

Abstract
RhoH is a hematopoietic-specific, GTPase-deficient member of the Rho GTPase family that functions as a regulator of thymocyte development and T-cell receptor signaling by facilitating localization of ZAP70 to the immunological synapse. Here we investigated the role of RhoH in the B-cell lineage. B-cell receptor (BCR) signaling was intact in Rhoh−/− mice. Since RhoH interacts with ZAP70, which is a prognostic factor in B-cell chronic lymphocytic leukemia (CLL), we analyzed the mRNA levels of RhoH in primary human CLL cells and demonstrated a 2.3-fold higher RhoH expression compared to normal B cells. RhoH expression in CLL positively correlated with the protein levels of ZAP70. Deletion of Rhoh in a murine model of CLL (Eμ-TCL1Tg mice) significantly delayed the accumulation of CD5+IgM+ leukemic cells in peripheral blood and the leukemic burden in the peritoneal cavity, bone marrow and spleen of Rhoh−/− mice compared with their Rhoh+/+ counterparts. Phosphorylation of AKT and ERK in response to BCR stimulation was notably decreased in Eμ-TCL1Tg;Rhoh−/− splenocytes. These data suggest that RhoH plays a role in the progression of CLL in a murine model and shows RhoH expression is altered in human primary CLL samples.
Keywords: RhoH GTPase, Chronic Lymphocytic Leukemia, TCL1, B cells, lymphopoiesis
Introduction
B-cell chronic lymphocytic leukemia (CLL) represents the most common leukemia in adults and a disease for which there are no curative treatments available(1). The disease is characterized by the clonal expansion of small mature-appearing CD19+CD5+CD23+ B cells in the peripheral blood, bone marrow, lymph nodes and other lymphatic tissues. The absence of somatic mutations in the immunoglobulin heavy chain variable region (IGHV) genes and expression of the zeta-chain-associated protein kinase 70 (ZAP70) are strongly associated with an aggressive clinical course(2, 3). ZAP70, a non-receptor tyrosine kinase that plays a central role in T-cell receptor (TCR) signal transduction, is abnormally expressed in aggressive CLL and has been shown to enhance B-cell receptor (BCR) signaling in CLL cells(4-6). These and other data suggest that signaling through the BCR plays an important role in CLL pathogenesis(1).
Rho-GTPases represent a family of proteins which play key functions in intracellular signaling and regulate cellular morphology, motility and proliferation(7). While most Rho-GTPases cycle between GTP-bound active and GDP-bound inactive states, RhoH, an hematopoietic-specific member of the RhoE subfamily, is GTPase-deficient and therefore remains in the active GTP-bound state(8). We and others have previously described the critical interaction of ZAP70 with the small GTPase RhoH in TCR function(9, 10). In T-cells, RhoH functions as a critical regulator of thymocyte development and TCR signaling by mediating recruitment of ZAP70 to the immunological synapse. RhoH is required for phosphorylation of CD3ζ, membrane translocation of ZAP70 and subsequent activation of ZAP70-mediated pathways(9). RhoH serves as an adaptor molecule for ZAP70 and Lck in TCR signaling (Chae, et al., manuscript under review) and contains functional immunoreceptor tyrosine-based activation (ITAM)-like motifs(9). Thus RhoH is unique among Rho GTPases in that the cellular activity is dependent on expression levels and post-translational modification of the protein.
Several lines of evidence link dysregulation of RhoH to the development of malignancies. The RHOH gene was first identified in a non-Hodgkin's lymphoma cell line harboring a t(3;4)(q27;p11) translocation(11) and rare chromosomal alterations involving the RHOH gene have been detected in cases of follicular lymphoma and multiple myeloma(12). Moreover, similar to other well-characterized protooncogenes (BCL6, MYC, PAX5), the 5’-noncoding region of the RHOH gene is a target of somatic hypermutation in a variety of B lymphoid malignancies such as diffuse large B-cell lymphomas(13, 14), AIDS-related non-Hodgkin lymphomas(15) and primary central nervous system lymphomas(16). While these studies have suggested a possible role of RhoH dysregulation in malignant transformation, a direct demonstration of the involvement of RhoH in cancer initiation or progression has not been provided.
Here we examined the role of RhoH in physiologic and malignant B-cell lineages. While the development of B-cells is only minimally perturbed in the absence of RhoH, an analysis of primary human CLL cells revealed significantly higher RhoH expression levels than in normal B-cells. Moreover, the expression level of RhoH correlates with the protein levels of ZAP70. To further investigate the role of RhoH in the development and progression of CLL, we utilized a genetic mouse model of CLL, the Eμ-TCL1 transgenic mouse(17). In this model, the expression of the TCL1 oncogene is targeted to the B-cell lineage, resulting in the expansion of the B-1 cell compartment and the development of CD5+ B-cell leukemia resembling the aggressive form of human CLL(17, 18). Deletion of Rhoh in the Eμ-TCL1 background results in a significant delay of disease progression, highlighting the critical role of RhoH in CLL pathogenesis.
Materials and Methods
Patients and cell samples
The human samples for this study were obtained after informed consent and with approval of the Institutional Review Boards of the Cincinnati Children's Hospital Medical Center and the Ohio State University Comprehensive Cancer Center. Peripheral blood samples were collected from 29 patients with known diagnosis of CLL. Peripheral blood mononuclear cells were separated by centrifugation on Histopaque-1083 (Sigma, St. Louis, MO) and CLL cells were obtained by Rosette-Sep purification (StemCell Technology, London, UK). The purity of the samples was checked by flow cytometry and was >90% for CD19+CD5+ cells. Cell samples were cryopreserved in Iscove's Modified Dulbecco's Medium (IMDM; Invitrogen, Carlsbad, CA) containing 10% fetal bovine serum (FBS) and 10% dimethyl sulfoxide (DMSO; Sigma-Aldrich) until further use. Normal peripheral blood B lymphocytes from 6 healthy donors were isolated with CD19 Microbeads (Miltenyi Biotec, Auburn, CA), following the manufacturer's instructions.
RhoH mRNA measurement by quantitative real time PCR
Total RNA from CLL and normal human B cell samples was isolated with the RNeasy Mini Kit (Qiagen, Valencia, CA) following the manufacturer's instructions. Aliquots of 1 μg total RNA were reverse transcribed using the Protoscript First Strand cDNA synthesis kit (New England Biolabs, Ipswich, MA).
Quantitative real-time PCR was performed using SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA) in a ABI Prism 7700 Sequence Detector System (Applied Biosystems). Primer sequences for RhoH amplification were: 5’-TTGATTTCCGGAGTCAGTCA-3’ (forward) and 5’-GTTCTCCACGGCTTCAGTTT-3’ (reverse). Phosphoglycerate kinase 1 (PGK1) was amplified as an endogenous control, using the following primers: 5’-GGGAAAAGATGCTTCTGGGAA-3’ (forward) and 5’-TTGGAAAGTGAAGCTCGGAAA-3’ (reverse).
Mice
The generation of Rhoh−/− mice has been previously described(9). The 129/Sv Rhoh−/− strain was backcrossed on the C57BL/6J background. Mice used were derived from more than 20 backcross generations and were 7-14 weeks of age.
In order to investigate the role of RhoH in a murine model of CLL, Eμ-TCL1Tg/Tg mice on a mixed B6C3 background(17) were crossed with Rhoh−/− mice. The resulting F1 mice were either crossed with Rhoh−/− mice, yielding Eμ-TCL1-transgenic mice nullizygous or heterozygous for Rhoh (Eμ-TCL1Tg;Rhoh−/− or Eμ-TCL1Tg;Rhoh+/−) and their non-transgenic littermates (Rhoh−/−, Rhoh+/−), or were crossed with wild-type (WT) C57BL/6J mice yielding Eμ-TCL1–transgenic mice with normal Rhoh alleles (Eμ-TCL1Tg;Rhoh+/+) and their non-transgenic littermates (Rhoh+/+). All experiments involving animals were approved by the Institutional Animal Care and Use Committee of the Cincinnati Children's Research Foundation (Cincinnati, OH).
Flow cytometry
For flow cytometry analysis, the following antibody conjugates were used: B220-APC-Cy7 (RA3-6B2), CD19-PE-Cy7 (1D3), CD43-PE (S7), IgM-PerCP-Cy5.5 (R6-60.2), IgM-APC (II/41), IgD-FITC (11-26c.2a), CD3ε-PE(145-2C11), CD21/CD35-PE (7G6), CD69-FITC (H1.2F3; all from BD Biosciences, San Jose, CA); CD93-APC (AA4.1) and CD5-PE-Cy7 (53-7.3; both from eBioscience, San Diego, CA). Red blood cells were lysed by incubation in BD PharmLyse (BD Biosciences) for 10 min, and the resulting cell suspensions were incubated at 4°C for 30 min with a 1:100 dilution of the appropriate conjugates. After staining, cells were washed and resuspended in phosphate buffered saline (PBS). In some assays, 7-AAD (2 μg/ml) was added to the resuspension solution for dead cell exclusion. For apoptosis assays, cells were stained with 7-AAD and Annexin V-FITC (BD Biosciences). Cells were analyzed on a FACSCanto flow cytometer (BD Biosciences).
Immunophenotyping of normal murine B cell populations
Single cell suspensions were prepared from bone marrow, spleen, lymph nodes, peripheral blood and peritoneal cavity of WT and Rhoh−/− mice. Cells were stained using fluorescently labeled antibodies and analyzed by flow cytometry. B-cell differentiation stages of normal B cells in the bone marrow were defined as follows: pro-B (B220+CD19+CD43+CD93+IgM−IgD−), pre-B (B220+CD19+CD43−CD93+IgM−IgD−), immature (B220+CD19+CD43−CD93+IgM+IgD−) and mature B cells (B220+CD19+CD43−CD93+IgM+IgD+). In secondary lymphoid organs, B cells were classified as transitional (B220+CD93+), B-2/follicular (B220+CD93−IgDhiCD21lo) and marginal zone (MZ) B cells (B220+CD93−IgDloCD21hi). Peritoneal B-1 cells were defined as B220loIgM+CD5+.
Immunophenotyping of murine leukemic cells
The peripheral blood immunophenotype of Eμ-TCL1Tg mice was monitored longitudinally in a cohort of mice (Eμ-TCL1Tg;Rhoh+/+, Eμ-TCL1Tg;Rhoh+/−, Eμ-TCL1Tg;Rhoh−/− with n=5-19 mice per time point, and their respective non-transgenic littermate controls). Starting at 2 months of age, peripheral blood was obtained monthly from the tail veins of the mice. Additional mice were sacrificed at 6 months of age to investigate the degree of leukemic infiltration of the bone marrow, spleen and peritoneal cavity at a defined time point. CLL cells were defined as CD3−CD5+IgM+. The leukemic burden was expressed as the total number of CD3−CD5+IgM+ cells at each anatomical site.
Isolation of normal murine splenic B cells
Resting B cells were purified from splenic single cell suspensions by negative selection using mouse CD43 Microbeads and LD columns (Miltenyi Biotec, Auburn, CA), following the manufacturer's recommendations. In all experiments, a pool of cells from 2-3 mice of each genotype was used. The purity of the negative fractions was checked by flow cytometry (B220+) and was typically higher than 95%. For subfractionation of B-2 and MZ B cells, CD43-depleted fractions were stained with FITC-conjugated anti-IgD and PE-conjugated anti-CD21/CD35 and sorted on a FACSVantage flow cytometer. B-2 cells were defined as IgDhiCD21lo and MZ B cells as IgDloCD21hi.
In vitro activation and proliferation of murine B cells
For in vitro assays, purified splenic B cells were plated in 96-well plates at a concentration of 2×106 cells/ml in IMDM containing 10% FBS (Omega Scientific, Tarzana, CA), penicillin/streptomycin (Hyclone), 2 mM L-glutamine (Invitrogen, Carlsbad, CA), and the appropriate stimuli. These included lipopolysaccharide (LPS) from E. coli (2 or 20 μg/ml; Sigma), F(ab’)2 goat anti-mouse IgM (1 or 10 μg/ml; Jackson Immunoresearch; West Grove, PA), affinity purified anti-mouse CD40 (2 μg/ml; clone 1C10; eBioscience), murine recombinant IL-4 (Peprotech, Rocky Hill, NJ), or combinations thereof.
B-cell activation was measured by flow cytometric detection of the activation antigen CD69 after 20 h of stimulation. Proliferation was determined by [3H]-thymidine incorporation. Briefly, splenic B cells were cultured in the presence of 2 or 20 μg/ml LPS for 48 h in triplicates, and then pulsed with 1 μCi of [3H]-thymidine (GE Healthcare, Buckinghamshire, England) for 12 h. Incorporation of 3H-thymidine was measured in a scintillation counter (Beckman, Fullerton, CA). The production of IgM in the culture supernatant was quantified by ELISA after 48 hours of stimulation.
Chemotaxis assays
Splenic B cells (1×106 cells in 100 μl complete medium) were seeded in triplicate in the upper chamber of 24-well, 5-μm Transwell plates (Corning, Corning, NY) in which the lower chamber contained 600 μl of complete medium supplemented with CXCL12 (Peprotech) or CXCL13 (R&D Systems, Minneapolis, MN) and incubated for 4 h at 37°C. Cells that migrated to the lower chamber were collected and counted using a hemocytometer. The results were expressed as the percentage of migrated cells, relative to the total number of input cells.
ELISA
The concentration of different immunoglobulin isotypes (IgM, IgG1, IgG3) in serum or in culture supernatants were determined by ELISA using isotype-specific antibodies (Mouse IgM, IgG1, and IgG3 quantitation kits; Bethyl Laboratories, Montgomery, TX), following the manufacturer's recommendations.
Analysis of BCR signaling
For the analysis of protein phosphorylation, prewarmed normal splenic B cells or leukemic splenocytes from mice 6-15 months of age were stimulated with 25 μg/ml F(ab’)2 goat anti-mouse IgM (Jackson Immunoresearch) for 1 or 5 min at 37°C, washed in cold PBS, lysed and analyzed by western blotting.
Intracellular calcium flux was measured by flow cytometry. Cells (3×106) were stained with 2.5 μM Fluo-3 (Invitrogen) in Tyrode's salt solution (Sigma) at 37°C for 45 min, washed twice, resuspended in 450 μl of Tyrode's buffer and incubated at 37°C for another 30 min. Baseline fluorescence (FITC channel) was measured for 30 s before the addition of 2.5 or 25 μg/ml F(ab’)2 goat anti-mouse IgM (Jackson Immunoresearch), after which data acquisition was continued for 5-10 min.
Immunoblotting
For western blotting, cells were lysed in RIPA buffer (10 mM TrisHCl pH 7.4, 130 mM NaCl, 1% Triton X-100, 0.1% sodium dodecyl sulfate (SDS), 0.5% sodium deoxycholate) containing protease and phosphatase inhibitors (5 mM EDTA, 5 mM EGTA, 10 mM NaF, 10 mM β-glycerophosphate, 1 mM sodium ortovanadate, and Complete protease inhibitor cocktail, Roche Applied Science, Indianapolis, IN). The following primary antibodies were used (all of them at a 1:1000 dilution): ZAP70 (99F2); phospho-p44/p42 MAPK (Thr202/Tyr204; 197G2), p44/p42 MAPK, phospho-JNK (Thr183/Tyr185), JNK, phospho-p38 (Thr180/Tyr182; 28B10), p38, phospho-AKT (Ser473), AKT; phospho-SYK (Tyr525/526), SYK (all from Cell Signaling Technology, Danvers, MA); β-actin (AC-15; Sigma; 1:10,000 dilution) was used as a loading control.
Statistical analysis
Differences between CLL and normal B cells with respect to RhoH expression, and differences in leukemic cell counts between Eμ-TCL1Tg;Rhoh+/+ and Eμ-TCL1Tg;Rhoh−/− mice at 6 months of age were tested by exact Wilcoxon tests. The correlation between ZAP70 and RhoH expression was estimated by the Pearson correlation coefficient. The difference between Eμ-TCL1Tg;Rhoh+/+, Rhoh+/− and Rhoh−/− groups with respect to the logarithm of cell count profiles over time was tested via comparisons of linear random effects models(19). Tests and models were fit using the univariate, corr, and mixed procedures of SAS 9.1(20). All p-values were two-sided. Data with p values lower than 0.05 were considered significant.
Results
Characterization of the B-cell compartment of Rhoh−/− mice
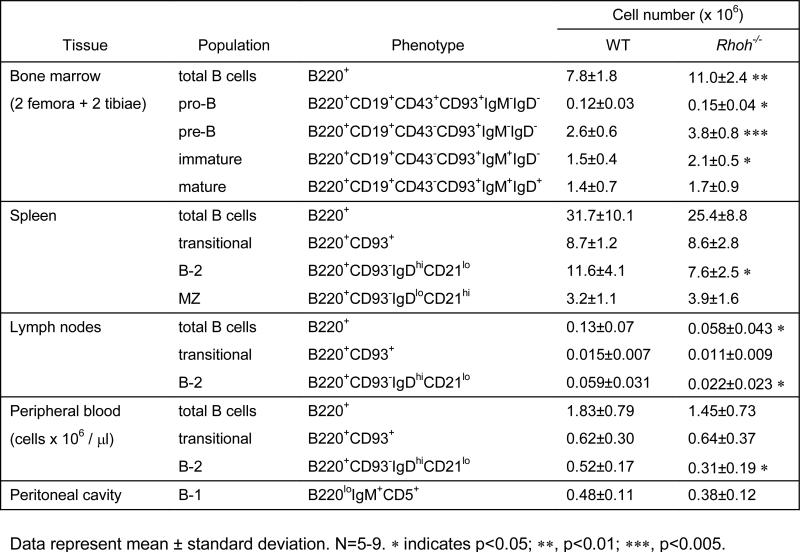
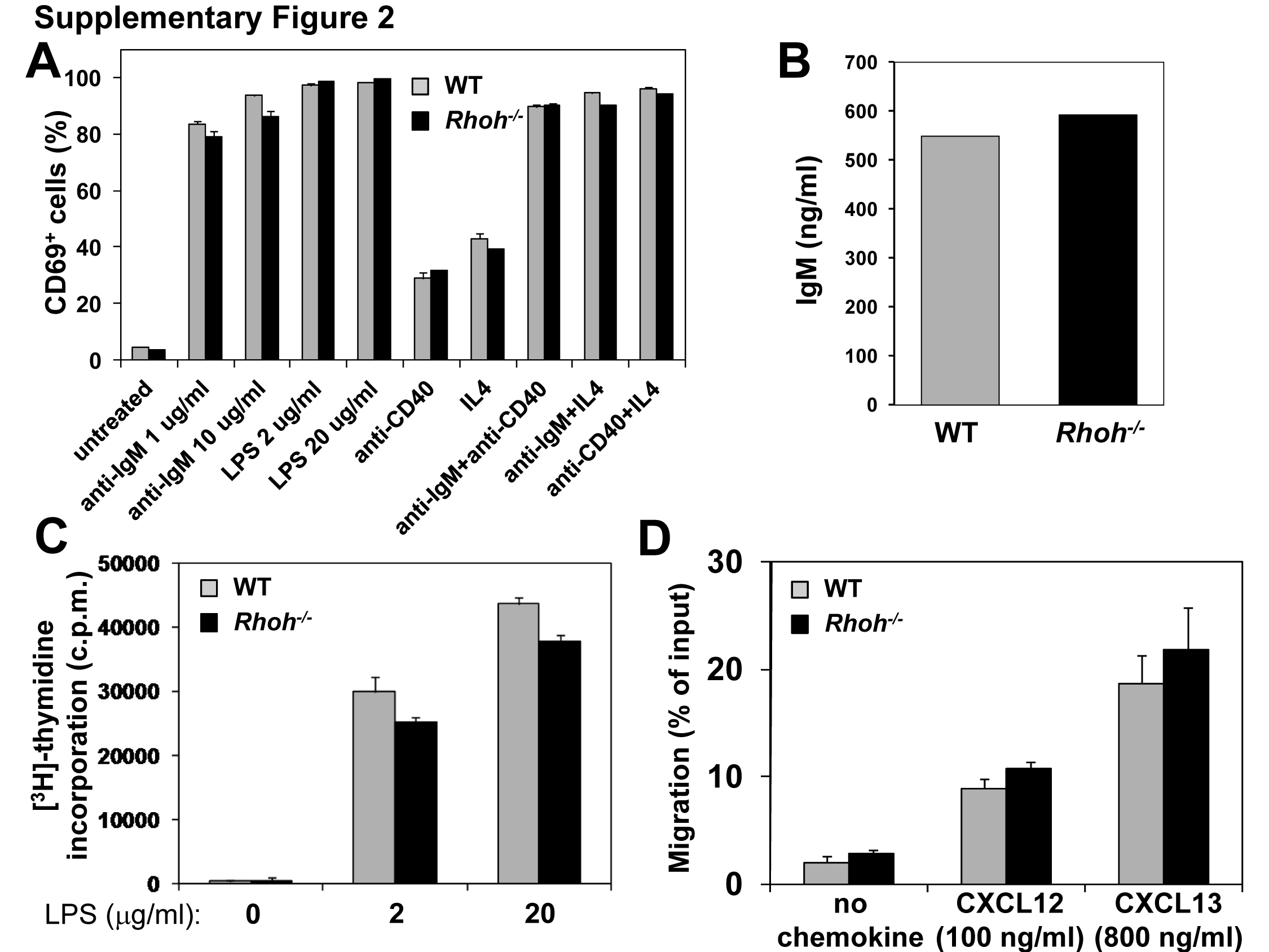
We performed a detailed analysis of B-cell subsets and differentiation stages in Rhoh−/−mice. Rhoh−/− mice showed modestly elevated B-cell numbers in the bone marrow (7.8±1.8 × 106 vs 11.0±2.4 × 106 cells/2 tibias and 2 femurs, WT vs Rhoh−/−; p<0.05). There was an increase in the number of pro-B (B220+CD19+CD43+CD93+IgM−IgD−), pre-B (B220+CD19+CD43−CD93+IgM−IgD−) and immature B cells (B220+CD19+CD43-CD93+IgM+IgD−) (Figure 1A; Table 1). In the spleen, Rhoh−/− mice showed a reduced number of B-2 cells (B220+CD93−IgDhiCD21lo) (65% of WT; Figure 1B). The B-2 compartment was also significantly diminished in peripheral blood (Figure 1C) and lymph nodes (Supplementary Figure 1A). The number of B-1 cells (B220loIgM+CD5+) in the peritoneal cavity of Rhoh−/− mice was comparable to that in WT animals (Supplementary Figure 1B). Analysis of the serum concentrations of immunoglobulin isotypes revealed dramatically decreased levels of IgG1 (p<0.001) (Figure 1D). The in vitro activation of splenic B cells using anti-IgM, LPS, anti-CD40, or IL-4 as agonists and the in vitro proliferation in response to LPS was not significantly different between WT and Rhoh−/−mice (Supplementary Figure 2A-C). Chemotaxis in response to CXCL12 (100 ng/ml) and CXCL13 was similar between WT and Rhoh−/− B cells (Supplementary Figure 2D) although Rhoh−/− cells appeared to show decreased migration at higher concentrations of CXCL12 (data not shown). These data suggest a modest effect of RhoH on B cell development. Given the normal in vitro proliferative and chemotaxis responses to multiple B cell agonists, and previously reported effects of RhoH deficiency on T cell function, B cell abnormalities in vivo may reflect T cell dependent alterations.
Figure 1. B cell subsets in WT and Rhoh−/− mice.

Analysis was by multiparameter flow cytometry, and absolute cell numbers were calculated using the total cellularity of each organ. (A) B cell differentiation stages in the bone marrow. (B, C) B cell numbers (B220+) and subsets in the spleen and peripheral blood. Data represent mean ± standard deviation. N=9 mice/analysis. * indicates p<0.05; **, p<0.005. (D) Serum concentrations of IgM, IgG1 and IgG3 in WT and Rhoh−/− mice, determined by ELISA. N=6 mice/genotype. * indicates p<0.001. The differences in IgM and IgG3 were close to statistical significance (p=0.07 and 0.09, respectively. Note logarithmic scale in the y-axis). (E) Analysis of BCR signaling. Sorted populations of B-2 and MZ B cells were stimulated with 25 μg/ml anti-IgM for 1 min and analyzed by immunoblotting for SYK and ERK phosphorylation. WT, wild-type; imm, immature B cells; mat, mature B cells; tr, transitional B cells; MZ, marginal zone B cells.
Table 1.
Composition of B cell subsets in Rhoh−/− mice.
|
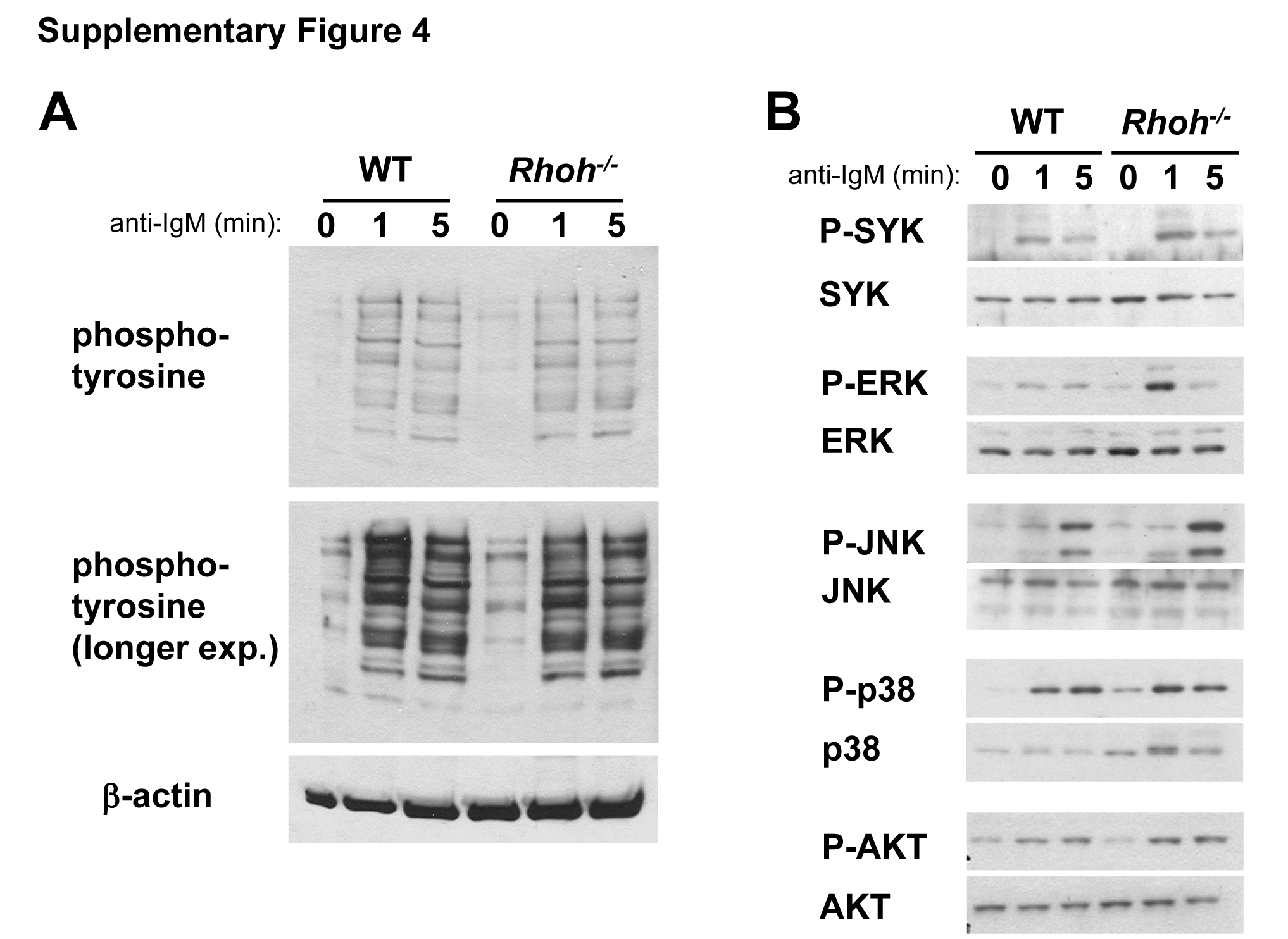
We have previously documented the critical role of RhoH in TCR signal transduction(9). In contrast, we did not observe major perturbations in signaling in Rhoh−/− B cells in response to BCR agonist. Rhoh−/− B cells appeared to show increased intracellular calcium flux following weak BCR stimulation, but this difference was not observed at higher concentration of anti-IgM (Supplementary Figure 3). Loss of RhoH did not impair protein tyrosine phosphorylation after BCR ligation (Supplementary Figure 4A). Moreover, several downstream components of the BCR pathway (SYK, ERK, JNK, p38) appeared to be hyperphosphorylated in the absence of RhoH when the analysis was performed on total splenic B cells (Supplementary Figure 4B). However, the phosphorylation of spleen tyrosine kinase (SYK) or extracellular signal-regulated kinase (ERK) in sorted populations of B-2 (IgDhiCD21lo) and MZ (IgDloCD21hi) B cells from Rhoh−/− and WT mice did not differ in response to BCR crosslinking (Figure 1E), implying that the observed differences can be explained by the different cellular composition of the B cell compartment (B-2 vs MZ B cells). In summary, Rhoh−/− B cells showed only modest variations of subtype distribution and no significant alteration of BCR signaling. Taken together, these data are consistent with a mild in vivo B-cell differentiation impairment with no obvious B cell-intrinsic signaling defect.
Elevated RhoH expression in primary human CLL cells
We have previously described the interaction between RhoH and ZAP70 in the TCR signaling pathway(9). ZAP70 is expressed in human CLL, is a negative prognostic factor in this disease(2, 3), and regulates BCR signaling in CLL cells(4-6). Therefore, we next investigated the potential role of RhoH in CLL. The mRNA expression levels of RhoH in primary human CLL cells (CD19+CD5+), as determined by real-time polymerase chain reaction (PCR), were significantly higher than in CD19+ peripheral blood B cells obtained from healthy donors (40.48±22.28 vs 18.22±7.95, CLL (n=29) vs normal B cells (n=6), p<0.05) (Figure 2A). No correlation was observed between RhoH expression and disease stage or leukocyte count. In order to determine whether RhoH levels were related to ZAP70 expression, the protein levels of ZAP70 in CLL samples were analyzed by immunoblotting. There was a significant, positive correlation between ZAP70 and RhoH expression (Pearson correlation coefficient of 0.42, p<0.05) (Figure 2B). Together, these observations imply that RhoH expression is elevated in human CLL and may be associated with the aggressive form of the disease.
Figure 2. RhoH expression in human CLL.

(A) The ratio of RhoH to PGK1 mRNA levels, measured by quantitative real-time PCR, is shown for primary human CLL cells and for peripheral blood B-cells from healthy donors. Dots represent individual patients. Horizontal bar represents the mean. * indicates p<0.05, normal vs. CLL cells. (B) Correlation between RhoH and ZAP70 expression in primary human CLL cells. ZAP70 protein expression was determined by immunoblotting and the densitometry readout of the ZAP70 bands was normalized with that of β-actin. RhoH mRNA expression was measured by quantitative real-time PCR and normalized with PGK expression. A calibrator sample was used to allow normalization across different PCR plates and western blot membranes. Pearson correlation, r=0.42, p<0.05.
RhoH involvement in the development of CLL in the Eμ-TCL1 transgenic CLL model
To further delineate the potential role of RhoH in CLL, we crossed Rhoh−/− mice with Eμ-TCL1 transgenic mice(17). These mice express the TCL1 oncogene in a B-cell specific fashion and accumulate abnormal numbers of CD5+IgM+ B cells progressively in multiple organs leading to death from tumor infiltration. Moreover, leukemic clones in these mice display very low levels of Ig somatic hypermutation, recapitulating the aggressive, unmutated form of human CLL(21).
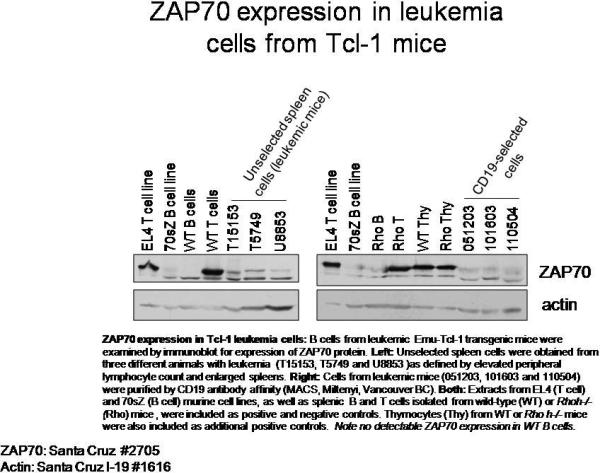
Starting at 2 months of age, the number of CD5+IgM+ CLL-like cells in peripheral blood was monitored in Eμ-TCL1-transgenic, Rhoh+/+, Rhoh+/− and Rhoh−/− mice (Figure 3A). In Eμ-TCL1 transgenic mice with normal RhoH alleles (Eμ-TCL1Tg;Rhoh+/+) or with one Rhoh allele (Eμ-TCL1Tg;Rhoh+/−), abnormal accumulation of CD5+IgM+ B cells was observed at 4 months, and the number of these cells increased dramatically thereafter. In contrast to normal murine B cells, splenic B cells from Eμ-TCL1Tg;Rhoh+/+ mice demonstrated easily detectable ZAP-70 by immunoblot (data not shown). The number of CD5+IgM+ B cells was significantly lower in Eμ-TCL1Tg;Rhoh+/− compared with Eμ-TCL1Tg;Rhoh+/+ mice, suggesting a gene-dosage effect of RhoH expression. In contrast, Eμ-TCL1Tg mice nullizygous for Rhoh (Eμ-TCL1Tg;Rhoh−/−) showed no significant increase in the number of CD5+IgM+ B cells until 5 months of age, and the disease burden remained significantly lower than in RhoH-expressing counterparts throughout the entire follow-up time (8 months). The three leukemic cell count profiles (Eμ-TCL1Tg;Rhoh+/+, Rhoh+/− and Rhoh−/−) over time were significantly different (p < 0.001). As expected, no increase of CD5+IgM+ B cells was observed in the non-transgenic controls (Rhoh+/+, Rhoh+/−, and Rhoh−/−; data not shown).
Figure 3. Loss of RhoH delays development of CLL in Eμ-TCL1 transgenic mice.

(A) RhoH deficiency delays the accumulation of CD5+IgM+ cells over time in peripheral blood of Eμ-TCL1 transgenic, Rhoh+/+, Rhoh+/− or Rhoh−/− mice. Data represent mean ± standard error of the mean (SEM). N=5-19 per time point. t-test, * indicates p<0.05 (Eμ-TCL1Tg;Rhoh+/+ vs Eμ-TCL1Tg/+;Rhoh−/−); #, p<0.05 (Eμ-TCL1Tg;Rhoh+/− vs Eμ-TCL1Tg;Rhoh−/−). Globally, the three leukemic cell count profiles (Eμ-TCL1Tg;Rhoh+/+, Rhoh+/− and Rhoh−/−) over time were significantly different (p < 0.001). All pairwise comparisons were also significantly different: Rhoh+/+ versus Rhoh+/− (p = 0.006), Rhoh+/+ versus Rhoh−/− (p < 0.001), and Rhoh+/− versus Rhoh−/− (p = 0.017). (B) Number of CD5+IgM+ cells in the spleen, bone marrow (BM) and peritoneal cavity (PC) of Eμ-TCL1Tg;Rhoh+/+ and Eμ-TCL1Tg;Rhoh−/− analyzed at 6 months of age. Data represent mean ± SEM. N=4-7. Differences were statistically significant for BM (*, p < 0.05) and showed a trend in the same direction for spleen (p = 0.11) and peritoneal cavity (p = 0.31).
To determine if these differences in peripheral blood represent abnormal distribution of cells or reduced production or survival of these cells, we assessed the number of CD5+IgM+ cells in peritoneal cavity, spleen and bone marrow in a cohort of mice at 6 months of age (N=4-7; Figure 3B). Leukemic burden was significantly decreased in the bone marrow of Eμ-TCL1Tg;Rhoh−/− compared to Eμ-TCL1Tg;Rhoh+/+ mice (5.4-fold reduction, p < 0.05) and showed a clear trend in the same direction in the spleen (2.4-fold reduction) and peritoneal cavity (2.4-fold reduction), although it did not reach statistical significance.
In order to determine whether the decreased leukemic burden was associated with reduced cell survival, we measured the levels of apoptosis in B-1/CLL cells in Eμ-TCL1Tg;Rhoh+/+ and Eμ-TCL1Tg;Rhoh−/− mice. The fraction of apoptotic, CD5+IgM+ peritoneal cells at 2 months of age did not significantly differ between Eμ-TCL1Tg;Rhoh+/+ and Eμ-TCL1Tg;Rhoh−/− mice (10.25±2.54% vs 12.85±3.43% annexin V+7-AAD− cells, n=4, p=0.27). However, CD5+IgM+ splenocytes in 6-8 month-old Eμ-TCL1Tg;Rhoh−/− showed higher levels of apoptosis (21.3±7.95% annexin V+7-AAD− cells, n=4) compared to Eμ-TCL1Tg;Rhoh+/+ mice (10.5%, n=2).
Impaired BCR-induced AKT and ERK phosphorylation in Eμ-TCL1Tg; Rhoh−/− splenocytes
In order to gain mechanistic insight into the impaired leukemia development in the absence of RhoH, the activation of several components of the BCR signal transduction pathway was analyzed in splenocytes from 3 Eμ-TCL1Tg;Rhoh+/+, 2 Eμ-TCL1Tg;Rhoh+/−and 4 Eμ-TCL1Tg;Rhoh−/− mice (Figure 4). In contrast to non-transgenic Rhoh−/− B cells, which showed normal AKT activation after BCR cross-linking (Supplementary Figure 4B), basal and agonist-induced phosphorylation of AKT (Ser473) was clearly diminished in Eμ-TCL1Tg;Rhoh−/− cells compared to their Rhoh+/+ counterparts (Figures 4A and B). Also, BCR-induced phosphorylation of ERK was clearly reduced in the absence of Rhoh (Figures 4A and 4C).
Figure 4. Impaired BCR-induced AKT phosphorylation in Eμ-TCL1Tg;Rhoh−/− splenocytes.

(A) Analysis of BCR signaling in splenocytes from Eμ-TCL1Tg;Rhoh+/+ and Eμ-TCL1Tg;Rhoh−/− mice. Cells were stimulated with 25 μg/ml anti-IgM for 1 or 5 min and analyzed by immunoblotting. (B) Densitometric quantification of AKT (Ser473) phosphorylation in splenocytes from Eμ-TCL1Tg;Rhoh+/+ (n=3), Eμ-TCL1Tg;Rhoh+/− (n=2) and Eμ-TCL1Tg;Rhoh−/− mice (n=4). Values were normalized with total AKT expression. (C) Densitometric quantification of ERK (Thr202/Tyr204) phosphorylation in splenocytes from Eμ-TCL1Tg;Rhoh+/+ (n=3), Eμ-TCL1Tg;Rhoh+/− (n=2) and Eμ-TCL1Tg;Rhoh−/− mice (n=4). Values were normalized with total ERK expression. In (B) and (C), one of the samples was used as a calibrator to allow normalization across different gels. ‡, not determined.
Discussion
RhoH is a hematopoietic-specific, GTPase-deficient member of the Rho GTPase family with high expression in lymphoid tissues(8, 22). RhoH has been shown to play an essential role in thymocyte development and TCR signaling(9, 10). In spite of the fact that RhoH is also expressed in B cells (data not shown), deletion of murine Rhoh does not substantially alter physiologic B-cell development in vivo, although changes in B cell numbers are seen in Rhoh−/− mice, probably related to T cell compartment abnormalities previously reported(9). Supporting this view, RhoH does not seem to play an essential role in B-cell activation, chemotaxis or BCR signaling in vitro. In T cells, RhoH functions as a facilitator of ZAP70 recruitment to the TCR and the immunological synapse(9) (Chae, et al., manuscript under review). Normal B cells express lower levels of ZAP70 compared to T cells(23) and BCR signal transduction is thought to be mainly mediated by the related protein tyrosine kinase, SYK(24, 25). SYK is recruited to the phosphorylated ITAM motifs of the activated BCR complex where it is activated by Src-family kinases(24), thus playing a somewhat parallel role to that of ZAP70 in TCR signaling. The different requirement of RhoH in T and B cell antigen receptor complexes likely reflects structural or biochemical dissimilarities between ZAP70 and SYK and/or differences in regulation of the two signaling complexes. For instance, SYK has much higher intrinsic tyrosine kinase activity than ZAP70(26), and can initiate immunoreceptor signaling in the absence of Src-related kinases(27).
In contrast to normal B-cells, ZAP70 is aberrantly expressed in the malignant cells of CLL with unmutated IGHV status and is associated with poor prognosis(2, 3). The reported observations that RhoH interacts with ZAP70(9) and that genetic alterations of RHOH, similar to those affecting bona fide protooncogenes (chromosomal translocations and aberrant somatic hypermutation), occurring in B-lymphoid malignancies(12, 13, 15, 16), prompted an examination of the role of RhoH in CLL. Indeed, RhoH was overexpressed in a subset of primary human CLL and the expression of RhoH was correlated to the protein levels of ZAP70 suggesting that RhoH might play a role in the pathogenesis of the disease. We did not observe significant differences in the expression of RhoH between CLL patients with mutated and unmutated IGHV genes (data not shown).
To directly investigate the role of RhoH in CLL in vivo, we used the Eμ-TCL1 transgenic mouse(17, 18) as a model of the aggressive, IGHV-unmutated subtype of CLL, which in humans correlates with high ZAP70 expression. Although TCL1 is not genetically altered in human CLL, it is expressed in the majority of the cases, and high levels of TCL1 correlate with ZAP70 expression and unmutated IGHV genes (28, 29). Inactivation of Rhoh in the Eμ-TCL1 genetic background significantly delayed disease progression as measured by the accumulation of CD5+IgM+ cells in peripheral blood and lymphoid tissues. As implied by the analysis of non-transgenic mice, this was not due to the absence of the putative target cell (the B-1 cell compartment) or to a general defect in the B cell lineage in the absence of RhoH. Rather, it suggests a specific defect in the survival or proliferation of the leukemic cell, associated with an alteration in a biochemical pathway regulated by RhoH. Thus, in our model, CD5+IgM+ splenocytes in Eμ-TCL1Tg;Rhoh−/− mice showed increased levels of apoptosis compared to their Eμ-TCL1Tg;Rhoh−/− counterparts.
Consistent with this hypothesis, activation of AKT (measured by Ser473 phosphorylation) and ERK in response to BCR crosslinking was significantly decreased in Eμ-TCL1Tg; Rhoh−/− splenocytes compared to their RhoH-expressing counterparts. This is in contrast to non-transgenic, normal splenic B cells, where activation of AKT and ERK downstream the BCR was unaffected by the loss of RhoH. The mechanism underlying this observation, and whether it is dependent on ZAP70, is currently unknown. Although TCL1 is a positive regulator of AKT activation(30), it seems unlikely that this is due to a direct regulation of TCL1 by RhoH, because the mechanism by which TCL1 activates AKT does not depend on AKT phosphorylation at Ser473 and seems to occur at the level of nuclear translocation of AKT(30).
Regardless of the specific mechanism, these findings appear to be relevant to the pathogenesis of CLL, taking into consideration that: (1) AKT is a critical regulator of cell survival, and accumulation of CLL cells has typically been attributed to their ability to escape apoptosis; (2) BCR signaling has been implicated in the pathogenesis of CLL, and this disease has been hypothesized to be initiated and driven by antigen stimulation (reviewed in(1)).
Taken together, our data strongly suggest that RhoH plays a role in the initiation and/or progression of CLL. This report provides direct experimental evidence implicating RhoH in a malignant process and defines the role of a member of the Rho GTPase family in CLL. The fact that RhoH inactivation impairs B-cell leukemogenesis, while not substantially affecting normal B cell development and function, makes RhoH a potentially attractive target for the treatment of this disease.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 5.
Acknowledgments
This work was supported by grants from the National Institutes of Health (grant CA113969 to DAW), the Deutsche Forschungsgemeinschaft (RA 1705/1-1 to IR), the St. Baldrick's Foundation (LUWM), and the Leukemia and Lymphoma Society (JCB). Patient samples and IGHV sequence data were obtained through the CLL Research Consortium (CRC).
Footnotes
The authors declare no competing financial interests.
Supplementary Information accompanies the paper on the Leukemia website (http://www.nature.com/leu).
References
- 1.Chiorazzi N, Rai KR, Ferrarini M. Chronic lymphocytic leukemia. The New England journal of medicine. 2005 Feb 24;352(8):804–815. doi: 10.1056/NEJMra041720. [DOI] [PubMed] [Google Scholar]
- 2.Crespo M, Bosch F, Villamor N, Bellosillo B, Colomer D, Rozman M, et al. ZAP-70 expression as a surrogate for immunoglobulin-variable-region mutations in chronic lymphocytic leukemia. The New England journal of medicine. 2003 May 1;348(18):1764–1775. doi: 10.1056/NEJMoa023143. [DOI] [PubMed] [Google Scholar]
- 3.Rassenti LZ, Huynh L, Toy TL, Chen L, Keating MJ, Gribben JG, et al. ZAP-70 compared with immunoglobulin heavy-chain gene mutation status as a predictor of disease progression in chronic lymphocytic leukemia. The New England journal of medicine. 2004 Aug 26;351(9):893–901. doi: 10.1056/NEJMoa040857. [DOI] [PubMed] [Google Scholar]
- 4.Chen L, Widhopf G, Huynh L, Rassenti L, Rai KR, Weiss A, et al. Expression of ZAP-70 is associated with increased B-cell receptor signaling in chronic lymphocytic leukemia. Blood. 2002 Dec 15;100(13):4609–4614. doi: 10.1182/blood-2002-06-1683. [DOI] [PubMed] [Google Scholar]
- 5.Chen L, Apgar J, Huynh L, Dicker F, Giago-McGahan T, Rassenti L, et al. ZAP-70 directly enhances IgM signaling in chronic lymphocytic leukemia. Blood. 2005 Mar 1;105(5):2036–2041. doi: 10.1182/blood-2004-05-1715. [DOI] [PubMed] [Google Scholar]
- 6.Chen L, Huynh L, Apgar J, Tang L, Rassenti L, Weiss A, et al. ZAP-70 enhances IgM signaling independent of its kinase activity in chronic lymphocytic leukemia. Blood. 2008 Mar 1;111(5):2685–2692. doi: 10.1182/blood-2006-12-062265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Williams DA, Zheng Y, Cancelas JA. Rho GTPases and regulation of hematopoietic stem cell localization. Methods Enzymol. 2008;439:365–393. doi: 10.1016/S0076-6879(07)00427-2. [DOI] [PubMed] [Google Scholar]
- 8.Li X, Bu X, Lu B, Avraham H, Flavell RA, Lim B. The hematopoiesis-specific GTP-binding protein RhoH is GTPase deficient and modulates activities of other Rho GTPases by an inhibitory function. Molecular and cellular biology. 2002;22(4):1158–1171. doi: 10.1128/MCB.22.4.1158-1171.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gu Y, Chae H, Siefring J, Jast iJ, Hildeman D, Williams DA. RhoH, a GTPase recruits and activates Zap70 required for T cell receptor signaling and thymocyte development. Nat Immunol. 2006 Nov;7(11):1182–1190. doi: 10.1038/ni1396. [DOI] [PubMed] [Google Scholar]
- 10.Dorn T, Kuhn U, Bungartz G, Stiller S, Bauer M, Ellwart J, et al. RhoH is important for positive thymocyte selection and T-cell receptor signaling. Blood. 2007 Mar 15;109(6):2346–2355. doi: 10.1182/blood-2006-04-019034. [DOI] [PubMed] [Google Scholar]
- 11.Dallery E, Galiegue-Zouitina S, Collyn-d'Hooghe M, Quief S, Denis C, Hildebrand MP, et al. TTF, a gene encoding a novel small G protein, fuses to the lymphoma- associated LAZ3 gene by t(3;4) chromosomal translocation. Oncogene. 1995;10(11):2171–2178. [PubMed] [Google Scholar]
- 12.Preudhomme C, Roumier C, Hildebrand MP, Dallery-Prudhomme E, Lantoine D, Lai JL, et al. Nonrandom 4p13 rearrangements of the RhoH/TTF gene, encoding a GTP- binding protein, in non-Hodgkin's lymphoma and multiple myeloma. Oncogene. 2000;19(16):2023–2032. doi: 10.1038/sj.onc.1203521. [DOI] [PubMed] [Google Scholar]
- 13.Pasqualucci L, Neumeister P, Goossens T, Nanjangud G, Chaganti RS, Kuppers R, et al. Hypermutation of multiple proto-oncogenes in B-cell diffuse large-cell lymphomas. Nature. 2001 Jul 19;412(6844):341–346. doi: 10.1038/35085588. [DOI] [PubMed] [Google Scholar]
- 14.Hiraga J, Katsumi A, Iwasaki T, Abe A, Kiyoi H, Matsushita T, et al. Prognostic analysis of aberrant somatic hypermutation of RhoH gene in diffuse large B cell lymphoma. Leukemia. 2007 Aug;21(8):1846–1847. doi: 10.1038/sj.leu.2404717. [DOI] [PubMed] [Google Scholar]
- 15.Gaidano G, Pasqualucci L, Capello D, Berra E, Deambrogi C, Rossi D, et al. Aberrant somatic hypermutation in multiple subtypes of AIDS-associated non-Hodgkin lymphoma. Blood. 2003 Sep 1;102(5):1833–1841. doi: 10.1182/blood-2002-11-3606. [DOI] [PubMed] [Google Scholar]
- 16.Montesinos-Rongen M, Van Roost D, Schaller C, Wiestler OD, Deckert M. Primary diffuse large B-cell lymphomas of the central nervous system are targeted by aberrant somatic hypermutation. Blood. 2004 Mar 1;103(5):1869–1875. doi: 10.1182/blood-2003-05-1465. [DOI] [PubMed] [Google Scholar]
- 17.Bichi R, Shinton SA, Martin ES, Koval A, Calin GA, Cesari R, et al. Human chronic lymphocytic leukemia modeled in mouse by targeted TCL1 expression. Proceedings of the National Academy of Sciences of the United States of America. 2002 May 14;99(10):6955–6960. doi: 10.1073/pnas.102181599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Johnson AJ, Lucas DM, Muthusamy N, Smith LL, Edwards RB, De Lay MD, et al. Characterization of the TCL-1 transgenic mouse as a preclinical drug development tool for human chronic lymphocytic leukemia. Blood. 2006 Aug 15;108(4):1334–1338. doi: 10.1182/blood-2005-12-011213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Verbeke G, Molenberghs G. Linear mixed models for longitudinal data. Springer Verlag; New York: 2000. [Google Scholar]
- 20.SAS/STAT® user's guide. 1, 2, 3. SAS Institute, Inc.; Cary, NC: 2002. [Google Scholar]
- 21.Yan XJ, Albesiano E, Zanesi N, Yancopoulos S, Sawyer A, Romano E, et al. B cell receptors in TCL1 transgenic mice resemble those of aggressive, treatment-resistant human chronic lymphocytic leukemia. Proceedings of the National Academy of Sciences of the United States of America. 2006 Aug 1;103(31):11713–11718. doi: 10.1073/pnas.0604564103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gu Y, Jasti AC, Jansen M, Siefring JE. RhoH, a hematopoietic-specific Rho GTPase, regulates proliferation, survival, migration, and engraftment of hematopoietic progenitor cells. Blood. 2005 Feb 15;105(4):1467–1475. doi: 10.1182/blood-2004-04-1604. [DOI] [PubMed] [Google Scholar]
- 23.Scielzo C, Camporeale A, Geuna M, Alessio M, Poggi A, Zocchi MR, et al. ZAP-70 is expressed by normal and malignant human B-cell subsets of different maturational stage. Leukemia. 2006 Apr;20(4):689–695. doi: 10.1038/sj.leu.2404138. [DOI] [PubMed] [Google Scholar]
- 24.Law CL, Sidorenko SP, Chandran KA, Draves KE, Chan AC, Weiss A, et al. Molecular cloning of human Syk. A B cell protein-tyrosine kinase associated with the surface immunoglobulin M-B cell receptor complex. J Biol Chem. 1994 Apr 22;269(16):12310–12319. [PubMed] [Google Scholar]
- 25.Chan AC, van Oers NS, Tran A, Turka L, Law CL, Ryan JC, et al. Differential expression of ZAP-70 and Syk protein tyrosine kinases, and the role of this family of protein tyrosine kinases in TCR signaling. J Immunol. 1994 May 15;152(10):4758–4766. [PubMed] [Google Scholar]
- 26.Latour S, Chow LM, Veillette A. Differential intrinsic enzymatic activity of Syk and Zap-70 protein-tyrosine kinases. J Biol Chem. 1996 Sep 13;271(37):22782–22790. doi: 10.1074/jbc.271.37.22782. [DOI] [PubMed] [Google Scholar]
- 27.Zoller KE, MacNeil IA, Brugge JS. Protein tyrosine kinases Syk and ZAP-70 display distinct requirements for Src family kinases in immune response receptor signal transduction. J Immunol. 1997 Feb 15;158(4):1650–1659. [PubMed] [Google Scholar]
- 28.Narducci MG, Pescarmona E, Lazzeri C, Signoretti S, Lavinia AM, Remotti D, et al. Regulation of TCL1 expression in B- and T-cell lymphomas and reactive lymphoid tissues. Cancer Res. 2000 Apr 15;60(8):2095–2100. [PubMed] [Google Scholar]
- 29.Herling M, Patel KA, Khalili J, Schlette E, Kobayashi R, Medeiros LJ, et al. TCL1 shows a regulated expression pattern in chronic lymphocytic leukemia that correlates with molecular subtypes and proliferative state. Leukemia. 2006 Feb;20(2):280–285. doi: 10.1038/sj.leu.2404017. [DOI] [PubMed] [Google Scholar]
- 30.Pekarsky Y, Koval A, Hallas C, Bichi R, Tresini M, Malstrom S, et al. Tcl1 enhances Akt kinase activity and mediates its nuclear translocation. Proceedings of the National Academy of Sciences of the United States of America. 2000 Mar 28;97(7):3028–3033. doi: 10.1073/pnas.040557697. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.