β isoforms in murine palatogenesis. Development. 1990;109:585–595. doi: 10.1242/dev.109.3.585. [DOI] [PubMed] [Google Scholar]
- Ferguson MWJ. Palate development. Development. 1988;(suppl):41–60. doi: 10.1242/dev.103.Supplement.41. [DOI] [PubMed] [Google Scholar]
- Ferguson MWJ, Honig LS, Slavkin HC. Differentiation of cultured palatal shelves from alligator, chick and mouse embryos. Anat Rec. 1984;209:231–149. doi: 10.1002/ar.1092090210. [DOI] [PubMed] [Google Scholar]
- Gato A, Martinez ML, Tudela C, Alonso I, Moro JA, Formoso MA, Ferguson MW, Martinez-Alvarez C. TGF-beta(3)-induced chondroitin sulphate proteoglycan mediates palatal shelf adhesion. Dev Biol. 2002;250:393–405. [PubMed] [Google Scholar]
- Gorlin RJ, Cohen MM, Hennekam RCM. Syndromes of the head and neck. 4th. Oxford University Press; New York: 2001. [Google Scholar]
- Greene RM, Kochhar DM. Surface coat on the epithelium of developing palatine shelves in the mouse as revealed by electron microscopy. J Embryol Exp Morphol. 1974;31:683–692. [PubMed] [Google Scholar]
- Hayward AF. The permeability of the epithelium of the skin of fetal rats demonstrated with a lanthanum-containing solution. J Anat. 1983;136:379–788. [PMC free article] [PubMed] [Google Scholar]
- Holbrook KA, Odland F. The fine structure of developing human epidermis: light, scanning, and transmission electron microscopy of the periderm. J Invest Dermatol. 1975;65:16–38. doi: 10.1111/1523-1747.ep12598029. [DOI] [PubMed] [Google Scholar]
- Humphrey T. Palatopharyngeal fusion in a human fetus and its relation to cleft formation. Ala J Med Sci. 1970;7:398–429. [PubMed] [Google Scholar]
- Jiang R, Lan Y, Chapman HD, Shawber C, Norton CR, Serreze DV, Weinmaster G, Gridley T. Defects in limb, craniofacial, and thymic development in Jagged2 mutant mice. Genes Dev. 1998;12:1046–1057. doi: 10.1101/gad.12.7.1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaartinen V, Cui XM, Heisterkamp N, Groffen J, Shuler CF. Transforming growth factor-beta3 regulates transdifferentiation of medial edge epithelium during palatal fusion and associated degradation of the basement membrane. Dev Dyn. 1997;209:255–260. doi: 10.1002/(SICI)1097-0177(199707)209:3<255::AID-AJA1>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Kaartinen V, Voncken JW, Shuler C, Warburton D, Bu D, Heisterkamp N, Groffen J. Abnormal lung development and cleft palate in mice lacking TGF-beta 3 indicates defects of epithelial-mesenchymal interaction. Nat Genet. 1995;11:415–421. doi: 10.1038/ng1295-415. [DOI] [PubMed] [Google Scholar]
- Kadesch T. Notch signaling: the demise of elegant simplicity. Curr Opin Genet Dev. 2004;14:506–512. doi: 10.1016/j.gde.2004.07.007. [DOI] [PubMed] [Google Scholar]
- King CTG. Teratogenic effects of meclozine-hydrochloride on the rat. Science. 1963;141:353–355. doi: 10.1126/science.141.3578.353. [DOI] [PubMed] [Google Scholar]
- Koziol CA, Steffek AJ. Acid phosphatase activity in palates of developing normal and chlorcyclizine treated rodents. Arch Oral Biol. 1969;14:317–321. doi: 10.1016/0003-9969(69)90234-9. [DOI] [PubMed] [Google Scholar]
- Lai EC. Notch signaling: control of cell communication and cell fate. Development. 2004;131:965–973. doi: 10.1242/dev.01074. [DOI] [PubMed] [Google Scholar]
- Lan Y, Kingsley PD, Cho ES, Jiang R. Osr2, a new mouse gene related to Drosophila odd-skipped, exhibits dynamic expression patterns during craniofacial, limb, and kidney development. Mech Dev. 2001;107:175–179. doi: 10.1016/s0925-4773(01)00457-9. [DOI] [PubMed] [Google Scholar]
- Lanford PJ, Lan Y, Jiang R, Lindsell C, Weinmaster G, Gridley T, Kelley MW. Notch signaling pathway mediates hair cell development in mammalian cochlea. Nat Genet. 1999;21:289–292. doi: 10.1038/6804. [DOI] [PubMed] [Google Scholar]
- Lechler T, Fuchs E. Assymetric cell divisions promote stratification and differentiation of mammalian skin. Nature. 2005;437:275–280. doi: 10.1038/nature03922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MH, Kopan R. Long-range, nonautonomous effects of activated-Notch1 on tissue homeostasis in the nail. Dev Biol. 2003;263:343–359. doi: 10.1016/j.ydbio.2003.07.007. [DOI] [PubMed] [Google Scholar]
- Lissemore JL, Starmer WT. Phylogenetic analysis of vertebrate and invertebrate Delta/Serrate/LAG-2 (DSL) proteins. Mol Phylogenet Evol. 1999;11:308–319. doi: 10.1006/mpev.1998.0588. [DOI] [PubMed] [Google Scholar]
- Martinez-Alvarez C, Blanco MJ, Perez R, Rabadan MA, Aparicio M, Resel E, Martinez T, Nieto MA. Snail family members and cell survival in physiological and pathological cleft palates. Dev Biol. 2004;265:207–218. doi: 10.1016/j.ydbio.2003.09.022. [DOI] [PubMed] [Google Scholar]
- Martinez-Alvarez C, Bonelli R, Tudela C, Gato A, Mena J, O'Kane S, Ferguson MW. Bulging medial edge epithelial cells and palatal fusion. Int J Dev Biol. 2000;44:331–335. [PubMed] [Google Scholar]
- Mato M, Uchiyama Y. Ultrastructures of glosso-palatal fusion after treatment of meclozine-hydrochloride. Virchows Arch A Path Anat And Histol. 1975;369:7–17. doi: 10.1007/BF00432457. [DOI] [PubMed] [Google Scholar]
- McGowan KM, Coulombe PA. Onset of keratin-17 expression coincides with the definition of major epithelial lineages during skin development. J Cell Biol. 1998;143:469–486. doi: 10.1083/jcb.143.2.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meller SM, Barton HL. Extracelluar coat in developing human palatal process: electron microscopy and ruthenium red binding. Anat Rec. 1978;190:223–232. doi: 10.1002/ar.1091900206. [DOI] [PubMed] [Google Scholar]
- Meller SM, De Paola DP, Barton LH, Mandella RD. Secondary palatal development in the New Zealand white rabbit: a scanning electron microscopic study. Anat Rec. 1980;198:229–244. doi: 10.1002/ar.1091980210. [DOI] [PubMed] [Google Scholar]
- Mustonen T, Tummers M, Mikami T, Itoh N, Zhang N, Gridley T, Thesleff I. Lunatic fringe, FGF, and BMP regulate the Notch pathway during epithelial morphogenesis of teeth. Dev Biol. 2002;248:281–293. doi: 10.1006/dbio.2002.0734. [DOI] [PubMed] [Google Scholar]
- Nawshad A, LaGamba D, Hay ED. Transforming growth factor beta (TGFbeta) signalling in palatal growth, apoptosis and epithelial mesenchymal transformation (EMT) Arch Oral Biol. 2004;49:675–689. doi: 10.1016/j.archoralbio.2004.05.007. [DOI] [PubMed] [Google Scholar]
- Nguyen AV, Pollard JW. Transforming growth factor β3 induces cell death during the first stage of mammary gland involution. Development. 2000;127:3107–3118. doi: 10.1242/dev.127.14.3107. [DOI] [PubMed] [Google Scholar]
- Opperman LA, Adab K, Gakunga PT. Transforming growth factor-beta 2 and beta 3 regulate fetal rat cranial suture morphogenesis by regulating rates of cell proliferation and apoptosis. Dev Dyn. 2000;219:237–247. doi: 10.1002/1097-0177(2000)9999:9999<::AID-DVDY1044>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- Pelton RW, Hogan BL, Miller DA, Moses HL. Differential expression of genes encoding TGFs β1, β2, and β3 during murine palate formation. Dev Biol. 1990;141:456–460. doi: 10.1016/0012-1606(90)90401-4. [DOI] [PubMed] [Google Scholar]
- Polakowska RR, Piacentini M, Bartlett R, Goldsmith LA, Haake AR. Apoptosis in human skin development: morphogenesis, periderm, and stem cells. Dev Dyn. 1994;199:176–188. doi: 10.1002/aja.1001990303. [DOI] [PubMed] [Google Scholar]
- Pourtois M. Onset of the acquired potentiality for fusion in the palatal shelves of rats. J Embryol Exp Morphol. 1966;16:171–182. [PubMed] [Google Scholar]
- Proetzel G, Pawlowski SA, Wiles MV, Yin M, Boivin GP, Howles PN, Ding J, Ferguson MW, Doetschman T. Transforming growth factor-beta 3 is required for secondary palate fusion. Nat Genet. 1995;11:409–414. doi: 10.1038/ng1295-409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice R, Spencer-Dene B, Connor EC, Gritli-Linde A, McMahon AP, Dickson C, Thesleff I, Rice DP. Disruption of Fgf10/Fgfr2b-coordinated epithelial-mesenchymal interactions causes cleft palate. J Clin Invest. 2004;113:1692–1700. doi: 10.1172/JCI20384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeter EH, Kisslinger JA, Kopan R. Notch-1 signalling requires ligand-induced proteolytic release of intracellular domain. Nature. 1998;393:382–386. doi: 10.1038/30756. [DOI] [PubMed] [Google Scholar]
- Schupbach PM, Chamberlain JG, Schroeder HE. Development of the secondary palate in the rat: a scanning electron microscopic study. J Craniofac Genet Dev Biol. 1983;3:159–177. [PubMed] [Google Scholar]
- Scott WJ, Jr, Nau H, Wittfoht W, Merker HJ. Ventral duplication of the autopod: chemical induction by methoxyacetic acid in rat embryos. Development. 1987;99:127–136. doi: 10.1242/dev.99.1.127. [DOI] [PubMed] [Google Scholar]
- Shah RM. Palatomandibular and maxillo-mandibular fusion, partial aglossia and cleft palate in a human embryo. Report of a case. Teratology. 1977;15:261–272. doi: 10.1002/tera.1420150308. [DOI] [PubMed] [Google Scholar]
- Shapiro BL, Sweney L. Electron microscopic histochemical examination of oral epithelial-mesenchymal interaction (programmed cell death) J Dent Res. 1969;48:652–660. doi: 10.1177/00220345690480050801. [DOI] [PubMed] [Google Scholar]
- Shawber C, Boulter J, Lindsell CE, Weinmaster G. Jagged2: a serrate-like gene expressed during rat embryogenesis. Dev Biol. 1996;180:370–376. doi: 10.1006/dbio.1996.0310. [DOI] [PubMed] [Google Scholar]
- Sidow A, Bulotsky MS, Kerrebrock AW, Bronson RT, Daly MJ, Reeve MP, Hawkins TL, Birren BW, Jaenisch R, Lander ES. Serrate2 is disrupted in the mouse limb-development mutant syndactylism. Nature. 1997;389:722–725. doi: 10.1038/39587. [DOI] [PubMed] [Google Scholar]
- Souchon R. Surface coat of the palatal shelf epithelium during palatogenesis in mouse embryos. Anat Embryol. 1975;147:133–142. doi: 10.1007/BF00306728. [DOI] [PubMed] [Google Scholar]
- Sun D, Vanderburg CR, Odierna GS, Hay ED. Tgfbeta3 promotes transformation of chick palatal medial edge epithelium to mesenchyme in vitro. Development. 1998;125:95–105. doi: 10.1242/dev.125.1.95. [DOI] [PubMed] [Google Scholar]
- Taya Y, O'Kane S, Ferguson MW. Pathogenesis of cleft palate in TGF-beta3 knockout mice. Development. 1999;126:3869–3879. doi: 10.1242/dev.126.17.3869. [DOI] [PubMed] [Google Scholar]
- Vaziri Sani F, Hallberg K, Harfe BD, McMahon AP, Linde A, Gritli-Linde A. Fate-mapping of the epithelial seam during palatal fusion rules out epithelial-mesenchymal transformation. Dev Biol. 2005;285:490–495. doi: 10.1016/j.ydbio.2005.07.027. [DOI] [PubMed] [Google Scholar]
- Waterman RE, Ross LM, Meller SM. Alterations in the epithelial surface of A/Jax mouse palatal shelves prior to and during palatal fusion: a scanning electron microscopic study. Anat Rec. 1973;176:361–376. doi: 10.1002/ar.1091760311. [DOI] [PubMed] [Google Scholar]
- Wilkie AO, Morriss-Kay GM. Genetics of craniofacial development and malformation. Nat Rev Genet. 2001;2:458–468. doi: 10.1038/35076601. [DOI] [PubMed] [Google Scholar]
- Zschabitz AR, Biesalski HK, Krahn V, Gabius HJ, Weiser H, Khaw A, Hemmes C, Stofft E. Distribution patterns in glycoconjugate expression during the development of the rat palate. Histochem J. 1994;26:705–720. doi: 10.1007/BF00158203. [DOI] [PubMed] [Google Scholar]