

Abstract
Substantial progress in RNA biology highlights the importance of RNAs (e.g., microRNAs) in diseases and the potential of targeting RNAs for drug discovery. However, the lack of RNA-specific modeling techniques demands for development of new tools for RNA-targeted rational drug design. Herein, we implemented integrated approaches of accurate RNA modeling and virtual screening for RNA inhibitor discovery with the most comprehensive evaluation to date of five docking and 11 scoring methods. For the first time, statistical analysis was heavily employed to assess the significance of our predictions. We found that GOLD:GOLD Fitness and rDock:rDock_solv could accurately predict the RNA ligand poses, and ASP rescoring further improved the ranking of ligand binding poses. Due to the weak correlations (R2<0.3) of existing scoring with experimental binding affinities, we implemented two new RNA-specific scoring functions, iMDLScore1 and iMDLScore2, and obtained better correlations with R2=0.70 and 0.79, respectively. We also proposed a multi-step virtual screening approach and demonstrated that rDock:rDock_solv together with iMDLScore2 rescoring obtained the best enrichment on the flexible RNA targets, whereas GOLD:GOLD Fitness combined with rDock_solv rescoring outperformed other methods for rigid RNAs. This study provided practical strategies for RNA modeling and offered new insights into RNA-small molecule interactions for drug discovery.
Keywords: RNA-ligand interactions, molecular docking, scoring function, virtual screening, statistical analysis
Introduction
The recent substantial progress in RNA biology underscores the importance of RNA in normal and aberrant cellular functions. It also highlights the potential of targeting RNA for treatment of a multitude of diseases including bacterial/viral infection 1, 2 and cancer 3, 4. RNAs can form well-defined tertiary structures, such as double helices, hairpins, bulges, and pseudoknots, which offer structural basis for designing therapeutic agents. As a matter of fact, some structured RNAs, including bacterial 16S ribosomal RNAs (rRNA), HIV-1 TAR RNAs and small non-coding microRNAs (miRNAs), exhibit attractive structural and functional characteristics similar to proteins 5. We have linked miRNAs to different diseases including cancer 6, 7, and recently embarked on the discovery of small molecule inhibitors targeting miRNAs (SMIR) 8, 9. Druggable RNA targets are largely unexplored 10, and RNA inhibitors, such as aminoglycosides which are for the most well-defined RNA target -- 16S rRNA A-site, usually have poor selectivity or oral bioavailability. There have been several reports in targeting prokaryotic rRNA A-site 11-13, HIV-1 TAR RNA 14-16, and riboswitches 17-19 with small molecules. Researchers are also exploring new generations of drug-like molecules targeting pathogenic or human disease-related RNAs including CUG- or CCUG-repeated mRNA 20-22, miRNA 23, 24, and internal ribosome entry site (IRES) 25, 26. These studies provided proof of principle that RNAs can be specifically targeted for antiviral or anticancer therapeutic development.
Structure-based modeling techniques, such as molecular docking, have been widely used in the field of protein-targeted drug discovery; however, most of these in silico methodologies were developed for proteins. Mature tools specific for RNAs (e.g. virtual screening for RNA inhibitor identification) are lacking 27. Thus, there is an unmet need to exploit current computational tools and implement new ones for RNA modeling such as fast screening of small molecules against RNA targets. To this end, Li et al. evaluated two docking programs (GOLD and Glide) and concluded that they are helpful in RNA-based drug discovery 28. DOCK6 with implicit solvent models, i.e. GB/SA and PB/SA models, was used to model RNAs and obtained low root mean square deviations (RMSD) 10. AutoDock4 was also modified to dock RNA targets with flexible grids and achieved some degree of success 29, 30. The knowledge-based DrugScoreRNA was parameterized with 670 RNA-ligand and -protein complexes but only a fair correlation with the experimental binding affinities was observed for a limited set of 15 RNA-ligand complexes 31. RiboDock (currently as rDock) has proved to be successful in several drug design cases 12, 32, 33. The latest version of rDock employed a fast weighted solvent accessible surface areas (WSAS) to approximate the solvent accessible areas 32. MORDOR (molecular recognition with a driven dynamics optimizer) 34 was implemented with induced-fit algorithms for flexible RNA docking. However, as MORDOR is computationally expensive, it is not feasible to screen a large chemical database in an efficient manner. Furthermore, we found that the docking parameters widely used in proteins are not always applicable to RNA systems. For instance, the electrostatic interactions between RNA phosphates and ligands can be overestimated 10, 27, 35, while the desolvation term also needs to be modified 29.
To date, due to the challenge of RNA modeling and less developed programs in this field, there have been no reports that provided feasible approaches to address the three critical issues in docking: the accuracy of binding mode prediction, the ranking performance in virtual screening, and the accurate scoring 36. Our study aims to identify cost-effective combinations of existing techniques and develop new docking strategies for RNAs. Therefore, we comprehensively evaluated five popular docking programs, including GOLD 5.0.1 37, Glide 5.6 38, Surflex 2.415 39, AutoDock 4.1 40, 41 and rDock 2006.2 32, along with 11 scoring functions to explore their capability in RNA docking. Based on our study, we proposed the workflow for structure-based modeling for RNA-targeted drug discovery. As illustrated in Fig. 6, appropriate docking/scoring combinations need to be used to obtain the best virtual screening performance based on the flexibility of binding sites. To maximize the modeling accuracy and improve the enrichment, different modeling strategies can be rationally applied at three critical quality-control steps: near-native pose coverage, pose selection, and hit selection. For the first time, intensive statistical analysis was employed in such studies, and we also implemented RNA-specific scoring functions with the ever largest high-quality RNA-ligand binding affinity dataset. Moreover, our study provided new insights of RNA-small molecule interactions, and this helped us explore the best computational strategies for RNA modeling in drug discovery and development.
Figure 6.

The suggested workflow for structure-based virtual screening for RNA-targeted inhibitor discovery.
Results
Reproduction of experimental binding modes
We first examined whether the current protein-ligand derived docking/scoring software could reproduce the ligand binding poses similar to the experimental structures. Ideally, a “good” RNA docking program should be able to sample all the conformational space and identify at least one near-native pose. Table 1 showed that, if we arbitrarily employed C(5, 3.0Å) (the top 5 ranked pose includes at least one near-native pose with RMSD<3.0Å) to define a successful docking case, GOLD:GOLD Fitness and rDock:rDock_solv outperformed others, both with 73.21% success rate. Additionally, GOLD:ChemScore, GOLD:ASP, Glide:GlideScore(SP), Glide:Emodel(SP) and rDock:rDock obtained more than 50% docking success rate. In contrast, the success rate for Glide:GlideScore(XP), Glide:Emodel(XP), Surflex and AutoDock4.1 (default) were low, ranging from 30.36% to 44.64%. All programs, especially AutoDock4.1 and Surflex, had weak performance (<60%) on flexible and extensively-charged aminoglycosides. When stringent cutoffs such as C(3, 1.5Å) were used, the accuracy decreased but GOLD:GOLD Fitness and rDock:rDock_solv kept as the best (>40%). Results for other programs are available in Table 1.
Table 1.
Successful binding mode reproduction of the 56 RNA-ligand complexes using different docking/scoring combinations with arbitrary cutoffs. The values in the brackets indicated the total number of complexes in that category. The values before the parentheses results satisfying C(5, 3.0Å), and the values in the parentheses were derived with the stringent criterion C(3, 1.5Å) (described in the Materials and Methods). The Glide XP mode could not obtain enough data with the stringent restraints, thus the VUS values and R2 were not available.
| GOLD 5.0.1 | AutoDock 4.1 | Surflex 2.415 | Glide 5.6 | rDock 2006.2 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
|
|
|
|
|||||||
| GOLD Fitness | ChemScore | ASP | Autodock4.1 Score | Surflex-dock Score | GlideScore (SP) | Emodel (SP) | GlideScore (XP) | Emodel (XP) | rDock | rDock_solv | |
| Aminoglycoside [26] | 18 (9) | 13 (3) | 15 (9) | 1 (1) | 4 (2) | 12 (3) | 13 (3) | 4 (2) | 4 (2) | 13 (6) | 16 (8) |
| Small Molecule [30] | 23 (15) | 17 (10) | 22 (15) | 16 (9) | 21 (13) | 18 (13) | 18 (13) | 16 (11) | 15 (11) | 21 (13) | 25 (15) |
| X-ray crystal [36] | 29 (19) | 26 (13) | 29 (21) | 13 (8) | 17 (11) | 20 (9) | 21 (9) | 12 (8) | 12 (8) | 24 (16) | 29 (17) |
| NMR [20] | 12 (5) | 4 (0) | 8 (3) | 4 (2) | 8 (4) | 10 (7) | 10 (7) | 8 (5) | 7 (5) | 10 (3) | 12 (6) |
| Total [56] | 41 (24) | 30 (13) | 37 (24) | 17 (10) | 25 (15) | 30 (16) | 31 (16) | 20 (13) | 19 (13) | 34 (19) | 41 (23) |
| Overall Success Rate % | 73.21 (42.86) | 53.57 (23.21) | 66.07 (42.86) | 30.36 (17.86) | 44.64 (26.79) | 53.57 (28.57) | 55.36 (28.57) | 35.71 (23.21) | 33.93 (23.21) | 60.71 (33.93) | 73.21 (41.07) |
| VUS % | 78.11 | 65.48 | 70.17 | 43.30 | 55.22 | 65.41 | 66.01 | NA | NA | 63.09 | 73.13 |
| Score-binding affinity correlation R2 a | 0.25 | 0.03 | 0.29 | 0.22 | 0.05 | 0.10 | 0.14 | NA | NA | 0.15 | 0.18 |
excluding 1TOB, 2TOB and 1LVJ when calculating Pearson correlation coefficient
As expected, we observed that the average docking accuracy on crystal structures was higher than that on NMR structures for all of 11 current docking/scoring combinations (58.84% versus 42.27%, p=0.06). Not surprisingly, the pose reproduction performance on small-molecule RNA ligands was remarkably better than that on flexible aminoglycosides with high statistical significance (64.55% versus 39.51%, p<0.01). Among the failed cases (defined as “two or less docking programs are able to reproduce the near-native structure (RMSD<3.0Å) among top 5 scored poses”), five are crystal structures (2O3V, 2BE0, 2FD0, 2PWT and 2Z75) and seven are NMR structures (1UUD, 1LVJ, 1TOB, 1AKX, 1EI2, 1KOD and 1QD3). We found that the current methods were usually less accurate on RNA complexes containing large aminoglycosides (e.g. lividomycin, paromomycin, etc.), weak RNA binders (e.g. arginine and citrulline), or phosphate-containing hydrophilic ligands (glucosamine 6-phosphate). Because the negatively charged moieties can form specific interactions with RNA phosphates in the presence of metal ions acting as the “metal bridge” 42, such as 2GDI and 2Z74, we tried docking with consideration of metal ions. As expected, we could significantly improve the pose prediction of the diphosphate tail of thiamine diphosphate in 2GDI when the Mg2+ ion was taken into account as part of RNA targets. When compared with rDock:rDock_solv, the GOLD:GOLD Fitness combination achieved better performance for the pose reproduction on aminoglycosides-RNA complexes such as 1J7T, 2FCZ, 2BE0, 1NEM and 2TOB, whereas rDock:rDock_solv was more accurate for drug-like ligands including 2Z74, 2Z75, 1EHT and 1AKX. The detailed results (scores, RMSD and statistics) are available in Supplementary Table S3-S4.
To better demonstrate the relation of the docking accuracy with RMSD and ranking, we illustrated our results with Fig. 1A, in which the heavy-atom RMSD and the ranking of pose were considered at the same time. The volume under the surface (VUS) represents the overall capacity of reproducing the near-native binding modes. It showed that GOLD:GOLD Fitness achieved the best VUS (78.11%), while rDock:rDock_solv was the second best (Table 1 and Fig. 1A). We proposed to employ the contour of 50% success rate to guide the pose selection in RNA docking: if one aims to cover at least one near-native pose (RMSD<3.0Å) in 50% of the ligands, at least the top five poses should be kept when using GOLD:GOLD Fitness. In contrast, we should keep at least top 20 poses to achieve 50% success for Surflex and AutoDock 4.1 (Fig. 1B). From these assessments, we suggest that GOLD:GOLD Fitness and rDock:rDock_solv be the best methods for pose predictions in RNA docking.
Figure 1.
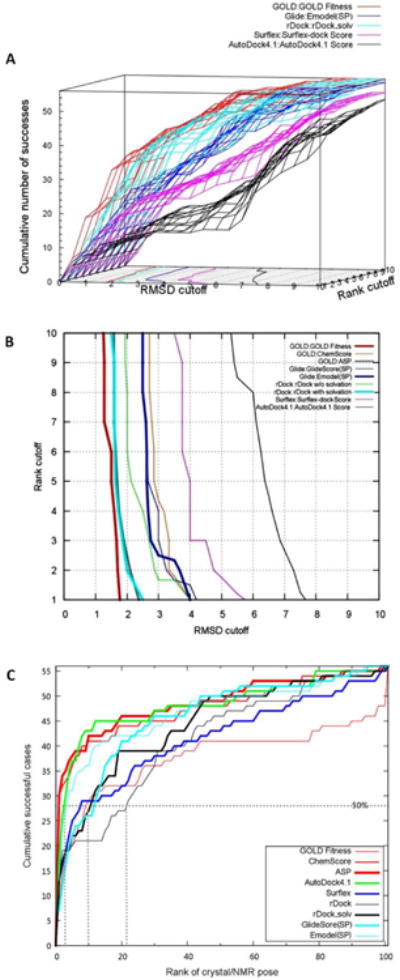
The 3D cumulative success rate from the binding mode reproduction experiments. (A). The cumulative success rate in 3D representation was calculated based on a series of C(X, YÅ). Only scoring functions which obtained the highest VUS values for each docking program were selected for illustration. The contour on the XY (RMSD-Rank) plane represented the 50% (Z=28) success rate (the binding mode can be reproduced for 50% of RNA-ligand complexes); (B). The 50% success contour (Z=28) for all available scoring functions (GlideScore (XP) and Emodel (XP) were not included due to the unavailability of VUS values). (C). The cumulative success rate for 56 RNA-ligand complexes based on the ranking of X-ray/NMR determined poses against 100 decoys. The 50% success line and the corresponding rankings to achieve 50% success were shown as dots.
Rescoring to improve near-native binding pose ranking
To improve the pose ranking accuracy with rescoring, we assessed the scoring function capability of differentiating the ligand crystal/NMR structures from their decoy poses. This was done by investigating two parameters: the ranking of native poses, and the Spearman's correlation between scores and RMSDs.
Generally, an ideal scoring function should rank the crystal/NMR and very near-native poses statistically higher than other decoys. Since GOLD:GOLD Fitness outperformed other docking programs on the coverage of the near-native poses as aforementioned, it was utilized to generate 100 decoys for each target in the hope to cover a wide range of poses, from near-native to decoys. We investigated whether a given scoring function could obtain the highest rankings for experimentally determined ligand poses. Analogous to IC50 (in assessing biological activity), we used 50% success rate to evaluate the performance of different docking/scoring methods. As demonstrated in Fig. 1C, the 50% success rate line (dashed) clustered these scoring functions into three groups: ASP, ChemScore, AutoDock4.1 Score and Emodel (SP) were the first group; the second group included other scoring functions, except rDock which ranked the lowest as the third group. Fig. 1C indicated that GOLD Fitness has 50% of possibility to rank the native ligand conformation within top 10% of the predicted poses, whereas for ASP, ChemScore, AutoDock4.1 Score and Emodel (SP), this value was reduced to top 5%. The native pose ranking performance for different docking/scoring schemes varied with different RNA target structures. For example, most programs performed significantly better for crystal structures than NMR structures (69.14% versus 38.89%, p<0.01) with the top 10 as the cutoff to define a successful ranking case. Surprisingly, ASP was remarkably better in crystal structure ranking, in which only two targets (2O3V and 3DIL) failed, while AutoDock4.1 outperformed others on ranking NMR structures. Taken together, these data suggested that RNA targets with different structural resolutions should be rescored with respective appropriate scoring functions (e.g., ASP or AutoDock4.1) after the initial step of docking with GOLD:GOLD Fitness or rDock:rDock_solv. Detailed results of this ranking study are available in Supplementary Table S5.
The score-RMSD correlation is another parameter related to the ranking capability. Here we investigated this parameter using Spearman's rank correlation because it is known that RMSD is not linearly correlated with docking scores 29. Our study showed that in most cases RMSD and docking scores were positively correlated, as desired, but the correlations varied for different RNA targets. Thus, we grouped the 56 RNA targets based on the strength of correlation for each scoring function (see Materials and Methods). ASP, GlideScore (SP) and Emodel (SP) were the best three scoring functions which had most cases with moderate or strong correlation between RMSD and score (Table 2). rDock, rDock_solv and Surflex-dock scores obtained fair performance, which could derive weak or strong correlations for more than 1/3 of cases. Surprisingly, GOLD Fitness could not achieve satisfactory performance to enrich the near-native ligand conformations (44 cases obtained the weak correlations) (Table 2). Combined with the native pose ranking analysis, these results demonstrated that other scoring functions such as ASP could enrich the near-native poses when applied to decoy poses generated by GOLD:GOLD Fitness.
Table 2.
Score-RMSD Spearman's rank correlations for various scoring functions. The values indicated the number of RNA-ligand complexes fit in each correlation category (Weak: ρ<0.3, Moderate: 0.3≤ρ<0.5, Strong: ρ≥0.5).
| Weak | Moderate | Strong | |
|---|---|---|---|
| GOLD Fitness | 44 | 5 | 7 |
| ChemScore | 41 | 7 | 8 |
| ASP | 33 | 15 | 8 |
| GlideScore (SP) | 31 | 15 | 10 |
| Emodel (SP) | 29 | 14 | 13 |
| Surflex-dock Score | 38 | 12 | 6 |
| AutoDock4.1 Score | 40 | 5 | 11 |
| rDock | 35 | 12 | 9 |
| rDock_solv | 36 | 12 | 8 |
As we have identified ASP as the best scoring function for ranking RNA ligand poses, we wanted to further study whether it could improve the identification of the near-native binding poses generated by GOLD:GOLD Fitness. Supplementary Table S4 showed that when all top 10 poses generated by GOLD:GOLD fitness were rescored by the ASP scoring function, the number of RNA targets satisfying C(5, 3.0Å) increased from 41 to 44, while this number for C(3, 1.5Å) increased from 24 to 30, compared to original GOLD:GOLD Fitness performance. Specifically, we observed that the best RMSD in top 5-scored docking conformations of 2GDI, 2Z74, 2PWT and 1ZZ5 was significantly reduced (below 3.0Å) after ASP rescoring (Supplementary Figure 1 and Supplementary Table S4). In contrast, GOLD:GOLD Fitness alone failed to identified the near-native conformation for these targets. Furthermore, the VUS increased from 78.11% to 79.18%. Compared with the docking accuracy using GOLD:GOLD Fitness alone, the average RMSD for the top-scored conformations was further reduced to 2.61±0.38Å after ASP rescoring (Supplementary Table S4). Combined with native pose ranking and RMSD-score correlation results, our results confirmed that ASP has the best ability for pose ranking, and ASP rescoring can significantly enrich the near-native decoys generated by GOLD:GOLD Fitness for pose reproduction purpose in RNA docking.
Development of new RNA-specific scoring functions for RNA-ligand binding affinity prediction
As most of the scoring functions used to predict RNA-ligand binding affinity were optimized for protein-ligand interactions, it demands for careful evaluation of existing scoring schemes and implementation of new RNA-specific methods. Since the determination of RNA-ligand binding affinities depends on multiple factors (e.g., temperature, assay method, etc.), we carefully curated 45 RNA complexes with reported binding affinities (Kd) (Supplementary Table S2). The score-binding affinity correlations were calculated for the selected scoring functions. As expected, the correlation coefficients (R2) for all evaluated scoring functions were low (R2<0.3). The values were in Table 1, and the score-binding affinity plots for the best three scoring functions (ASP, GOLD Fitness and AutoDock4.1 Score (default)) are available in Supplementary Figure 2.
To improve the result, we developed scoring functions with our large RNA-ligand dataset with available binding affinities. This was done by optimizing AutoDock4.1 scoring terms using multi-linear regression (MLR) methods. During the implementation, only four terms were optimized because 1. The charge-based desolvation energy function employed by AutoDock4.1 was based on Wesson & Eisenberg's model 43, which was entirely trained by protein targets; 2. The solvent accessible surface area calculation in this model was simplified by using inter-atomic contact radius 41. Upon MLR optimization, the contributions of those scoring terms are 0.1460 for vdW, 0.0745 for hbond, 0.0559 for electrostatic, and 0.3073 for torsions. This new score functions was named as iMDLScore1. Since the training set for iMDLScore1 includes diverse RNA binders, varying from tiny hypoxanthines to large aminoglycosides, we observed a significant contribution of vdW interactions. As illustrated in Fig. 2A, we achieved a much better correlation (R2 = 0.70) between docking scores and binding affinities after optimizing the coefficients of AutoDock4.1 scoring terms. When iMDLScore1 was further validated against an external test set consisting of eight complexes, the correlation coefficient between the score and binding affinity was 0.82, and the root-mean-square error (RMSE) of prediction was as low as 4.09kJ/mol.
Figure 2.

Correlation between docking scores and experimental binding affinity for iMDLScore1 and iMDLScore2.
A known challenge in RNA virtual screening is how to enrich the active compounds from a focused library with charged molecules because most RNA binders are potentially positively charged 27. To overcome this problem, we derived a second scoring function, iMDLScore2, with a dataset containing 18 complexes with positively charged ligands. In iMDLScore2 the contribution of the electrostatic term to the docking scores was over 10%. Interestingly, R2 and Q2 (leave-one-out cross validation R2) for the training set reached 0.79 and 0.62, respectively (Fig. 2B), and R2 for the test set was 0.76. Additionally, the RMSE of prediction was comparable to that of iMDLScore1 (4.35kJ/mol). Q2, R2 and RMSE of prediction indicated the good predictive capability of RNA-ligand binding affinities by iMDLScore2. The new coefficients in iMDLScore2 were 0.1634 (vdW), 0.2436 (hbond), 0.2311 (electrostatic), and 0.2212 (torsion). Obviously, the contributions of electrostatic and hbond were increased. This result indicated that polar non-bonded interactions are critical attributes to accurately predict the relative binding affinity for charged ligands. All results and parameters for the new scoring functions could be found in Table 3. We evaluated the performance of these new scoring functions in the virtual screening section.
Table 3.
Contributions of AutoDock4.1 energetic terms and associated R2, Q2 and RMSE of prediction.
| Parameter | Default | iMDLScore1 | iMDLScore2 |
|---|---|---|---|
| vdW | 0.1662 | 0.146 | 0.1634 |
| hbond | 0.1209 | 0.07451 | 0.2436 |
| electrostatic | 0.1406 | 0.05593 | 0.2311 |
| desolvation | 0.1322 | 0 | 0 |
| torsion | 0.2983 | 0.3073 | 0.2212 |
|
| |||
| Number of complexes as training set | NA | 25 | 18 |
| R2 (training set) | 0.22 | 0.70 | 0.79 |
| LOO-Q2 (training set) | NA | 0.44 | 0.62 |
| R2 (test set) | NA | 0.82 | 0.76 |
| RMSE of prediction (kJ/mol, test set) | NA | 4.09 | 4.35 |
Identification of rescoring scheme to improve the cross-docking performance
We employed cross-docking study to explore the best docking/scoring strategy to identify the cognate ligands for their corresponding RNA targets. Among the 56 RNA complexes, we selected a diversity subset of 16 crystal structure and 7 NMR structures for which we obtained good pose reproduction rates. As anticipated, no single docking program could lead to satisfactory enrichment in term of the average rankings of cognate ligands (e.g. average ranking below 5 out of 23). Glide:GlideScore (SP) achieved best average ranking of the cognate ligands (6.70±2.63), whereas GOLD-associated scoring functions had similar performances: 7.22±2.45 for GOLD Fitness, 7.13±3.01 for ChemScore, and 7.17±2.71 for ASP (Fig. 3). Glide:GlideScore (SP) acquired the best recovery AUC of cross-docking (74.66%), but the performance of Glide:Emodel (SP) was the least ideal (AUC=58.61%).
Figure 3.
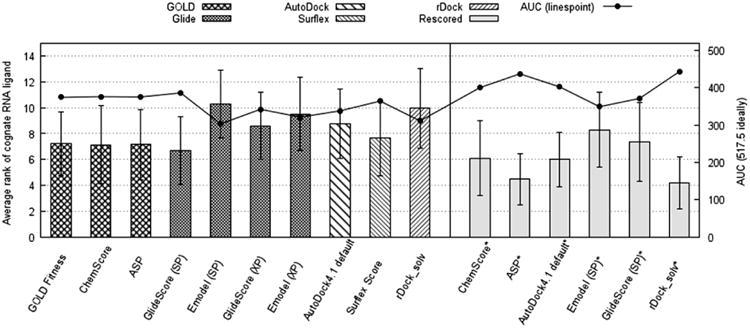
The average ranking of the cognate ligands and AUC in the cross-docking study. The height of boxes represented the average ranking of the cognate ligands of the 23 RNA targets, while the linespoints are for the AUC. The AUC was calculated from the cumulative number of RNA targets in which the ranking of the cognate ligands was below different cutoffs. The error bars represented the 95% confidence interval from the 23 cross-docking cases. The “*” represented the method of rescoring based on the top 10 predicted poses by GOLD:GOLD Fitness. Ideally, AUC is 517.5 if all cognate ligands are ranked on as the top 1.
To improve the cross-docking performance, we rescored the top 10 poses generated by GOLD:GOLD Fitness with ChemScore, ASP, AutoDock4.1 Score, Emodel (SP), GlideScore (SP) and rDock_solv, because these scoring functions performed well in ranking poses (Fig. 1C). Fig. 3 and Supplementary Table S6 demonstrated that rDock_solv rescoring achieved the best average ranking of cognate ligands (4.2±2.0) and recovery AUC (85.69%). It ranked 16 cognate ligands on top 3; As a comparison, Glide:GlideScore (SP), the best cross-docking program above, could only rank 10 cognate ligands on top 3. When compared with the initial result from GOLD:GOLD Fitness, rDock_solv rescoring improved the ranking of 17 cognate ligands. The ASP rescoring was the second best (after rDock_solv) with average ranking of 4.4±1.9 and recovery AUC=84.53%. It improved the ranking of cognate ligands in 15 cases. Therefore, as expected, we confirmed that rescoring could significantly improve both average ranking of cognate ligands and recovery AUC.
When comparing the average rankings for low-resolution structures with high-resolution ones, we could group these scoring functions into two categories: soft-core scoring and hard-core scoring. For instance, we think the AutoDock4.1 scoring function is relatively “soft”, which obtained better cognate ligand ranking for low-resolution RNA structures that may contain unfavorable structural errors (e.g. clashes) 44. In contrast, ASP and rDock_solv statistically achieved significantly better performance (p<0.05) on high-resolution crystal structures, indicating that they are hard-core scoring methods capable of penalizing structural defects (Supplementary Table S6). These characterizations were consistent to the results from our above native pose ranking evaluation, where AutoDock4.1 also achieved better performance for NMR structures while ASP was more accurate for ranking high-resolution crystal structures. Moreover, these findings are critical for selection of proper scoring functions for RNA virtual screening based on the RNA structural flexibility (see below).
Implementation of a two-step scheme for virtual screening
Receptor flexibility remains a challenge in structure-based virtual screening 45. This has been partially addressed by applying soft potentials, rotamer libraries, and molecular dynamic simulations during/after docking 46. Herein, we aim to identify the most rational combination of docking/scoring/rescoring strategies for RNA virtual screening. We utilized two different RNA targets: the bacterial 16S rRNA A-site and the lysine riboswitch, representing two typical kinds of RNA targets: bulge-containing A-form helix and aptamer, respectively.
We first quantitatively characterize the flexibility 16S rRNA A-site (PDB ID: 1J7T) by comparing the B-factors of the active site residues (4Å around paromomycin) with other non-terminal residues. Supplementary Figure 4A showed that the B-factors of active site were statistically higher than other part of the RNA (p=0.002), indicating that rRNA A-site is a flexible target. Furthermore, normal mode analysis with oGNM 47 confirmed this local flexibility because significant fluctuation of the A-site residues (arrow highlighted) could be observed within five lowest-frequency modes (low-frequency motions are expected to have larger contribution to the conformational changes 48). In particular, four critical A-site residues (A16, A17, A38 and A39, equivalent to A1492 and A1493 based on E. coli rRNA nucleotide numbering) were predicted to have the largest atomic fluctuations (Supplementary Figure 4B). Our analyses agreed with the finding that great conformational changes occur upon paromomycin binding and that A1492 and A1493 nucleosides can fluctuate between extra- and intra-helical states in molecular dynamic studies 49, 50.
The virtual screening against the 16S rRNA A-site showed that, for 31 aminoglycoside mimetics and 11 aminoglycosides, both GOLD:GOLD Fitness and rDock:rDock_solv achieved extremely excellent virtual screening enrichment against MayBridge drug-like decoys (ROC AUC=1.0, Table 4). However, they could not obtain reasonable results for the Foloppe dataset 12. rDock:rDock_solv obtained the best enrichment with only AUC=0.61, while for GOLD:GOLD Fitness the AUC is 0.58 (Table 4). Therefore, we concentrated on exploring novel strategies to improve the enrichment of virtual screening of drug-like, positively charged compounds (Foloppe dataset). Surprisingly, GOLD:GOLD Fitness coupled with rDock_solv or ASP rescoring did not improve the results (Fig. 4A and Table 4). However, the enrichment was significantly increased by rescoring either rDock:rDock_solv or GOLD:GOLD Fitness generated poses using our new scoring function iMDLScore2 with AUC=0.74 and 0.69, respectively (Fig. 4A and Table 4). The AUC difference between one-step and two-step virtual screening indicated that, if the binding modes are well covered (e.g., by GOLD:GOLD Fitness or rDock:rDock_solv), rescoring (e.g., by iMDLScore2) could significantly affect the enrichment in virtual screening. As demonstrated in Fig. 4C and Table 4, we also found that the enrichment of virtual screening against 16S rRNA A-site was positively correlated with the weight of polar interactions (Welec + Whbond) when iMDLScore2 was used for rescoring. Therefore, we suggested that a soft-core rescoring function that favors electrostatic and hydrogen bond interactions (e.g. iMDLScore2) may be good for a flexible target such as the rRNA A-site.
Table 4.
ROC AUC for different docking and scoring function combinations in RNA virtual screening study.
| Initial docking & scoring function | Rescoring function | Bacterial rRNA A-site (1J7T) | Lysine riboswitch (3DIL) | |||
|---|---|---|---|---|---|---|
|
|
|
|||||
| Aminoglycosides 1 | Zhou dataset 1 | Foloppe dataset 1 | 7 known inhibitors 1 | 7 known inhibitors 2 | ||
| GOLD:GOLD Fitness | None | 1.0 | 1.0 | 0.58 | 0.97 | 0.82 |
| rDock:rDock_solv | None | 1.0 | 1.0 | 0.61 | 0.999 | 0.86 |
| GOLD:GOLD Fitness | ASP | NA | NA | 0.50 | 0.98 | 0.51 |
| GOLD:GOLD Fitness | rDock_solv | NA | NA | 0.68 | 0.998 | 0.86 |
| GOLD:GOLD Fitness | AutoDock4.1 Score | NA | NA | 0.64 | 0.77 | NA |
| GOLD:GOLD Fitness | iMDLScore1 | NA | NA | 0.58 | 0.66 | NA |
| GOLD:GOLD Fitness | iMDLScore2 | NA | NA | 0.69 | 0.92 | 0.51 |
| rDock:rDock_solv | AutoDock4.1 Score | NA | NA | 0.67 | 0.46 | NA |
| rDock:rDock_solv | iMDLScore1 | NA | NA | 0.61 | 0.33 | NA |
| rDock:rDock_solv | iMDLScore2 | NA | NA | 0.74 | 0.81 | 0.51 |
The decoy set was MayBridge dataset
The decoy set was seven known lysine analogs inactive to lysine riboswitch
Figure 4.
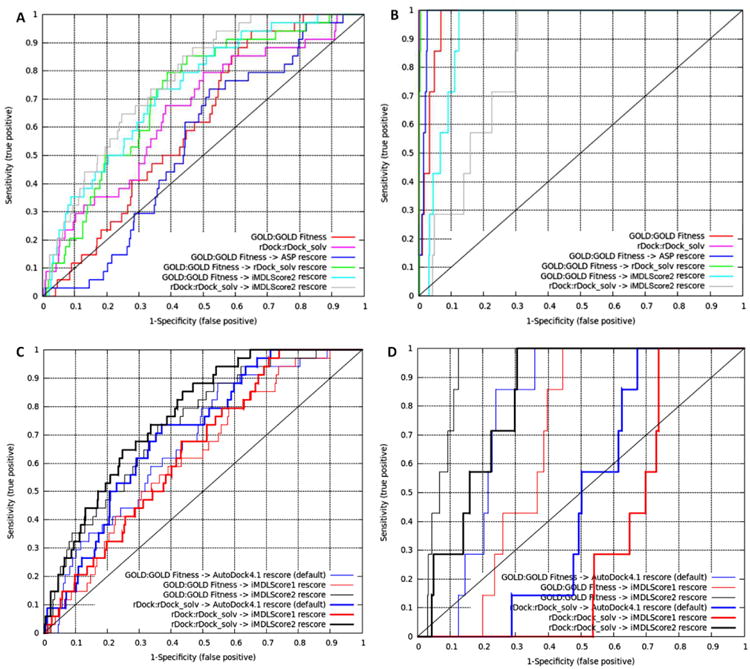
ROC curves of the virtual screening experiments with various docking/scoring combinations. (A). Virtual screening against the 16S rRNA A-site using the Foloppe dataset. (B). Virtual screening against the lysine riboswitch using 7 known active compounds. (C-D). ROC comparison of the virtual screening performances of AutoDock4.1 and iMDLScore1/iMDLScore2 scoring functions with rRNA A-site (C). and lysine riboswitch (D). GOLD:GOLD Fitness dockings were in thin lines, while rDock:rDock_solv dockings were in thick lines. AutoDock4.1 default scoring function, iMDLScore1 and iMDLScore2 were colored red, blue and black, respectively.
In contrast, the results from virtual screening against rigid and closed lysine riboswitch were different. Based on the crystal structure, the ligand (lysine) is completely enveloped in the rigid binding pocket of lysine riboswitch, and only the small molecules which can sterically fit the pocket can be accommodated 51, 52. B-factor analysis demonstrated that lysine-binding pocket was statistically less flexible than other residues (Supplementary Figure 4A). Normal mode analysis further confirmed the rigidity of this pocket as no significant atomic fluctuation of the active site residues (arrow highlighted) could be observed within five normal modes with the lowest frequencies (Supplementary Figure 4C). Our virtual screening showed that rDock:rDock_solv and GOLD:GOLD Fitness had comparable enrichments (Fig. 4B). GOLD:GOLD Fitness coupled with ASP or rDock_solv rescoring could improve the enrichment. Unfortunately, all AutoDock4.1 related scoring functions (default, iMDLScore1 and iMDLScore2) could not obtained as good enrichment (AUC<0.85) as other rescoring schemes (AUC>0.95). This might be due to the failure of assigning enough penalties to the steric clashes so that the big molecules were docked into the small and closed lysine-binding pocket (Fig. 4B). Additionally, we investigated whether any computational strategies could differentiate the seven known lysine riboswitch inhibitors from the seven experimentally validated lysine-analog decoys (very low chemical diversity compared to the above experiment with MayBridge decoys). Based on this dataset, we found that GOLD:GOLD Fitness combined with rDock_solv rescoring achieved the best enrichment (AUC=0.86) and ranked all seven active compounds on the top eight. As expected, iMDLScore2 rescoring achieved low enrichment (AUC=0.51) (Table 4).
When comparing our iMDLScore1/iMDLScore2 with the original AutoDock4.1 scoring function, we found that rescoring by iMDLScore2, which has the least vdW contribution but most electrostatic and hbond contributions, attained the best enrichment against 16S rRNA A-site with AUC=0.69 (based on GOLD:GOLD Fitness docking) and 0.74 (based on rDock:rDock:solv docking) (Table 4 and Fig. 4C). In contrast, the results from iMDLScore1 rescoring were not ideal. We observed the similar trend (iMDLScore2 (black) > AutoDock4.1 default (blue) > iMDLScore1 (red)) with ROC AUC analysis for the lysine riboswitch, as demonstrated in Table 4 and Fig. 4D. In summary, we identified the best combinations for RNA virtual screening: rDock:rDock_solv coupled with iMDLScore2 rescoring for flat, open and flexible binding sites of RNAs, while GOLD:GOLD Fitness combined with rDock_solv rescoring could be appropriate for closed and rigid RNA targets.
Application of ensemble docking/scoring for structural flexibility of RNAs
RNAs are flexible and ligand binding can significantly alter the backbone dihedral angles, which may in turn change the sugar puckering, narrow grooves, and introduce kinks or intercalation 53. For example, when different ligands bind to HIV-1 TAR RNA, the sugar puckering of A22 can switch from C3′-endo (1UUD) to O4′-endo (1UUI), while U23 switch from C3′-endo (1UUD) to C1′-endo (1UUI). The technique of ensemble docking has been widely used in the field of protein to model flexibility with a limited number of discrete conformations 54. The ensemble structures can be obtained from different x-ray crystal structures, NMR models or normal mode analysis 16, 55. To investigate whether ensemble docking could also improve the docking/scoring accuracy in the RNA system, we collected five RNA targets and their alternative conformations in Table 5 for native pose ranking and cross-docking experiments. We found that, when RNA flexibility was modeled by structural ensemble, the results of the native pose ranking were significantly improved using rDock_solv scoring functions for 1LVJ, 1AM0 and 2Z74. As aforementioned, without considering RNA flexibility, we could not obtain reasonable results for these three targets in native pose ranking study. However, with other methods, except ASP, only slight improvement of ranking (<10) were observed. We found that the dihedral angle ε (C4′-C3′-O3′-Pi+1) of U23 (critical for inhibitor binding) in 1LVJ was statistically correlated with the native pose rankings from rDock_solv based on the Spearman's rank correlation (p<0.05) (Supplementary Table S8). It was probably because when ε increased, the phosphate of C24 had a better orientation to interact with the positively charge amine group of the ligands. Such interactions were correctly estimated by the rDock_solv scoring functions and thus obtained better rankings. In the cross-docking experiment, when the RNA flexibility was considered, the rankings of cognate ligand were significantly improved using GOLD:GOLD Fitness coupled with rDock_solv rescoring for both 1LVJ and 1UUD. Unfortunately, the ranking got slightly worse for 1LVJ and 1YKV when GOLD:GOLD Fitness coupled with AutoDock4.1 rescoring was used. Detailed comparative data are available in Supplementary Table S7. Our study implied that the consideration of RNA flexibility by ensemble docking could be beneficial to docking/scoring accuracy with hard-core scoring function (e.g. rDock_solv); however, it also depends on the RNA target and the docking/scoring methods used for modeling.
Table 5.
PDBs used to assess the ensemble docking. Primary RNA targets, which had been studied in native pose ranking and cross-docking, were underlined. Apo structures or the structures containing different ligand with the primary target were given in italic.
| RNA | Ligand type | PDB ID |
|---|---|---|
| HIV-1 TAR RNA | PMZ (intercalator) | 1LVJ (12 conformer models) |
| AMP aptamer | AMP | 1AM0 (8 conformer models) |
| HIV-1 TAR RNA | P14 (guanidine-containing ligands) | 1UUD, 1UUI, 1AKX |
| Diels-Alder ribozyme | DAI | 1YKV, 1YLS, 1YKQ |
| T. tengcongensis glmS ribozyme | glucose 6-phosphate | 2Z74, 2HO7, 2H0Z, 2Z75, 2GCV, 2H0W, 2GCS |
Discussion
RNA represents a type of important targets for therapeutic development. In this study, we identified novel strategies for RNA-ligand modeling and virtual screening through a thorough and comprehensive evaluation with statistical analysis heavily used. We provided novel insights into four aspects associated with in silico structure-based molecular design against RNAs. First, GOLD:GOLD Fitness and rDock:rDock_solv, found as the best pose predictors for RNA targets, were appropriate for the initial binding mode generation. However, in order to accurately identify the cognate ligands for RNA targets or improve the virtual screening enrichment, an extra step of rescoring of the predicted binding modes is necessary. Second, we found that ASP and AutoDock4.1 scoring functions, rather than GOLD Fitness, were able to enrich the native or near-native poses. This was based on the observation that ASP rescoring could significantly improve the docking accuracy using GOLD:GOLD Fitness generated poses. Third, soft-core potentials (e.g. iMDLScore1 and iMDLScore2) and hard-core potentials (rDock_solv and ASP) should be properly employed based on the structural resolution and flexibility of the binding sites of RNA targets. Hard-core scoring functions could produce more accurate results for virtual screening using RNA ensemble structures. Finally, implementation of RNA-specific scoring function (e.g. iMDLScore2) improved the virtual screening enrichment as well as the accuracy of RNA-ligand binding affinity prediction. In summary, the suggested workflow for structure-based modeling for RNA-targeted drug discovery was illustrated in Fig. 6.
Consistent to other docking evaluation reports 56, we suggest that good performance in binding mode reproduction do not guarantee the success in virtual screening for RNA inhibitor identification. Thereby we proposed a two-step docking/scoring strategy for RNA virtual screening. Although some scoring functions, such as iMDLScore2, can be used in the rescoring (the second step scoring) but are not ideal for the initial pose generation (the first step docking/scoring). We found when iMDLScore2 was used for the initial pose generation, positively charged groups, such as guanidium and amine, were biased to form interactions with the RNA phosphate moieties (Supplementary Figure 3). Based on our evaluation, no existing docking program achieved satisfactory performances on both pose prediction and hit identification. Our two-step strategy performed well by separating conformation-wise pose selection and ligand-wise hit selection using different docking and scoring functions during virtual screening.
The false positives in the selected hits are an overall consequence of incorrectly predicted binding modes and scoring inaccuracy. Here we highlighted the importance of sampling enough conformational space for the final rescoring, as illustrated in Fig. 1B. However, virtual screening involves not only the coverage of near-native conformations for each ligand, but also the enrichment of the true binders. Accordingly, we observed that retaining too many poses for rescoring might accumulate noises. For example, Fig. 5 illustrated the trend that, only when we picked up the top 3 poses based on GOLD:GOLD Fitness docking, could we achieve the best AUC with iMDLScore2 rescoring. The AUC gradually declined if the number of picked top poses were more than 3. The similar trend was also observed for rDock:rDock_solv docking, but the turning point of the number of picked poses was 6. We anticipate that by using GOLD:GOLD Fitness or rDock:rDock_solv as the initial pose generators, retaining top 10 poses for rescoring should be appropriate to cover at least one near-native binding pose without significantly compromising (< 3%) the virtual screening enrichment (Fig. 1B and Fig. 5).
Figure 5.

The ROC AUC (Y axis) against number of candidate poses (X axis) selected for iMDLScore2 rescoring against 16S rRNA A-site. (x025BE)represents the number of picked poses corresponding to the best ROC AUC (turning point).
Similar to protein-ligand interactions which are now considered as non-linear dynamic and cooperative processes 46, distal thermodynamic changes may also alter the binding affinity in RNA systems significantly. For example, in SAM-I riboswitch, the mutations on three base pairs (3G×7) only slightly affect the binding modes, but significantly decrease the binding affinity (∼300 fold) (Supplementary Table S2). Existing scoring functions failed to reflect such dramatic change in binding affinity quantitatively according to the slight change of the structure. A better estimation of binding affinity via in silico approach may require refinement of RNA-specific force field, more advanced atom typing, and consideration of structural flexibility. For instance, an RNA-specific WSAS desolvation model was developed instead of inheriting Wesson & Eisenberg's model 43, 57. In addition, the electrostatic potential in RNAs frequently is not dominated by the negatively charged phosphates because these phosphates can be masked by counterions and waters 27. Therefore, we suggest that advanced scoring functions treat phosphate-ligand and base-ligand interactions differently. In particular, the H-bond formed between RNA bases and aromatic moieties should be considered as more favorable than that formed with phosphate. Rewarding short-range electrostatic and π-related interactions such as aromatic stacking, as rDock does 32, with base atoms could also be beneficial.
Finally, unlike proteins, RNA flexible docking is currently limited by the availability of rotamer libraries of nucleosides. Therefore, RNA flexibility are usually addressed by applying soft grids/potentials 58, using ensembles with molecular dynamic simulation or various NMR models 16, or performing post-docking local optimization 34. Since the force field-based optimization algorithms (e.g., MORDOR) are computationally expensive, more studies are needed to reduce the computational cost, probably by either accelerating the force field calculation or alternatively developing novel algorithms for more efficient sampling of ligand-bound conformations. For instance, Rohs et al. utilized Monte Carlo simulation to model flexible DNA-ligand interactions efficiently without any prior binding-site selections 59, and this might be applied to RNAs as well. The successful rational design of HIV-1 TAR RNA inhibitors by using structure ensembles from normal mode analysis represented a good example for the ensemble docking 16. However, the performance of RNA ensemble docking varies depending on targets, scoring functions and other factors, in agreement with our assessment here. Sometimes it is possible to cause more noise rather than signals when flexibility is introduced 54. Further exploration of RNA flexible docking/scoring will be necessary when more RNA structural data or more RNA flexibility-oriented docking programs become available.
Materials and Methods
Datasets
Most of the currently published datasets are either too small or lack target diversity 10, 28, 29, 31, 32, 34. Based on these datasets, we compiled our own dataset of high-resolution RNA-ligand complex structures by removing those low-resolution, redundant structures as well as those structures with critical structural defects (e.g. 1AJU). We also included additional structures by mining Protein Data Bank (PDB) and literature to identify RNA-ligand complexes with available binding affinities. This resulted in a unique set of 56 RNA-ligand complex structures with 36 high-resolution (<3.0Å) crystal and 20 NMR structures. Another issue of the published datasets was that over 65% of the ligands were aminoglycosides or low-affinity binders (e.g. spermine) 28. To avoid the potential problems of statistically overestimating the weight of any typical RNA ligands, we reduced the number of aminoglycosides and low-affinity binders, but increased the number of high-affinity small molecules. Our curation encompassed a variety of known RNA targets including: RNA aptamers, prokaryotic and eukaryotic rRNA A-sites, ribozymes, riboswitches, and viral RNAs (TAR RNA, HCV IRES domain, etc.). These RNAs are listed in Supplementary Table S1.
RNA docking and decoy generation
In this article, we used “A:B” to represent the strategy that “docking using A program and B scoring function”. RNA molecules and ligands were prepared using Protein Preparation Wizard in Maestro. Briefly, all water molecules, ions, and cofactors were removed. The hydrogen atoms were minimized in the ligand-bound complexes using default parameters. The average structure of NMR models for each complex was minimized and employed for our studies. All of the phosphates in RNAs were deprotonated. The ligands were protonated/deprotonated using Epik (Schrödinger) 60 at PH 7.0. If an RNA has duplicate binding sites/ligands (e.g. 1J7T), the region with the lowest B-factors was used. The ligands were energetically minimized, and molecular docking and rescoring were performed using the similar approaches as previously described 28, 29, 32, 61. Briefly, we employed five docking programs (GOLD 5.0.1, Glide 5.6, Surflex v2.415, AutoDock 4.1 and rDock 2006.2) combined with their native scoring functions to generate 10 poses using the default parameters, but with some modifications to ensure the sufficient conformational sampling. In order to ensure the high diversity and quality of the conformational decoys, we employed GOLD:GOLD Fitness to generate 100 conformational decoys for each RNA target with the tuned genetic algorithm parameters. Detailed docking parameters are available in Supplementary Material.
Reproduction of experimental structures with docking
In this study, both RMSD between experimental structures and predicted docking poses and pose ranking were considered. To simplify the expression, we define C(X, YÅ) as the criterion that “at least one near-native binding mode (<Y Å RMSD) was predicted within the top X poses”. To evaluate the overall ability of docking/scoring programs to reproduce experimental structures, we implemented a new parameter, namely volume under the surface (VUS), to describe the overall performance of binding mode reproduction. As Fig. 1a demonstrated, the surface was based on a series of discrete grids. VUS was calculated as the sum of the volume of all triangular prisms under this surface. Briefly, a series of coordinates were obtained based on their RMSD cutoff (the X dimension), ranking cutoff (the Y dimension), and the number (the Z dimension) of successfully reproduced structures satisfying C(X, YÅ). The spacing of RMSD cutoff was 0.5Å, and it was 1 for the ranking cutoff. The surface was made by connecting any two adjacent points and then partitioned into a series of triangles. Any of these triangles and their projections on the XY plane was used to define the triangular prism unit. Detailed calculation of the volume of each triangular prism unit and VUS were demonstrated in the Supplementary Material. The ideal VUS was calculated as 10(RMSD cutoff)×9(rank cutoff)×56(number of targets). Different from most of previous similar studies, if not all, statistical significance (p-value) was always considered for the analysis throughout this report. Briefly, p-values calculated from student t-test were employed to determine the statistical significance when comparing two different groups.
Docked/native pose ranking
For each ligand, we generated 100 decoys to the corresponding RNA target. Therefore, together with the crystal/NMR ligand structure, we obtained 101 RMSD-docking score data points for each cognate RNA-ligand pair. For native pose ranking study, we scored these 101 poses using different docking programs as aforementioned. The ranking of native poses for 56 targets were calculated, and the recovery curves were made as the ranking cutoffs (X axis) against the cumulative number of targets (Y axis) in which the ranking of native pose cutoff was smaller than the ranking cutoff. The Spearman's rank correlation coefficient was used to evaluate the ranking capability. To make the docking scores positively correlated with RMSD (the higher the scores, the higher the RMSD), we used the negative value of GOLD Fitness, ChemScore, ASP and Surflex-dock scores. If a pose was assigned a score with the absolute value more than 1000 (outliers), this RMSD-score pair will be excluded. The Spearman's rank correlation coefficient (ρ) was computed from Equation (1), where rRMSD,i and rscore,i are the ranks of the RMSD and score for the pose i, and we took the average of the ranks for the tied values. and are the average ranks of RMSD and score for 101 poses. We classified the resulted 56 ρ values (calculated from 56 RNA-ligand complexes) for each scoring function into three groups based on the widely-used criteria: weak correlation: ρ<0.3, moderate correlation: 0.3≤ρ<0.5, strong correlation: ρ≥0.5.
| Equation (1) |
Cross-docking
The dataset used for cross-docking was a subset of our overall collection. In order to improve the robustness of cross-docking, we only choose 23 complexes with unique RNAs and a limited number of non-selective ligands (e.g. arginine and aminoglycoside). The RNA-ligand complexes selected for cross-docking are available in Supplementary Table S6. Cross-docking was performed by docking all 23 ligands in this subset to each RNA targets using the methods described in “RNA docking and decoy generation”. Rescoring was performed based on the top 10 scored poses for each docking method. Cross-docking performance was evaluated by average ranking of cognate ligand calculated from these 23 RNA targets and AUC of the recovery curve based on the average ranking of cognate ligands (similar to the recovery curve used in “Docking pose ranking” section).
Virtual Screening for RNA inhibitor identification
Two different targets were assessed, namely bacterial 16S rRNA A-site (representing open and flexible binding site) and lysine riboswitch (representing closed and rigid binding site). Bacterial 16S rRNA A-site and lysine riboswitch structures were obtained from PDB as 1J7T and 3DIL, respectively. We collected 75 known rRNA inhibitors including 34 drug-like small molecules from the Foloppe dataset 12 and 31 aminoglycoside mimetics from the Zhou dataset 13. Additionally, we obtained 11 aminoglycoside inhibitors which have the crystal structures in complex with the bacterial rRNA A-site (1J7T, 1YRJ, 2F4T, 2ET8, 1LC4, 1MWL, 2BE0, 2G5Q, 2ESI, 2PWT and 1BYJ). For virtual screening against lysine riboswitch, we collected 14 compounds including 7 known inhibitors and 7 experimentally validated inactives 17. In order to avoid artificial enrichment 62, a focused library containing 942 drug-like and positively charged decoys was generated from MayBridge database. We assumed this randomly constructed decoy library does not include or include very few active compounds as previous studies did 28. The receiver operating characteristic (ROC) curve along with the area under the curve (AUC) was used to assess the overall performance (e.g., recovery rate, enrichment, etc.) of our protocols.
Docking score-binding affinity correlation
Although it is known that current docking scores are usually poorly correlated with experimental binding affinity 56, we are still interested to evaluate the performance of our protocols on RNA targets and try to learn how to improve the scoring. To this end, dissociation constant (Kd) values were carefully collected from literature (values and references are available in Supplementary Table S2), and we compared them with PDBBind database (http://www.pdbbind-cn.org/) and other reports/databases to ensure the consistency of data. If the collected Kd values for each RNA-ligand pair are within 10-fold difference, the average values were used. Of note, we used 2μM as the Kd of gentamicin C1a-rRNA A-site complex (1BYJ) for studies because this is the Kd under room temperature, instead of 0.01μM (Kd under 4°C) 63. Additionally, Kd for neomycinB-HIV-1 TAR RNA complex (1QD3) should be 5.9±4μM. The Kd values used in some previous studies were actually from the U24C mutant 29, 32, 64. The binding free energy were calculated as ΔG = RTln(Kd) under 300K. The Pearson correlation coefficients (R2) between these docking scores and their corresponding binding affinities were obtained. Three common outliers, 1LVJ, 1TOB and 2TOB, were excluded to avoid noise, as they contained many unfavorable steric clashes.
RNA-specific scoring function optimization
The weak correlation between docking scores and binding affinity might be because most of the current scoring functions were derived from protein-ligand interactions. To implement new RNA-specific scoring functions, we optimized the energetic coefficients in AutoDock4.1 scoring function using our largest ever RNA-ligand dataset with known experimental binding affinities. This empirical scoring function was shown as Equation (2) 41. The parameters (A, B, C, D, S, V) were obtained from default AutoDock4 scoring function, as previously described 41. With our RNA-ligand dataset, we derived a new set of coefficients of five terms (WvdW, Whbond, Welec, Wsol and Wtors) with multiple linear regression methods. Besides R2, we calculated leave-one-out (LOO) cross-validation correlation coefficients (Q2) and validated against an external test set consisting of eight complexes to evaluate the predictive power of our new schemes. The PDBs employed to train and validate iMDLScore1 and iMDLScore2 are listed in Supplementary Table S2.
| Equation (2) |
Supplementary Material
Acknowledgments
Special thanks to CCDC teams, in particular Dr. John Liebeschuetz, for helpful discussion on RNA docking with GOLD. This work was partially supported by the US Department of Defense Concept Awards (BC085871), the University Cancer Foundation via the Institutional Research Grant program at the University of Texas M.D. Anderson Cancer Center, M.D. Anderson Prostate SPORE Career Award (P50 CA140388 SPORE), and Grant #IRG-08-061-01 from the American Cancer Society. G.A.C is the Alan M. Gewirtz Leukemia & Lymphoma Society Scholar, and he is supported in part by the CLL Global Research Foundation. We also thank the free academic licenses from the rDock and AutoDock development teams. Special thanks to Ms. Xiaofei Xiong for helping curation of RNA-ligand complexes.
Footnotes
Supporting Information: Supplementary methods are the details of docking parameters and volume under the surface calculation. Supplementary table S1-S8 are the PDB codes, RNA-ligand binding affinity data, and the detailed data for binding mode reproduction, native pose ranking, cross-docking and RNA ensemble docking. Supplementary figure 1-4 are the pose comparisons, score-binding affinity plots and binding site flexibility analysis. These materials are available free of charge via the internet.
References
- 1.Bannwarth S, Gatignol A. HIV-1 TAR RNA: the target of molecular interactions between the virus and its host. Curr HIV Res. 2005;3:61–71. doi: 10.2174/1570162052772924. [DOI] [PubMed] [Google Scholar]
- 2.Hermann T. Aminoglycoside antibiotics: old drugs and new therapeutic approaches. Cell Mol Life Sci. 2007;64:1841–52. doi: 10.1007/s00018-007-7034-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shay JW, Wright WE. Telomerase therapeutics for cancer: challenges and new directions. Nat Rev Drug Discov. 2006;5:577–84. doi: 10.1038/nrd2081. [DOI] [PubMed] [Google Scholar]
- 4.Nicoloso MS, Spizzo R, Shimizu M, Rossi S, Calin GA. MicroRNAs--the micro steering wheel of tumour metastases. Nat Rev Cancer. 2009;9:293–302. doi: 10.1038/nrc2619. [DOI] [PubMed] [Google Scholar]
- 5.Thomas JR, Hergenrother PJ. Targeting RNA with small molecules. Chem Rev. 2008;108:1171–224. doi: 10.1021/cr0681546. [DOI] [PubMed] [Google Scholar]
- 6.Spizzo R, Nicoloso MS, Croce CM, Calin GA. SnapShot: MicroRNAs in Cancer. Cell. 2009;137:586–586 e1. doi: 10.1016/j.cell.2009.04.040. [DOI] [PubMed] [Google Scholar]
- 7.Fabbri M, Bottoni A, Shimizu M, Spizzo R, Nicoloso MS, Rossi S, Barbarotto E, Cimmino A, Adair B, Wojcik SE, Valeri N, Calore F, Sampath D, Fanini F, Vannini I, Musuraca G, Dell'Aquila M, Alder H, Davuluri RV, Rassenti LZ, Negrini M, Nakamura T, Amadori D, Kay NE, Rai KR, Keating MJ, Kipps TJ, Calin GA, Croce CM. Association of a microRNA/TP53 feedback circuitry with pathogenesis and outcome of B-cell chronic lymphocytic leukemia. JAMA. 2011;305:59–67. doi: 10.1001/jama.2010.1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Melo S, Villanueva A, Moutinho C, Davalos V, Spizzo R, Ivan C, Rossi S, Setien F, Casanovas O, Simo-Riudalbas L, Carmona J, Carrere J, Vidal A, Aytes A, Puertas S, Ropero S, Kalluri R, Croce CM, Calin GA, Esteller M. Small molecule enoxacin is a cancer-specific growth inhibitor that acts by enhancing TAR RNA-binding protein 2-mediated microRNA processing. Proc Natl Acad Sci U S A. 2011;108:4394–9. doi: 10.1073/pnas.1014720108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang S, Chen L, Jung EJ, Calin GA. Targeting microRNAs with small molecules: from dream to reality. Clin Pharmacol Ther. 2010;87:754–8. doi: 10.1038/clpt.2010.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lang PT, Brozell SR, Mukherjee S, Pettersen EF, Meng EC, Thomas V, Rizzo RC, Case DA, James TL, Kuntz ID. DOCK 6: combining techniques to model RNA-small molecule complexes. RNA. 2009;15:1219–30. doi: 10.1261/rna.1563609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen Q, Shafer RH, Kuntz ID. Structure-based discovery of ligands targeted to the RNA double helix. Biochemistry. 1997;36:11402–7. doi: 10.1021/bi970756j. [DOI] [PubMed] [Google Scholar]
- 12.Foloppe N, Chen IJ, Davis B, Hold A, Morley D, Howes RA. structure-based strategy to identify new molecular scaffolds targeting the bacterial ribosomal A-site. Bioorg Med Chem. 2004;12:935–47. doi: 10.1016/j.bmc.2003.12.023. [DOI] [PubMed] [Google Scholar]
- 13.Zhou Y, Gregor VE, Ayida BK, Winters GC, Sun Z, Murphy D, Haley G, Bailey D, Froelich JM, Fish S, Webber SE, Hermann T, Wall D. Synthesis and SAR of 3,5-diamino-piperidine derivatives: novel antibacterial translation inhibitors as aminoglycoside mimetics. Bioorg Med Chem Lett. 2007;17:1206–10. doi: 10.1016/j.bmcl.2006.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Du Z, Lind KE, James TL. Structure of TAR RNA complexed with a Tat-TAR interaction nanomolar inhibitor that was identified by computational screening. Chem Biol. 2002;9:707–12. doi: 10.1016/s1074-5521(02)00151-5. [DOI] [PubMed] [Google Scholar]
- 15.Filikov AV, Mohan V, Vickers TA, Griffey RH, Cook PD, Abagyan RA, James TL. Identification of ligands for RNA targets via structure-based virtual screening: HIV-1 TAR. J Comput Aided Mol Des. 2000;14:593–610. doi: 10.1023/a:1008121029716. [DOI] [PubMed] [Google Scholar]
- 16.Stelzer AC, Frank AT, Kratz JD, Swanson MD, Gonzalez-Hernandez MJ, Lee J, Andricioaei I, Markovitz DM, Al-Hashimi HM. Discovery of selective bioactive small molecules by targeting an RNA dynamic ensemble. Nat Chem Biol. 2011;7:553–9. doi: 10.1038/nchembio.596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Blount KF, Wang JX, Lim J, Sudarsan N, Breaker RR. Antibacterial lysine analogs that target lysine riboswitches. Nat Chem Biol. 2007;3:44–9. doi: 10.1038/nchembio842. [DOI] [PubMed] [Google Scholar]
- 18.Daldrop P, Reyes FE, Robinson DA, Hammond CM, Lilley DM, Batey RT, Brenk R. Novel ligands for a purine riboswitch discovered by RNA-ligand docking. Chem Biol. 2011;18:324–35. doi: 10.1016/j.chembiol.2010.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mulhbacher J, Brouillette E, Allard M, Fortier LC, Malouin F, Lafontaine DA. Novel riboswitch ligand analogs as selective inhibitors of guanine-related metabolic pathways. PLoS Pathog. 2010;6:e1000865. doi: 10.1371/journal.ppat.1000865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pushechnikov A, Lee MM, Childs-Disney JL, Sobczak K, French JM, Thornton CA, Disney MD. Rational design of ligands targeting triplet repeating transcripts that cause RNA dominant disease: application to myotonic muscular dystrophy type 1 and spinocerebellar ataxia type 3. J Am Chem Soc. 2009;131:9767–79. doi: 10.1021/ja9020149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arambula JF, Ramisetty SR, Baranger AM, Zimmerman SC. A simple ligand that selectively targets CUG trinucleotide repeats and inhibits MBNL protein binding. Proc Natl Acad Sci U S A. 2009;106:16068–73. doi: 10.1073/pnas.0901824106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wong CH, Fu Y, Ramisetty SR, Baranger AM, Zimmerman SC. Selective inhibition of MBNL1-CCUG interaction by small molecules toward potential therapeutic agents for myotonic dystrophy type 2 (DM2) Nucleic Acids Res. 2011;39:8881–90. doi: 10.1093/nar/gkr415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Young DD, Connelly CM, Grohmann C, Deiters A. Small molecule modifiers of microRNA miR-122 function for the treatment of hepatitis C virus infection and hepatocellular carcinoma. J Am Chem Soc. 2010;132:7976–81. doi: 10.1021/ja910275u. [DOI] [PubMed] [Google Scholar]
- 24.Gumireddy K, Young DD, Xiong X, Hogenesch JB, Huang Q, Deiters A. Small-molecule inhibitors of microrna miR-21 function. Angew Chem Int Ed Engl. 2008;47:7482–4. doi: 10.1002/anie.200801555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Parsons J, Castaldi MP, Dutta S, Dibrov SM, Wyles DL, Hermann T. Conformational inhibition of the hepatitis C virus internal ribosome entry site RNA. Nat Chem Biol. 2009;5:823–5. doi: 10.1038/nchembio.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Seth PP, Miyaji A, Jefferson EA, Sannes-Lowery KA, Osgood SA, Propp SS, Ranken R, Massire C, Sampath R, Ecker DJ, Swayze EE, Griffey RH. SAR by MS: discovery of a new class of RNA-binding small molecules for the hepatitis C virus: internal ribosome entry site IIA subdomain. J Med Chem. 2005;48:7099–102. doi: 10.1021/jm050815o. [DOI] [PubMed] [Google Scholar]
- 27.Foloppe N, Matassova N, Aboul-Ela F. Towards the discovery of drug-like RNA ligands? Drug Discov Today. 2006;11:1019–27. doi: 10.1016/j.drudis.2006.09.001. [DOI] [PubMed] [Google Scholar]
- 28.Li Y, Shen J, Sun X, Li W, Liu G, Tang Y. Accuracy assessment of protein-based docking programs against RNA targets. J Chem Inf Model. 2010;50:1134–46. doi: 10.1021/ci9004157. [DOI] [PubMed] [Google Scholar]
- 29.Detering C, Varani G. Validation of automated docking programs for docking and database screening against RNA drug targets. J Med Chem. 2004;47:4188–201. doi: 10.1021/jm030650o. [DOI] [PubMed] [Google Scholar]
- 30.Moitessier N, Westhof E, Hanessian S. Docking of aminoglycosides to hydrated and flexible RNA. J Med Chem. 2006;49:1023–33. doi: 10.1021/jm0508437. [DOI] [PubMed] [Google Scholar]
- 31.Pfeffer P, Gohlke H. DrugScoreRNA--knowledge-based scoring function to predict RNA-ligand interactions. J Chem Inf Model. 2007;47:1868–76. doi: 10.1021/ci700134p. [DOI] [PubMed] [Google Scholar]
- 32.Morley SD, Afshar M. Validation of an empirical RNA-ligand scoring function for fast flexible docking using Ribodock. J Comput Aided Mol Des. 2004;18:189–208. doi: 10.1023/b:jcam.0000035199.48747.1e. [DOI] [PubMed] [Google Scholar]
- 33.Pinto IG, Guilbert C, Ulyanov NB, Stearns J, James TL. Discovery of ligands for a novel target, the human telomerase RNA, based on flexible-target virtual screening and NMR. J Med Chem. 2008;51:7205–15. doi: 10.1021/jm800825n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guilbert C, James TL. Docking to RNA via root-mean-square-deviation-driven energy minimization with flexible ligands and flexible targets. J Chem Inf Model. 2008;48:1257–68. doi: 10.1021/ci8000327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lind KE, Du Z, Fujinaga K, Peterlin BM, James TL. Structure-based computational database screening, in vitro assay, and NMR assessment of compounds that target TAR RNA. Chem Biol. 2002;9:185–93. doi: 10.1016/s1074-5521(02)00106-0. [DOI] [PubMed] [Google Scholar]
- 36.Kirchmair J, Markt P, Distinto S, Wolber G, Langer T. Evaluation of the performance of 3D virtual screening protocols: RMSD comparisons, enrichment assessments, and decoy selection--what can we learn from earlier mistakes? J Comput Aided Mol Des. 2008;22:213–28. doi: 10.1007/s10822-007-9163-6. [DOI] [PubMed] [Google Scholar]
- 37.Verdonk ML, Cole JC, Hartshorn MJ, Murray CW, Taylor RD. Improved protein–ligand docking using GOLD. Proteins: Structure, Function, and Bioinformatics. 2003;52:609–623. doi: 10.1002/prot.10465. [DOI] [PubMed] [Google Scholar]
- 38.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE, Francis P, Shenkin PS. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem. 2004;47:1739–49. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 39.Jain AN. Surflex-Dock 2.1: robust performance from ligand energetic modeling, ring flexibility, and knowledge-based search. J Comput Aided Mol Des. 2007;21:281–306. doi: 10.1007/s10822-007-9114-2. [DOI] [PubMed] [Google Scholar]
- 40.Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. Journal of Computational Chemistry. 1998;19:1639–1662. [Google Scholar]
- 41.Huey R, Morris GM, Olson AJ, Goodsell DS. A semiempirical free energy force field with charge-based desolvation. J Comput Chem. 2007;28:1145–52. doi: 10.1002/jcc.20634. [DOI] [PubMed] [Google Scholar]
- 42.Mayaan E, Range K, York DM. Structure and binding of Mg(II) ions and di-metal bridge complexes with biological phosphates and phosphoranes. J Biol Inorg Chem. 2004;9:807–17. doi: 10.1007/s00775-004-0583-7. [DOI] [PubMed] [Google Scholar]
- 43.Wesson L, Eisenberg D. Atomic solvation parameters applied to molecular dynamics of proteins in solution. Protein Sci. 1992;1:227–35. doi: 10.1002/pro.5560010204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bindewald E, Skolnick J. A scoring function for docking ligands to low-resolution protein structures. J Comput Chem. 2005;26:374–83. doi: 10.1002/jcc.20175. [DOI] [PubMed] [Google Scholar]
- 45.Rabal O, Schneider G, Borrell JI, Teixido J. Structure-based virtual screening of FGFR inhibitors: cross-decoys and induced-fit effect. BioDrugs. 2007;21:31–45. doi: 10.2165/00063030-200721010-00005. [DOI] [PubMed] [Google Scholar]
- 46.Chen L, Morrow JK, Tran HT, Phatak SS, Du-Cuny L, Zhang S. From Laptop to Benchtop to Bedside: Structure-based Drug Design on Protein Targets. Curr Pharm Des. 2012 doi: 10.2174/138161212799436386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang LW, Rader AJ, Liu X, Jursa CJ, Chen SC, Karimi HA, Bahar I. oGNM: online computation of structural dynamics using the Gaussian Network Model. Nucleic acids research. 2006;34:W24–31. doi: 10.1093/nar/gkl084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dobbins SE, Lesk VI, Sternberg MJ. Insights into protein flexibility: The relationship between normal modes and conformational change upon protein-protein docking; Proceedings of the National Academy of Sciences of the United States of America; 2008; pp. 10390–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Romanowska J, McCammon JA, Trylska J. Understanding the origins of bacterial resistance to aminoglycosides through molecular dynamics mutational study of the ribosomal A-site. PLoS Comput Biol. 2011;7:e1002099. doi: 10.1371/journal.pcbi.1002099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Romanowska J, Setny P, Trylska J. Molecular dynamics study of the ribosomal A-site. J Phys Chem B. 2008;112:15227–43. doi: 10.1021/jp806814s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Garst AD, Heroux A, Rambo RP, Batey RT. Crystal structure of the lysine riboswitch regulatory mRNA element. J Biol Chem. 2008;283:22347–51. doi: 10.1074/jbc.C800120200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Serganov A, Huang L, Patel DJ. Structural insights into amino acid binding and gene control by a lysine riboswitch. Nature. 2008;455:1263–7. doi: 10.1038/nature07326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Patel DJ, Suri AK, Jiang F, Jiang L, Fan P, Kumar RA, Nonin S. Structure, recognition and adaptive binding in RNA aptamer complexes. J Mol Biol. 1997;272:645–64. doi: 10.1006/jmbi.1997.1281. [DOI] [PubMed] [Google Scholar]
- 54.Korb O, Olsson TS, Bowden SJ, Hall RJ, Verdonk ML, Liebeschuetz JW, Cole JC. Potential and limitations of ensemble docking. J Chem Inf Model. 2012;52:1262–74. doi: 10.1021/ci2005934. [DOI] [PubMed] [Google Scholar]
- 55.Tran HT, Zhang S. Accurate Prediction of the Bound Form of the Akt Pleckstrin Homology Domain Using Normal Mode Analysis To Explore Structural Flexibility. Journal of Chemical Information and Modeling. 2012;51:2352–2360. doi: 10.1021/ci2001742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Warren GL, Andrews CW, Capelli AM, Clarke B, LaLonde J, Lambert MH, Lindvall M, Nevins N, Semus SF, Senger S, Tedesco G, Wall ID, Woolven JM, Peishoff CE, Head MS. A critical assessment of docking programs and scoring functions. J Med Chem. 2006;49:5912–31. doi: 10.1021/jm050362n. [DOI] [PubMed] [Google Scholar]
- 57.Wang J, Wang W, Huo S, Lee M, Kollman PA. Solvation Model Based on Weighted Solvent Accessible Surface Area. The Journal of Physical Chemistry B. 2001;105:5055–5067. [Google Scholar]
- 58.Krüger DM, Bergs J, Kazemi S, Gohlke H. Target Flexibility in RNA–Ligand Docking Modeled by Elastic Potential Grids. ACS Medicinal Chemistry Letters. 2011;2:489–493. doi: 10.1021/ml100217h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rohs R, Bloch I, Sklenar H, Shakked Z. Molecular flexibility in ab initio drug docking to DNA: binding-site and binding-mode transitions in all-atom Monte Carlo simulations. Nucleic Acids Res. 2005;33:7048–57. doi: 10.1093/nar/gki1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shelley JC, Cholleti A, Frye LL, Greenwood JR, Timlin MR, Uchimaya M. Epik: a software program for pK( a ) prediction and protonation state generation for drug-like molecules. J Comput Aided Mol Des. 2007;21:681–91. doi: 10.1007/s10822-007-9133-z. [DOI] [PubMed] [Google Scholar]
- 61.Holt PA, Chaires JB, Trent JO. Molecular Docking of Intercalators and Groove-Binders to Nucleic Acids Using Autodock and Surflex. Journal of Chemical Information and Modeling. 2008;48:1602–1615. doi: 10.1021/ci800063v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Verdonk ML, Berdini V, Hartshorn MJ, Mooij WT, Murray CW, Taylor RD, Watson P. Virtual screening using protein-ligand docking: avoiding artificial enrichment. J Chem Inf Comput Sci. 2004;44:793–806. doi: 10.1021/ci034289q. [DOI] [PubMed] [Google Scholar]
- 63.Yoshizawa S, Fourmy D, Puglisi JD. Structural origins of gentamicin antibiotic action. EMBO J. 1998;17:6437–48. doi: 10.1093/emboj/17.22.6437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Barbault F, Zhang L, Zhang L, Fan BT. Parametrization of a specific free energy function for automated docking against RNA targets using neural networks. Chemometrics and Intelligent Laboratory Systems. 2006;82:269–275. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.