

Abstract
This review examines characterization challenges inherently associated with understanding nanomaterials and the roles surface and interface characterization methods can play in meeting some of the challenges. In parts of the research community, there is growing recognition that studies and published reports on the properties and behaviors of nanomaterials often have reported inadequate or incomplete characterization. As a consequence, the true value of the data in these reports is, at best, uncertain. With the increasing importance of nanomaterials in fundamental research and technological applications, it is desirable that researchers from the wide variety of disciplines involved recognize the nature of these often unexpected challenges associated with reproducible synthesis and characterization of nanomaterials, including the difficulties of maintaining desired materials properties during handling and processing due to their dynamic nature. It is equally valuable for researchers to understand how characterization approaches (surface and otherwise) can help to minimize synthesis surprises and to determine how (and how quickly) materials and properties change in different environments. Appropriate application of traditional surface sensitive analysis methods (including x-ray photoelectron and Auger electron spectroscopies, scanning probe microscopy, and secondary ion mass spectroscopy) can provide information that helps address several of the analysis needs. In many circumstances, extensions of traditional data analysis can provide considerably more information than normally obtained from the data collected. Less common or evolving methods with surface selectivity (e.g., some variations of nuclear magnetic resonance, sum frequency generation, and low and medium energy ion scattering) can provide information about surfaces or interfaces in working environments (operando or in situ) or information not provided by more traditional methods. Although these methods may require instrumentation or expertise not generally available, they can be particularly useful in addressing specific questions, and examples of their use in nanomaterial research are presented.
INTRODUCTION
It is increasingly recognized that nanomaterials present a range of characterization challenges that have the potential to inhibit or delay the scientific and technological impact of nanoscience and nanotechnology. The incomplete characterization and under-reporting of data needed for reproducing and validating experimental findings1, 2, 3, 4, 5, 6 involving nanomaterials limits scientific understanding, slows the development of new technologies, and makes it difficult to reliably address important issues, such as product lifetimes and questions that are relevant to occupational and public health. Concerns related to adequate characterization of nanomaterials are not new, and a variety of issues have been highlighted by many research groups, multiorganizational teams of researchers, and the scientific press.2, 3, 7, 8, 9, 10, 11, 12 Challenges to appropriate characterization of many types of nanomaterials create frustration for researchers and engineers who would like to either use them for biomedical, energy, or other applications, as well as for regulatory bodies that need to understand their impact on human health and the environment.1 The incomplete characterization reports on many materials are viewed by some as a significant failure on the part of the scientific community.2
There are several reasons why complete nanomaterial characterization is not achieved, despite the inherent desire of most research groups to do high-quality research. First, nanomaterials present some unique preparation and analysis difficulties (described in following sections) that are new to some of the disciplines and researchers now involved in nanomaterials-related research. Second, because of the multidisciplinary nature of the field, not every research team has access to the range of characterization tools needed to obtain potentially important information. Third, the range of information needed to understand nanomaterials may require the application of tools and data analysis beyond the expertise of the research teams, sometimes leading to less-than-optimum application of important methods and/or incomplete understanding of the data produced. Nanoscience and nanotechnology are relatively immature fields, and the research community is just beginning to understand some of the limitations of analytical approaches that have been successfully applied in other areas.13 As understanding evolves, improvements to established methodologies, new technologies, and approaches are being developed. Thus, most of the sample preparation and characterization challenges can be addressed to differing degrees, especially when they are recognized at the outset. After exploring the nature and causes of some of the analytical challenges, we consider the ability of current- and next-generation surface analysis tools and developing capabilities to address some of the seemingly daunting materials characterization issues.
Although we focus much attention on engineered nanoparticles, many of the characterization challenges apply equally to other natural and engineered nanomaterials, as well as to areas such as atmospheric aerosols.14 In the first section, we highlight the need for the characterization of nanomaterials by showing the large and increasing impact of nanoscience and nanotechnology on science and technology around the world. The section after examines the challenges associated with nanomaterial characterization. In later sections, we explore the roles that traditional surface analysis methods can play in addressing analysis problems and show, by example, a few other methods that address questions related to characterizing nanomaterial interfaces in working environments (in situ) and determining the uniformity of coatings (intentional or accidental) on nanoparticles. The discussion and conclusion sections make recommendations, indicating approaches for overcoming synthesis and characterization challenges.
IMPACT OF NANOSCIENCE AND NANOTECHNOLOGY
The origin of nanotechnology usually is associated with a 1959 lecture by Richard Feyman.15 Possibly, the first use of the term “nanotechnology” in the scientific literature was in a 1974 paper involving “ion-sputter machining” by Taniguchi.16 The first nanotechnology journal, Nanotechnology, was launched in 1990 by the Institute of Physics in the United Kingdom. In 2000, then U.S. President Bill Clinton announced the U.S. National Nanotechnology Initiative (NNI), and Congress subsequently enacted the 21st Century Nanotechnology Research and Development Act in 2003, which significantly expanded the research focus and financial support in the area. A 2010 World Technology Evaluation Center (WTEC.org) and National Science Foundation (NSF) report, “Nanotechnology Research Directions for Societal Needs in 2020,”17 assessed the impacts of nanotechnology from 2000 to 2010 and looked toward the future. Quite appropriately, the report highlighted both the numbers of nanotechnology patent applications (growing from 224 in 1991 to nearly 13,000 in 2008) and the market size of products involving nanotechnology (approximately $91 billion in 2009). The report also notes that nanotechnology has penetrated “most production sectors of the economy” and is used in a wide range of commercial products, “including coatings, industrial chemicals, cosmetics, textiles, and magnetic storage devices, among many others.”17 The report concludes that future impacts will be both greater and more revolutionary than what has been observed to date.
As expected, the number of publications related to nanotechnology mirrors the historical time line. Results from a Web of Science search combining the terms “nanomaterial,” “nanoparticle,” and “nanostructure” show dramatic growth in published papers (Fig. 1), from less than 100 in 1990 to almost 45,000 in 2011. Similarly, nanotechnology plays a significant role in many technical societies and has spawned new ones, as well as several journals focused on all things “nano.” Nanotechnology within the American Vacuum Society (AVS) follows a similar time line. The AVS Nanoscience and Nanotechnology Division was established in 1993. Since 1995, program activities specifically identified as “nano” have had a continuous presence in the annual AVS Symposium. By 1995, 5% of the papers in the Journal of Vacuum Science and Technology (JVST) A and JVST B were topically related to “nano” in some way with about 2% of the papers related to nanomaterials. Nano-related work in JVST grew at a nearly linear rate until 2003 when new NNI funding began appearing in significant amounts, and growth accelerated. By the end of 2011, nearly 40% of all JVST publications were linked to the key word “nano” in some way with about 15% identified as nanomaterial related using the three search terms described earlier (Fig. 2).
Figure 1.
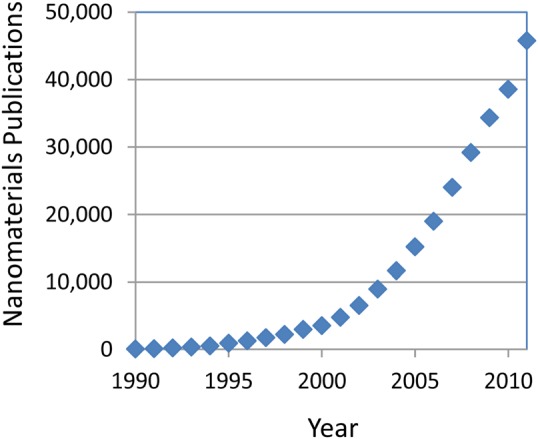
(Color online) All “nanomaterials” publications identified by a Web of Science topic search (including: nanomaterials AND nanoparticles AND nanostructure) by year.
Figure 2.
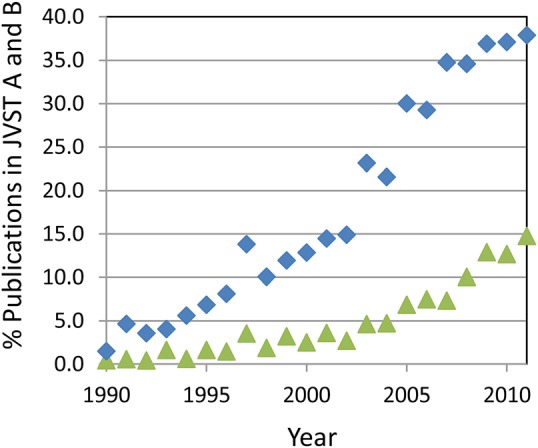
(Color online) Percentage of publications in JVST A and B that include “nano” as a topic, ♦ as identified in the Web of Science and those focused on nanomaterials ▲.
The focus on things “nano” in the AVS and other societies occurs not only because we now have the label “nano” and due to significant funding in areas labeled nanotechnology, but the work we now call “nanotechnology” is part of a natural progression in many different areas of science coupled with development of new tools that enable researchers to see, model, and control structures at the nanometer scale.18, 19, 20 Although, in many ways, nanotechnology is not new, experimental and computational tool development enables research to be done that was not previously possible. New approaches can be taken to address previously intractable problems. Therefore, nanoscience and nanotechnology do not represent a new discipline, but rather advancements within several disciples and a convergence of concepts and ideas across disciplines.19 As such, nanotechnology introduces important new ideas and cross-fertilization that enables scientific breakthroughs and development of various new technologies.
The productive cross-fertilization of multiple disciplines also places new demands on research teams by requiring an expanded range of scientific, analytical, and other skills that are not always readily available. In addition, the fundamental nature of nanomaterials introduces characterization issues that are not routinely addressed by many analytical tools.7, 11, 13 The same characteristics of nanomaterials that make them scientifically interesting actually cause some of the analysis challenges. Nanoparticles have a physical size characteristic of biological molecules,21 and, in some ways, they can be described as having protein-like properties.7, 22 Similar to biological molecules, nanoparticles also may change their structure and properties, depending upon the physical environment.7 Particles may undergo structural transformation, dissolve, agglomerate, or pick up coatings in different environments. These behaviors complicate the ability to identify and predict properties of these materials and diminish the ability to assess the impact of these materials on the environment or their health and safety implications.
These characterization challenges are readily noted in papers, such as “Common pitfalls in nanotechnology…,”23 “The characterization bottleneck,”1 and “Discriminating the states of matter in metallic nanoparticle transformations: What are we missing?,”24 and in scientific news articles, such as “Tiny traits cause big headaches….”2 The nature of some of these “headaches” is described in the next section.
NANOMATERIAL CHARACTERIZATION SURPRISES
Research on the synthesis and properties of a range of nanomaterials takes place in the Environmental Molecular Sciences Laboratory (EMSL), a national scientific user facility sponsored by the U.S. Department of Energy's Office of Biological and Environmental Research (DOE-BER) and located at Pacific Northwest National Laboratory (PNNL), and the authors of this review paper have been associated with EMSL as staff members, research partners, or users. We have found that appropriate characterization of nanomaterials can be more difficult and, in many circumstances, more interesting than expected. Because of our long-term interest in the interactions of iron-25, 26, 27, 28 and cerium-based29, 30, 31, 32 nanoparticles in a variety of environments, many of the examples shown herein are directly derived from our work on these materials. The issues that have impacted our research are similar to those identified in a variety of studies on other materials systems.
Nanomaterials can take a variety of forms, including free-standing nano-objects (such as nanoparticles and carbon nanotubes), materials with nano-sized holes (including porous films), and other types of materials with a variety of different nanoscale structures (such as nanometer-thick layered structures). Many of the examples in this paper focus on behaviors and properties of nanoparticles. However, the detailed characterization of other types of nanomaterials often has produced results that have surprised the researchers involved in this study (described in the next section). These and other examples from the literature are used to identify some of the common issues and challenges that should be considered whenever analyzing nanomaterials.
Inconsistent behaviors and disappearing particles—cerium oxide nanoparticles
Ceria (CeO2) is used in a variety of applications, including functioning as a catalyst and catalyst support,33 serving as an antioxidant in medical treatment,34, 35 and having good ion conductivity that might prove useful for solid oxide fuel cells.36 For some applications, one of ceria's important properties is the ease at which Ce can switch oxidation states between +3 and +4. Several studies suggest that as ceria particles decrease in size, the dominant chemical state of Ce in the particles changes from +4 to +3.37
Unfortunately, published reports regarding ceria nanoparticles report properties that often contradict one another.32 We were interested in the sources of variability in measured band gaps for particles of similar size32 and the range of conflicting reports on the toxicity of ceria in biological systems.30 As summarized herein, our studies have shown that particle properties can be changed by variations in environmental conditions, the synthesis routes used to create the particles, and even seemingly inconsequential changes in well-established synthesis or sample handling processes.
Environmental and time-dependent properties
Until recently, many reports regarding nanoparticle properties in the early literature assumed that their physicochemical properties remained relatively constant after they formed. In contrast, we found that the chemical properties of 3- to 10-nm ceria particles suspended in aqueous solution changed depending on the nature of the solution and aging time32 [shown in Fig. 3a]. This work demonstrated that the oxidation state of ceria particles responds to the environment, changing from +3 to +4 [Fig. 3b] when an oxidizing agent is added and slowly reverting back to +3 as the solution ages and the oxidizing condition of the solution changes. Furthermore, it was shown that the rate of change depended on the composition of the media in which the particles were synthesized or suspended. For example, in a study of the effects of polyhydroxyls added to an aqueous solution, the kinetics of switching of oxidation states in solution was observed to be slower in basic medium compared to acidic medium.29 Taken together, these results suggest that some of the inconsistencies in particle properties reported in the literature may be related to the differing synthesis, processing, and environmental conditions experienced by particles in the different studies.
Figure 3.
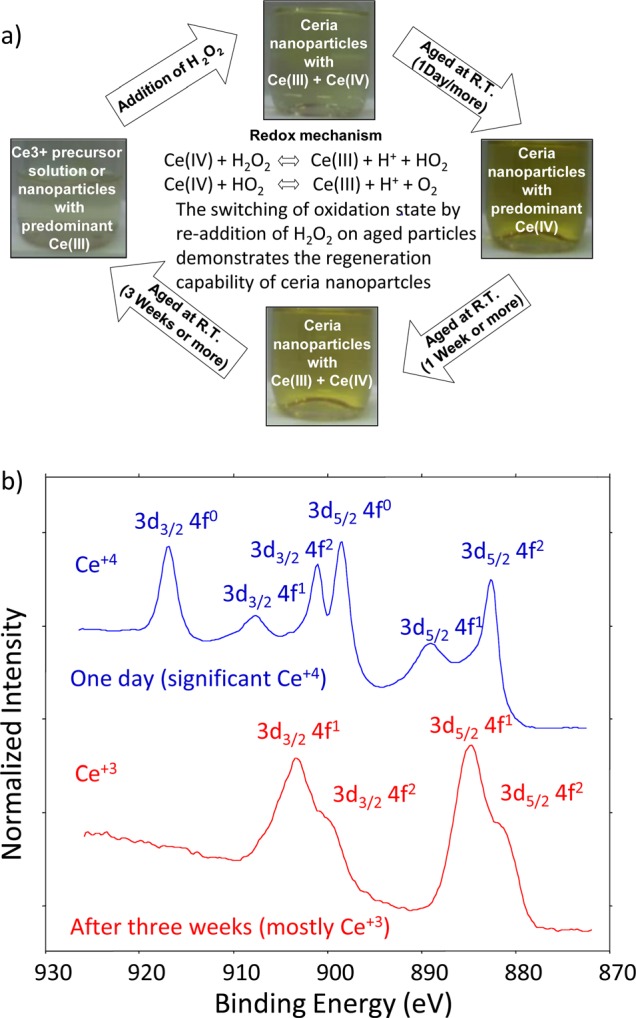
(Color online) (a) Schematic representation of the regenerative capability and oxidation state switching of ceria nanoparticles in an aqueous environment. The pictures of the bottles containing the nanoparticle suspension in DI water indicate color and oxidation state changes after different aging periods. (b) XPS Ce 3d spectra from particles removed from solution after one day and three weeks. Consistent with the optical measurements and solution color, the particles were mostly Ce+4 after one day and mostly Ce+3 after three weeks. When H2O2 was added to the aged nanoparticles in solution, they switched from Ce+3 back to Ce+4. Adapted with permission from Kuchibhatla et al., J. Phys. Chem. C 116, 14108 (2012). Copyright 2012, American Chemical Society.
Contradictory biological impacts
Different research reports in the literature indicate ceria nanoparticles to be oxidative or antioxidative in biological systems and to be toxic or benign depending on the particle or the biological test conducted. We examined the literature to explore the hypothesis that some of these apparently contradictory results might be due to particles differences. Although some of the published reports were too incomplete for an adequate evaluation, it was possible to determine that fundamentally different synthesis and processing routes were taken to produce the ceria particles used in different studies. Many of the ceria nanoparticles fit into one of three categories: (1) particles produced in solution at room temperature,34, 38, 39 (2) those produced by thermal hydrolysis (in the presence of various solvents/oxidizing conditions) involving heating of particles in solution,40, 41, 42 and (3) others synthesized by a variety of elevated temperature processes43, 44, 45 that involve either no solution or heating particles to elevated temperatures after synthesis (Fig. 4).
Figure 4.

Three categories of synthesis and processing approaches used to produce ceria particles: high-temperature processing (a)–(c) (Refs. 43, 44, 45), indirect heating of precursors or nanoparticles in solution (d)–(f) (Refs. 40, 41, 42), and room-temperature synthesis of nanoparticles (g)–(i) (Refs. 39, 38, 34). Adapted from Ref. 30.
When these synthesis routes were linked to the biological endpoints, it was found that in solution-based synthesis processes not involving heating produced particles that often were either nontoxic or antioxidative. Meanwhile, particles that had been heated to elevated temperatures were more likely to have undesirable effects (Fig. 5).30 Heating is likely to influence a variety of particle properties, including grain, particle size, and crystallinity. Although other factors, such as specific biological endpoint and particle coatings (deliberate or accidental), also will have impact, the literature survey suggests that synthesis route and particle history can significantly impact the biological properties of nanoparticles.
Figure 5.
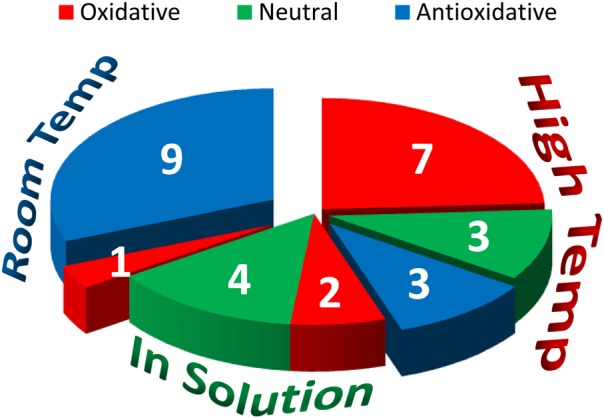
(Color online) Summary of the relationships among synthesis categories and biological impacts, showing that synthesis routes have a significant impact of biological outcomes. Adapted from Ref. 30.
Detailed observations made by a University of California, Los Angeles-led research team for a set of well-characterized silica nanoparticles also highlight differences produced by a high-temperature process route, showing toxicity, while silica nanoparticles produced by low-temperature colloidal synthesis were nontoxic in a variety of tests.46 Taken together, both studies strongly indicate that synthesis and processing history can have a major impact on nanoparticle behaviors and properties. Unfortunately, because processing history, storage time, and conditions, and other information sometimes are missing in reports in the literature, it is not always possible to resolve the sources of apparently conflicting results.
Disappearing particles
In some cases, low-temperature colloidal synthesis processes, as noted, tend to produce particles with nontoxic behaviors. In the case of ceria, antioxidative behaviors often have been observed.31, 34 A significant body of work has been established by Sudipta Seal's group at the University of Central Florida for particles synthesized by a room-temperature solution process.32 Particles synthesized by this process have been observed to remain relatively stable in solution for periods of more than a year. Oddly, in-house work with particles produced by some of the same people using the same process did not have the same long-term stability, and the particles disappeared from solution in less than 60 days.30 Follow-up studies showed that switching the storage vial from glass to plastic altered some properties of the solutions, the particle lifetime, and effective diameter. We also found that variations in the precursor chemicals [from the same vendor but with different x-ray diffraction (XRD) patterns] and using different sources of deionized (DI) water for the synthesis impacted the long-term stability of the particles.30 Regardless of the specific cause of the particles' behavior changes, we note that subtle, seemingly insignificant, or unintentional changes in precursor chemicals or processes can have a significant impact on the lifetime and properties of nanoparticles.
Surprise on the surface—copper oxide nanoparticles
Copper and copper oxide nanomaterials are useful in a range of commercial applications and products, including being used as catalysts, superconductors, and lithium–ion electrodes.47 Although several studies of Cu-based nanoparticles suggest they can be toxic,48 it is recognized that as these nanoparticles age in different environments, their properties will change as a function of time.47 To assess the impacts of different chemical states of Cu nanoparticles, studies have been undertaken that include measurements of the biological response to Cu-based nanoparticles, where the particles are delivered with a known surface chemistry.
In one such study, copper oxide nanoparticles (≈42 nm primary particle size) were generated using spark discharge (Palas Karlsrühe) between opposing high-purity (99.95%) Cu rods. The aim was to generate fresh Cu (II) oxide nanoparticles for inhalation exposures. Preliminary XRD measurements revealed that the particles were a mix of metallic Cu, Cu2O, and CuO. To further characterize the nature of the particle surfaces, the material was shipped to EMSL in an argon atmosphere for x-ray photoelectron spectroscopy (XPS) analysis. At EMSL, the container was opened, and the particles were mounted on a sample holder in a glove box that was connected directly to an XPS spectrometer. Although high-energy resolution spectra focused on the Cu 2p region could answer questions about the chemical nature of the Cu, consistent with recommended practice (but not universally applied by all),49 we routinely collect large energy regions (survey spectra) to verify the nature and quality of samples provided. The high-energy resolution analysis of the Cu 2p region indicated that the particle surfaces were mostly Cu(II) (apparently, including CuO and Cu(OH)2). However, the presence of fluorine in the wide-energy survey spectra [Fig. 6a] was not expected. A higher energy resolution spectrum of the F 1s photoelectron region [Fig. 6b] found peaks that were consistent with polytetrafluoroethylene (PTFE) and CuF2. Analysis of the C 1s region [Fig. 6d] showed several carbon peaks consistent with the presence of PTFE and PTFE breakdown products.50, 51 For comparison, a spectrum from undamaged PTFE also is shown [Fig. 6c]. Not only did the particles contain an element on the surface that was unplanned, but PTFE breakdown products are known to induce acute and severe lung inflammation.52In vivo assessment of the toxicological response to these particles as CuO nanoparticles could have been misleading because the particles also were coated with PTFE and damaged PTFE.
Figure 6.

(Color online) XPS spectra from Cu oxide nanoparticles and clean PTFE reference: (a) XPS survey spectra with unexpected F lines; (b) High-energy resolution F 1s region, showing photoelectron peaks consistent with the presence of PTFE and CuF2; (c) High-energy resolution spectrum C 1s region from clean PTFE, showing a photoelectron peak consistent with CF2 bonds in PTFE; and d) High-energy resolution C 1s, showing the presence of breakdown products from PTFE (e.g., CF3, CF2, CF2-CHF, CHF-CHF, along with CH from advantageous surface contamination; a small amount of C-O and C = O is possible but not included in this peak fit). XPS identified the presence of F that was not expected or desired on these particles.
Before XPS analysis was done on these particles, they had been examined by XRD and transmission electron microscopy (TEM), including energy-dispersive x-ray spectroscopy (EDS). Due to the likely thin surface coating and a noncrystalline nature of the F either in the particle or on the particle surfaces, the presence of F was not detected by these methods. Without the surface sensitive analysis (XPS, in this case), the presence of the contamination at the particle surface may have remained unrecognized. A careful evaluation of the synthesis process helped determine the source of the F and steps were made to successfully reduce the F contamination by replacing PTFE components in the generator with machinable glass. Tools with high surface sensitivity provide information regarding surface contamination that may not be detected by other methods. The XPS data provided important information critical to the development of the planned toxicology study by confirming and reinforcing the need for characterizing the surface of nanoparticles upon generation and also during exposure to properly interpret the in vivo responses.
It is easy for unexpected contamination to occur during the nanoparticle synthesis (as reinforced by another example later in this paper). Although researchers know to avoid or minimize contamination, the tools needed may not be readily available and/or are not always applied. If not detected and properly considered, the presence of contaminants in and on nanoparticles used for toxicology tests can introduce an uncontrolled variable. The toxicity of some carbon nanotubes, for example, was found to be associated with contaminants that remained from the synthesis process.53 Catalysts used in the growth of carbon nanotubes can remain as residues that make up as much as 40% of the final product weight.
Clean and dry?—surprises associated with sample processing and preparation for analysis
Many types of nanostructured materials and nanoparticles are synthesized, shipped, processed, cleaned, tested, or used in combination with solutions of various types. The interactions of nanoparticles with these solutions can have a variety of both intended and unintended consequences. Two examples from our work demonstrate related causes but differing effects.
Apparent changes in thickness of nanoporous films due to solvent treatment
In earlier work, nanoporous silica films were examined as a method to create low-k dielectric materials for advanced high-density integrated circuits. These films were synthesized, processed, and cleaned in a variety of ways. Ion sputter profiling and XPS analysis were applied to obtain an estimate of relative layer thickness. As part of these studies, we found that films exposed to isopropyl alcohol (IPA) appeared to be thicker than those not exposed to IPA. The samples exposed to IPA had been removed from solution, dried, and placed in vacuum for several hours before being subjected to Ar ion sputtering.54 Figure 7 shows the apparent changes in film thickness. Based upon the sputter time to expose the interface, the exposure to IPA appeared to thicken the films. Our initial assumption was that the IPA exposure altered the film in some way that likely altered the sputter rate and caused the apparently thickening. To explain the observation, several different hypotheses regarding changes in the film structure or chemistry and/or deposition of material within the porous structures were considered and tested before we concluded that some IPA was retained in the nanostructured films even after drying for several hours in air and vacuum. Thus, the apparent differences in thickness were due to additional mass being included in the film. Eventually, we learned that leaving the samples in vacuum for more than 48 h would remove the retained liquid, and the time to sputter through the films for unexposed and IPA-exposed films became consistent.54 Capillary forces were sufficiently strong to retain significant amounts of a highly volatile solvent after hours in air and vacuum.
Figure 7.
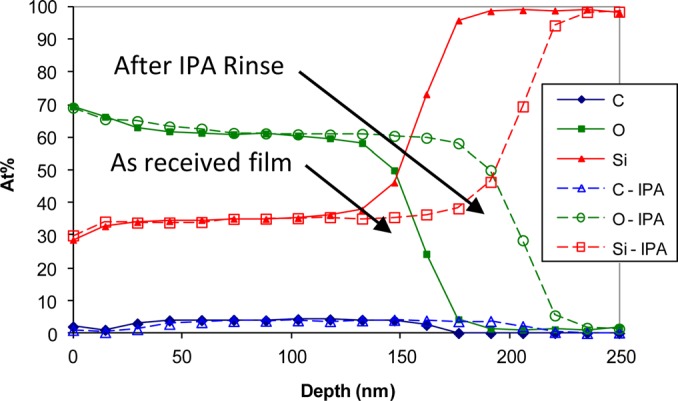
(Color online) Sputter depth profiles of plasma processed p-OSG film before and after exposure to IPA. Based on comparison to the sputter rate for SiO2 the apparent thickness was approximately 157 nm with no IPA exposure and 205 nm after IPA exposure. From Gaspar et al., Surf. Interface Anal. 37, 417 (2005), Copyright 2005, John Wiley & Sons, Ltd.
Removing nanoparticles from solution without destroying important information
For many types of detailed analyses, it is necessary to remove particles from synthesis, process, or testing solution prior to analysis. As outlined by Nurmi et al.,26 there are several factors that need to be taken into consideration including: (1) removing residual solutes to minimize deposition of salts or other solution species, (2) removing solvent and cosolvents in a manner that minimizes aggregation and interference with the measurement process, (3) eliminating nonstructural water or other solvent without significantly altering particle phases, (iv) minimizing erosion of original surface coatings by dissolution or abrasion, and (v) avoiding reactions with the medium or its contaminants that may occur upon exposure to oxygen and other potentially reactive species that will alter the samples either immediately or as a function of time. Two different particles extraction processes are described herein that highlight the need to tailor the extraction process to the particle type and analysis objectives.
A variety of extraction or immersion processes are used as reported in the literature. Techane et al.55 described the process they used to remove Au nanoparticles coated with a self-assembled monolayer (SAM) from the solution. In this case, the objective was to remove the particles from the solution where the SAM was grown on the particles, minimizing any organic residue from excess 16-mercaptohexadecanoic acid (C16-COOH) thiol remaining in the solution. The process for removing the particles from the AuNP C16 COOH-SAM solution and depositing the particles on a substrate for XPS analysis involved several steps, including dialyzing the solution three times, concentrating the particles by centrifugation, and multiple deposits of solution on a clean silicon substrate. The samples were stored under N2 gas until surface analysis measurements were conducted, usually within about 2 h.55 This procedure produced SAM-coated Au nanoparticles consistent with good-quality coatings found on Au flat surfaces.
Our research conducted on metal-core oxide-shell Fe nanoparticles (nano-Fe0), which focused on understanding the impact of variations in nanoparticle synthesis, composition, and structure on the reduction of contaminants in groundwater,27 had somewhat different analysis objectives and required a different sample preparation method. Because the particles were reacting with both the water and contaminants in the water, we needed to stop the reaction to obtain the desired analysis. In the work's early stages, a “flash” drying process was developed to extract particles from solution as part of a method to “quench” particles into a stable state at different times during the reaction process. The aim was to stop the reaction or aging process and minimize solution contaminants remaining on the particles. The drying process26 involved removing the particles from solution in a glove box using a standard vacuum filtration apparatus assembled with a 0.02-μm PTFE filter and a vacuum pump capable of producing −20 mm Hg. After pouring the nanoparticle suspension onto the filter, approximately 30 s were needed to remove the original solvent. Then, the filtrate was washed three times with acetone (or other hygroscopic solvent), each time using just enough to completely cover the filtered nanoparticles. After washing, the vacuum was maintained until the particle layer appeared dry (typically 5–10 min). At this stage, most types of nano-Fe0 were a loose powder that was readily transferred to a container for storage or testing.
Although the samples appeared dry using this procedure, electrochemical measurements made on particles showed that they continued to change as a function of time. A range of thermal analysis, mass spectrometry, and other measurements demonstrated that a variable amount of water was retained in the sample (even though the powder appeared dry). Slightly different types of particles retained variable amounts of moisture, in some cases up to nearly 20 wt. % of the sample. The thermal analysis helped the development of a revised drying procedure that involved simple but important changes. After the third and final wash with solvent, the top of the Büchner funnel was sealed with a silicone stopper [Fig. 8a], and a vacuum gauge was used to adjust the pressure inside the funnel to −20 mm Hg. This condition was maintained for 30 min as it was determined this was sufficient time for removal of most of the residual solvent [shown in Fig. 8b]. The vacuum was relieved without disrupting the layer of nanoparticles on the filter by slowly opening a needle valve on a stainless-steel tube that pierced the silicone stopper. The flash-dried powder usually was transferred to an amber vial and stored (loosely capped) in a vacuum desiccator containing a 50/50 mix of Drierite® and activated charcoal.26 This process removed most of the moisture from the samples (some additional moisture was removed by storage in the desiccator), effectively stopped the changes observed in the electrochemical studies due to storage time, and also increased the reproducibility of other experiments.
Figure 8.
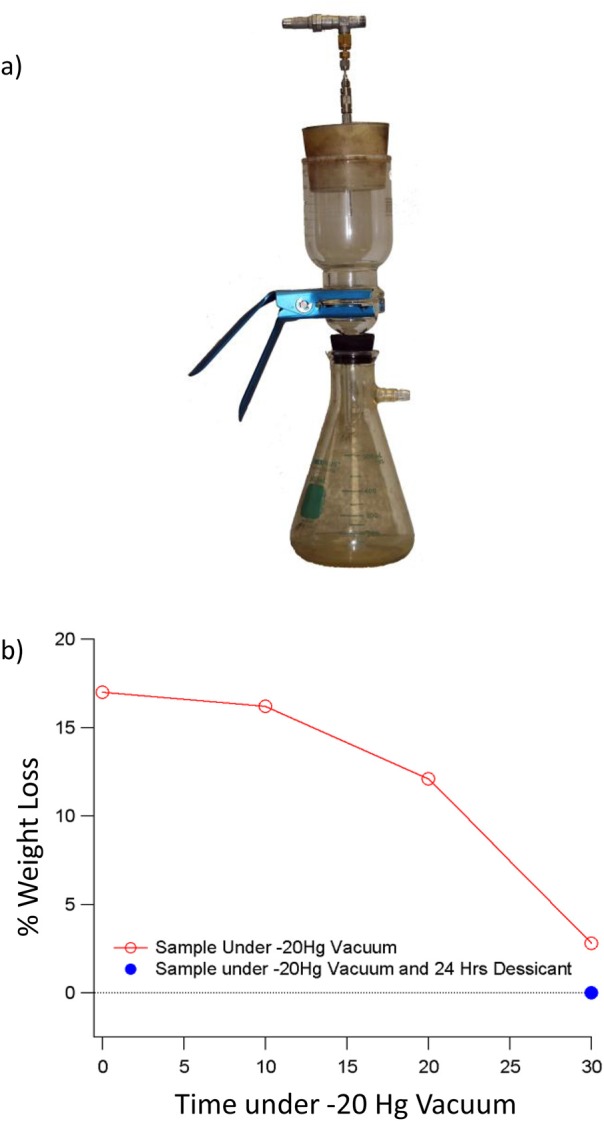
(Color online) (a) Image of filter and pump arrangement used to “flash dry” nanoparticles removed from aqueous solution for detailed. (b) Graph showing percent weight (moisture) loss as a function of drying time under −20 mm Hg vacuum (open circles). Without the vacuum assist, variable amounts of moisture often were retained in the collection of particles. As shown by the horizontal line, an additional 2 to 3% of moisture could be removed by storing the particles for 24 h in a desiccator filled with 50:50 anhydrous calcium sulfate and activated charcoal. Reprinted with the permission from Nurmi et al., J. Nanopart. Res. 13, 1937 (2010). Copyright 2010, Springer Science and Business Media.
Both the nanoporous film and nanoparticle immersion results highlight the fact that capillary forces can be quite effective at retaining a range of solvents within various types of nanostructures. For the nano-Fe0 studies, it was particularly important to effectively stop the changes taking place when the particles were in the solution to relate the physical behaviors of the particles to the particle reactivity and surface chemistry when ex situ measurements were conducted.28
NANOMATERIAL PROPERTIES AND CHARACTERIZATION CHALLENGES
The following summary highlights general areas or analysis challenges important for many types of nanomaterial characterization, including the importance of surfaces and interfaces; enhanced significance of synthesis, handling, processing, and storage conditions; and the dynamic nature of nanosized objects. A variety of effects have been grouped into topics as shown in Fig. 9 and Table Table I..
Figure 9.
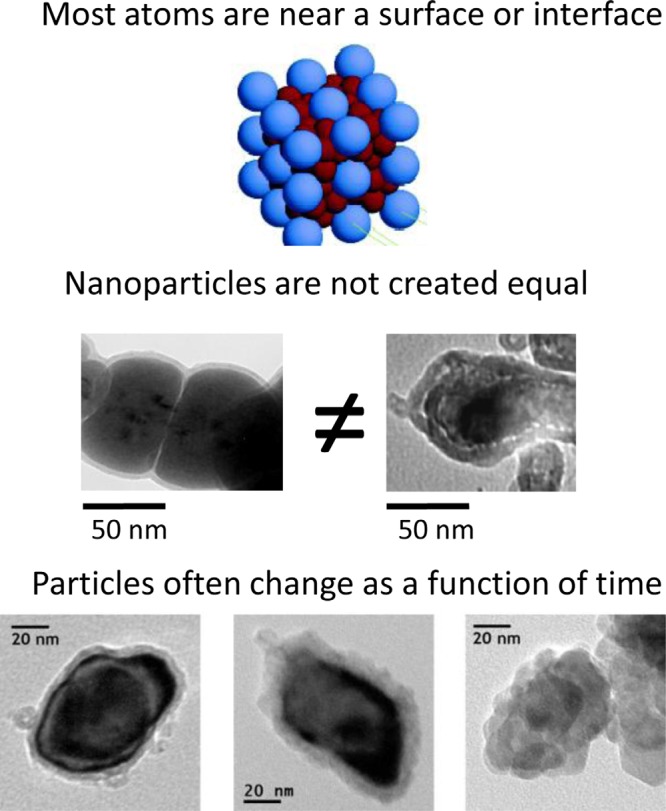
(Color online) Properties of nanoparticles that often introduce characterization challenges; also see Table Table I..
Table I.
Challenges associated with characterization of nanoparticles.
| Nanomaterial surfaces and interfaces control properties |
|
| “Equivalent” nanoparticles usually are not |
|
| Nano-sized objects are dynamic: they change with time and respond to their environment |
|
Nanomaterial surfaces and interfaces control many material properties
It is widely recognized that as particle size decreases, surfaces and interfaces begin to dominate and control the properties of nanostructured materials. However, the chemical and physical nature of these surfaces and interfaces often are not measured or reported. Grainger and Castner10 point out that over the past 40 years, surface scientists have obtained detailed knowledge about the behavior of surfaces, including the important role of deliberate and accidental surface layers. They note that the rigor needed to understand the chemistry of other surfaces also is required to understand and control properties of nanoparticles and other nanomaterials. Such “nanosurface” analysis10 has been extensively used to characterize supported nanoparticle catalysts but generally unused to characterize unsupported nanoparticles in biomedical and other applications. Because of the significant fraction of atoms or molecules associated with surfaces and interfaces of nanomaterials, surface impurities, surface enrichment or depletion, and surface contamination10 on nanomaterial properties can dominate material properties. For example, the electrical characteristics of Si nanowires are highly dependent on the nature of the environment and wire surface.56 The nature and impact of surfaces may depend upon the application and properties of interest. However, Karakoti and coworkers12 note that the importance of nanoparticle surface chemistry, especially as applied to toxicology, has been significantly underemphasized.
Others have noted57 that adsorption is a spontaneous process on high surface energy materials that reduces the free energy of nanoparticles. Consequently, thermodynamics drives the formation of many types of adlayers on particles during synthesis, processing, and storage. In relation to biological systems, Jones et al.57 observed that adsorbate layers on nanomaterials, not the virgin nanomaterial chemistry, are increasingly thought to be the primary surface that interacts with biological components. The segregation of components during synthesis or in different environments also can create materials for which the surface is significantly different from the bulk material. An excellent example where such segregation was unexpected has been observed during the formation of organic aerosols.58
As shown by the Cu oxide nanoparticle study, where F was identified by XPS, efforts must be made to characterize nanoparticle surfaces because the presence of unexpected contaminants or the unanticipated surface enrichment of a component species would make the nature of the particles significantly different than expected. In many circumstances, nanomaterials synthesis involves using surfactants to control particle shape59 during growth, and different types of chemicals are used during synthesis or processing. Residue from these processes23 may remain on particle surfaces or in the solutions in which many particles are stored and shipped. Often, the routinely applied analytical methods cannot identify such adlayers on particle surfaces. Consequently, lack of surface characterization contributes to the uncertainly and variability of results in the literature. Detailed reporting of synthesis, processing, and storage processes also can assist efforts to track down possible differences in particle behaviors.
“Equivalent” nanoparticles usually are not—nanoparticles are not created equal
When we compared the ability of two types of Fe metal-core oxide-shell nanoparticles (nano zero-valent iron) created by different synthesis processes to reduce CCl4, we found the reaction rates and reaction pathways to vary significantly.27, 60 In further work, we identified residue from different salts that could be used to synthesize particles61 by the “same” process significantly altered the properties of particles. As described previously, we found that ceria particles can behave differently, even when we have attempted to produce them using an identical process,30 and differences in the toxicity of particles that are the same material but have been processed in different ways now are well established.30, 46 Subtle differences, as well as not-so-subtle differences, in materials synthesis, processing, and storage (time, temperature, and light) may effectively make “similar” nanomaterials have different characteristics (some of which are not readily identified by most characterization methods).
Two observations from the Nanotechnology Characterization Laboratory (NCL) of the National Cancer Institute further emphasize some important issues.23, 62 Similar to our challenges to reproduce the formulation of ceria particles, they frequently observe batch-to-batch variation in particle properties. They report measurements on chemically attached polyethylene glycol, or PEGylated, gold nanoparticles, where a series of important measurements produced identical results, but only one batch produced highly inflammatory lung lesions. A highly detailed analysis eventually showed a difference in the degree of PEG coating between the batches, which impacted the degree of plasma protein binding.23, 62 Another important observation made by NCL staff over the years is that many researchers use commercial materials and the manufacturer specifications at face value without further testing.23 Because of history, shipping, storage, and other effects, this is a highly questionable practice. Particles made and characterized at one time and location are often not identical for the same material at a different time or location.
Properties of nanosized objects are dynamic—they often change with time and environment
In an article on metal nanoparticle catalysts, Somorjai and Park63 comment: “The restructuring of the surface coincides with the mobility of the adsorbate; thus the surface, both the metal and the adsorbate, has to be dynamic for catalytic reactions to occur.” Similar observations were made regarding the activity of catalysts associated with the growth of nanofibers.64, 65 It is possible to observe shape changes for nanoparticles during electron microscopy.66 It may be important to think of nanoparticles as dynamic, rather than static, objects. Many studies have shown the dynamic nature of natural and engineered nanoparticles, especially for particles smaller than 10 nm,67, 68 which has many different types of manifestations and implications. In particular, they can change with time, may be altered by proximity to other particles or surfaces, can be easily damaged during analysis, and frequently change in response to their environment.
Particle shapes can change
In reviewing structure and shape changes for nanosized particles, Yacaman et al.66 noted that different configurations for a nanoparticle have very similar energy, thereby making shape transformation easy and common. The types of energies we commonly apply during analysis will provide energy adequate to stimulate such changes.7 It may be appropriate to consider the dynamic nature of particles an essential characteristic that gives rise to important chemical properties as previously noted.63, 65
Particles dissolve, grow, and adsorb material from the environment
The high surface energy of many types of particles is lowered by growing57 or dissolving.69 Particles also may assemble into larger units57, 70 that may be easy (soft agglomerates) or difficult (aggregates) to break apart. As noted, to reduce surface energy, most particles naturally adsorb material from their environment.57 When nanoparticles are placed into biological media, a protein coating (corona) forms around the particles that may be tens of nm thick.71 The protein corona forms quickly, and the composition is influenced by the size of the nanoparticles.72 In many environments, even single-material nanoparticles quickly become core-shell particles, either by adsorbing material from the environment or reacting with that environment (e.g., oxidation/corrosion).28
Environmentally induced changes in structure and chemistry (including sample preparation and processing)
In addition to forming surface coatings, the environment can alter either physical structure or chemistry of a nanoparticle. Changes in the chemical state of ceria nanoparticles have been found to vary due to the presence of peroxide in the solution in which they are suspended.32 The crystalline nature of ZnS nanoparticles was observed to change when the particles' environment varied from wet to dry.73 As expected, oxide-shell metal-core iron nanoparticles oxidize in water, impacting their ability to reduce contaminants such as CCl4.28
The impact of environmentally induced particle changes has implications in at least two different ways: impacts on sample preparation and analysis and how they may transform in their “native” or working environments. Many surface analysis methods require placing samples in environments (often vacuum) that differ from those in which the samples are normally found. This can alter the particles and needs to be considered as part of the analysis and sample preparation process. Issues associated with sample handling and removal of samples from the natural environment to the analysis environment also apply here. The deliberate need to stop a process in the environment of interest was the focus on the flash-drying study noted earlier.26 The need to remove components from the solution environment that were not associated with the particles was a focus of both the flash-drying process and the extraction process of the SAM-coated Au nanoparticles.55
There also are broader implications of environmental-induced transformation, both for application and aspects of analysis. Wiesner et al.67 commented on the “Ubiquity and Mutability of Nano-Sized Materials in the Environment.” It is important to realize that nanomaterials (especially nanoparticles) may change in some ways if their local environment changes during incorporation into a product, they are introduced into a biological system, or they are released into the natural environment. Cohen et al.74 noted the importance of comprehending particle–media interaction to understand the particle dose delivered to cells for either toxicological impacts or pharmaceutical purposes. The need to understand particles and their transformations in the native environment underscores the demand for various types of in situ real-time measurements.
The nature of sample preparation must be linked directly to analysis and the testing to be conducted. To examine the reaction intermediates during a catalysis reaction, Krier et al.75 conducted a study that combined both of the two preceding issues. They noted that organic ligands typically are used to stabilize nanoparticles during synthesis, and these remain on the particles after synthesis. However, the presence of an organic capping agent may block a significant fraction of active sites when the particles are intended for catalysis studies. Their specific study focused on the application of sum frequency generation-vibrational spectroscopy (SFG-VS). They observed that the capping layer produced a strong signal that inhibited measurement of the reaction intermediates of interest. They tested both solvent cleaning and ultraviolet (UV) cleaning for removing the capping layers and found that both cleaning processes enabled the collection of the needed SFG signals. They further noted that the solvent cleaning process did not remove the entire capping layer, while the UV cleaning enabled complete removal. However, the solvent cleaning produced particles that were stable under reaction conditions, while the UV-cleaned particles had significant aggregation and were effectively unstable, actually complicating the study objectives. In this case, the “partial” cleaning produced the most useful material and stable material.
Proximity effects
The properties of individual or isolated nanoparticles may change as they are in contact or near surfaces or other nanoparticles.7, 22 This behavior can be used to create materials with tuned properties76 or as the physical basis of a nanoscale ruler.77 Implications of proximity effects related to sample mounting78, 79 include substrate interactions, particle charging, coupling of quantum states, and spacing impacts on electronic and magnetic properties of the effective composite. We have found, for example, that the chemical state observed by XPS may depend upon the concentration of particles loaded onto a substrate as shown in Fig. 10. The ceria particles examined were deposited on a silicon wafer by placing a drop of solution on the substrate in a glove box and letting the solution evaporate. After the solvent evaporated, a nonuniform distribution of particles formed on the surface. XPS analysis of portions of the deposit with a low density indicated Ce+3 [Fig. 10b], while the portion of the deposit with a higher density contained at least some Ce+4 [Fig. 10a]. When we first observed this behavior, we asked a few others if they had ever observed similar behaviors. One group agreed that they had observed something very similar on a different substrate, but they thought something had gone wrong with the measurement. We think this behavior is a proximity or environmental effect. In the region of higher particle density, the particles were interacting with each other (ceria nanoparticles with other ceria nanoparticles). Meanwhile, for the region of lower particle density, ceria nanoparticles had greater interaction with the substrate.
Figure 10.
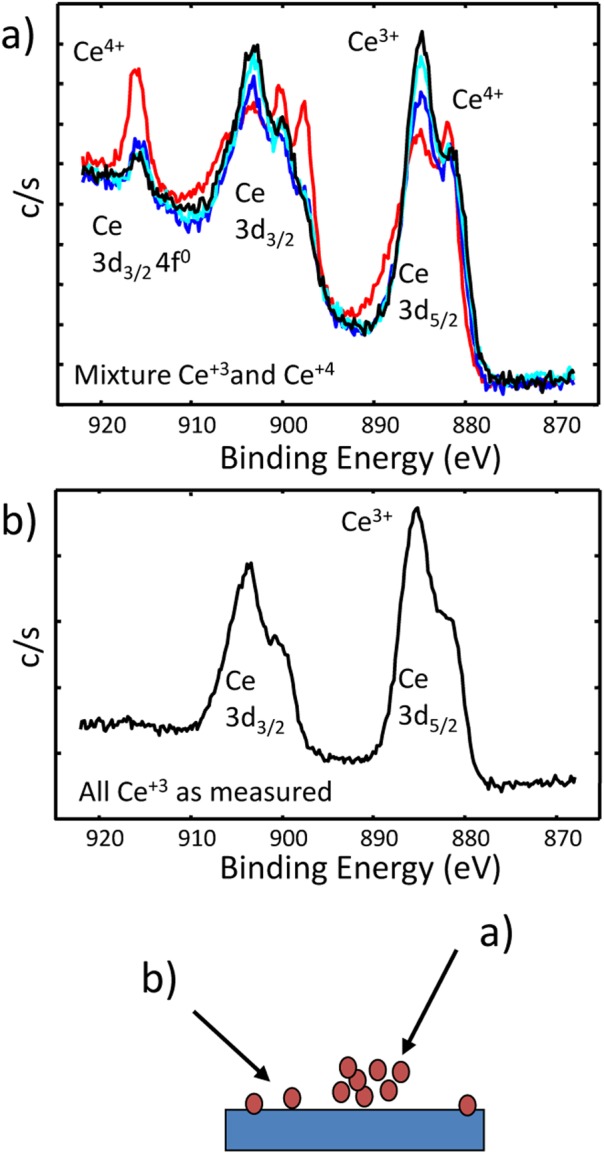
(Color online) Ce 3d XPS spectra from ceria nanoparticles deposited on a Si substrate. Based on optical data, we would expect the ceria to be mostly Ce+3. The deposition process produced regions of high (a) and low (b) particle density. The XPS photoelectron spectrum from the higher density of particles differed (some Ce+4) from the lower density region (only Ce+3).
Ease of sample damage
The energies associated with chemical binding, particle bending or fracture, and charge buildup become roughly equal and less than those associated with analysis methods for nanoparticles.7, 21 One consequence of the similar energies is that particles are easily damaged during analysis or upon exposure to the environmental conditions of the analysis method. Samples have been observed to melt upon exposure to an electron beam,80 change oxidation upon x-ray81 or electron beam exposure,82 and sputter at enhanced rates.83
During XPS analysis of ceria nanoparticles, we discovered that the type of damage observed could change depending on the synthesis process used to create the particles. For the higher particle density, multiple spectra are shown in Fig. 10a. This set of spectra is presented again in Fig. 11a, showing that although the Ce+4 signal never totally went away, it decreased upon x-ray exposure.81 In contrast, a set of spectra in Fig. 11b shows an example of the Ce+4 increasing with time. We believe that the different damage processes are due to differences in the synthesis of the particles. In the first example showing damage-related reduction, the sample synthesis included the use of toluene, where a residue on the particle surface produced a locally reducing environment upon x-ray exposure. The second set of particles was synthesized in an aqueous solution containing no added organics, and an oxidizing environment was produced upon radiation.
Figure 11.
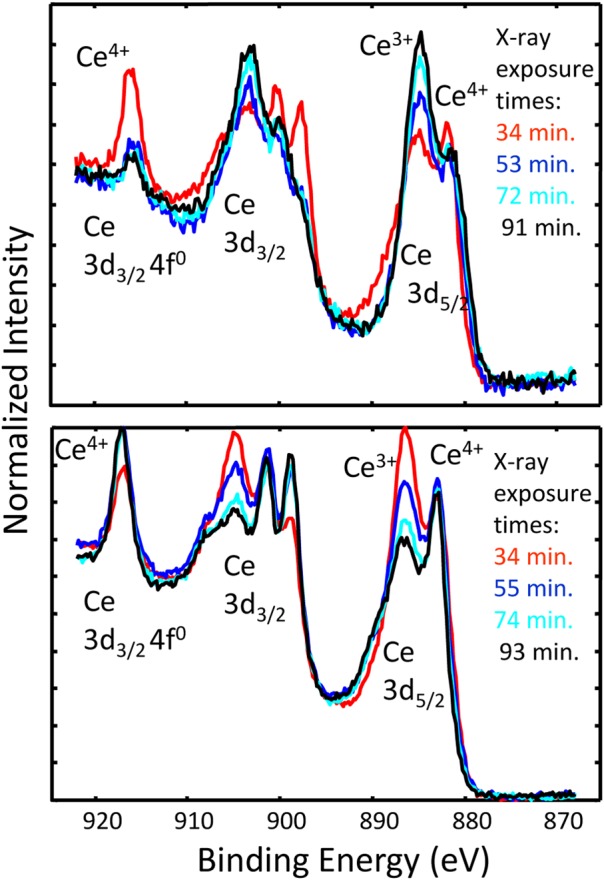
(Color online) Changes in the Ce 3d XPS spectra collected as a function of time for (a) particles formed in an aqueous solution, containing some amount of organic (toluene) (after Ref. 81), or (b) particles synthesized in a solution with no added organic. The 3–5 nm diameter particles produced in the solution with toluene (a) tended to become reduced upon x-ray exposure, while 10–14 nm particles formed in aqueous solution without added organics (b) tended to become oxidized upon x-ray exposure.
These and many other observations7 highlight that nanomaterials in various forms are easily damaged during analysis, and the nature of the damage may be altered by the specific nature of the particles and the environment where the damage takes place.
Time-dependent behaviors
For all of the preceding reasons, the properties and characteristics of nanoparticles frequently alter with time, a process sometimes called “aging.”28, 32, 47 This time dependence can be influenced by the synthesis process, exposure to light, storage conditions, temperature, and other aspects of the sample environment. Consequently, true characterization of nanoparticles might be considered a four-dimensional challenge60 with the axes being space, composition or structure, time, and environment. Although this might seem to present an overwhelming challenge for nanoparticle analysis and application, the rate of change is sufficiently slow in many cases as to be of no concern, and, in other circumstances, aging of nanoparticles presents an opportunity to tune particles for desired properties and lifetime or life cycle. Because the time scales of change can range from fractions of seconds to many years, the impact of any changes depends on the particle, environment, and nature of the study or application being considered.
Understanding these and other characteristics of nanoparticles and other nanomaterials enables researchers to design the types of analyses and characterization needed for specific research activities. It is clear, for example, that characterization of objects months or years before application or use may not provide useful information,4 unless the reliability of that information is confirmed. Understanding the changes a particle will undergo in particular environmental conditions is useful for determining the time frame and handling needed for a specific experiment. Keeping and reporting detailed records of the date of synthesis, storage times, and conditions, and times of analyses will facilitate the understanding of similarities and differences between studies. Because of environmental impact on particle properties and behaviors, it is important to monitor particles in the actual environment of interest, to the extent possible.11 Because the information available from such in situ measurements is often incomplete and less detailed than desired, it usually is important to combine real-time in situ measurements with a range of ex situ measurements, where sample handling has been carefully planned and appropriate integration of results is undertaken (we have attempted such integration in our Fe metal-core oxide-shell work26, 27, 28).
ROLE OF SURFACE ANALYSIS IN SUITE OF INFORMATION NEEDS
Incomplete characterization impacts several aspects of nanomaterials research, including the reproducibility of research results, the process of scaling production to meet manufacturing needs, and human health and environmental risk assessment.4, 10, 57, 84 Several different research, regulatory, economic development, and standards-related communities have worked to identify the minimum measurements needed to adequately characterize nanomaterials. Stefaniak et al.4 have examined 28 of these lists to understand the most critical needs related to metrological challenges and the development of nanoscale reference materials. As might be expected, particle size, surface area, particle composition (bulk), surface chemistry, agglomeration/aggregation, surface charge, and surface reactivity are prominent on many lists. Additional features, such as particle stability, solubility in biological media, and surface structure, are critical but less commonly discussed.
In a perspective article, Grainger and Castner10 observed that the information and experience obtained from surface science often is not integrated into studies of nanomaterials. In an impressive review of stabilization and functionalization of iron oxide nanoparticles for biomedical applications, Amstad et al.85 summarized the information needs and wide range of tools that have been applied to gather relevant data. Although they note the value of XPS for certain types of adsorbate measurements, surface sensitive methods generally play a minor role in these studies. It is our view that surface analysis methods of various types are underused as important components in the suite of tools needed for appropriate nanomaterial characterization. Although we have applied a range of methods to characterize the Fe metal-core oxide-shell nanoparticles,27 XPS became one of the few core methods applied in almost every circumstance to assure consistency and verify that contamination or other changes in the particles surface had not taken place.
The underuse of surface analysis methods for characterization of nanomaterials may have multiple sources. First, because the most common surface analysis tools have surface sensitivity in only one dimension and usually do not have the resolution to examine individual particles, some researchers do not consider applying the tools. A second possible reason is that these tools can require instrument access, as well as appropriate care in sample preparation and analysis, that may exceed the expertise available to prepare samples, collect data, and understand results. If applied or interpreted inappropriately, incorrect, inconsistent, or misleading results may be produced. A variety of research groups are demonstrating the application and value of these tools, as well as expanding types of information that can be obtained from these tools.
Information available from common surface analysis methods
The range of useful information about nanomaterials that can be obtained from data collected using traditional surface analysis methods exceeds what generally is expected by many analysts. The information available from several surface analysis methods is summarized in Table Table II.. These surface sensitive analysis methods can be applied in two somewhat different modes. For example, in many circumstances, it is highly useful to simply recognize the presence or absence of contaminants or to confirm the chemical state of elements present in the nanomaterial. Such uses dominate the literature, and their importance cannot be understated. However, with additional thoughtful analysis, an expanded range of information can be extracted—even from what was initially collected as qualitative data. In earlier reviews and summaries focused on XPS,78 nanoparticles, carbon nanotubes,60, 79 and nanomaterials,7, 86, 87 we provided many examples of how the most available surface analysis methods can be applied for nanomaterial characterization. Here, we highlight important underlying concepts, discuss some recent applications.
Table II.
Characteristics of common surface analysis methods and types of information available for nanomaterials.
| Surface analysis methods | Information available | Probe | Detected | Lateral resolution | Information depth | Depth resolution |
|---|---|---|---|---|---|---|
| Electron Spectroscopies | ||||||
| X-ray photoelectron spectroscopy |
|
X-rays | Photoelectrons | ≈2 mm System dependent | ≈10 nm | ≈2 nm |
| Auger electron spectroscopy |
|
Electrons (∼3 to 20 kV) | Auger electrons | ≈10 nm | ≈10 nm | ≈2 nm |
| Incident Ion Methods | ||||||
| Secondary ion mass spectrometry |
|
Ions (∼3 – 20 kV) | Sputtered ions | ≈50 nm (inorganic)> 200 nm (organic) | ≈1 nm | ≈1 nm (inorganic)≈10 nm (organic) |
| Low-energy ion scattering |
|
Ions(∼2 to 10 kV) | Elastically scattered ions | ≈100 mm | ≈10 nm | ≈0.1 nm |
| Medium-energy ion scattering |
|
Ions(50–200 kV) | Elastically scattered ions | 10 mm (TOF)1 mm (ESA) | ∼10 s nm | ≈0.1 nm |
| Scanning Probe Microscopies | ||||||
| Scanning tunneling microscopy |
|
Stylus | Tunneling current | ≈1 nm | ≈10 nm | |
| Atomic force microscopy |
|
Stylus | Force or displacement | ≈1 nm | ≈10 nm | |
X-ray photoelectron spectroscopy
Because evidence suggests that XPS is the most common surface analysis method applied to nanomaterials,88 we will highlight several examples of its application. It is important to recognize that although XPS is highly used and can provide valuable information, as normally applied in the laboratory, it requires placing a sample in ultrahigh vacuum and involves the analysis of many particles supported on a substrate. Strengths and limitations of XPS and other methods are noted in Table Table III..
Table III.
Strengths and limitations of primary methods discussed in this review.
| Technique | Strengths | Limitations or issues |
|---|---|---|
| XPS |
|
|
| AES |
|
|
| SIMS |
|
|
| LEIS |
|
|
| MEIS |
|
|
| AFM |
|
|
| STM |
|
|
| SFG-VS |
|
|
| NMR |
|
|
The nanostructure of a material impacts the peak intensities, peak energies, and structure (including background signals) of x-ray-excited photoelectron peaks.78 It is possible to invert the process and use details of the XPS peak shape and intensity to learn about the nanostructure of the materials being examined. The surface and structural sensitivity in XPS comes from the short distances that the excited photoelectrons can travel before losing the energy that identifies them as photoelectrons. The contribution to the detected signal from a depth z into the material is attenuated by material covering the layer closer to the surface. Although the equation is only approximately valid, the relationship between depth into a sample and signal strength can be usefully expressed as
| (1) |
where dIz is the intensity of the detected signal at depth z, I1 is the intensity that would have been produced if the layer were at z = 0 (the outer surface), L is the photoelectron attenuation lengths, θ is the angle of the detected electron relative to the surface normal, and dz is the thickness of the layer. The information depths from which detectable signal intensities can be extracted are typically in the range of 1–20 nm. Because L depends on energy, Eq. 1 demonstrates both the energy and angle dependence of the signal intensity from depth z. Applying the surface sensitivity of XPS, including the energy dependences of L and the impact of angle, a range of information can be extracted from XPS spectra,78 including surface composition and chemical state, presence and nature of functional groups, enrichment or depletion of elements at the surface, presence and thickness of coatings55, 89, 90 or contaminants (and reaction rates),91 and average nanoparticle size (if less than 10 nm).92, 93 It is also possible to use XPS to determine the electrical characteristics of particles or coatings and the distribution of Lewis and Brønsted acid sites on zeolite surfaces.79
A variety of methods can be used to extract the thickness of coatings on particles or flat films from XPS data—all effectively based on Eq. 1.89, 90, 91, 94, 95, 96 Computer algorithms using different approaches are available for layer thickness and structure determination, including MultiQuant™,95, 96, 97, 98 which looks at the impact of different particle shape and surface layer models on photoelectron peak signal strength, and quases,94, 99, 100, 101, 102 which considers the impacts of the surface nanostructure on signal strength and the related background signals around photoelectron peaks. Using simulation of electron spectra for surface analysis (SESSA)103 and Topofactor90 approaches to determine the thickness of coating on nanoparticles is discussed in more detail in an example to follow. The impact of particle shape depends somewhat on size. For those larger than 30 or 40 nm in diameter, the particles are effectively objects with curved surfaces, and the analytical approach to determining coating thickness will vary with particle shape but is independent of particle size. For smaller particles, for which photoelectrons can traverse the whole particle, it is necessary to know the particle size and shape to accurately relate signal strength to a layer or coating structure.78, 90
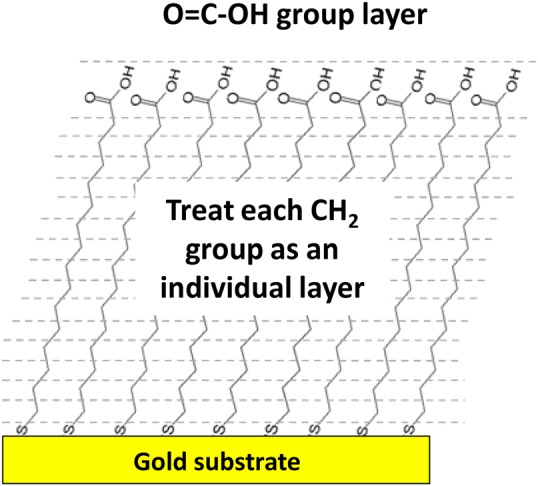
One of the recent tools for assisting the application of Auger electron spectroscopy (AES) and XPS is SESSA.103 In the initial form, the program provides important physical information to allow simulation of AES and XPS spectra from flat surfaces and (nano)-layered structures. Although a new version explicitly incorporating three-dimensional nanostructures, such as particles and wires, is being developed, Techane et al.55 modeled a C16 COOH-SAM adsorbed onto Au nanoparticles (AuNPs) using the flat surface version. This was done by constructing a “model” spherical nanoparticle made up of concentric cylinders with “flat” surfaces with a SAM coating (Fig. 12) at varying angles with respect to the axis of the analyzer as shown in Fig. 13.
Figure 12.
(Color online) Schematic model for the carboxylic-terminated SAM on a flat gold surface that was used in the SESSA calculations. Reprinted with permission from Techane et al., Anal. Chem. 83, 6704 (2011). Copyright 2011, American Chemical Society.
Figure 13.

(Color online) For application of SESSA to predict the signal strengths from SAM-coated Au nanoparticles, the particles were modeled as multiconcentric cylinders, where each cylinder surface has an average photoelectron take-off angle of ai. The XPS detector is positioned at 0° from the central axis of the AuNP. (a) The sphere is divided into nine concentric cylinders. (b) The end of each cylinder is modeled as a flat surface tiled relative to the axis of the spectrometer with infinite thickness of gold, and (c) the surface composition of each flat Au sample is weighted by its geometric factor then summed together to find the AuNP surface composition. Reprinted with permission from Techane et al., Anal. Chem. 83, 6704 (2011). Copyright 2011, American Chemical Society.
The study involved experimental measurements and SESSA modeling of XPS photoelectron signal strengths from two systems: (1) the SAM layer on a flat Au surface as a function of electron takeoff angle and (2) SAM-coated nanoparticles. SESSA was used to simulate both the angle variation of signals from the flat surface and the signals from spherical nanoparticles. In the initial form, SESSA can calculate signal strengths from a layered surface and the layer structure for the SAM on a flat Au surface. Figure 12 shows the SAM structure used in SESSA. The SESSA calculations as a function of angle then were compared to those observed experimentally. Appropriate agreement between the model and experiment was obtained when a small amount of surface contamination was included in addition to the SAM layer and Au substrate. As indicated in Fig. 13, a nanoparticle was approximated as a series of cylinders where the detected photoelectron signals will have different takeoff angles. For both the flat surface and nanoparticle, the SAM thickness and relative surface roughness (RSA) in SESSA were optimized to determine the best agreement between simulated and experimental surface composition. As summarized by Techane et al.,55 a SAM thickness of 0.11 nm/CH2 group, an RSA of 1.05, and a 0.15-nm CH2-contamination overlayer (total film thickness = 2.15 nm) provided the best agreement with the experimental XPS data. After applying the appropriate area weighting factors and summing the SESSA flat surface compositions, the most applicable results for the C16 COOH-SAM thickness and surface roughness on the AuNPs were determined to be 0.09 nm/CH2 group and 1.06 RSA with a 0.15-nm CH2-contamination overlayer (total film thickness = 1.85 nm). The 0.3 nm difference in SAM thickness between the flat Au and AuNP surfaces suggests that the SAM's alkyl chains may be slightly more tilted or disordered on the AuNP surfaces. SESSA and other detailed analysis approaches can provide a remarkable amount of information about the details of nanoparticles89, 104 and are especially useful when this type of information is required. However, these analysis methods are not yet easy to apply to “routine” samples. Sometimes, less rigorous methods can provide useful information as well.
Shard has developed a simple approach for analyzing XPS data to obtain the thickness of coatings on nanoparticles.90, 105 The method involves measuring peak intensity ratios (Fig. 14), knowing the general structure and approximate particle diameters. It can be applied using either analytical formulas or a simple graphical method. Shard has applied the method105 to the data from Techane et al.55 (as analyzed using SESSA) using the ratio for the C 1s and Au 4f photoelectron peak ratios and relevant photoelectron attenuation lengths. The coating thickness calculated by the simple approach is 1.74 nm. As the simple calculation does not include the contributions of the O- and S-containing layers at the top and bottom of the SAM, it can be concluded that the 1.74-nm thickness compares quite well with the 1.85-nm total shell thickness determined by detailed calculations of Techane et al.55 using SESSA. The advantage of the simple method is that it is fast and simple to use. However, for cases when more detail is required, such as core–shell particles with multilayers or an outer contaminant layer, numerical simulations still are preferred.
Figure 14.
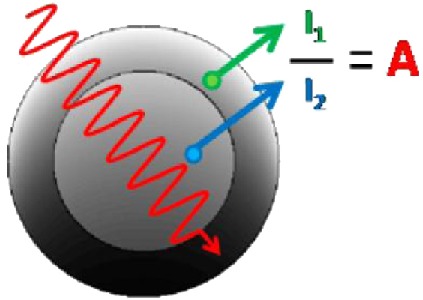
(Color online) Schematic diagram showing the relationships of peak intensity ratios (a) and shell thickness' of nanoparticles based upon knowledge of the radius of the nanoparticles. Reprinted with permission from Shard, J. Phys. Chem. C 116, 16806 (2012). Copyright 2012, American Chemical Society.
We have used the procedure described by Shard105 to examine Ag nanoparticles that were produced for a National Institute of Environmental Health Sciences (NIEHS) Consortium that is examining the toxicology of nanoparticles. During the course of the initial research with the particles, we learned the Ag particles had been formed around 7- to 8-nm Au cores. The presence of Au also had been identified in the XPS survey spectra. Au was observed by XPS in particles nominally 20 nm in diameter but was not observed in particles exceeding 100 nm in diameter. Before we could obtain any microscopy evidence of the Au cores, we used Shard's simple method to check the consistency of the XPS results with the assumption of the presence of a 7- to 8-nm Au core within ∼20-nm Ag particles.
Based on a standard atomic percent calculation, the apparent atom ratio of Ag to Au for the 20-nm particles was approximately 64. This value becomes parameter A in Shard's approach.105 The second important factor in determining the coating thickness is the ratio of electron attenuation lengths for Au and Ag photoelectrons in the particle shell. For the Ag-shell Au-core particles, B = L(Ag in Ag)/L(Au in Ag), where L(Ag in Ag) is the attenuation length of Ag photoelectrons in the Ag shell (overlayer) and L(Au in Ag) is the attenuation length for Au (from the substrate) in the Ag shell. It is possible to calculate an approximate value using B ≈ (Eo/Es)0.75 as noted in earlier work,90, 106 but attenuation lengths and electron mean free path lengths can be calculated using several methods and sources, including National Institute of Standards and Technology databases107 and the formulas contained in computer codes, such as quases.100 Using the experimental value of A and the calculated value of B, Eq. (6) of Ref. 105 was applied to calculate the thickness of the overlayer normalized by the attenuation length (L(Ag in Ag)), assuming the data was from a flat sample (see Table Table IV.). The conversion from the flat film calculation to the spherical nanoparticle geometry was done using the average Topofactor of 0.67 that applies to particles significantly larger than L(Ag in Ag). Multiplying the corrected (but normalized) thickness by L(Ag in Ag) produces the estimate of the particle shell's thickness. The shell thickness determined by this calculation was 5.5 nm. Assuming an 8-nm Au core surrounded by a 5.5-nm-thick shell produces a particle size of ≈19 nm—quite consistent with a nominal size of 20. A later set of scanning TEM (STEM) measurements (Fig. 15) confirmed the presence of the Au core in most particles.
Table IV.
Parameters used to calculate the shell thickness for Ag-shell Au-core nanoparticles using XPS signal strengths.
| Parameter | Description or unit | Value |
|---|---|---|
| A | Ag/Au atom ratio | 64 |
| B | Attenuation length ratio | 0.82 |
| T | Normalized flat surface thickness | 5.1 |
| Topo | Topofactor | 0.67 |
| L | Attenuation length (nm) | 1.6 |
| Shell thickness | nm | 5.5 |
| Particle diameter (assuming 8 nm core) | nm | 19 |
Figure 15.
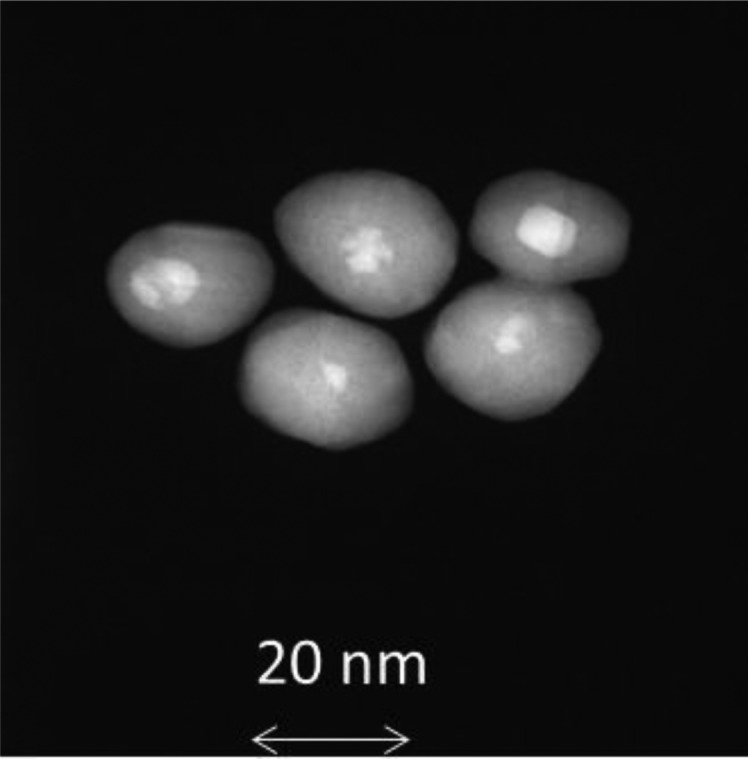
STEM dark field images of Ag-shell Au-core nanoparticles clearly show the presence of Au cores in most particles.
The analysis depth of XPS included in Table Table II. is based upon the most common x-ray sources for laboratory-based instruments. By using alternative energies, particularly those associated with synchrotrons, it is possible to extend the depth of analysis and to use x-ray energy as a method of tuning the analysis depth. Haverkamp et al.108 used this approach to understand the complex core-shell structure of IrO2, RuO2, Sb2O5, and SnO2 electrocatalyst nanoparticles.
It is useful to note that accurate knowledge of attenuation lengths is likely the most important limitation on the ability to accurately determine layer thicknesses of both films and on nanoparticles. The accuracy of these lengths is likely no better than 10% at one standard deviation.107, 109 Furthermore, the attenuation length information is normally applied to thin films (“nano” in one dimension), not for objects of “nano” size in three dimensions. The uncertainty in attenuation lengths limits overall accuracy of the analysis but still allows a great deal of information to be extracted.
Auger electron spectroscopy
Because both XPS and electron-induced AES involve detection of electrons of similar energies, many of the considerations described for XPS apply to AES. However, as generally defined, AES involves an incident electron beam with a spatial resolution that can sometimes be used to collect information about individual nanoparticles. It can be of importance to recognize that because AES involves electron excitation, issues of electron backscattering and transmission through a particle mean some additional parts of the sample may be stimulated to produce Auger electrons, which will limit the actual spatial resolution.110 Analysts have a tendency to look at a scanning electron image available in an AES instrument and assume, when collecting AES data from a feature identified in the image, that the entire signal is derived only from the area where the electron beam strikes. When the electron beam penetrates through a nanoparticle or other nanosized feature below or into the surrounding material, the Auger signals produced and detected usually come from both the nanofeature and surrounding region.
When the potential issues are understood, AES can be quite effective in providing information about nanostructured material, and it has been particularly useful in combination with other methods. Liang et al.111 were interested in the formation of what they called “Cu2O nanodots” for possible chemical or photochemical applications. AES was used to examine the nature of the nanodots formed on a SrTiO3 substrate after deposition using oxygen plasma assisted molecular beam epitaxy. In the AES instrument, it was possible to obtain secondary electron images of the nanodots, along with AES maps for Cu, Ti, and O (shown in Fig. 5 from Ref. 79). One objective of the AES analysis was to determine if a Cu2O wetting layer was observed between the Cu2O nanodots. This wetting layer was not observed. The secondary electron images and AES maps showed that nanodots could be formed with differing shapes. This process was also examined as a function of the amount of material deposited combining AES with atomic force microscopy (AFM) (Fig. 6 of Ref. 79).
As another example, AES was teamed with scanning electron microscopy (SEM) and TEM to examine the location of a composite organic–inorganic nanoparticle on leukemia cells. The combined technique provided reliable, high-resolution information about the nanoparticles and how they bind to cell surface antigens.79, 112
Secondary ion mass spectrometry
During secondary ion mass spectrometry (SIMS), measurements, primary ion beams with energies between 3 and 20 keV are incident on the surface, and the ions removed (sputtered) from the surface are detected. Surface selectivity arises from the depth of the region where the sputtered ions arise. Using cluster ion beams has enhanced the surface sensitivity and, for some materials, minimized the region of damage, allowing sputter depth profiles of organic layers.113, 114 To extract surface molecular information, SIMS is used in a “static” mode that involves a low density and low total dose of ions, so the surface damage and alteration are minimized. Both atomic and molecular secondary ions are used to extract the surface information.115 Like the electron spectroscopies, SIMS is useful for obtaining information about surface layers, functional groups added to the surface, and contamination. Two differences between the electron spectroscopies and SIMS are: (1) the high sensitivity (HS) of time-of-flight SIMS (TOF-SIMS) to many trace elements and functional groups and (2) the changes (damage) induced to the surface due to ion sputtering. The functional group sensitivity has been usefully applied in many ways. For example, TOF-SIMS has been used to examine peptides conjugated to Au nanoparticles as part of a protein kinase assay116 and to examine a multilayer, plasma-produced organic coating deposited on alumina nanoparticles.117 For relatively large nanoparticles produced during welding, SIMS with sputter profiling has been used to examine the complex layers that form on the particles.118 Unlike XPS, which often infers chemistry indirectly from binding energy shifts, SIMS has the advantage of measuring molecular fragments directly.
Because SIMS depends on the sputtering process, it inherently damages a nanostructured material under study, and this must be taken into consideration. Although many nanomaterials and nanoparticles have been successfully examined using SIMS, it is now established that nanoparticles sputter at different rates than flat surfaces or films.119, 120, 121 Recent work shows that in several circumstances, nanoparticles melt or evaporate after direct or grazing impact from many types of primary ions.122 Thus, it has been demonstrated that SIMS can be a useful and important tool for characterization of nanomaterials, but appropriate consideration should be given to potential implications of enhanced sputter rates or sample melting.
Scanning probe microscopy
Unlike other surface analysis tools, the nanocharacterization credentials of scanning probe microscopy (SPM) are widely recognized, and there are many variations of SPM-based methods: scanning tunneling microscopy (STM) and AFM are the most common examples. AFM can provide three-dimensional imaging/visualization of nanoparticles distributed on a flat surface. It also can provide qualitative and, in some cases, quantitative information on physical properties of nanoparticles, including size, morphology, surface texture, and roughness. With appropriate care in dealing with possible tip artifacts, AFM-generated images can be used to extract particle-size distributions and even particle volumes.123 Although most high-value measurements of nanoparticles are made for isolated particles on flat surfaces, efforts are being made to increase the range of applicability. Klapetek et al.124 have developed an approach to do AFM analysis of nanoparticles on rough surfaces and with overlapping particles. Because a variety of tip artifacts can influence the accuracy of the measurements, international standards committees (ASTM E42 and ISO TC201) have subcommittees working on relevant standards and guides to address these effects.125 Braga and Ricci devoted a book chapter to recognizing and avoiding artifacts in AFM imaging.126 The fact that AFM can be conducted in vacuum, under ambient conditions, within liquids, or other environments127 makes it a valuable tool for in situ studies.
The relatively low cost of AFM systems has allowed many research groups to extend the applicability in a variety of creative ways. Using nanoparticles (or molecules) attached to scanning probe tips, it is possible to measure interactions of individual nanoparticles (or molecules) with either a flat surface or to other nanoparticles.128 These measurements provide information about interaction forces and enable these forces to be examined in various environments. AFM also has been used to determine the roughness of nanoparticle surfaces. This is highly valuable as most methods for measuring surface roughness cannot be used on such small, curved surfaces. AFM also has been combined with TEM to investigate the impact of synthesis processes and particle size on the surface roughness of ceria nanoparticles.129
APPLICATIONS OF LESS COMMON METHODS, NEW DEVELOPMENTS, AND A SYNTHESIS PROBLEM-SOLVING EXAMPLE
Although important information can be obtained by using the most common surface analysis tools and each of these methods has strengths and limitations, some critical questions cannot be fully answered using these methods. Two of several important topics that challenge many tools are: (1) testing the uniformity of particle coatings at the atomic level and (2) measurements in the particles' native or working environments (in situ tools).11 Even before “nanotechnology” was a commonly used term, Dick Brundle frequently observed that as feature size gets smaller, the distinction between bulk and surface analysis disappears.130 Certainly, the distinctions between bulk (core) and surface analysis tend to get blurred for nanoparticles and other nanomaterials. Methods that are not part of the traditional set of surface analysis tools are providing highly valuable information about surfaces and thin layers. For example, it is possible to observe individual atoms move across particle surfaces using STEM, and it can be highly useful in examining the atomic distribution in nanoparticles, including core-shell structure.
In this section, we examine four methods that provide information not as easily available from the more common surface analysis techniques. In addition to the surface composition that can be obtained with the aforementioned methods, two focus more on characterization of the surface molecular structure and binding interactions. SFG is inherently sensitive to interfaces, and improved technology and the application of theoretical modeling now enable SFG to provide information about the binding of molecules on nanoparticle surfaces in liquid environments. Although not a traditional surface method, nuclear magnetic resonance (NMR) can be used to obtain information about interactions on particles in liquid and gaseous environments. Low energy and medium energy ion scattering (MEIS) can provide information about the outer layers and layer structure of particles, and recent technology developments increase their sensitivity and potential applicability to nanomaterials.
These methods are not among those commonly applied to nanomaterials. They may involve unique or uncommon instrumentation or require an expertise well beyond that generally available to many members of the research community. Nonetheless, it is useful to know that they can provide information not available through the most commonly available methods. Through the use of user facilities and collaboration, the research community may gain access to such instrumentation to address specific questions.
We (and others) often discuss nanomaterial characterization as if the only objective were adequate characterization of the material. In many cases, characterization is part of a feedback loop related to obtaining material of the desired composition, structure, size, or property (function). Some level of characterization may be needed to initiate the process, but the whole effort usually involves identification of characteristics or problems and a cycle of synthesis, characterization, process checks, and other steps. Often, multiple passes are required to produce the product or property of interest. This section will conclude with one such example.
Use of sum frequency generation to determine molecular binding states on nanoparticle surfaces
Developed in the 1980s, SFG spectroscopy is a nonlinear laser spectroscopy method that involves mixing two laser beams at a surface or interface that generate an output beam with a frequency equal to the sum of the two input frequencies. SFG is a more generalized version of second harmonic generation (SHG), where the initial frequency of one laser is doubled at an interface. The interface selectivity of SHG and SFG comes from the symmetry requirement for the second-order nonlinear processes that only molecules at the surface or interface of an amorphous or isotropic material or liquid can contribute to the SHG or SFG signal. Because it is an optical method, it can be used in a variety of environments (in situ) to deduce the composition, orientation, and some structural information of molecules at gas–solid, gas–liquid, and liquid–solid interfaces. With a well-designed instrument and setup, SFG can have sub-monolayer sensitivity. Due to the ability to tune one of the incoming laser beams, vibrational spectra can be obtained (SFG-VS), providing a highly interfacially sensitive version of infrared spectroscopy.131, 132
Work in the 1990s demonstrated that SHG could be readily applied to sub-micron and nanoparticles.133, 134 Although there now are many publications involving SFG characterization of nanoparticles, SFG-VS studies on nanoparticles mostly have been conducted in the dry environment, focusing on either the hydrocarbon chains135, 136, 137, 138 or carbon monoxide.139, 140 A few SFG-VS experiments on nanoparticles have been conducted in the presence of liquid, usually probing the C–H stretching vibrations (2800–3000 cm−1).141, 142
In a recent study, Lu et al.143 used in situ SFG-VS to study the binding of deuterated acetic acid on ceria nanoparticles with different oxidation states in aqueous solution.143 The objectives were to directly measure ligand binding vibrational spectra signatures on ceria nanoparticle surfaces in solution and understand how the oxidation state of ceria can influence the type of molecular bonding at the nanoparticle surface. In the experiment, ceria nanoparticles were deposited on the flat surface of a CaF2 hemisphere incorporated into an experimental cell that allowed contact with acetic acid solutions (shown in Fig. 16). Ceria nanoparticles deposited on the bottom (flat surface) of the CaF2 hemisphere were exposed to solutions containing acetic acid. Analysis of peaks in the spectra shown in Fig. 17 illustrates that neutral acetic acid at relatively low pH was deprotonated before being adsorbed on oxidized (Ce+4) and mixed oxidation state ceria (+3 and +4) nanoparticles. From the polarization dependence in the SFG spectra, the two peaks can only be attributed to the symmetric stretch modes of bidentate binding structures. The two major peaks then are identified as bidentate bridging and chelating species of roughly equal intensity for the partially reduced particles. The measurements also suggest that the chelated species is decreased for the fully oxidized particles. The direct observation on the coexistence of the bidentate chelating and bridging ligand binding, as well as their dependence on the surface oxidation states, is encouraging for characterization and understanding of the surface structure and reactivities of nanoparticles. These types of measurements, in combination with modeling of adsorbates on model ceria clusters, suggest that SFG-VS can be a powerful spectroscopic tool for in situ characterization of binding on nanoparticles surfaces.
Figure 16.
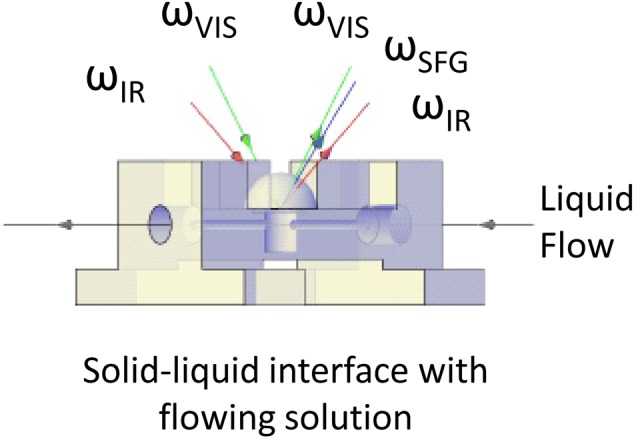
(Color online) Illustration of the SFG liquid cell and experimental geometry. The cell body was made of Teflon. The nanoparticles were deposited on the flat bottom of a CaF2 1 in. diameter hemisphere, which served as the optical window. The liquid flowed through ports sealed by Teflon plugs for studies not requiring the following liquid. The visible and infrared beams were propagated through the CaF2 hemisphere and overlap at the center of the flat surface of the CaF2 hemisphere. The SFG signals were collected in a reflective geometry.
Figure 17.
(Color online) SFG-VS spectra (2-cm−1-step scan) of (a) partially reduced ceria nanoparticles and (b) oxidized ceria nanoparticles in contact with CD3CO2H solutions. The vertical dashed lines reveal the shift of peak positions. The two major peaks are identified as bidentate bridging and chelating species of deprotonated acetic acid adsorbed on the particle surfaces. The relative ratio of the type of bonding varies for reduced and oxidized surfaces. There are two lines for each set of data related to the polarization combinations of the incident and SFG signals. The series ssp identifies s-polarizations for the SFG and visible beams and p-polarization for the IR beam, and ppp indicates p-polarizations for the SFG, visible, and IR beams.
Reactivity of anatase nanoparticles with ethanol measured by nuclear magnetic resonance
NMR is well-known as a nondestructive characterization method based on the excitation, evolution, and detection of spin coherences between nuclear magnetic substates. In the proper circumstances, it can be used directly or indirectly to probe the surfaces of nanoparticles.
Typically, NMR is performed by placing a sample containing nuclei with nuclear spin quantum numbers greater than zero within a static magnetic field followed by irradiation of these nuclei with radiofrequency magnetic fields at or near to their Larmor frequencies.144, 145, 146 The evolution frequencies of spin coherences detected in an NMR experiment are typically reported in values of parts-per-millions (ppm) of a shift from the evolution frequencies detected from a known standard, and these shifts depend on the local electromagnetic environment of the nuclei.147 Hence, NMR shifts report on local structure (bond lengths, number of bonds, identity of nearby atoms, bonding electron polarization, nearby unpaired electrons, etc.) and dynamics (through averaging of these local features), so it has become an indispensable tool for scientists and engineers seeking to understand, monitor, and control local structure. NMR is also applicable to samples in various physical and chemical states, with wide-ranging reports from samples in gaseous, liquid, and solid phases.
Although not a common method for characterizing nanoparticle surfaces, in many cases, the nanoparticle surfaces can be probed directly or indirectly through chemical functionalization,148, 149 and there are a number of reports describing NMR's utility for nanoparticle studies (see Ref. 150 and references therein). Given ideal conditions, the high surface area of a collection of nanoparticles makes the study of their surfaces more straightforward than for larger particulates. However, when the surface atoms contain so-called “quadrupolar nuclei” (those with nuclear spin quantum numbers greater than ½), additional couplings between the electric quadrupole moment of the nonspherical nuclei and local gradients in the electric field can cause additional line broadening,151, 152 resulting in decreased spectral sensitivity and resolution. However, reports of direct, surface-sensitive NMR spectroscopy or the use of probe molecules to indirectly survey local surface structure describe the use of isotropic chemical shift as the predominant interaction for structural elucidation.148
In work at EMSL, we have been interested in the evaluation of reactive sites on the surfaces of materials, including nanoparticles. Such studies are important for understanding bonding within composite materials, where nanoparticles or other materials are added to, for example, polymer systems to change physical or chemical behavior. In our fundamental studies, small organic molecules, such as alcohols and organic acids, are particularly well-suited for evaluating various bonding environments on the surfaces of nanoparticles. Because the chemical reactivities of these molecules typically are associated with the –CR2OH and –COOH functionalities, 13C-containing versions of small organic molecules labeled at these terminal functional sites often are used.
Measurements of ethanol's reactivity with TiO2 nanoparticles provide an example of NMR measurements to determine the nature of small molecule binding on nanoparticle surfaces. Anatase TiO2 nanoparticles initially were heated to 400 °C under vacuum for 4 h to remove surface waters. Then, the particles were exposed to 1–13C-ethanol vapor for 3 h in a dosing apparatus kept at 100 °C. After evacuating the remaining gas phase ethanol, the dosing apparatus was sealed and moved to a dry glove box, where the sample was transferred to an NMR sample holder. Magic angle spinning (MAS) 13C NMR was performed on these systems using a cross-polarization pulse sequence with high-field 1H decoupling.153 This MAS-NMR method provides high resolution for the 13C on the solid surfaces and also benefits from the transfer of polarization from highly polarized 1H spins that are nearby to the 13C being detected. These experiments were conducted using a spectrometer with a 17.6 Tesla magnetic field strength (corresponding to resonance frequencies of 750 MHz for 1H and 188 MHz for 13C) to take advantage of the gains in both sensitivity and resolution obtained when moving to such high-field strengths.
As shown in Fig. 18, two major resonance lines are observed for labeled ethanol reacted with the anatase nanoparticle surfaces, indicating ethanol molecules attached in two distinct bonding environments. The two resonance lines at 60 and 70 ppm [shift from a tetramethylsilane (TMS) reference sample] arise from 13C nuclei originally attached to the –OH group on the 1-13C-ethanol. An additional, smaller resonance from the naturally abundant 13C in the methyl groups was detected at lower frequency (approximately 17 ppm). Under these treatment and loading conditions, the more highly populated environment, detected as a resonance at 60 ppm, arises from ethanol molecules that are physisorbed to the titania surface. The chemical shift of this peak is close to that of neat ethanol (57.0 ppm), and this resonance presumably represents ethanol that is hydrogen-bonded to surface hydroxyls or other species present on the titania nanoparticle surfaces. The second broader resonance peak at 70 ppm represents the ethanol molecules that have formed C–O–Ti ether linkages with the surface of the nanoparticles.
Figure 18.
(Color online) 1H/13C cross-polarization MAS NMR spectrum of 1-13C-ethanol dosed onto a sample of titania (anatase) nanoparticles, exhibiting two bonding environments for the ethanol molecules reacted with the anatase surface. Data were obtained at a 13C resonance frequency of 188.657 MHz, and a 1H resonance frequency of 750.198 MHz with TPPM 1H decoupling after a 2 ms cross-polarization contact time, using a 1H 90° pulse of 6.5 μs and a 5 s recycle delay. A total of 8192 transients were collected, and the data processing included 50 Hz of Lorentzian broadening.
Low and medium energy ion scattering measurements of nanoparticle surfaces
For more than 40 years, low, medium, and high energy ion scattering have been important tools for the characterization and modification of materials surfaces.154 Recent advances in both low energy ion scattering (LEIS) and MEIS enhance their ability to provide more routine and useful information about the outer layers of nanoparticles. In these featured examples, both LEIS and MEIS are used to understand the nature of bi- and trimetal catalysts and are being developed to maintain high catalytic activity while lowering the cost by decreasing the amount of precious metals required.
LEIS and nanoparticle surface characterization
LEIS involves the elastic scattering for 1 to 10 kV ions (often helium) from surfaces. Also known as ion scattering spectrometry (ISS), LEIS is a well-established but not widely used method. The amount of energy lost by the ion during this scattering process is used to determine the identity of the elements present in the outermost atomic layer of the material under analysis. Only species heavier than the primary (incident) ion are detected, and the mass resolution (ability to distinguish different elements) depends on the primary ion and the mass of the species being analyzed. Recent developments and the availability of a fully integrated commercial instrument have made it particularly useful because of the high sensitivity to the outermost atomic layers of a sample.
This high surface specificity has been demonstrated for Pt3Fe alloys by showing that the outer atomic layer of these alloys contains no Fe after annealing.155 The LEIS for the annealed alloy (Fig. 19) shows an elastically scattered peak for only Pt, while the same surface after some sputtering shows the presence of both Fe and Pt. This high surface selectivity provides important information about the true outer nature of surfaces. This useful surface sensitivity applies equally to nanoparticles, and, with some assumptions and calculations, LEIS can readily be used to determine the size of single metal nanoparticles and the surface enrichment of a species in a bimetal catalyst.156
Figure 19.
(Color online) Surface characterization of a Pt3Fe alloy using 1 keV Ne+ LEIS to collect data from mildly sputtered and annealed surfaces. As indicated by the schematic model, no Fe is observed for the annealed surface, indicating the formation of a thin Pt skin, which is destroyed upon even very mild sputtering. Reprinted with permission from Stamenkovic et al., Nat. Mater. 6, 241 (2007). Copyright 2007, Macmillan Publishers Nature Publishing Group.
Although most of the LEIS signal comes from the very outer surface layer, it also is possible to extract information about the thickness and atom profile of layers a few monolayers thick. Rafati et al.157 used high sensitivity LEIS (HS-LEIS) to determine the thickness of the SAM layer on Au nanoparticles discussed earlier. The LEIS determined thickness was 1.8 nm, which lies between the SESSA-determined value of 1.85 nm and 1.74 nm obtained by Shard's simplified approach.90
MEIS and nanoparticle characterization
MEIS involves elastic scattering of ions with energies between 50 and 200 kV. Because of the higher energy, it is sensitive to a thicker layer of material than LEIS, but, with high energy resolution, it also can sense a single atom layer. MEIS using electrostatic analyzers (ESA) typically involves mm-sized beams and considerable time for data collection. A new-generation of TOF analyzers has increased sensitivity and affords analysis of smaller areas. A web presentation by Grande158 highlights some of the recent advances in MEIS, including several examples of application to nanoparticles. Because MEIS signals are themselves influenced by nanoparticle shapes and sizes, the resultant data can be used to determine nanoparticle shapes and sizes, as well as the profile of elements within particles. To minimize confusion, profiles of nanoparticle-layered structures require a nearly single layer of nanoparticles on a support. By comparing data to a model for CdSe-core ZnS-shell nanoparticles, MEIS determined the core (Cd0.65 Se0.35) and shell (Zn0.41S0.59) stoichiometries, core diameter (5 nm), and shell thickness (0.6 nm).158
Using MEIS and XPS to identify the source of contamination on soft-landed nanoparticles generated by magnetron sputtering
In an effort to learn more about the distribution of elements in multi-component particles supported on a substrate, MEIS has been applied to Pt/V/Cu nanoparticles. The data obtained highlight the value of applying a surface sensitive analysis method (MEIS) and demonstrate the challenges associated with preparing and characterizing nanoparticles. In addition, it is shown that even nanoparticles prepared using extremely well-controlled physical synthesis methods may suffer from unexpected and undesirable contamination, and multiple analytical techniques may be needed to confidently identify the presence and confirm the removal of unwanted additional materials.
The Pt-catalyzed oxygen reduction reaction (ORR) that occurs at the cathode in proton exchange membrane fuel cells (PEMFCs) has been widely examined.159 For PEMFCs to compete commercially with conventional engines, the Pt mass activity at the cathode needs to be improved by a factor of four.160 Alloying of Pt with one or two other metals has been demonstrated to produce more active and less costly catalysts for the ORR.161 Studies of bulk surfaces have shown that bimetallic Pt3M (M = Fe, Co, Ni) alloys exhibit improved catalytic activity toward the ORR compared to pure Pt.162 Consequently, supported bi- and trimetallic alloy nanoparticles are promising candidates for use as ORR catalysts in PEMFCs. Because it is difficult to use solution chemistry to form well-defined binary and ternary alloy particles, a variety of physical synthesis techniques163, 164, 165, 166, 167, 168, 169 are being developed to generate bare alloy nanoparticles of exact size and composition and deposit them onto surfaces at controlled coverage. Such well-defined supported nanoparticles then may be investigated to determine which properties need to be adjusted to maximize their catalytic activity.
Soft landing of mass-selected ions is a novel method that enables precise control over the size, composition, and surface density of complex molecules, clusters, and nanoparticles delivered to substrates.170, 171, 172, 173, 174, 175 Ion soft landing combined with magnetron sputtering of a metal in a high-pressure inert gas has been demonstrated as an effective technique for generating and depositing well-defined nanoparticles across a range of sizes and compositions.164, 165, 168, 176, 177, 178 Direct current (DC) magnetron sputtering of up to three independent targets with Ar in the same vacuum region has been used to generate Pt-alloy nanoparticles containing one and two additional metals.179 Using this multitarget sputtering approach, we have generated ternary Pt-alloy, core–shell, and cluster-in-cluster nanoparticles. The anionic nanoparticles were size-selected, employing a quadrupole mass filter,180 and soft landed onto the surface of highly oriented pyrolytic graphite (HOPG) and Si(100) substrates at low kinetic energy. Following soft landing, the substrates were analyzed using a combination of TOF-MEIS, AFM, and XPS to determine the size, composition, and coverage of the supported nanoparticles.
Precise control of the process parameters during magnetron sputtering coupled with mass-selection of the anionic nanoparticles prior to soft landing enables substrates to be coated with accurate amounts of monodispersed nanoparticles. The well-defined nature and precise surface coverage of the soft-landed nanoparticles makes them particularly amenable to analysis by TOF-MEIS. When 8 × 1012 particles are deposited on Si(100), AFM images show the nanoparticles are predominately isolated and distributed evenly across the surface. Figure 20a shows the 90-keV He+ ion scattering spectrum of 8 × 1012 6 nm diameter Pt/V/Cu nanoparticles soft landed onto a Si(100) substrate. The peak for Pt, the primary component of the particles, is clearly resolved, while the peaks for Cu and V are weaker and superimposed. Unfortunately, the large background peak for Si makes it difficult to detect other low energy peaks, such as oxygen.
Figure 20.

(a) TOF-MEIS spectrum of 6 nm Pt/V/Cu nanoparticles soft landed onto Si(100). The spectrum shows the composition of the particles, but the large peak associated with Si makes it difficult to detect low energy peaks from the particles, such as oxygen. (b) TOF-MEIS spectrum of 4 nm Pt/V/Cu nanoparticles soft landed onto RF plasma cleaned HOPG. The new peak appearing after deposition on the HOPG substrate was from Mo that was unintentionally deposited on the substrate during the cleaning process.
To eliminate the large background signal at low energy associated with Si in the TOF-MEIS measurement, HOPG surfaces were used as the soft-landing substrate. Topography measurements obtained using AFM reveal that at low coverage, the soft-landed nanoparticles stick preferentially along the step edges of the HOPG surface, creating linear chains. Unfortunately, at the higher coverages needed to obtain an adequate signal-to-noise ratio for the TOF-MEIS measurements, the nanoparticles begin to pile on top of each other, forming a dense aggregated layer. Therefore, an intermediate coverage, where the majority of nanoparticles remained isolated yet with sufficient material to provide a good signal-to-noise ratio, was needed.
To create defect sites that could pin or stabilize the particles deposited on the surface, plasma sputtering of the HOPG surfaces was attempted. The HOPG surfaces were subjected to low power (30 W) radio frequency (RF) plasma sputtering in Ar for 10 min prior to soft landing of nanoparticles. The TOF-MEIS spectrum from 5 × 1012 4 nm diameter Pt/V/Cu nanoparticles deposited onto RF plasma, pretreated HOPG is shown in Fig. 20b. Because the peaks for Pt, Cu, and V were more intense on HOPG compared to Si(100) and as the large Si background peak is not present on HOPG, it was possible to measure the oxygen peak at 55 keV. However, the most striking difference between the two TOF-MEIS spectra was the appearance of an additional intense peak between Pt and Cu on the plasma-treated HOPG surface that is not present on Si(100).
Because there was no obvious explanation for the additional peak in the TOF-MEIS spectrum of the nanoparticles deposited on the plasma-treated HOPG, a series of experiments were conducted to determine the contaminant source. Two freshly cleaved HOPG surfaces were mounted in the sample holder used for the previous uncontaminated samples then introduced into the soft landing instrument. The first surface was allowed to reside in vacuum for the duration of a typical soft landing experiment (90 min) and removed. The second surface was RF plasma cleaned, following the same procedure described, and allowed to reside in vacuum for 90 min. Both surfaces then were examined using XPS.
Figure 21a depicts the XPS spectrum of the bare HOPG surface allowed to reside in the deposition instrument for 90 min. The C 1s peak is dominant, and there are no other significant features in the spectrum. In contrast, the results for the HOPG surface that was RF plasma sputtered reveal the presence of intense peaks assigned as Mo, C, and O [Fig. 21b]. The O peak could be explained as a result of the oxidation of the defects created on the sputtered HOPG surface following its removal from vacuum. However, the Mo peak source was more subtle. The sample holder used to mount the substrates in the instrument for RF plasma cleaning and soft landing of nanoparticles is made of Mo. Therefore, during the effort to create more defects and functional groups on the surface of HOPG to pin isolated nanoparticles, the surface was inadvertently covered with the sample holder element, Mo. These findings are particularly relevant as RF plasma sputtering is often recommended by instrumentation manufacturers as a way to clean surfaces prior to further modification by deposition or chemical synthesis.
Figure 21.

(a) XPS spectrum of HOPG placed in vacuum for 90 min. (b) XPS spectrum of HOPG RF plasma cleaned in Ar for 10 min. XPS data demonstrates that Mo was introduced into the soft landing process during the sputter cleaning of the HOPG substrate.
Soft landing is a unique and powerful approach for creating highly controlled, well-defined nanoparticle (and large biological molecule) layers on substrates. However, as is all too common, when one part of the sample preparation process was altered, a surprise element was introduced. Only via the application of multiple methods sensitive to thin surface layers was it possible to determine how this minor variation in the soft landing procedure resulted in nanoparticle contamination. MEIS and XPS now are being used to determine the elemental profile of metals in the alloy nanoparticles without the unwanted contamination.
DISCUSSION
This review is written in the context of what some researchers consider a crisis regarding the adequate characterization and underreporting of nanomaterial information. We have highlighted a range of issues, challenges, and opportunities associated with nanomaterials synthesis and characterization. In many ways, nanoscience and nanotechnology are immature, developing fields that have made remarkable progress. Although there are important analysis and characterization challenges, there also are major advances and exciting new scientific and significant technological developments. The community is increasingly recognizing the need to pay additional attention to characterization of nanomaterials, and the tools described or mentioned in this paper and others allow most of the analysis issues to be addressed. Somewhat less recognized is the importance of recording and reporting details of the processes and history of nanomaterials used in a variety of studies. Such information cannot be ignored or cited as trivial in the case of nanomaterials.
There is a close relationship between systematically developing a reliable nanoparticle synthesis process and characterization methodology. Herein, we make some general recommendations related to particles synthesis, preparation of particles for analysis, and convey an approach for selecting the tools to be applied for “routine” characterization of nanoparticles.
Synthesis, processing, and documentation
The core of nanomaterials research, with many implications for applications, starts with controlled synthesis of materials that exhibit the desired and, often, tunable properties. We have shown examples of how surface sensitive analysis methods can be important tools for developing reliable synthesis methods and examining the consistency and reproducibility of these processes. However, we also have discovered that minor changes in synthesis steps and inadvertent introduction of minor contaminants, sometimes below the level that can readily be detected, can alter material behaviors. In other words, we cannot always characterize our way out of difficulties introduced during synthesis. Therefore, it can be important to take extreme care in handling and keep thorough records that can identify procedural or other steps in a synthesis process that may introduce unintended changes in nanomaterial properties.
Based on experiences among our research teams, some guidelines for nanoparticles synthesis are listed (as follows). These may be especially useful to enhance repeatable synthesis of nanoparticles and maintain stable properties for nanoparticles produced by wet chemical and other particle generating methods.
-
(1)
Plan and organize the experiment to minimize (or hold constant) the number of parameters that can vary in the process. Without adequate planning and records, the process and particle properties usually will change (however slightly) with each different person and over time.
-
(2)
Emphasis should be given to the cleaning of glassware or other synthesis components (from instruments or autoclave, etc.). Several oxidizing and/or reducing agents are available commercially for cleaning glassware. If the synthesis is carried out on routine basis, it is advisable to purchase dedicated glassware and instruments for a particular type of nanomaterial to avoid cross contamination.
-
(3)
In common practice, a master batch of nanoparticles is stored separately, and small aliquots are taken out from the batch when required for characterization. However, the repeated opening of the master batch of nanoparticles suspension/powder to atmospheric condition is undesired and can be a source of contamination. Repeated use of a spatula or pipette tips in the master batch also should be avoided to minimize contamination. It is suggested that instead of preparing and storing the nanoparticles sample in one master batch, the material should be stored in several small, pre-cleaned vials. This will minimize repeated exposure of the entire sample to the external environment. If surface oxidation or chemical state information is important, once exposed to the environment, a vial should not be used for surface characterization studies, although bulk characterization and property measurements might still be done on the same sample.
-
(4)
Seemingly harmless instruments, such as pH/conductivity probes, temperature measurement probes, etc., should be cleaned thoroughly before placing in contact with the batch of nanoparticles. The exposure to a master batch of nanoparticles should be avoided (if possible), and smaller aliquots taken out of the master batches should be used for measurement.
-
(5)
The storage conditions for nanoparticles should also be considered. Our experience suggests that storage conditions, such as light, dark, temperature (room temperature, refrigerated, or frozen), humidity, and duration of storage, can affect the stability and reactivity of nanomaterials. For example, on several occasions, we have observed that solutions of well-dispersed nanoparticles cannot be resuspended after they have been frozen once.
-
(6)
Researchers synthesizing nanomaterials for biological applications should treat the nanoparticles as a biological sample. It is advised that the materials and biological scientists work together to understand the nuances of process parameters and how it can influence the biological end points. All nanomaterials used in biological studies should be handled analogous to live biological samples, and the contamination from various sources, such as bare hands, glassware, hoods, working benches, etc., should be minimized. All glassware and work benches should be sterilized, and the researchers should use good laboratory practices, sterilized tips and glassware, and personal protective equipment for materials synthesis.
-
(7)
Select an appropriate core set of characterization tools (including a surface sensitive method) to verify the repeatability and consistency of the synthesis approach.
-
(8)
Records related to chemicals used in synthesis, dates of synthesis, and means of storage should be kept and linked to samples, as well as to batches of samples, so the parentage and history of materials can be maintained and possible similarities and sources of differences can be tracked if they are identified. Times between synthesis and use or characterization should be recorded and reported.
Sample preparation
Several of the examples described in this paper have noted the importance and sometimes difficulties of preparing samples for analysis. There are two major components to sample preparation. The first is to collect the sample (e.g., removing particles from the master batch of solution or powder or extracting the sample from a test environment). Two methods of removing nanoparticles from solution for detailed analysis were discussed earlier. Another challenge is preparing material for analysis without either destroying the desired information26 or having that information masked by components from the solution.55 The specific information requirements for the analysis will determine the requirements related to sample handling and minimizing the loss of information. Obtaining the desired information may require mounting samples in anaerobic environments, using techniques that can obtain information in the working environment, or even freezing a specimen.
In addition, each analysis method will have a set of sample requirements. For example, XPS requires enough sample material within the analysis area (tens to hundreds of square micrometers) such that it is usually desirable to have enough material deposited on the supporting substrate to totally block the substrate signal. These piles of particles need to be appropriately considered in quantitative analysis of coatings on particles.90 In contrast, for MEIS, it is desirable to have a single layer of nanoparticles distributed/supported on a substrate that will produce only a small background signal. For MEIS, a “pile” of particles would complicate the data analysis. Because the information needs and method requirements will vary, there will be no universally “correct” method of sample preparation and mounting.
Consideration of analysis needs, including the nature of the question, required speed of analysis, number or specimens to be analyzed, and amount of material available, will impact choices and options for sample preparation. As described for sample synthesis and processing, understanding discrepancies and differences among results can be minimized by documenting and reporting sample preparation steps. Both ASTM E42 and ISO TC201 have guides for sample preparation and handling181, 182 with many of the practices appropriate for nanomaterials. In addition, TC201 has a working group developing a guide specifically for preparation and mounting of nanomaterials for surface analysis.
Characterization—what type and how much?
A range of characterization issues or challenges have been identified and discussed in other parts of this review, and the value of surface sensitive analysis methods has been noted. Although the challenges of adequate characterization may seem daunting, by focusing on a few important issues and gaining an understanding of the nature of the material involved, many of the challenges can be effectively addressed.
Surfaces, coatings, and time
Although important aspects of the characterization for a nanomaterial must be dictated by the function and purpose of the material, the nature of the surface of a nanomaterial impacts how the material will interact with other materials and its surrounding environment.
Because interactions of a material with its environment usually alter the material, information about the rate of any changes (including the formation and alteration of surface coatings) in relevant environments is important for fundamental understanding, as well as for assessing material reliability and predicting environmental (or biological) impacts. Consequently, there is a need for increased attention to surface and coating characterization and for advancing the understanding of the kinetics of particle change (surface and “bulk”).
Selecting “core” characterization tools
The essential characterization needs for a specific nanomaterial are not always clear, and unnecessary characterization can be costly while providing little added value. In our experience with iron metal-core oxide-shell nanoparticles, we found that we needed to obtain a general understanding of the nanoparticles we were studying, and this initial understanding required the application of a variety of tools.27 However, once the nature of the particles (and how they change) was understood, a routine core set of tools was adequate, in most circumstances, to provide the information we needed to assess the nature of a particular set of particles and how they changed with time.28 In many types of studies, a routine set of measurements can help assure that the materials being examined have properties consistent with those tested or applied at earlier times. For example, particle size and surface potential measured by a dynamic light scattering instrument may be an important check on material consistency just prior to in vitro biological studies.
It seems unlikely that there will be a unique set of essential characterization tools that apply to all or most nanomaterials. The critical measurements will be material and application dependent. However, we have discovered that we almost always need to collect some of the data that appear on many of the characterization lists,8 including particle size, shape, structure (TEM and XRD), and surface composition (XPS). The need for multimethod analysis is particularly valuable in addressing issues related to the reliable synthesis of materials and obtaining an initial understanding about how the materials may change with time or in an altered environment.
Jensen et al.183 observed that need for multitechnique analysis for adequate characterization of new materials is well recognized but not always put into practice. In their study of a set of papers reporting on new carbon nanotube-templated thin layer chromatography plates,183 they found that half of the papers analyzed used three or fewer techniques to characterize new materials, whereas for the specific materials they were examining, the application of five or more methods was used to provide the needed information in more complete studies. As discussed, there is no specific number of methods required to characterize a material, the needed testing will depend on the material and application, as well as how well the material is understood. Nonetheless, it appears that many research papers would benefit from considering of a wider range of characterization tools, at least during early stages of research. The knowledge gained by forming a self-consistent picture of a nanomaterial's bulk, surface, and interface structure, and composition can be used to identify the core set of analysis methods needed for routine characterization during a research project.
The tools we have discussed in relation to surface analysis each have strengths and limitations as summarized in Table Table IV.. The strength column does not include all of the types of information that can be obtained by the techniques, but it highlights information that might be most easily obtained. Because of the importance of surface properties, some type of surface sensitive characterization method should be used to characterize most nanomaterials. However, the selection of the method to be used can be based on the accessibility, ease of use, and type of information needed.79, 86, 87 We have found that XPS can be quick and easily applied, particularly for screening various types of compositional or chemical state surprises.78
The relative amount of application of a variety of analysis methods for characterization of nanomaterials is shown in Fig. 22. This use frequency was obtained by combining the search terms used for Fig. 1 (nanomaterial AND nanoparticle AND nanostructure) with terms related to the specific technique (full technique name OR the common identifier). The most commonly applied methods are XRD, TEM, and SEM. Although both AFM and XPS are the most commonly used surface analysis tools, we have argued that their importance warrants increased application. The data also suggests that the application of MEIS, LEIS, and SFG to nanomaterials is currently very uncommon.
Figure 22.
(Color online) Comparison of the relative rates of application of a wide range of analysis tools to nanomaterials based on a Web of Science search as described in the text. The most widely used surface analysis tools are XPS and AFM. Techniques not previously discussed in this paper include: Fourier transform infrared (FTIR) spectroscopy, dynamic light scattering (DLS), ultraviolet–visible spectroscopy (UV-VIS), x-ray adsorption spectroscopy (XAS), extended x-ray adsorption fine structure (EXAFS), and x-ray absorption near edge structure (XANES).
Combining in situ real-time and ex situ measurements
Because particles may change as the environment around them is altered, it is useful to obtain information in the environment of interest and have the ability to make measurements as a function of time in that environment, or conduct in situ and in situ real-time measurements.11 In many circumstances, a combination of in situ real-time and ex situ measurements may be needed to obtain the appropriate understanding of a system, as we discovered in our iron metal-core oxide-shell nanoparticle work.28 Depending on the nature of the system, there are many different types of methods that could be used to provide in situ and real-time data. Although they are not discussed in detail, Table Table V. provides an indication of methods that might be used and some of the types of information that can be obtained. When used in combination with other surface and “bulk” analysis methods, they can provide a “picture” of how nanomaterials are behaving in realistic environments and how they alter with time. In some cases, a simple measurement, such as zeta potential, corrosion current, or gas evolution, can provide time-relevant information easily and at low cost. When simple in situ tools are appropriately combined with ex situ methods, mechanistic information can be obtained.
Table V.
Examples of measurements that can be used to collect in situ or in situ real-time information about nanomaterials. The importance depends on the property of interest. Techniques not otherwise discussed in this paper include: attenuated total reflectance FTIR (ATR-FTIR), optical fluorescence, scanning x-ray transmission microscopy (STXM), and infrared-scattering scanning near-field optical microscopy (IR-sSNOM).
| Method | Type of information | Comments |
|---|---|---|
| AFM | Surface topography and particle shape with nm resolution | Gas and liquid environment, including high pressure |
| Electrochemical | Electrochemical currents and corrosion potentials | Can sometimes see product formation |
| Optical | Requires appropriate environmental transparency | |
| FTIR | Molecular vibrations in analysis region | ATR-FTIR has enhanced interface sensitivity |
| SHG | Selectively probe non-resonant or electronic responses from molecular or atomic interfaces | Excellent in observing changes but data usually cannot tell chemical identity |
| SFG | Chemical identity, molecular structure and interactions at surfaces and interfaces | Data modeling useful to understand environmental effects on molecular vibrations |
| Fluorescence | Tagged particle fluorescence can provide particle or reaction locations in a complex environment including living cells | |
| VU-VIS | Size of metal nanoparticles and chemical state of some oxide particles | Commonly used for particles in solution |
| Tip-enhanced IR-sSNOM | Vibrational information with high spatial resolution | Can be done in various gaseous environments |
| DLS | Common method for sizing particles in solution | Often done in combination with zeta potential measurements |
| X-ray | ||
| XRD | Structures present and grain size | Microbeams and synchrotrons allow liquid and gas environment analysis |
| XAS | Localized structure and chemical state | Environment around specific atoms |
| STXM | XAS information with 10 s nm resolution | Gas or liquid environments now possible |
| XPS | Surface composition chemical state | Differentially pumped systems allow measurements to be made in some gaseous environment |
| NMR | Molecular structure and phase information | Gas or liquid environments, needs careful planning |
| Transmission Electron Microscopy | Morphology and structure with high resolution | Can now be done in gas and liquid environment and conducted on designed systems such as model batteries |
There are several environments where important information cannot yet be readily obtained. For example there is a need to obtain additional information from “buried” interfaces, such as the solid–liquid and solid–solid interfaces involving nanomaterials such as those occurring in nanocomposites. Such specific needs provide opportunities for creative development and application of new methods and analysis approaches.
Qualitative versus quantitative
Although the most common application of surface analysis of nanoparticles is qualitative in the sense that the presence of contamination, nature of surface coatings, or chemical state of the surface can be readily determined, a good deal of additional information also can be obtained by combining the surface information with knowledge about particle size or what is known about layered structures. To determine the nature of nanostructured materials, several models and computer codes have been developed for interpreting data from XPS and AES.90, 94, 95, 96, 97, 101, 103, 105 These codes can provide quantitative information about the enrichment or depletion of elements at a surface, the thickness of surface layers on particles of various shapes, and island formation. Evolving surface analysis tools will rely heavily on links between computation modeling and experiment. There is more valuable information in surface analysis data than commonly used, and the value of surface analysis methods increases when quantitative information is extracted.
SUMMARY AND CONCLUSIONS
There is growing recognition in the research community that in too many circumstances, nanomaterials are incompletely characterized. This can be a major handicap to both advancing science and development of reliable technologies. However, because of traits inherent in their nature, the characterization of nanomaterials often is significantly more complex than recognized by parts of the research community, as well as by those charged with regulating their health and safety. Nonetheless, tools are currently available to answer many of the important questions. In many cases, application of an expanded set of tools will provided the needed information.
The time-dependent and environmentally responsive nature of nanomaterials complicates determination of their essential properties. In fact, understanding the response of nanomaterials to changes in their native or working environments is central to understanding and predicting their behaviors and long-term impacts (in vitro and in vivo for biological studies) and on their physicochemical properties for almost any application. The need to follow these materials and their interactive surfaces as a function of time in native environments places a premium on the development of real-time in situ analysis methods.
Because surfaces and interfaces play predominant roles in determining properties of nanomaterials, appropriate analysis of these surfaces is critical to both science and technology of nanomaterials. Both qualitative and quantitative application of common surface analysis methods (as described in this paper) can help address some of these analysis needs. In the view of the current authors, these tools are underused. Although simple qualitative use of surface tools can provide critical information, such as the presence of contaminants or confirmation of some aspect of chemistry, needs and opportunities exist for more advanced modeling and interpretation of the experimental data. This requires both education for analysts and development of easier-to-use analysis software.
One of the major challenges associated with using surface analysis methods (perhaps, any analysis method) involves the appropriate preparation of materials for analysis. The environmental sensitivity of many nanomaterials and the need to remove materials from solution without introducing artifacts require the appropriate development of procedures and protocols for samples handling and appropriate analysis conditions. Some efforts to deal with these issues are being addressed by ISO, ASTM, and other standards organizations.181, 182 It is important for an analyst to consider if moving a sample from the “native” environment to the environment of the analysis (vacuum, in many cases) alters the sample and compromises the ability to collect the desired information. In many circumstances, the nature and extent of such effects can be understood and minimized, or alternative approaches identified.
Analysis tools constantly are being developed that expand the ability to obtain important information and afford a variety of in situ measurements. In most cases, the information that can be extracted from in situ measurements—indeed, almost any single measurement—is incomplete without integration with information from other analysis methods. The expanded abilities of SFG-VS offer one example of the increasing ability and need to combine advanced experimental measurements and detailed theoretical modeling, both to understand information for the measurements and to predict behaviors of the nanomaterials.
Despite any synthesis, reproducibility, and characterization challenges, the opportunities and current successes of nanoscience and nanotechnology are significant. Nanotechnology offers many potential routes for addressing the energy, environmental, and medical problems we face as a society and individuals. The apparent challenges present great opportunities for the next generation of scientists.
AUTHOR BIOS
 Don Baer is a Laboratory Fellow at PNNL and Lead Scientist for Interfacial Chemistry EMSL, a U.S. Department of Energy national scientific user facility located at PNNL. He received a B.S. in physics in 1969 from Carnegie-Mellon University and a Ph.D. in experimental physics in 1974 from Cornell. He was a postdoc at the Materials Research Center and Department of Physics at the University of Illinois before joining what is now PNNL in 1976. He is a fellow of the American Vacuum Society (AVS) and the American Association for the Advancement of Science and holds affiliate positions in physics at Washington State University, Tri-Cities and chemistry at the University of Washington. Dr. Baer has served as an Associate Editor of JVST, is an Associate Editor of Surface Science Spectra, and is the Reviews Editor of Surface and Interface Analysis. Most of his research has explored the impact of surfaces and interfaces on material properties and has required the use a variety of surface sensitive analysis tools. Research topics have included understanding the behaviors of nanoparticles in aqueous and biological systems, determining the role of interface chemistry on stress corrosion cracking, and examining various surface properties of oxides and minerals. He has more than 250 publications and has served as Chair of the ASTM Committee E42 on Surface Analysis, Chair of the Applied Surface Science Division of the AVS, and leads a working group on surface characterization nanomaterials for ISO Committee TC201 on Surface Chemical Analysis. He has received the Albert Nerken Award from the AVS, the John Rivière Award of the UK Surface Analysis Forum, and has been a member of teams receiving a Federal Laboratory Consortium Technology Transfer award, a Microscopy Today Innovation award, and a DOE Division of Materials Sciences Outstanding Scientific Accomplishment award.
Don Baer is a Laboratory Fellow at PNNL and Lead Scientist for Interfacial Chemistry EMSL, a U.S. Department of Energy national scientific user facility located at PNNL. He received a B.S. in physics in 1969 from Carnegie-Mellon University and a Ph.D. in experimental physics in 1974 from Cornell. He was a postdoc at the Materials Research Center and Department of Physics at the University of Illinois before joining what is now PNNL in 1976. He is a fellow of the American Vacuum Society (AVS) and the American Association for the Advancement of Science and holds affiliate positions in physics at Washington State University, Tri-Cities and chemistry at the University of Washington. Dr. Baer has served as an Associate Editor of JVST, is an Associate Editor of Surface Science Spectra, and is the Reviews Editor of Surface and Interface Analysis. Most of his research has explored the impact of surfaces and interfaces on material properties and has required the use a variety of surface sensitive analysis tools. Research topics have included understanding the behaviors of nanoparticles in aqueous and biological systems, determining the role of interface chemistry on stress corrosion cracking, and examining various surface properties of oxides and minerals. He has more than 250 publications and has served as Chair of the ASTM Committee E42 on Surface Analysis, Chair of the Applied Surface Science Division of the AVS, and leads a working group on surface characterization nanomaterials for ISO Committee TC201 on Surface Chemical Analysis. He has received the Albert Nerken Award from the AVS, the John Rivière Award of the UK Surface Analysis Forum, and has been a member of teams receiving a Federal Laboratory Consortium Technology Transfer award, a Microscopy Today Innovation award, and a DOE Division of Materials Sciences Outstanding Scientific Accomplishment award.
 Mark Engelhard is a Senior Research Scientist in the Interface Spectroscopy and Diffraction Facility in EMSL, a Department of Energy national scientific user facility located at PNNL. Since 1978, he has specialized in the use of surface sensitive techniques to study surface and interphase chemistry. His considerable experience in surface analysis methods has facilitated collaborative research in both fundamental and applied studies. He has served as principal investigator, and project manager on various research projects funded by governmental and industrial clients and as a facility cognizant space manager in EMSL. Engelhard has managed and coordinated scientific collaborations involving the application of XPS, ultraviolet photoelectron spectroscopy (UPS), AES, and TOF-SIMS techniques. These techniques have been applied in numerous studies encompassing physical, chemical, geological, and biological sciences. Mark's work, much of it performed using XPS as a primary technique, is reported in over 290 peer-reviewed scientific publications and three book chapters. His publications have been cited over 6000 times in the literature and his H-index is currently 42. He serves on the AVS Publications Committee, Vice Chair of the Pacific Northwest Chapter of the AVS, Associate Editor of the AVS journal Surface Science Spectra, Secretary of ISO TC-201 Surface Chemical Analysis SC-2 General Procedures, Executive Committee Member of ASTM International E42 Surface Science, and subcommittee Chair of ASTM E42-08 Ion Beam Sputtering. Engelhard received the George T. Hanyo Award from the American Vacuum Society in 1997; the DOE Outstanding Scientific Accomplishment in Materials Chemistry Award; the Pacific Northwest National Laboratory Director's Award for Excellence; and the EMSL Director's Award in 2007 and 2008.
Mark Engelhard is a Senior Research Scientist in the Interface Spectroscopy and Diffraction Facility in EMSL, a Department of Energy national scientific user facility located at PNNL. Since 1978, he has specialized in the use of surface sensitive techniques to study surface and interphase chemistry. His considerable experience in surface analysis methods has facilitated collaborative research in both fundamental and applied studies. He has served as principal investigator, and project manager on various research projects funded by governmental and industrial clients and as a facility cognizant space manager in EMSL. Engelhard has managed and coordinated scientific collaborations involving the application of XPS, ultraviolet photoelectron spectroscopy (UPS), AES, and TOF-SIMS techniques. These techniques have been applied in numerous studies encompassing physical, chemical, geological, and biological sciences. Mark's work, much of it performed using XPS as a primary technique, is reported in over 290 peer-reviewed scientific publications and three book chapters. His publications have been cited over 6000 times in the literature and his H-index is currently 42. He serves on the AVS Publications Committee, Vice Chair of the Pacific Northwest Chapter of the AVS, Associate Editor of the AVS journal Surface Science Spectra, Secretary of ISO TC-201 Surface Chemical Analysis SC-2 General Procedures, Executive Committee Member of ASTM International E42 Surface Science, and subcommittee Chair of ASTM E42-08 Ion Beam Sputtering. Engelhard received the George T. Hanyo Award from the American Vacuum Society in 1997; the DOE Outstanding Scientific Accomplishment in Materials Chemistry Award; the Pacific Northwest National Laboratory Director's Award for Excellence; and the EMSL Director's Award in 2007 and 2008.
 Dr. Grant E. Johnson received his B.S. degree in chemistry from the University of Delaware Honors Program in 2002. He was employed as a chemist with the United States Army at the Edgewood Chemical Biological Center in Edgewood, Maryland, from 2002 to 2004. He received his Ph.D. in chemistry from the Pennsylvania State University under Professor A.W. Castleman, Jr., in 2009. He was awarded a National Science Foundation Central Europe Summer Research Institute fellowship in 2006 to study with Professor Vlasta Bonacic-Koutecky at the Humboldt University in Berlin, Germany. He received a Department of Energy fellowship to attend the 2006 meeting of Nobel Laureates in chemistry in Lindau, Germany. He was awarded a Linus Pauling Distinguished Postdoctoral fellowship at the Pacific Northwest National Laboratory from 2010 to 2012. He is currently a scientist in the Separations, Detection, and Analysis program working with Laboratory Fellow Dr. Julia Laskin.
Dr. Grant E. Johnson received his B.S. degree in chemistry from the University of Delaware Honors Program in 2002. He was employed as a chemist with the United States Army at the Edgewood Chemical Biological Center in Edgewood, Maryland, from 2002 to 2004. He received his Ph.D. in chemistry from the Pennsylvania State University under Professor A.W. Castleman, Jr., in 2009. He was awarded a National Science Foundation Central Europe Summer Research Institute fellowship in 2006 to study with Professor Vlasta Bonacic-Koutecky at the Humboldt University in Berlin, Germany. He received a Department of Energy fellowship to attend the 2006 meeting of Nobel Laureates in chemistry in Lindau, Germany. He was awarded a Linus Pauling Distinguished Postdoctoral fellowship at the Pacific Northwest National Laboratory from 2010 to 2012. He is currently a scientist in the Separations, Detection, and Analysis program working with Laboratory Fellow Dr. Julia Laskin.
 Julia Laskin is a Laboratory Fellow at PNNL. She received her M.Sc. in Physics from the Leningrad Polytechnical Institute (1990) and her Ph.D. in Physical Chemistry from the Hebrew University of Jerusalem (1998). She was a postdoctoral fellow at the University of Delaware (1998–1999) and at PNNL (2000–2002). She became a research scientist at PNNL in 2002. Dr. Laskin's research is focused on obtaining a fundamental understanding of the interactions of complex ions and molecules with surfaces for improved identification of large molecules using mass spectrometry and for selective modification of substrates using beams of mass-selected ions (ion soft-landing). Another area of Dr. Laskin's research is related to characterization of the chemical composition of organic aerosols (OA). In particular, she is interested in understanding the effect of organic oligomers in OA on physical properties of aerosols relevant to climate change. Finally, she is involved in the development of imaging and analysis capabilities of the nanospray desorption electrospray ionization (nano-DESI) mass spectrometry for imaging of fully hydrated biological samples in their native environment and for analysis of complex mixtures directly from solid substrates. Dr. Laskin has been honored with several prestigious awards including Presidential Early Career Award (PECASE) in 2007, ASMS Biemann Medal in 2008, Honor issue of JASMS in 2009, and other. She served as a treasurer of the ASMS in 2006–2008. She is a member of the Editorial Board of JASMS, Frontiers in Microbiology, and Russian Journal of Mass Spectrometry and a member of the Advisory Board of Analyst. She is an editor of a book "Principles of Mass Spectrometry Applied to Biomolecules" published by John Wiley & Sons in 2006. She is an author and co-author of more than 140 peer-reviewed publications.
Julia Laskin is a Laboratory Fellow at PNNL. She received her M.Sc. in Physics from the Leningrad Polytechnical Institute (1990) and her Ph.D. in Physical Chemistry from the Hebrew University of Jerusalem (1998). She was a postdoctoral fellow at the University of Delaware (1998–1999) and at PNNL (2000–2002). She became a research scientist at PNNL in 2002. Dr. Laskin's research is focused on obtaining a fundamental understanding of the interactions of complex ions and molecules with surfaces for improved identification of large molecules using mass spectrometry and for selective modification of substrates using beams of mass-selected ions (ion soft-landing). Another area of Dr. Laskin's research is related to characterization of the chemical composition of organic aerosols (OA). In particular, she is interested in understanding the effect of organic oligomers in OA on physical properties of aerosols relevant to climate change. Finally, she is involved in the development of imaging and analysis capabilities of the nanospray desorption electrospray ionization (nano-DESI) mass spectrometry for imaging of fully hydrated biological samples in their native environment and for analysis of complex mixtures directly from solid substrates. Dr. Laskin has been honored with several prestigious awards including Presidential Early Career Award (PECASE) in 2007, ASMS Biemann Medal in 2008, Honor issue of JASMS in 2009, and other. She served as a treasurer of the ASMS in 2006–2008. She is a member of the Editorial Board of JASMS, Frontiers in Microbiology, and Russian Journal of Mass Spectrometry and a member of the Advisory Board of Analyst. She is an editor of a book "Principles of Mass Spectrometry Applied to Biomolecules" published by John Wiley & Sons in 2006. She is an author and co-author of more than 140 peer-reviewed publications.
 Jinfeng Lai is an Associate Scientist in the Research Center of the Phillips 66 company. He received his B.S. degree in Chemistry in 1999 and M.S. degree in Inorganic Chemistry in 2002, both from the Xiamen University. He received a Ph.D. degree in Chemistry from the University of California, Riverside in 2009. From 2011 to 2012, he was a Postdoctoral Research Associate at the EMSL at Pacific Northwest National Laboratory. His research interests include Nuclear Magnetic Resonance spectroscopy as a probe of chemical structure and dynamics. He has published over 20 journal and proceeding papers. He is a reviewer for over 20 journals, including the Journal of the American Chemical Society.
Jinfeng Lai is an Associate Scientist in the Research Center of the Phillips 66 company. He received his B.S. degree in Chemistry in 1999 and M.S. degree in Inorganic Chemistry in 2002, both from the Xiamen University. He received a Ph.D. degree in Chemistry from the University of California, Riverside in 2009. From 2011 to 2012, he was a Postdoctoral Research Associate at the EMSL at Pacific Northwest National Laboratory. His research interests include Nuclear Magnetic Resonance spectroscopy as a probe of chemical structure and dynamics. He has published over 20 journal and proceeding papers. He is a reviewer for over 20 journals, including the Journal of the American Chemical Society.
 Karl Mueller is a Laboratory Fellow within the EMSL at the PNNL. He received a B.S. in chemistry in 1985 from the University of Rochester and a Ph.D. in chemistry in 1991 from the University of California at Berkeley. He was a postdoc at the University of British Columbia, where he was awarded a Killam Memorial Postdoctoral Fellowship and an NSERC International Postdoctoral Fellowship. Prior to his arrival at PNNL, Dr. Mueller spent 18 years on the faculty of Penn State University, where he advanced to the position of Professor of Chemistry and mentored over twenty graduate students through completion of their doctoral degrees. He currently holds a dual appointment between PNNL and Penn State. He has published over ninety research papers, focusing on the development and implementation of novel methods of solid-state nuclear magnetic resonance spectroscopy. In his current role as the Lead Scientist for Magnetic Resonance at EMSL, he and his staff work at the forefront of applications and development of magnetic resonance techniques, both in sponsored research projects and as collaborators with many visiting users of the magnetic resonance facilities at EMSL. Dr. Mueller is the recipient of an Arnold and Mabel Beckman Foundation Young Investigator Award and an Alfred P. Sloan Research Fellowship, and the Research Corporation for Science Advancement has named him as a Cottrell Scholar. In 2012, he was inducted as a Fellow of the American Association for the Advancement of Science.
Karl Mueller is a Laboratory Fellow within the EMSL at the PNNL. He received a B.S. in chemistry in 1985 from the University of Rochester and a Ph.D. in chemistry in 1991 from the University of California at Berkeley. He was a postdoc at the University of British Columbia, where he was awarded a Killam Memorial Postdoctoral Fellowship and an NSERC International Postdoctoral Fellowship. Prior to his arrival at PNNL, Dr. Mueller spent 18 years on the faculty of Penn State University, where he advanced to the position of Professor of Chemistry and mentored over twenty graduate students through completion of their doctoral degrees. He currently holds a dual appointment between PNNL and Penn State. He has published over ninety research papers, focusing on the development and implementation of novel methods of solid-state nuclear magnetic resonance spectroscopy. In his current role as the Lead Scientist for Magnetic Resonance at EMSL, he and his staff work at the forefront of applications and development of magnetic resonance techniques, both in sponsored research projects and as collaborators with many visiting users of the magnetic resonance facilities at EMSL. Dr. Mueller is the recipient of an Arnold and Mabel Beckman Foundation Young Investigator Award and an Alfred P. Sloan Research Fellowship, and the Research Corporation for Science Advancement has named him as a Cottrell Scholar. In 2012, he was inducted as a Fellow of the American Association for the Advancement of Science.
 Prabhakaran Munusamy is a materials scientist/polymer specializing in nanotechnology. He received his bachelors in chemical engineering First Class from University of Madras, India. He received his master's degree in Applied Polymer Science from Martin Luther University in Germany, where he also had the opportunity to work in several reputed labs such as the Max Planck Institute and Fraunhofer Institute and where he specialized in porous nanomaterials fabrication. After coming to the United States, Munusamy received his Ph.D. from Virginia Polytechnic Institute and State University while working with Professor Gary Pickrell' s group in the Materials Science and Engineering Department. His Ph.D. focused on nanomaterials for biomedical applications. He developed a novel nanoparticle-based drug delivery system using inorganic nanoparticles. Munusamy's Ph.D. research was supported by Institute for Critical Technology and Applied Sciences predoctoral funding. He also received Tau Beta Pi engineering honor during his Ph.D. program. He then joined Pacific Northwest National Laboratory as a postdoctoral research associate in EMSL, a Department of Energy national scientific user facility. At PNNL, Munusamy's research has primarily focused on synthesis and characterization of engineered nanoparticles for nanotoxicological studies. Since 2003, he has published over 15 journal and proceeding papers with a total of 200 citations. His area of expertise and interest are template synthesis of porous nanomaterials fabrication, semiconductor and luminescent nanostructures, and engineered nanoparticle formulation for targeted drug delivery applications.
Prabhakaran Munusamy is a materials scientist/polymer specializing in nanotechnology. He received his bachelors in chemical engineering First Class from University of Madras, India. He received his master's degree in Applied Polymer Science from Martin Luther University in Germany, where he also had the opportunity to work in several reputed labs such as the Max Planck Institute and Fraunhofer Institute and where he specialized in porous nanomaterials fabrication. After coming to the United States, Munusamy received his Ph.D. from Virginia Polytechnic Institute and State University while working with Professor Gary Pickrell' s group in the Materials Science and Engineering Department. His Ph.D. focused on nanomaterials for biomedical applications. He developed a novel nanoparticle-based drug delivery system using inorganic nanoparticles. Munusamy's Ph.D. research was supported by Institute for Critical Technology and Applied Sciences predoctoral funding. He also received Tau Beta Pi engineering honor during his Ph.D. program. He then joined Pacific Northwest National Laboratory as a postdoctoral research associate in EMSL, a Department of Energy national scientific user facility. At PNNL, Munusamy's research has primarily focused on synthesis and characterization of engineered nanoparticles for nanotoxicological studies. Since 2003, he has published over 15 journal and proceeding papers with a total of 200 citations. His area of expertise and interest are template synthesis of porous nanomaterials fabrication, semiconductor and luminescent nanostructures, and engineered nanoparticle formulation for targeted drug delivery applications.
 Dr. Suntharampillai Thevuthasan is a Staff Scientist and manager of the Interfacial Spectroscopy and Diffraction group at Environmental Molecular Sciences Molecular Sciences Laboratory (EMSL), a Department of Energy national scientific user facility located at PNNL. He received a B.Sc. (Honors) in Physics from the University of Peredeniya, Sri Lanka, and M.Sc. in Energy Technology from the Asian Institute of Technology, Thailand. He received his Ph.D. in Physics (Surface Science) from the University of Maine in 1989 and started his postdoctoral research at the University of Florida. Before joining PNNL in 1993, he was a postgraduate researcher at University of California-Davis and a guest scientist at Lawrence Berkeley National Laboratory for three years. At PNNL, he has been developing experimental capabilities associated with research in surface science, material synthesis and characterization including nanomaterials, and buried interface analysis in thin films, and nanomaterials. He made significant contributions to the study of surface structures of pure and adsorbate-covered single crystal surfaces, thin film and interfacial characterizations using XPS/diffraction, photoelectron holography and high energy ion beam techniques. Thevuthasan further contributed to the understanding of synthesis and characterization of nanomaterials, ionic transport processes in single- and multilayer oxide thin film electrolytes, growth and characterization of oxide thin films, and understanding radiation effects in ceramics and oxides. He has authored more than 225 peer reviewed research papers in these areas. He has organized several symposia and workshops as a part of international conferences. Thevuthasan is a Fellow of AVS. He has received PNNL's awards and honors include multiple Outstanding Team Performance awards, the Chester L. Cooper Mentor of the Year Award and the Fitzner-Eberhardt Award for mentoring junior scientists.
Dr. Suntharampillai Thevuthasan is a Staff Scientist and manager of the Interfacial Spectroscopy and Diffraction group at Environmental Molecular Sciences Molecular Sciences Laboratory (EMSL), a Department of Energy national scientific user facility located at PNNL. He received a B.Sc. (Honors) in Physics from the University of Peredeniya, Sri Lanka, and M.Sc. in Energy Technology from the Asian Institute of Technology, Thailand. He received his Ph.D. in Physics (Surface Science) from the University of Maine in 1989 and started his postdoctoral research at the University of Florida. Before joining PNNL in 1993, he was a postgraduate researcher at University of California-Davis and a guest scientist at Lawrence Berkeley National Laboratory for three years. At PNNL, he has been developing experimental capabilities associated with research in surface science, material synthesis and characterization including nanomaterials, and buried interface analysis in thin films, and nanomaterials. He made significant contributions to the study of surface structures of pure and adsorbate-covered single crystal surfaces, thin film and interfacial characterizations using XPS/diffraction, photoelectron holography and high energy ion beam techniques. Thevuthasan further contributed to the understanding of synthesis and characterization of nanomaterials, ionic transport processes in single- and multilayer oxide thin film electrolytes, growth and characterization of oxide thin films, and understanding radiation effects in ceramics and oxides. He has authored more than 225 peer reviewed research papers in these areas. He has organized several symposia and workshops as a part of international conferences. Thevuthasan is a Fellow of AVS. He has received PNNL's awards and honors include multiple Outstanding Team Performance awards, the Chester L. Cooper Mentor of the Year Award and the Fitzner-Eberhardt Award for mentoring junior scientists.
 Dr. Hongfei Wang is a Chief Scientist at the EMSL of the U.S. Department of Energy's PNNL at Richland, Washington. He received his B.S. degrees in chemical physics and conducted M.S. studies in laser chemistry, both at the University of Science and Technology of China (USTC) at Hefei, China in 1988 and 1991. He then received his Ph.D. degree in Chemistry in 1996 from Columbia University in the City of New York. He was a postdoc at the DuPont Marshall Laboratory at Philadelphia, Laboratory for Research of Structure of Matter (LRSM) and the Department of Chemistry at the University of Pennsylvania until 1999 when he became a full research professor at the Molecular reaction Dynamics Laboratory (MRDLab) at the Institute of Chemistry, Chinese Academy of Sciences (ICCAS) in Beijing. Before he joined PNNL in 2009, he served as the director of the MRDLab at ICCAS (2000–2004), and also served as the Executive Associated Editor of the Chinese Journal of Chemical Physics (CJCP) of the Chinese Physical Society (2006–2009). He was the recipient of the Chinese Academy of Sciences's Hundred Talent Program (1999–2002), and also a Distinguished Young Scholar of the National Natural Science Foundation of China (NSFC) (2005–2008). He has published over 50 peer-reviewed articles on surface nonlinear spectroscopy, and on spectroscopy and structure of molecular interfaces. In recognition of his research, he was elected as fellow of the American Physical Society in 2012.
Dr. Hongfei Wang is a Chief Scientist at the EMSL of the U.S. Department of Energy's PNNL at Richland, Washington. He received his B.S. degrees in chemical physics and conducted M.S. studies in laser chemistry, both at the University of Science and Technology of China (USTC) at Hefei, China in 1988 and 1991. He then received his Ph.D. degree in Chemistry in 1996 from Columbia University in the City of New York. He was a postdoc at the DuPont Marshall Laboratory at Philadelphia, Laboratory for Research of Structure of Matter (LRSM) and the Department of Chemistry at the University of Pennsylvania until 1999 when he became a full research professor at the Molecular reaction Dynamics Laboratory (MRDLab) at the Institute of Chemistry, Chinese Academy of Sciences (ICCAS) in Beijing. Before he joined PNNL in 2009, he served as the director of the MRDLab at ICCAS (2000–2004), and also served as the Executive Associated Editor of the Chinese Journal of Chemical Physics (CJCP) of the Chinese Physical Society (2006–2009). He was the recipient of the Chinese Academy of Sciences's Hundred Talent Program (1999–2002), and also a Distinguished Young Scholar of the National Natural Science Foundation of China (NSFC) (2005–2008). He has published over 50 peer-reviewed articles on surface nonlinear spectroscopy, and on spectroscopy and structure of molecular interfaces. In recognition of his research, he was elected as fellow of the American Physical Society in 2012.
 Nancy Washton is a Research Scientist within the EMSL at the PNNL, where she is also the Capability Lead for Magnetic Resonance. She received a B.S. degree in chemistry in 2001 from the University of Nevada, Las Vegas and a Ph.D. in chemistry in 2007 from Penn State University. Prior to her arrival at PNNL, Dr. Washton spent two years as a Senior Research Chemist in Fiberglass Science and Technology at PPG. She was also an Assistant Professor of Chemistry at the Community College of Baltimore County from 2007 to 2008. Her main research has focused on the interactions of water and other small molecules at oxide surfaces, including the characterization of reactive surface area in the environment. In her current role at EMSL, she manages a suite of 13 NMR spectrometers, ranging in magnetic field strength from low (for paramagnetic-containing samples) to ultrahigh (for inorganic solids, biomolecular samples, and energy systems).
Nancy Washton is a Research Scientist within the EMSL at the PNNL, where she is also the Capability Lead for Magnetic Resonance. She received a B.S. degree in chemistry in 2001 from the University of Nevada, Las Vegas and a Ph.D. in chemistry in 2007 from Penn State University. Prior to her arrival at PNNL, Dr. Washton spent two years as a Senior Research Chemist in Fiberglass Science and Technology at PPG. She was also an Assistant Professor of Chemistry at the Community College of Baltimore County from 2007 to 2008. Her main research has focused on the interactions of water and other small molecules at oxide surfaces, including the characterization of reactive surface area in the environment. In her current role at EMSL, she manages a suite of 13 NMR spectrometers, ranging in magnetic field strength from low (for paramagnetic-containing samples) to ultrahigh (for inorganic solids, biomolecular samples, and energy systems).
 Alison Elder, Associate Professor of Environmental Medicine at the University of Rochester, is an inhalation toxicologist with research interests that include the pulmonary, cardiovascular, and central nervous system inflammatory and oxidative stress-related effects of engineered nanomaterials and ambient air particulate matter and the physicochemical properties of the particles that are linked to response outcomes. Particle biokinetics and the impacts of age and other underlying vulnerabilities on response are also of interest. Dr. Elder has authored numerous research papers in the field, as well as review articles and book chapters. She is an editorial board member of four journals and is deputy Editor-in-Chief of Nanotoxicology. She also serves on the Threshold Limit Value-Chemical Substances committee of the American Conference of Governmental Industrial Hygienists.
Alison Elder, Associate Professor of Environmental Medicine at the University of Rochester, is an inhalation toxicologist with research interests that include the pulmonary, cardiovascular, and central nervous system inflammatory and oxidative stress-related effects of engineered nanomaterials and ambient air particulate matter and the physicochemical properties of the particles that are linked to response outcomes. Particle biokinetics and the impacts of age and other underlying vulnerabilities on response are also of interest. Dr. Elder has authored numerous research papers in the field, as well as review articles and book chapters. She is an editorial board member of four journals and is deputy Editor-in-Chief of Nanotoxicology. She also serves on the Threshold Limit Value-Chemical Substances committee of the American Conference of Governmental Industrial Hygienists.
 Brittany Baisch is a Ph.D. student in the Toxicology program at the University of Rochester in the laboratory of Dr. Alison Elder, where she studies the role of deposited dose rate in nanoparticle-induced inflammatory responses in respiratory tract target cells. She graduated summa cum laude with a triple B.A. in Chemistry, Biochemistry and Spanish from Western Connecticut State University in 2008. Prior to graduate school, Brittany was a formulations chemist at Advanced Technology Materials, Inc., where she coauthored 2 patents for green formulas that clean logic and microelectronic devices. In 2011, she earned her M.S. in Toxicology from the University of Rochester, and is expected to complete her Ph.D. in 2013. Brittany has earned several awards for her research, including an internship at Bristol-Myers Squibb (2009), Colt Foundation Travel Award for the Nanotoxicology Conference in Edinburgh, Scotland (2010), 1st Place Outstanding Graduate Student Award for the Nanotoxicology Specialty Section (2011), the Young Scholar Award for an Outstanding Student Presentation at the Nanotoxicology Conference in Beijing, China (2012), and 2nd place in Multi-Cellular/Organismal Studies by the University of Rochester's Graduate Student Society (2013). She has been a member of the Society of Toxicology (SOT) since 2009 and is a member of SOT's Nanotoxicology and Inhalation & Respiratory Specialty Sections. She is the current student representative for SOT's Committee on Diversity Initiatives and the Women in Toxicology Special Interest Group.
Brittany Baisch is a Ph.D. student in the Toxicology program at the University of Rochester in the laboratory of Dr. Alison Elder, where she studies the role of deposited dose rate in nanoparticle-induced inflammatory responses in respiratory tract target cells. She graduated summa cum laude with a triple B.A. in Chemistry, Biochemistry and Spanish from Western Connecticut State University in 2008. Prior to graduate school, Brittany was a formulations chemist at Advanced Technology Materials, Inc., where she coauthored 2 patents for green formulas that clean logic and microelectronic devices. In 2011, she earned her M.S. in Toxicology from the University of Rochester, and is expected to complete her Ph.D. in 2013. Brittany has earned several awards for her research, including an internship at Bristol-Myers Squibb (2009), Colt Foundation Travel Award for the Nanotoxicology Conference in Edinburgh, Scotland (2010), 1st Place Outstanding Graduate Student Award for the Nanotoxicology Specialty Section (2011), the Young Scholar Award for an Outstanding Student Presentation at the Nanotoxicology Conference in Beijing, China (2012), and 2nd place in Multi-Cellular/Organismal Studies by the University of Rochester's Graduate Student Society (2013). She has been a member of the Society of Toxicology (SOT) since 2009 and is a member of SOT's Nanotoxicology and Inhalation & Respiratory Specialty Sections. She is the current student representative for SOT's Committee on Diversity Initiatives and the Women in Toxicology Special Interest Group.
 Dr. Ajay S. Karakoti is a Research Scientist at the Battelle Science and Technology Pvt. Ltd. India. He completed B.S (1999) and M.S. (2001) in Chemistry from Delhi University in India and another M.S. in Corrosion Science and Engineering from Indian Institute of Technology Bombay, India in 2003. Thereafter he worked as a trainee in for one year in India increasing his knowledge in particulate materials. He received his Ph. D in Materials Science and Engineering from University of Central Florida in the year 2010 where he worked with Prof. Sudipta Seal on nanoparticulate materials. He received UCF. Presidential Doctoral Fellowship to pursue his Ph.D and was also a winner of the Dorothy M and Earl Hoffman Fellowship as a “top level graduate student” from the AVS in the year 2008. He joined Pacific Northwest National Lab in 2010 for his postdoctoral studies where he looked at the characterization challenges in nanomaterials at the Environmental and Molecular Sciences Laboratory. He was awarded the M. T. Thomas award for “Outstanding Postdoctoral Achievement” in the year 2012. Ajay joined Battelle Science and Technology Pvt. Ltd, India in September 2012. He has published over 45 research articles, two book chapters and filed for 4 patent applications (one granted). He has presented his research in several national and international conferences on the topics related to nanomaterials.
Dr. Ajay S. Karakoti is a Research Scientist at the Battelle Science and Technology Pvt. Ltd. India. He completed B.S (1999) and M.S. (2001) in Chemistry from Delhi University in India and another M.S. in Corrosion Science and Engineering from Indian Institute of Technology Bombay, India in 2003. Thereafter he worked as a trainee in for one year in India increasing his knowledge in particulate materials. He received his Ph. D in Materials Science and Engineering from University of Central Florida in the year 2010 where he worked with Prof. Sudipta Seal on nanoparticulate materials. He received UCF. Presidential Doctoral Fellowship to pursue his Ph.D and was also a winner of the Dorothy M and Earl Hoffman Fellowship as a “top level graduate student” from the AVS in the year 2008. He joined Pacific Northwest National Lab in 2010 for his postdoctoral studies where he looked at the characterization challenges in nanomaterials at the Environmental and Molecular Sciences Laboratory. He was awarded the M. T. Thomas award for “Outstanding Postdoctoral Achievement” in the year 2012. Ajay joined Battelle Science and Technology Pvt. Ltd, India in September 2012. He has published over 45 research articles, two book chapters and filed for 4 patent applications (one granted). He has presented his research in several national and international conferences on the topics related to nanomaterials.
 Satyanarayana V. N. T. Kuchibhatla In his current role, as a Research Scientist and group leader, Satya works with an interdisciplinary team of scientists to provide innovative solutions to client specific, energy, environment, and materials related problems. His interests include nanoscale particulate materials, thin films and coatings, materials characterization, ceramics and powder metallurgy. Before joining Battelle India, in October 2011, Satya Kuchibhatla was a senior research scientist in EMSL, PNNL, Richland, WA, USA. He received his doctoral degree in 2008 from University of Central Florida, Orlando, Florida, USA. His research is broadly focused on surfaces and interfaces, understanding/controlling/tuning the characteristics of materials having impact on nuclear energy, electrolytes for SOFC, gas sensors, self-healing, radiation protection materials, and nanoparticles for biomedical applications. This task is primarily achieved through an intelligent combination of various spectroscopy, diffraction and high-resolution microscopy tools. Specifically, influence of dopant and vacancy interactions in ceria/zirconia electrolytes, crystalline quality of the electrolyte on the oxygen ion conduction, radiation tolerance of single and polycrystalline ceria thin films, influence of aging and environment on the size, shape, self-assembly and chemistry of ceria nanoparticles were studied with the help of experiments and atomistic calculations over last several years. Satya contributed to 30 peer reviewed publications, delivered five invited talks and contributed to 100+ presentations at various international conferences.
Satyanarayana V. N. T. Kuchibhatla In his current role, as a Research Scientist and group leader, Satya works with an interdisciplinary team of scientists to provide innovative solutions to client specific, energy, environment, and materials related problems. His interests include nanoscale particulate materials, thin films and coatings, materials characterization, ceramics and powder metallurgy. Before joining Battelle India, in October 2011, Satya Kuchibhatla was a senior research scientist in EMSL, PNNL, Richland, WA, USA. He received his doctoral degree in 2008 from University of Central Florida, Orlando, Florida, USA. His research is broadly focused on surfaces and interfaces, understanding/controlling/tuning the characteristics of materials having impact on nuclear energy, electrolytes for SOFC, gas sensors, self-healing, radiation protection materials, and nanoparticles for biomedical applications. This task is primarily achieved through an intelligent combination of various spectroscopy, diffraction and high-resolution microscopy tools. Specifically, influence of dopant and vacancy interactions in ceria/zirconia electrolytes, crystalline quality of the electrolyte on the oxygen ion conduction, radiation tolerance of single and polycrystalline ceria thin films, influence of aging and environment on the size, shape, self-assembly and chemistry of ceria nanoparticles were studied with the help of experiments and atomistic calculations over last several years. Satya contributed to 30 peer reviewed publications, delivered five invited talks and contributed to 100+ presentations at various international conferences.
 DaeWon Moon is a DGIST fellow and a professor of Department of New Biology at DGIST. He is a member of Korean Academy of Science and Technology. He received a B.S degree in chemistry from Seoul National University in 1975, a M.S. degree in chemistry from KAIST in 1977 and a Ph.D. degree in chemistry from Pennsylvania State University in 1984. After his postdoctoral training in Princeton University, he moved to KRISS in 1985 as a principal researcher. His main research areas are nano and surface analysis of thin films. In his early career, his primary research interest was to understand the composition and structure of the interfaces of semiconductor films at the nano-scale mainly using ion beams such as SIMS and medium-energy ion scattering (MEIS). Recently, he developed a TOF MEIS system with ∼10 μm spatial resolution, which facilitates the atomic scale analysis of nanobio materials. Since 2003, he became interested in applying nanosurface analysis tools for bioimaging to understand the biointerfaces of cells and tissues at the nanoscale using his previous experience in semiconductor nanosurface science. His contribution to this research field was the development of three label-free nanobioimaging techniques: multiplex coherent anti-Stokes Raman scattering, surface plasmon resonance imaging ellipsometry, and bio-SIMS. He has published over 170 articles and holds 19 patents. He has moved to DGIST as of September 1, 2012. DGIST is a brand-new university supported by the Korean government as one of the strategic convergence science and technology-oriented universities. In DGIST, he takes more challenges to develop noble nanobio imaging tools for innovative biomedical science.
DaeWon Moon is a DGIST fellow and a professor of Department of New Biology at DGIST. He is a member of Korean Academy of Science and Technology. He received a B.S degree in chemistry from Seoul National University in 1975, a M.S. degree in chemistry from KAIST in 1977 and a Ph.D. degree in chemistry from Pennsylvania State University in 1984. After his postdoctoral training in Princeton University, he moved to KRISS in 1985 as a principal researcher. His main research areas are nano and surface analysis of thin films. In his early career, his primary research interest was to understand the composition and structure of the interfaces of semiconductor films at the nano-scale mainly using ion beams such as SIMS and medium-energy ion scattering (MEIS). Recently, he developed a TOF MEIS system with ∼10 μm spatial resolution, which facilitates the atomic scale analysis of nanobio materials. Since 2003, he became interested in applying nanosurface analysis tools for bioimaging to understand the biointerfaces of cells and tissues at the nanoscale using his previous experience in semiconductor nanosurface science. His contribution to this research field was the development of three label-free nanobioimaging techniques: multiplex coherent anti-Stokes Raman scattering, surface plasmon resonance imaging ellipsometry, and bio-SIMS. He has published over 170 articles and holds 19 patents. He has moved to DGIST as of September 1, 2012. DGIST is a brand-new university supported by the Korean government as one of the strategic convergence science and technology-oriented universities. In DGIST, he takes more challenges to develop noble nanobio imaging tools for innovative biomedical science.
ACKNOWLEDGMENTS
This article has evolved from research programs, research conducted as part of the EMSL User Program (http://www.emsl.pnnl.gov/emslweb/), and interactions with colleagues from around the world. DRB in particular thanks Justin Teeguarden, Joel Pounds, and Brian Thrall and the other members of the NIEHS U19 consortium, as well as colleagues from ASTM Committee E42 on Surface Analysis and ISO Committees TC201 Surface Chemical Analysis and TC229 Nanotechnology. Portions of this work were performed using EMSL, a national scientific user facility sponsored by DOE-BER and located at PNNL. Aspects of the work have been supported by the DOE's offices of Basic Energy Science (BES) and BER and the NIEHS under Center grants U19 ES019544 and P30 ES01247, as well as a training grant (T32 ES07026). G.E.J. acknowledges support from the Linus Pauling Fellowship and PNNL's Laboratory Directed Research and Development Program.
References
- Richman E. K. and Hutchison J. E., ACS Nano 3, 2441 (2009). 10.1021/nn901112p [DOI] [PubMed] [Google Scholar]
- Cressey D., Nature 467, 264 (2010). 10.1038/467264b [DOI] [PubMed] [Google Scholar]
- Stuart C., Small Times 6, (2006); http://www.electroiq.com/articles/stm/2006/03/particle-size-matters-studies-fail-to-include-basics-for-assessing-toxicity.html [Google Scholar]
- Stefaniak A. B.et al. , Nanotoxicology (2012). 10.3109/17435390.2012.739664 [DOI]
- Nat. Nano 7, 545 (2012).
- See: http://www.ilsi.org/NanoCharacter/Pages/NanoCharacter.aspx for NanoCharacter: A collaborative project to improve comparability in how nanomaterials are characterized, University of Michigan Risk Science Center (2012).
- Baer D. R. et al. , Surf. Interface Anal. 40, 529 (2008). 10.1002/sia.2726 [DOI] [Google Scholar]
- Beverhof D. R. and David R. M., Anal. Bioanal. Chem. 396, 953 (2010). 10.1007/s00216-009-3103-3 [DOI] [PubMed] [Google Scholar]
- Finke R. G., in Metal Nanoparticles, edited by Feldheim D. L. and Foss D. C. A. (Marcel Dekker, New York, 2002), Chap. 2, pp. 17–54. [Google Scholar]
- Grainger D. W. and Castner D. G., Adv. Mater. 20, 867 (2008). 10.1002/adma.200701760 [DOI] [Google Scholar]
- Grassian V. H., J. Phys. Chem. C 112, 18303 (2008). 10.1021/jp806073t [DOI] [Google Scholar]
- Karakoti A. S., Hench L. L., and Seal S., JOM 58, 77 (2006). 10.1007/s11837-006-0147-0 [DOI] [Google Scholar]
- Billinge S. J. L. and Levin I., Science 316, 561 (2007). 10.1126/science.1135080 [DOI] [PubMed] [Google Scholar]
- Canagaratna M. R. et al. , Mass. Spectrom. Rev. 26, 185 (2007). 10.1002/mas.20115 [DOI] [PubMed] [Google Scholar]
- See: www.foresight.org/nano/history.html for a short history of nanotechnology, Foresight Institute (2013).
- Taniguchi N., in Proceedings International Conference on Product Engineering Tokyo, Part II (Japan Society of Precision Engineering, Tokyo, 1974), 5 pp.
- Roco M. C., Mirkin C. A., and Hersam M. C., Nanotechnology Research Directions for Societal Needs in 2020, Retrospective and Outlook (Springer, Berlin, 2010). [Google Scholar]
- See: http://www.nano.gov/nanotech-101/what/definition for What is nanotechnology?, National Nanotechnology Initiative (2013).
- Baer D. R., Burrows P. E., and El-Azab A. A., Prog. Org. Coat. 47, 342 (2003). 10.1016/S0300-9440(03)00127-9 [DOI] [Google Scholar]
- Li J. G., Li W. X., Xu J. Y., Cai X. O., Liu R. L., Li Y. J., Zhao Q. F., and Li Q. N., Environ. Toxicol. 22, 415 (2007). 10.1002/tox.20270 [DOI] [PubMed] [Google Scholar]
- Phillips R. and Quake S. R., Phys. Today 59, 38 (2006). 10.1063/1.2216960 [DOI] [Google Scholar]
- Pelaz B. et al. , ACS Nano 6, 8468 (2012). 10.1021/nn303929a [DOI] [PubMed] [Google Scholar]
- Crist R. M., Grossman J. H., Patri A. K., Stern S. T., Dobrovolskaia M. A., Adiseshaiah P. P., Clogston J. D., and McNeil S. E., Integr. Biol. 5, 66 (2013). 10.1039/c2ib20117h [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettibone J. M., Gigault J., and Hackley V. A., ACS Nano 7, 2491 (2013). 10.1021/nn3058517 [DOI] [PubMed] [Google Scholar]
- Baer D. R., Tratnyek P. G., Qiang Y., Amonette J. E., Linehan J. C., Sarathy V., Nurmi J. T., Wang C. M., and Antony J., in Environmental Applications of Nanomaterials: Synthesis, Sorbents, and Sensors, edited by Fryxell G. and Cao G. (Imperial College Press, London, 2007), Chap. 3, pp. 49–86. [Google Scholar]
- Nurmi J. T., Sarathy V., Tratnyek P. T., Baer D. R., Amonette J. E., and Karkamkar A., J. Nanopart. Res. 13, 1937 (2011). 10.1007/s11051-010-9946-x [DOI] [Google Scholar]
- Nurmi J. T. et al. , Environ. Sci. Technol. 39, 1221 (2005). 10.1021/es049190u [DOI] [PubMed] [Google Scholar]
- Sarathy V., Tratnyek P. G., Nurmi J. T., Baer D. R., Amonette J. E., Chun C. L., Penn R. L., and Reardon E. J., J. Phys. Chem. C 112, 2286 (2008). 10.1021/jp0777418 [DOI] [Google Scholar]
- Karakoti A. S., Kuchibhatla S., Babu K. S., and Seal S., J. Phys. Chem. C 111, 17232 (2007). 10.1021/jp076164k [DOI] [Google Scholar]
- Karakoti A. S. et al. , Surf. Interface Anal. 44, 882 (2012). 10.1002/sia.5006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karakoti A. S., Singh S., Kumar A., Malinska M., Kuchibhatla S., Wozniak K., Self W. T., and Seal S., J. Am. Chem. Soc. 131, 14144 (2009). 10.1021/ja9051087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuchibhatla S. V. N. T., Karakoti A. S., Baer D. R., Samudrala S., Engelhard M. H., Amonette J. E., Thevuthasan S., and Seal S., J. Phys. Chem. C 116, 14108 (2012). 10.1021/jp300725s [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell C. T. and Peden C. H. F., Science 309, 713 (2005). 10.1126/science.1113955 [DOI] [PubMed] [Google Scholar]
- Das M., Patil S., Bhargava N., Kang J. F., Riedel L. M., Seal S., and Hickman J. J., Biomaterials 28, 1918 (2007). 10.1016/j.biomaterials.2006.11.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karakoti A. S., Monteiro-Riviere N. A., Aggarwal R., Davis J. P., Narayan R. J., Self W. T., McGinnis J., and Seal S., JOM 60, 33 (2008). 10.1007/s11837-008-0029-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao Z. P. and Haile S. M., Nature 431, 170 (2004). 10.1038/nature02863 [DOI] [PubMed] [Google Scholar]
- Wu L., Wiesmann H. J., Moodenbaugh A. R., Klie R. F., Zhu Y., Welch D. O., and Suenaga M., Phys. Rev. B 69, 125415 (2004). 10.1103/PhysRevB.69.125415 [DOI] [Google Scholar]
- Ma J. Y., Zhao H., Mercer R. R., Barger M., Rao M., Meighan T., Schwegler-Berry D., Castranova V., and Ma J. K., Nanotoxicology 5, 312 (2011). 10.3109/17435390.2010.519835 [DOI] [PubMed] [Google Scholar]
- Perez J. M., Asati A., Nath S., and Kaittanis C., Small 4, 552 (2008). 10.1002/smll.200700824 [DOI] [PubMed] [Google Scholar]
- Hardas S. S. et al. , Toxicol. Sci. 116, 562 (2010). 10.1093/toxsci/kfq137 [DOI] [PubMed] [Google Scholar]
- Pierscionek B. K., Li Y. B., Yasseen A. A., Colhoun L. M., Schachar R. A., and Chen W., Nanotechnology 21, 035102 (2010). 10.1088/0957-4484/21/3/035102 [DOI] [PubMed] [Google Scholar]
- Safi M., Sarrouj H., Sandre O., Mignet N., and Berret J. F., Nanotechnology 21, 145103 (2010). 10.1088/0957-4484/21/14/145103 [DOI] [PubMed] [Google Scholar]
- Eom H. J. and Choi J., Toxicol. Lett. 187, 77 (2009). 10.1016/j.toxlet.2009.01.028 [DOI] [PubMed] [Google Scholar]
- Yokel R. A. et al. , Nanotoxicology 3, 234 (2009). 10.1080/17435390902974496 [DOI] [Google Scholar]
- Zhang H. F., He X. A., Zhang Z. Y., Zhang P., Li Y. Y., Ma Y. H., Kuang Y. S., Zhao Y. L., and Chai Z. F., Environ. Sci. Technol. 45, 3725 (2011). 10.1021/es103309n [DOI] [PubMed] [Google Scholar]
- Zhang H. et al. , J. Am. Chem. Soc. 134, 15790 (2012). 10.1021/ja304907c [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mudunkotuwa I. A., Pettibone J. M., and Grassian V. H., Environ. Sci. Technol. 46, 7001 (2012). 10.1021/es203851d [DOI] [PubMed] [Google Scholar]
- Karlsson H. L., Cronholm P., Gustafsson J., and Möller L., Chem. Res. Toxicol. 21, 1726 (2008). 10.1021/tx800064j [DOI] [PubMed] [Google Scholar]
- Castle J. E. and Powell C. J., Surf. Interface Anal. 36, 225 (2004). 10.1002/sia.1678 [DOI] [Google Scholar]
- Schulze M., Bolwin K., Gülzow E., and Schnurnberger W., Fresen J. Anal. Chem. 353, 778 (1995). 10.1007/BF00321370 [DOI] [Google Scholar]
- Hoflund G. B. and Everett M. L., J. Phys. Chem. B 108, 15721 (2004). 10.1021/jp048975j [DOI] [Google Scholar]
- Johnston C. J., Finkelstein J. N., Gelein R., Baggs R. and Oberdörster G., Toxicol. Appl. Pharm. 140, 154 (1996). 10.1006/taap.1996.0208 [DOI] [PubMed] [Google Scholar]
- Pulskamp K., Diabaté S., and Krug H. F., Toxicol. Lett. 168, 58 (2007). 10.1016/j.toxlet.2006.11.001 [DOI] [PubMed] [Google Scholar]
- Gaspar D. J., Engelhard M. H., Henry M. C., and Baer D. R., Surf. Interface Anal. 37, 417 (2005). 10.1002/sia.2031 [DOI] [Google Scholar]
- Techane S. D., Baer D. R., and Castner D. G., Anal. Chem. 83, 6704 (2011). 10.1021/ac201175a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jie J., Zhang W., Peng K., Yuan G., Lee C. S., and Lee S.-T., Adv. Funct. Mater. 18, 3251 (2008). 10.1002/adfm.200800399 [DOI] [Google Scholar]
- Jones C. F., Castner D. B., and Grainger D. W., in Handbook of Immunological Properties of Engineered Nanomaterials (World Science, London, 2012), 117 pp. [Google Scholar]
- Vaden T. D., Song C., Zaveri R. A., Imre D., and Zelenyuk A., Proc. Natl. Acad. Sci. U.S.A. 107, 6658 (2010). 10.1073/pnas.0911206107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scher E. C., Manna L., and Alivisatos A. P., Philos. Trans. Roy. Soc. A 361, 241 (2003). 10.1098/rsta.2002.1126 [DOI] [PubMed] [Google Scholar]
- Baer D. R., J. Surf. Anal. 17, 163 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore K., Forsberg B., Baer D. R., Arnold W. A., and Penn R. L., J. Environ. Eng.-ASCE 137, 889 (2011). 10.1061/(ASCE)EE.1943-7870.0000407 [DOI] [Google Scholar]
- Adiseshaiah P. P., Hall J. B., and McNeil S. E., Nanomed. Nanobiol. 2, 99 (2010). 10.1002/wnan.66 [DOI] [PubMed] [Google Scholar]
- Somorjai G. A. and Park J. Y., Top. Catal. 49, 126 (2008). 10.1007/s11244-008-9077-0 [DOI] [Google Scholar]
- Ajayan P. M., Nature 427, 402 (2004). 10.1038/427402a [DOI] [PubMed] [Google Scholar]
- Helveg S., Lopez-Cartes C., Sehested J., Hansen P. L., Clausen B. S., Rostrup-Nielsen J. R., Abild-Pedersen F., and Norskov J. K., Nature 427, 426 (2004). 10.1038/nature02278 [DOI] [PubMed] [Google Scholar]
- Yacaman M. J., Ascencio J. A., Liu H. B., and Gardea-Torresdey J., J. Vac. Sci. Technol. B 19, 1091 (2001). 10.1116/1.1387089 [DOI] [Google Scholar]
- Wiesner M. R., Lowry G. V., Casman E., Bertsch P. M., Matson C. W., Di Giulio R. T., Liu J., and Hochella M. F., ACS Nano 5, 8466 (2011). 10.1021/nn204118p [DOI] [PubMed] [Google Scholar]
- Pennycook T. J., McBride J. R., Rosenthal S. J., Pennycook S. J., and Pantelides S. T., Nano Lett. 12, 3038 (2012). 10.1021/nl3008727 [DOI] [PubMed] [Google Scholar]
- Hochella M. F., Earth Planet Sc. Lett. 203, 593 (2002). 10.1016/S0012-821X(02)00818-X [DOI] [Google Scholar]
- Hotze E. M., Phenrat T., and Lowry G. V., J. Environ. Qual. 39, 1909 (2010). 10.2134/jeq2009.0462 [DOI] [PubMed] [Google Scholar]
- Lynch I., Cedervall T., Lundqvist M., Cabaleiro-Lago C., Linse S., and Dawson K. A., Adv. Colloid. Interface 134–135, 167 (2007). 10.1016/j.cis.2007.04.021 [DOI] [PubMed] [Google Scholar]
- Zhang H. et al. , Proteomics 11, 4569 (2011). 10.1002/pmic.201100037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H. Z., Gilbert B., Huang F., and Banfield J. F., Nature 424, 1025 (2003). 10.1038/nature01845 [DOI] [PubMed] [Google Scholar]
- Cohen J., DeLoid G., Pyrgiotakis G., and Demokritou P., Nanotoxicology 7, 417 (2013). 10.3109/17435390.2012.666576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krier J. M., Michalak W. D., Baker L. R., An K., Komvopoulos K., and Somorjai G. A., J. Phys. Chem. C 116, 17540 (2012). 10.1021/jp303363m [DOI] [Google Scholar]
- Frankamp B. L., Boal A. K., Tuominen M. T., and Rotello V. M., J. Am. Chem. Soc. 127, 9731 (2005). 10.1021/ja051351m [DOI] [PubMed] [Google Scholar]
- Reinhard B. M., Siu M., Agarwal H., Alivisatos A. P., and Liphardt J., Nano Lett. 5, 2246 (2005). 10.1021/nl051592s [DOI] [PubMed] [Google Scholar]
- Baer D. R. and Engelhard M. H., J. Electron. Spectrosc. 178–179, 415 (2010). 10.1016/j.elspec.2009.09.003 [DOI] [Google Scholar]
- Baer D. R., Gaspar D. J., Nachimuthu P., Techane S. D., and Castner D. G., Anal. Bioanal. Chem. 396, 983 (2010). 10.1007/s00216-009-3360-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J. P., Chen Z. Y., Cai X. J., and Rabalais J. W., J. Vac. Sci. Technol. B 24, 1104 (2006). 10.1116/1.2188410 [DOI] [Google Scholar]
- Baer D. R., Engelhard M. H., Gaspar D. J., Matson D. W., Pecher K. H., Williams J. R., and Wang C. M., J. Surf. Anal. 12, 101 (2005). [Google Scholar]
- Wang C. M., Baer D. R., Amonette J. E., Engelhard M. H., Antony J. J., and Qiang Y., Ultramicroscopy 108, 43 (2007). 10.1016/j.ultramic.2007.03.002 [DOI] [PubMed] [Google Scholar]
- Gaspar D. J., Laskin A., Wang W., Hunt S. W., and Finlayson-Pitts B. J., Appl. Surf. Sci. 231, 520 (2004). 10.1016/j.apsusc.2004.03.046 [DOI] [Google Scholar]
- Kingshott P., Andersson G., McArthur S. L., and Griesser H. J., Curr. Opin. Chem. Biol. 15, 667 (2011). 10.1016/j.cbpa.2011.07.012 [DOI] [PubMed] [Google Scholar]
- Amstad E., Textor M., and Reimhult E., Nanoscale 3, 2819 (2011). 10.1039/c1nr10173k [DOI] [PubMed] [Google Scholar]
- Baer D. R., Surf. Interface. Anal. 44, 1305 (2012). 10.1002/sia.4938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Organization for Standards, “Surface chemical analysis—Characterization of nanostructured materials,” ISO/TR 14187:2011 (2011).
- Powell C. J., J. Vac. Sci. Technol. A 21, S42 (2003). 10.1116/1.1599862 [DOI] [Google Scholar]
- Frydman A., Schmal M., Castner D. B., and Campbell C. T., J. Catal. 157, 133 (1995). 10.1006/jcat.1995.1274 [DOI] [Google Scholar]
- Shard A. G., Wang W. J., and Spencer S. J., Surf. Interface Anal. 41, 541 (2009). 10.1002/sia.3044 [DOI] [Google Scholar]
- Yang D. Q., Gillet J. N., Meunier M., and Sacher E., J. Appl. Phys. 97, 024303 (2005). 10.1063/1.1835566 [DOI] [Google Scholar]
- Yang D. Q., Meunier M., and Sacher E., Appl. Surf. Sci. 173, 134 (2001). 10.1016/S0169-4332(00)00895-3 [DOI] [Google Scholar]
- Yang D. Q. and Sacher E., “ X-ray photoelectron spectroscopy characterization of Nanoparticles (NPs): I. Dimensional effects,” 2008, http://www.scribd.com/doc/2194883/Nano-XPS-nanost-1
- Tougaard S., Microsc. Microanal. 11, 676 (2005). 10.1017/S143192760550022924017209 [DOI] [Google Scholar]
- Mohai M., Surf. Interface Anal. 36, 828 (2004). 10.1002/sia.1775 [DOI] [Google Scholar]
- Mohai M. and Bertoti I., Surf. Interface Anal. 36, 805 (2004). 10.1002/sia.1769 [DOI] [Google Scholar]
- Mohai M., Kiss E., Toth A., Szalma J., and Bertoti I., Surf. Interface Anal. 34, 772 (2002). 10.1002/sia.1408 [DOI] [Google Scholar]
- Mohai M., XPS-MultiQuant, http://www.chemres.hu/aki/XMQpages/XMQhome.htm (2013).
- McIntyre N. S., Nie H. Y., Grosvenor A. P., Davidson R. D., and Briggs D., Surf. Interface Anal. 37, 749 (2005). 10.1002/sia.2075 [DOI] [Google Scholar]
- quases, www.quases.com
- Hajati S., Coultas S., Blornfield C., and Tougaard S., Surf. Interface Anal. 40, 688 (2008). 10.1002/sia.2633 [DOI] [Google Scholar]
- Hajati S., Zaporojtchenko V., Faupel F., and Tougaard S., Surf. Sci. 601, 3261 (2007). 10.1016/j.susc.2007.06.001 [DOI] [Google Scholar]
- Smekal W., Werner W. S. M., and Powell C. J., Surf. Interface Anal. 37, 1059 (2005). 10.1002/sia.2097 [DOI] [Google Scholar]
- Frydman A., Castner D. B., Schmal M., and Campbell C. T., J. Catal. 152, 164 (1995). 10.1006/jcat.1995.1070 [DOI] [Google Scholar]
- Shard A. G., J. Phys. Chem. C 116, 16806 (2012). 10.1021/jp305267d [DOI] [Google Scholar]
- Cumpson P. J., Surf. Interface Anal. 29, 403 (2000). [DOI] [Google Scholar]
- NIST Standard Ref. Database 82—NIST Electron Effective-Attenuation-Length Database, U.S. Department of Commerce, National Institute of Standards and Technology (2011); http://www.nist.gov/srd/nist82.cfm
- Haverkamp R. G., Marshall A. T., and Cowie B. C. C., Surf. Interface Anal. 43, 847 (2011). 10.1002/sia.3644 [DOI] [Google Scholar]
- Seah M. P., Surf. Interface Anal. 44, 1353 (2012). 10.1002/sia.5033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell C. J., App. Surf. Sci. 230, 327 (2004). 10.1016/j.apsusc.2004.01.073 [DOI] [Google Scholar]
- Liang Y., Lea A. S., McCready D. E. and Meethunkij P., in State-of-the-Art Application of Surface and Interface Analysis Methods to Environmental Material Interactions: In Honor of James E. Castle's 65th edited by Baer D. R., Clayton C. R., Davis G. D. and Halada G. P. (The Electrochemical Society, Washington DC, 2001), Vol. 2001-5, pp. 125. [Google Scholar]
- Koh A. L., Shachaf C. M., Elchuri S., Nolan G. P., and Sinclair R., Ultramicroscopy 109, 111 (2008). 10.1016/j.ultramic.2008.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ninomiya S., Ichiki K., Yamada H., Nakata Y., Seki T., Aoki T., and Matsuo J., Rapid Commun. Mass Spectrosc. 23, 3264 (2009). 10.1002/rcm.4250 [DOI] [PubMed] [Google Scholar]
- Smentkowski V. S., Zorn G., Misner A., Parthasarathy G., Couture A., Tallarek E., and Hagenhoff B., J. Vac. Sci. Technol. A 31, 030601 (2013). 10.1116/1.4793730 [DOI] [Google Scholar]
- Ratner B. D., Castner D. G., Brison J., Barnes C., and Daneshcvar R., http://www.semineedle.com/system/files/BuddyRatner_5-14-09.pdf?snc=5963
- Kim Y. P., Oh E., Oh Y. H., Moon D. W., Lee T. G., and Kim H. S., Angew. Chem. Int. Edit. 46, 6816 (2007). 10.1002/anie.200701418 [DOI] [PubMed] [Google Scholar]
- Shi D., Zhou Y., Wang S. X., Van Ooij W. J., Wang L. M., and Zhao J. G., MRS Symp. 635, C4.28.1 (2001). [Google Scholar]
- Borade R., Sayari A., Adnot A., and Kaliaguine S., J. Phys. Chem. 94, 5989 (1990). 10.1021/j100378a068 [DOI] [Google Scholar]
- Gaspar D. J., Laskin A., Wang W., Hunt S. W., and Finlayson-Pitts B. J., Appl. Surf. Sci. 231, 520 (2004). [Google Scholar]
- Seah M. P., Surf. Interface Anal. 44, 208 (2012). 10.1002/sia.3798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L., Seah M. P., Anstis E. H., Gilmore I. S., and Lee J. L. S., J. Phys. Chem. C 116, 9311 (2012). 10.1021/jp300900j [DOI] [Google Scholar]
- Kersting R., Breitenstein D., Hagenhoff B., Fartmann M., Heller D., Grehl T., Brüner P., and Niehuis E., Surf. Interface Anal. 45, 503 (2013). 10.1002/sia.5117 [DOI] [Google Scholar]
- Scalf J. and West P., “Part I: Introduction to nanoparticle characterization with AFM,” www.nanoparticles.org/pdf/Scalf-West.pdf
- Klapetek P., Valtr M., Necas D., Salyk S., and Dzik P., Nanoscale Res. Lett. 6, 514 (2011). 10.1186/1556-276X-6-514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- ASTM, ASTM E2382-04 Guide to Scanner and Tip Related Artifacts in Scanning Tunneling Microscopy and Atomic Force Microscopy (ASTM, West Conshohocken, PA, 2004). [Google Scholar]
- Braga P. C. and Ricci D., Atomic Force Microscopy (Humana Press, New York, 2004). [Google Scholar]
- Serry F. M., Microsc. Microanal. 11, 380 (2005). 10.1017/S143192760550991724017062 [DOI] [Google Scholar]
- Vakarelski I. U., Brown S. C., Moudgil B. M., and Higashitani K., Adv. Powder Tech. 18, 605 (2007). 10.1163/156855207782514905 [DOI] [Google Scholar]
- Gupta S., Brouwer P., Bandyopadhyay S., Patil S., Briggs R., Jain J., and Seal S., J. Nanosci. Nanotechnol. 5, 1101 (2005). 10.1166/jnn.2005.151 [DOI] [PubMed] [Google Scholar]
- Baer D. R., Surf. Interface Anal. 39, 283 (2007). 10.1002/sia.2508 [DOI] [Google Scholar]
- Eisenthal K. B., Chem. Rev. 96, 1343 (1996). 10.1021/cr9502211 [DOI] [PubMed] [Google Scholar]
- Shen Y. R., Nature 337, 519 (1989). 10.1038/337519a0 [DOI] [Google Scholar]
- Eisenthal K. B., Chem. Rev. 106, 1462 (2006). 10.1021/cr0403685 [DOI] [PubMed] [Google Scholar]
- Wang H., Yan E. C. Y., Borguit E., and Eisenthal K. B., Chem. Phys. Lett. 259, 15 (1996). 10.1016/0009-2614(96)00707-5 [DOI] [Google Scholar]
- Kawai T., Neivandt D. J., and Davies P. B., J. Am. Chem. Soc. 122, 12031 (2000). 10.1021/ja002785e [DOI] [Google Scholar]
- Weeraman C., Yatawara A. K., Bordenyuk A. N., and Benderskii A. V., J. Am. Chem. Soc. 128, 14244 (2006). 10.1021/ja065756y [DOI] [PubMed] [Google Scholar]
- Wang C. Y., Groenzin H., and Shultz M. J., J. Phys. Chem. B 108, 265 (2004). 10.1021/jp0356463 [DOI] [Google Scholar]
- Frederick M. T., Achtyl J. L., Knowles K. E., Weiss E. A., and Geiger F. M., J. Am. Chem. Soc. 133, 7476 (2011). 10.1021/ja200466z [DOI] [PubMed] [Google Scholar]
- Baldelli S., Eppler A. S., Anderson E., Shen Y. R., and Somorjai G. A., J. Chem. Phys. 113, 5432 (2000). 10.1063/1.1290024 [DOI] [Google Scholar]
- Somorjai G. A., Beaumont S. K., and Alayoglu S., Angew. Chem. Int. Edit. 50, 10116 (2011). 10.1002/anie.201008214 [DOI] [PubMed] [Google Scholar]
- Roke S., Roeterdink W. G., Wijnhoven J., Petukhov A. V., Kleyn A. W., and Bonn M., Phys. Rev. Lett. 91, 258302 (2003). 10.1103/PhysRevLett.91.258302 [DOI] [PubMed] [Google Scholar]
- Buchbinder A. M., Ray N. A., Lu J., Van Duyne R. P., Stair P. C., Weitz E., and Geiger F. M., J. Am. Chem. Soc. 133, 17816 (2011). 10.1021/ja2067274 [DOI] [PubMed] [Google Scholar]
- Lu Z., Karakoti A. S., Velarde L., Wang W., Yang P., Thevuthasan S., and Wang H., “ Dissociative binding of carboxylic acid ligand on nano ceria surface in aqueous solution: A joint in situ characterization and first-principles study,” J. Phys. Chem. C (submitted).
- Bloch F., Phys. Rev. 70, 460 (1946). 10.1103/PhysRev.70.460 [DOI] [Google Scholar]
- Purcell E. M., Bloembergen N., and Pound R. V., Phys. Rev. 70, 988 (1946). 10.1103/PhysRev.70.988 [DOI] [Google Scholar]
- Levitt M. H., Spin Dynamics: Basics of Nulcear Magnetic Resonance (Wiley, Chichester, 2001). [Google Scholar]
- Fyfe C. A., Solid State NMR for Chemists (CFC Press, Guelph, 1984). [Google Scholar]
- Fiurasek P. and Reven L., Langmuir 23, 2857 (2007). 10.1021/la0629781 [DOI] [PubMed] [Google Scholar]
- Sadasivan S., Khushalanib D., and Mann S., J. Mater. Chem. 13, 1023 (2003). 10.1039/b300851g [DOI] [Google Scholar]
- Zhang B. and Yan B., Anal. Bioanal. Chem. 396, 973 (2010). 10.1007/s00216-009-2996-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abragam A., Principles of Nuclear Magnetism (Clarendon Press, Oxford, 1961). [Google Scholar]
- Das T. P. and Hahn E. L., Nuclear Quadrupole Resonance Spectroscopy (Academic Press, New York, 1964). [Google Scholar]
- Stejskal E. O. and Schaefer J., J. Magn. Reson. 28, 105 (1977). [Google Scholar]
- Baer D. R.et al. , in Handbook of Deposition Technologies for Films and Coatings, edited by Martin P. M. (William Andrew Press, Oxford, UK, 2009). [Google Scholar]
- Stamenkovic V. R., Mun B. S., Arenz M., Mayrhofer K. J. J., Lucas C. A., Wang G., Ross P. N., and Markovic N. M., Nat. Mater. 6, 241 (2007). 10.1038/nmat1840 [DOI] [PubMed] [Google Scholar]
- Jansen W. P. A., Harmsen J. M. A., Denier v.d. Gon A. W., Hoebink J. H. B. J., Schouten J. C., and Brongersma H. H., J. Catal. 204, 420 (2001). 10.1006/jcat.2001.3391 [DOI] [Google Scholar]
- Rafati A., ter Veen R., and Castner D. G., Surf. Interface Anal. 10.1002/sia.5315 [DOI] [PMC free article] [PubMed]
- Grande P. L., “Recent advances of MEIS for near surface analysis,” http://www.slideshare.net/Engenharia.de.Superficies/recent-advances-of-meis-for-near-surface-analysis (2011).
- Wang Y., Chen K. S., Mishler J., Cho S. C., and Adroher X. C., Appl. Energy 88, 981 (2011). 10.1016/j.apenergy.2010.09.030 [DOI] [Google Scholar]
- Mani P., Srivastava R., and Strasser P., J. Phys. Chem. C 112, 2770 (2008). 10.1021/jp0776412 [DOI] [Google Scholar]
- Stephens I. E. L., Bondarenko A. S., Gronbjerg U., Rossmeisl J., and Chorkendorff I., Energy Environ. Sci. 5, 6744 (2012). 10.1039/c2ee03590a [DOI] [Google Scholar]
- Wang C. et al. , J. Phys. Chem. Lett. 3, 1668 (2012). 10.1021/jz300563z [DOI] [PubMed] [Google Scholar]
- Milani P., Piseri P., Barborini E., and Iannotta S., Mater. Sci. Forum 195, 43 (1995). 10.4028/www.scientific.net/MSF.195.43 [DOI] [Google Scholar]
- Pratontep S., Carroll S. J., Xirouchaki C., Streun M., and Palmer R. E., Rev. Sci. Instrum. 76, 045103 (2005). 10.1063/1.1869332 [DOI] [Google Scholar]
- Heiz U., Vanolli F., Trento L., and Schneider W. D., Rev. Sci. Instrum. 68, 1986 (1997). 10.1063/1.1148113 [DOI] [Google Scholar]
- Ayesh A. I., Qamhieh N., Ghamlouche H., Thaker S., and El-Shaer M., J. Appl. Phys. 107, 034317 (2010). 10.1063/1.3296131 [DOI] [Google Scholar]
- Majumdar A., Kopp D., Ganeva M., Datta D., Bhattacharyya S., and Hippler R., Rev. Sci. Instrum. 80, 095103 (2009). 10.1063/1.3213612 [DOI] [PubMed] [Google Scholar]
- Haberland H., Karrais M., Mall M., and Thurner Y., J. Vac. Sci. Technol. A 10, 3266 (1992). 10.1116/1.577853 [DOI] [Google Scholar]
- Klipp B., Grass M., Muller J., Stolcic D., Lutz U., Gantefor G., Schlenker T., Boneberg J., and Leiderer P., Appl. Phys. A 73, 547 (2001). 10.1007/s003390100947 [DOI] [Google Scholar]
- Franchetti V., Solka B. H., Baitinger W. E., Amy J. W., and Cooks R. G., Int. J. Mass Spectrom. 23, 29 (1977). [Google Scholar]
- Cyriac J., Pradeep T., Kang H., Souda R., and Cooks R. G., Chem. Rev. 112, 5356 (2012). 10.1021/cr200384k [DOI] [PubMed] [Google Scholar]
- Johnson G. E., Priest T., and Laskin J., ACS Nano 6, 573 (2012). 10.1021/nn2039565 [DOI] [PubMed] [Google Scholar]
- Johnson G. E., Wang C., Priest T., and Laskin J., Anal. Chem. 83, 8069 (2011). 10.1021/ac202520p [DOI] [PubMed] [Google Scholar]
- Johnson G. E., Hu Q. C., and Laskin J., Annu. Rev. Anal. Chem. 4, 83 (2011). 10.1146/annurev-anchem-061010-114028 [DOI] [PubMed] [Google Scholar]
- Johnson G. E., Priest T., and Laskin J., J. Phys. Chem. C 116, 24977 (2012). 10.1021/jp308795r [DOI] [Google Scholar]
- Claeyssens F., Pratontep S., Xirouchaki C., and Palmer R. E., Nanotechnology 17, 805 (2006). 10.1088/0957-4484/17/3/032 [DOI] [Google Scholar]
- Kaden W. E., Wu T. P., Kunkel W. A., and Anderson S. L., Science 326, 826 (2009). 10.1126/science.1180297 [DOI] [PubMed] [Google Scholar]
- Wepasnick K. A., Li X., Mangler T., Noessner S., Wolke C., Grossmann M., Gantefoer G., Fairbrother D. H., and Bowen K. H., J. Phys. Chem. C 115, 12299 (2011). 10.1021/jp202165u [DOI] [Google Scholar]
- Martinez L., Diaz M., Roman E., Ruano M., Llamosa D., and Huttel Y., Langmuir 28, 11241 (2012). 10.1021/la3022134 [DOI] [PubMed] [Google Scholar]
- Baker S. H., Thornton S. C., Keen A. M., Preston T. I., Norris C., Edmonds K. W., and Binns C., Rev. Sci. Instrum. 68, 1853 (1997). 10.1063/1.1147957 [DOI] [Google Scholar]
- ASTM, E1078-09, Standard Guide for Specimen Preparation and Mounting in Surface Analysis (2006).
- International Organization for Standards, “Surface chemical analysis—Guidelines for preparation and mounting of specimens for analysis,” ISO 18116:2005 (2005).
- Jensen D. S. et al. , Surf. Interface Anal. 45, 1273 (2013). 10.1002/sia.5268 [DOI] [Google Scholar]